
Abstract
Long-term elevated atmogenic deposition (~5 g m−2 year−1) of reactive nitrogen (N) causes N saturation in forests of subtropical China which may lead to high nitrous oxide (N2O) emissions. Recently, we found high N2O emission rates (up to 1,730 μg N2O–N m−2 h−1) during summer on well-drained acidic acrisols (pH = 4.0) along a hill slope in the forested Tieshanping catchment, Chongqing, southwest China. Here, we present results from an in situ 15N–NO3 − labeling experiment to assess the contribution of nitrification and denitrification to N2O emissions in these soils. Two loads of 99 at.% K15NO3 (equivalent to 0.2 and 1.0 g N m−2) were applied as a single dose to replicated plots at two positions along the hill slope (at top and bottom, respectively) during monsoonal summer. During a 6-day period after label application, we found that 71–100 % of the emitted N2O was derived from the labeled NO3 − pool irrespective of slope position. Based on this, we assume that denitrification is the dominant process of N2O formation in these forest soils. Within 6 days after label addition, the fraction of the added 15N–NO3 − emitted as 15N–N2O was highest at the low-N addition plots (0.2 g N m−2), amounting to 1.3 % at the top position of the hill slope and to 3.2 % at the bottom position, respectively. Our data illustrate the large potential of acid forest soils in subtropical China to form N2O from excess NO3 − most likely through denitrification.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Soils under natural vegetation in tropical and subtropical regions have been reported to be a major source of nitrous oxide (N2O), a potent greenhouse gas and decomposer of ozone in the stratosphere (Hirsch et al. 2006; Kort et al. 2011; IPCC 2007). N2O emission from tropical forest are high compared to temperate and boreal natural ecosystems (Dalal and Allen 2008), but the contribution of subtropical forest in densely populated SE Asia to global N2O emissions is unclear. Subtropical forests in South China receive increasing amounts of reactive nitrogen (Nr) by atmogenic deposition, including nitrogen oxides (NOx) and ammonia (NHx) (Liu et al. 2011), which could result in N-saturation (Chen and Mulder 2007a) and may stimulate N2O emissions. Recently, high N2O emissions have been reported from acidic subtropical forests in South China (4.4 kg N ha−1 year−1, Fang et al. 2009; 4.1–5.0 kg N ha−1 year−1, Zhu et al. 2013a), raising questions about the mechanisms involved in N2O production in these soils.
N2O in soil is predominately produced via ammonium (NH4 +) oxidation (nitrification), nitrite (NO2 −) reduction (nitrifier denitrification) and nitrate (NO3 −) reduction (denitrification and dissimilatory nitrate reduction to ammonium, DNRA) (Baggs 2011). These processes have been reported to occur simultaneously at different micro-sites in the soil (Templer et al. 2008). A common approach to quantify the relative contribution of different processes to N2O emission is 15N labeling of inorganic N pools, both in situ and ex situ. For example, based on in situ labeling in a Norway spruce and a beech forest in Germany at soil pH between 3.3 and 4.0, Eickenscheidt et al. (2011) found that N2O emission was mainly attributed to denitrification. However, the contribution of nitrification was not addressed in this study. In a laboratory incubation experiment with acidic soils from 11 different forest sites across Europe, Ambus et al. (2006) found that NO3 − contributed on average 62 % to N2O production while NH4 + contributed on average 34 %. Wolf and Brumme (2002) conducted an in situ 15N-labeling experiment throughout spring, summer and autumn in an acidic beech forest soil in Germany (pH 3.8) and found that N2O emitted during summer was derived mainly from denitrification. Incubating acidic soil (pH 4.2–4.5) from Chinese subtropical coniferous and broad-leaf mixed forests at about 40–50 % water filled pore space (WFPS) and 25 °C, Zhang et al. (2011a) found that denitrification was responsible for 53–56 % of N2O production. Several studies have shown that by increasing WFPS or by lowering soil pH, the proportion of N2O produced from denitrification was elevated (Baggs et al. 2010; Bateman and Baggs 2005; Wolf and Russow 2000; Khalil et al. 2004).
In a recent study, Larssen et al. (2011) hypothesized that denitrification, and possibly increased emission of N2O, may be an important pathway for N loss from sub-tropical forest ecosystems in south China, receiving high loads of atmogenic N. The hypothesis was based on N mass balances of four forested watersheds showing various degrees of N retention that could not be explained by plant N uptake. Among the above studied sub-tropical forest ecosystems in south China, the Tieshanping (TSP) catchment, dominated by acidic acrisols, was found to be N-saturated (Chen and Mulder 2007a). The occurrence of N-saturation at TSP is indicated by large NO3 − concentrations in soil water. However, NO3 − concentrations in stream water at TSP were significantly smaller, suggesting that overall N retention at TSP was high (Larssen et al. 2011). A recent laboratory incubation study using soils from TSP (Zhu et al. 2013b) confirmed high denitrification potentials despite the low pH, with highest potentials (0.29 g m−2 d−1) in the organic surface horizon of the mesic hill slopes. At the same time, the incubations showed that the ability of the denitrifier communities to reduce NO3 − all the way to N2 was low, suggesting that the acidic hill slope soils at TSP give rise to high N2O/(N2O + N2) product ratios during in situ denitrification. High N2O/(N2O + N2) product ratios of denitrification in low pH soils have been reported earlier (Simek and Cooper 2002) and seem to be associated with impaired assembly of the enzyme N2O reductase (Liu et al. 2010; Bergaust et al. 2010). Nitrification could be another possible pathway for N2O production, particularly since two-thirds of the N deposited at TSP enters as NH4 + (Larssen et al. 2011). Chen and Mulder (2007a) found that NH4 + disappears quickly within the first few centimeters of the acidic forest soil at TSP, while NO3 − concentrations increase, suggesting high nitrification potentials. Especially acidic soils have been reported to result in high apparent N2O/NO3 − ratios during nitrification (Mørkved et al. 2007), probably due to chemical decomposition of NO2 − (VanCleemput and Samater 1996).
In a previous study, we observed high N2O emission rates in summer on the well drained hill slope (HS) of TSP with WFPS and temperature being the main drivers (Zhu et al. 2013a). The studies at TSP indicate that denitrification could indeed be a significant sink for N deposited in the catchment, resulting in substantial N2O emissions that have to be taken into account in regional N budgets. To further explore this phenomenon, we conducted a 15N–NO3 − labeling study to assess the relative importance of nitrification and denitrification for N2O emissions in situ and to provide input for process-oriented models and regional N budgets. The main objectives of the present study were (1) to explore the relative contributions of nitrification and denitrification to N2O emission during summer at TSP, (2) to estimate gross nitrification rates and (3) to assess the proportion of freshly added NO3 − lost as N2O. To explore the role of the elevational gradient along a HS on source partitioning of N2O, the 15N–NO3 − labeling study was conducted at two distinct topographic positions (one at the top and one at the bottom of the HS), thus considering natural gradients in soil moisture and soil texture. At each position two application rates of 15N–NO3 − were used, equivalent to 4 and 20 % of the annual Nr deposition, respectively.
Materials and methods
Site description
The TSP catchment is located on a forested sandstone ridge, 450 m asl, about 25 km northeast of Chongqing city, SW China (29°38′N 104°41′E) (Fig .1a). Details on climate, vegetation, and soil characteristics as well as atmogenic Nr deposition rates can be found elsewhere (Larssen et al. 2004). Briefly, the area has a subtropical monsoonal climate with a mean annual precipitation of 1,028 mm and a mean annual temperature of 18.2 °C (three year average from 2001 to 2003); 75 % of the precipitation occurs during summer (April–September; 3 years average 2001–2003). Mean annual inorganic N deposition (from 2001 to 2003; estimated from throughfall samples) was about 4 g m−2, 61 % of which occurred as NH4 +–N (Chen and Mulder 2007b). Recent studies suggest further increases of atmogenic N deposition exceeding 5 g m−2 year−1 in 2009 and 2010 (L. Duan, pers. comm.). The vegetation is a coniferous-broadleaf mixed forest dominated by Masson pine (Pinus massoniana) with a well developed understory of evergreen shrubs. The dominant soil type is a clay-rich loamy yellow mountain soil (Haplic Acrisol; WRB 2006) developed from sandstone. Clay mineralogy is dominated by kaolinite. Due to warm and wet summers, the turnover rates of soil organic matter are high (Raich and Schlesinger 1992; Zhou et al. 2008), and little organic matter accumulates in the forest floor, resulting in a thin organic horizon (O horizon; 0–2 cm). Land use is naturally regenerated secondary forest after the original forest was cut during 1958–1962.
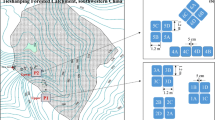
For the study we selected a north-east facing HS in a 4.6 ha sub-catchment at TSP (Fig. 1). Rainfall generates considerable interflow along the HS over the argic B horizon, due to its low hydraulic conductivity. In contrast, the O/A and AB horizons of the HS have a greater hydraulic conductivity and the larger pores are relatively quickly drained after rainfall (Sørbotten 2011). Due to the convergence of interflow at the foot of the HS, the soil stays wet after rainstorms for longer periods as compared with the soil at the top of HS. Hence, we selected two sites for the labeling experiment, one at the top of HS close to the watershed divide, between plot T1 and T2 (indicated as T1/T2) and the other at the bottom of HS (T5, Fig. 1b). Each site included triplicate experimental plots. Major soil characteristics of the sites are summarized in Table 1 and detailed information can be found in Zhu et al. (2013a).

Location of the Tieshanping (TSP) site, Chongqing, China (a); digital elevation model (DEM) of the catchment (b) showing the set-up of the six plots at each site (lower right, b). Diagram of the sampling area for N2O within each of the six 1 m × 1 m plots (upper right, b)
Experimental set-up and sampling
The labeling experiment was conducted from June 25, 2010 to July 1, 2010 and lasted 147 h. Six adjacent 1 m × 1 m plots were chosen at each of the two sites (T1/T2 and T5, respectively), and randomly assigned to two different treatments (triplicates) (Fig. 1b). Even though the major component of atmogenic inorganic N is NH4 +, we conducted the experiment with labeled NO3 − since NO3 − in soil water by far exceeds NH4 + and labeled NO3 − allows assessment of denitrification as the main mechanism of N2O production. The latter is only true if negligible dissimilatory reduction of NO3 −–NH4 + (DNRA) is assumed (see discussion). At the beginning of the experiment (day 1), three of the plots were labeled with 0.2 g NO3 −–N m−2 (99 at.% 15N) as KNO3 (low N load, amounting to about 5 % of annual N deposition). The other three plots were labeled with 1 g NO3 −–N m−2 (99 % at.% 15N) as KNO3 (high N load, amounting to about 20 % of annual N deposition). The labeling solutions (10 L, equivalent to 10 mm precipitation) were applied as a single dose within 0.5 h by spraying on the surface of the soil using a back-pack sprayer (holding the nozzle carefully underneath the ground vegetation). After the label addition, 0.5 L water (0.5 mm) without KNO3 was applied by spraying to wash off label which may have been intercepted by living and dead organic matter on the soil surface. Each of the 1 m2 plots was divided into designated zones for N2O fluxes measurements, soil pore water sampling and soil sampling (Fig. 1b). Two precipitation events occurred during the observation period. The plots were shielded with plastic foil installed 1 m above the soil surface during the first event (June 27, ~ 56 h after label application) in order to prevent dilution of the label. This was not done during the second event (June 29, ~ 98 h after label application).
Closed, vented zinc-coated iron chambers (30 cm diameter and 11 cm high) were used for N2O flux measurements (Hutchinson and Mosier 1981). The chambers were inserted firmly into the clayey A horizon (depth of about 1 cm) at the same position, marked at the soil surface, for all sampling occasions prior to each gas sampling while keeping the air pressure constant via a vent tube attached to the top of the chamber. For each flux sampling, gas samples were taken from the sampling port at the centre of the chamber top 1, 15, 30, 60 and 120 min after chamber deployment. A syringe was attached to the sampling port and the plunger of the syringe was pumped up and down several times to mix the gases in the chamber before taking a sample. The first three gas samples were collected in evacuated 120-mL flasks using Chromacol butyl septa. The flask was attached to the sampling port via a needle for more than 10 s in order to get pressure equilibrium between flask and chamber. Subsequently, an additional 20 mL sample was collected with the syringe from the chamber and injected into the sample flask to achieve overpressure preventing contamination during shipment. The last two samples were transferred to evacuated 12 mL vials using Chromacol butyl septa. The temperature in the inner chamber was measured manually before taking the first and after taking the last gas sample. Fluxes were sampled on in total nine sampling occasions (during about 6 days), 0.5, 4.4, 7.5, 22.0, 30.3, 51.8, 72.0, 99.8 and 147.2 h after label application. Gas samples were stored at room temperature and transported to the laboratory at the Norwegian University of Life Sciences for N2O (all five samples of each sampling occasion) and 15N–N2O (only the first three samples) analyses.
Soil pore water was sampled at each plot from three macrorhizons (Rhizolab, the Netherlands) by connecting an evacuated 120 mL serum flask to all three samplers. Macrorhizons were inserted permanently with a slant into the top soil before starting the experiment, allowing sampling of soil water between 0 and 5 cm depth. Soil water samples were collected 1 day prior to label application and simultaneously with the gas samplings. In addition, soil samples from O/A and AB horizons were taken at each sampling occasion using a soil auger (φ = 2.5 cm). Three replicates were taken from each horizon at each plot and mixed. Sampling holes were refilled with clay plugs as to avoid disturbance of the soil profile. Both, soil pore water and soil samples were frozen immediately. Within one month after sampling, soil samples were thawed and extracted using 10 g dw fresh soil in 50 mL 2 M KCl solution at the Chongqing Academy of Environmental Sciences. After shaking (1 h), the extracts were filtered using pre-washed filter paper (30–50 μm) and frozen before analyses for NH4 +–N and NO3 −–N. Both soil pore water samples and soil extracts were frozen and shipped in a foam box to the Norwegian University of Life Sciences, where they were stored in a freezer (−20 °C) before analysis.
Soil temperature and soil moisture (VM) in the upper 5 cm of the mineral soil were measured using a hand-held TDR (Hydraprobe; Stevens Water Monitoring Systems, Beaverton, OR, USA) at each of the nine sampling occasions at three randomly chosen positions within each plot. Soil WFPS was calculated using bulk density (BD) of the soil at a depth of 0–5 cm and assuming a soil particle density (PD) of 2.65 g cm−3, as
Analytical procedures
The concentration of N2O in the gas samples was analyzed within half a year after sampling. The analysis was carried out by gas chromatography (Model 7890A, Agilent, Santa Clara, CA, US) with an electron capture detector (ECD) after passing a packed Heysept column used for back-flushing of water and a Poraplot Q column run at 38 °C for separating N2O from CO2. Helium (He 5.0) was used as carrier gas and the ECD was run at 375 °C with 17 mL min−1 Argon/Methane (90/10 vol%) as make-up gas. The N2O emission rate (μg N m−2 h−1) was calculated based on the rate of change of the N2O concentration in the chamber as estimated by the slope of a linear or a second order polynomial interpolation, the chamber internal volume, the soil surface area and the temperature. Analysis of 15N in N2O was done using an isotope ratio mass spectrometer coupled with a pre-concentration unit (PreCon-GC-IRMS, Thermo Finnigan MAT, Bremen, Germany). The analysis was done within a year upon sampling and only the first three samples of each sampling occasion (the samples in 120 mL flasks) were analyzed for 15N in N2O. The last two samples were taken to measure any detectable 15N–N2 (data not presented here). Previous tests on blanks indicated that air freight and long storage times did not have a significant effect on the measured concentrations of the gases.
The NO3 − concentration (including NO2 −) in soil extracts and soil pore water was analyzed using the N-(1-naphthyl) ethylenediamine dihydrochloride method, (Norwegian Standard 4745) with a continuous flow injection analyzer (FIA, Tecator, Sweden). The NH4 + concentration was analyzed with the hypochlorite method using a spectrophotometer in the ultraviolet range (Norwegian Standard 4746).
For 15N analysis in NO3 −, a modification of the denitrifier method (Casciotti et al. 2002) was used. Briefly, Pseudomonas aureofaciens (ATCC 13985) was grown aerobically in NO3 −-free tryptic soya broth (pretreated anaerobically with P. denitrificans (ATCC : 19367) to 1.9–5.6 × 108 cells mL−1 and used to reduce sample NO3 − quantitatively to N2O while turning anoxic after He-washing. Compared to the original denitrifier method (Casciotti et al. 2002), the modified method does not need an anoxic pre-culture with external NO3 − as an electron acceptor, thus preparation time is decreased and the background of N2O in the medium is reduced (for details see supplementary material).
Calculations and statistics
Absolute isotope abundances for 15N–N2O and 15N–NO3 − [at.% absolute] were calculated as:
Excess isotope abundances for 15N–N2O and 15N–NO3 − [at.% excess] were expressed in absolute isotope abundances above background (natural abundance). Since no natural abundance of 15N–N2O was measured in this study, it was assumed to be the same as the natural abundance of the soil NO3 − pool.
The 15N–N2O abundances of N2O emitted from the soil were calculated using the Keeling plot approach (Yakir and Sternberg 2000) based on the samples taken at 1, 15, 30 min after chamber deployment. The cumulative N2O flux and 15N–N2O flux (both in mg N m−2) during the observation period were calculated by linear interpolation of the N2O emission rates and 15N abundances in N2O (μg N m−2 h−1 and μg 15N m−2 h−1, respectively) between the measurements.
Assuming negligible dissimilatory reduction of NO3 − to NH4 + (DNRA), only the soil NO3 − pool is labeled. Emitted N2O therefore is either from the soil NH4 + pool with 15N at natural abundance level via nitrification or from the labeled 15N–NO3 − pool via denitrification. A two end-member mixing model (Phillips 2001) was used to determine N2O source partitioning at each time point from the difference in the isotope values of 15N–N2O, the soil NO3 − pool (extractable NO3 − of the O/A horizon) and the soil NH4 + pool (extractable NH4 + of the O/A horizon), which was assumed to be the same as the natural abundance of soil extractable NO3 − of the O/A horizon.
The recovery of 15N from emitted N2O during the observation period was calculated as the percentage of cumulative 15N in N2O of applied 15N tracer assuming constant flux between the measurement dates.
Gross nitrification was estimated from the decline of 15N in NO3 − in soil extracts (pool dilution method; Barraclough 1995), excluding samples prior to 22 h after K15NO3 application to avoid confounding effects from 15NO3 − pool inhomogeneity after label application.
Minitab 16.1.1 (Minitab Inc.) was used for correlation analysis between N2O emission rates and ancillary parameters. The effects of treatments (low and high N loads) and sites (upper and lower) on soil WFPS, N2O emission rate, NO3 − concentration in pore water and extract solution, 15N signal of N2O and NO3 − as well as on the percentage of emitted N2O were tested using General Linear Models. The interaction of the factors (treatments and sites) and the repeated measures at each site on the sampling occasions were considered by General Linear Models. The level of significance was set to p < 0.05.
Results
Weather conditions during the experiment
From June 25 to June 29, 2010, the daily average air temperature varied from 22.9 to 29.9 °C, with a pronounced increase during the last two days (June 30 and July 1, 2010; Fig. 2). Daily average soil temperature was around 24 °C during most of the observation period while increasing to 28.1 °C towards the end (Fig. 3a). Soil temperature did not differ significantly between upper site T1/T2 and lower site T5. Prior to the start of the experiment, there were two precipitation events with 29 mm day−1 (June 19) and 13 mm day−1 (June 23), respectively (Fig. 2). Two precipitation events occurred during the experimental period but for both the volume of rainfall was small (5 mm day−1 on June 27 when the plots were covered with plastic foil and 8 mm day−1 on June 29) (Fig. 2). Soil WFPS was significantly greater at the lower T5 than at the upper T1/T2 site throughout the experiment (p = 0.000; Fig. 3b). WFPS at both sites was high after applying the label solution (10 mm) at the beginning of the experiment and decreased gradually before increasing after precipitation on June 27 (hour 58, Fig. 3b). Due to the plastic foil, WFPS increased only slightly at the upper site T1/T2, while the increase was more pronounced at the lower site T5, likely because this site received additional water through interflow from the HS. The larger precipitation event on June 29, when no plastic foil was used, caused a further increase in WFPS at both sites (Fig. 3b).

Hourly records of air temperature and precipitation from June 19, 2010 to July 1, 2010. K15NO3 was applied on June 25, 2010 with 10.5 mm solution (hour 0, upper x-axis)

Soil temperature (a) and soil moisture (b), both at 0–5 cm depth during the experiment. Values are means of three replicates for the two treatments at upper (T1/T2) and lower (T5) sites. The bars indicate the standard errors
Soil properties
Soils at both sites were strongly acidic with pH values well below 4.0 (Table 1) and pH in a given horizon did not differ between the sites. Values for TOC and TN were greater at the upper T1/T2 than the lower T5 site. Yet, the C/N ratios (about 20) were similar at both sites and for all horizons (Table 1).
Concentration and isotopic signature of NO3 −
Prior to the addition of 15N labeled KNO3, the concentrations of NO3 − in pore water at 5 cm soil depth (8–11 and 10–13 mg N L−1 at lower T5 and upper T1/T2 site, respectively; Fig. 4a) were slightly lower than average values typically observed in summer at the two sites (Zhu et al. 2013a). The addition of KNO3 caused an instantaneous increase in pore water NO3 − concentrations, followed by a rapid decrease at both sites and N addition rates. After about 10 h, the concentrations of NO3 − in pore water stabilized at values 10–30 and 70–100 % above those prior to N addition for plots with low and high N load, respectively. At each N addition level, the concentration of NO3 − in pore water was greater at the upper than at the lower site, probably due to smaller values of WFPS (Fig. 3b) and thus less dilution at the upper site.

a Concentration of NO3 − (mg N L−1) in soil pore water from soil of the top 5 cm depth; b concentration of NO3 − (μg N g−1 dw soil) in soil extracts (2 M KCl) from soil of O/A and AB horizons; c concentration of NH4 + (mg N L−1) in soil pore water from soil of top 5 cm depth; d concentration of NH4 + (μg N g−1 dw soil) in soil extracts (2 M KCl) from soil of O/A and AB horizons. Values are means of three replicates per treatment at upper (T1/T2) and lower (T5) sites during the experiment. The insert indicates the standard error of the means
Before addition of N, KCl-extractable NO3 − in the O/A horizon (8–9 μg N g−1, Fig. 4b) was considerably smaller than annual average values reported earlier for the sites (Zhu et al. 2013a). Similar to NO3 − in pore water, KCl-extractable NO3 − in O/A horizons increased sharply upon the addition of 1 g N m−2 as KNO3. By contrast, the addition of 0.2 g N m−2 had only a minor effect on extractable NO3 −–N in the O/A horizon. Although less pronounced than in pore water, KCl-extractable NO3 − in the O/A horizon was greater at the upper than at the lower site. Soil KCl extracts of AB horizons had significantly smaller NO3 − concentrations than O/A horizons (p = 0.000; Fig. 4b).
Prior to the addition of KNO3, the concentration of NH4 + in pore water was similar to average values in summer reported earlier (0.1 and 0.5 mg N L−1 at site T5 and T1/T2, respectively; Zhu et al. 2013a). Thus, before N addition, NH4 + contributed only 1–5 % to the total concentration of inorganic N in pore water. Addition of KNO3 caused an instantaneous increase in the NH4 + concentration in pore water, probably due to cation exchange with a minor proportion of the added K+ ions. This was followed by a decrease in concentration and stabilization from about 10 h onwards resulting in concentrations of 1.3 and 0.8 mg NH4 +–N L−1 (equivalent to about 5 % of the total inorganic N concentration in pore water) in response to high and low N addition, respectively, at the upper site. At the lower site the concentration of NH4 + rapidly decreased to values prior to N addition (about 0.1 mg NH4 +–N L−1, Fig. 4c).
Similar to the findings for NO3 −, KCl-extractable NH4 + in the O/A horizon, prior to N addition (17–15 μg N g−1 at the lower and the upper site, respectively; Fig. 4d) were smaller than annual average values (Zhu et al. 2013a). Neither in the O/A, nor in the AB horizon, did we see a pronounced effect of KNO3 addition, irrespective of its load, on KCl-extractable NH4 +. This is not surprising as KCl-extractable NH4 +, which includes both dissolved and exchangeable NH4 +, is not likely to be affected by the addition of K+. The larger values for KCl-extractable NH4 + in the soil at the upper site were in line with the larger NH4 + concentration in pore water and the greater content of TOC (Table 1), an important source of negative charge in surface soil horizons at TSP. By contrast to NH4 +–N concentrations in pore water, those in KCl extracts were of the same order of magnitude as NO3 −–N concentrations, reinforcing the importance of the exchangeable pool for soil NH4 +. However, in both horizons and at both sites, KCl-extractable NH4 +–N accounted at most for 4 mmolc kg−1 soil, which is only a small fraction of the cation exchange capacity of this soil (CEC; Table 1).
In response to the high 15N load, 15N at.% excess of NO3 − in pore water increased sharply from 0.35 at.% (natural abundance) to 36 and 42 at.% excess at the lower and upper site, respectively (Fig. 5a). Also the low N load caused an instantaneous increase in at.% 15N excess of NO3 − in pore water (to about 31 %) at both the upper and lower site. At both sites, the 15N signal decreased gradually at the low N addition plots, but not at the high N addition plots. After 6 days (about 147 h), the 15N abundance in pore water NO3 − decreased to 16 and 12 at.% at the low N addition plots at upper and lower site, respectively. Both 15N values in NO3 − and NO3 − concentrations (Fig. 4a) at the lower site were found to be smaller than at the upper site throughout the experiment, presumably due to stronger dilution at the higher WFPS at the lower site (Fig. 3b).

a at.% 15N excess of NO3 −–N in soil pore water from soil of top 5 cm depth; b at.% 15N excess of NO3 −–N in soil extracts (2 M KCl) from soil of O/A and AB horizons. Values are means of three replicates per treatment at upper (T1/T2) and lower (T5) sites during the experiment. The inset figure indicates the standard error of the means
Soil extracts of the AB horizons had significantly smaller 15N–NO3 − at.% excess than O/A horizons (p = 0.013; Fig. 5b). The 15N at.% excess of KCl extractable NO3 − in O/A and AB horizons was similar in magnitude to that of dissolved NO3 − in pore water, remaining relatively stable throughout the experiment (about 35–42 at.% excess, at the high N load). For the low N load, we found a 15N pattern in KCl-extractable NO3 − similar to that in pore water with a gradual decrease of 15N at.% excess to values between 9 and 17 % (Fig. 5a, 5b).
Nitrification rate
Gross nitrification rate was calculated from dilution of the 15N signal in KCl- extractable NO3 − for the low N addition treatment only, since no clear decline in 15N–NO3 − was found with the high N load (Table 2). Calculated rates of the gross nitrification in the O/A and AB horizons were 0.2–0.6 μg g−1 soil day−1, with the larger values for the lower site. Note however, that the standard deviations were large, making the gross nitrification rates not significantly different from zero.
N2O emission flux and isotopic signature
The N2O flux following the application of 15N was smaller at the upper than the lower site. This was true irrespective of the N addition level (Fig. 6a). Average N2O fluxes during the 147 h following the label addition were not affected by the N input level, neither at the upper site (79 ± 31 and 88 ± 3 μg N m−2 h−1 at low and high N load, respectively), nor at the lower (209 ± 52 and 257 ± 133 μg N m−2 h−1 at low and high N load, respectively). At the lower site, N2O emission rates increased rapidly in response to KNO3 addition, reaching a peak after about 7.5 h (Fig. 6a). Maximum N2O emission rates at the lower site were about 350 and 400 μg N m−2 h−1, at the low and high N addition plots, respectively. After reaching a peak, N2O emission rates decreased substantially. The N2O flux dynamics at the upper site differed from those at the lower site, in that the N2O emission rate did not peak until 30–40 h after N addition (both at low and high addition rates) reaching maximum values of about 100 μg N m−2 h−1 in both treatments. Next, the N2O emission rate remained relatively stable (Fig. 6a). Overall, the N2O emission was positively correlated with both soil temperature (p = 0.028) and WFPS (p = 0.006) but not with either the concentration of NO3 − in pore water (p = 0.264) or in the KCl extract (p = 0.392).

a N2O fluxes (μg N m−2 h−1); b at.% 15N excess of N2O–N fluxes. Values are means of three replicates per treatment at upper (T1/T2) and lower (T5) sites during the experiment. The insert indicates the standard errors of the means
Similar to the dynamics of the N2O flux, the at.% 15N excess of emitted N2O increased sharply in all plots, reaching highest values 30 h after the application of label, except for the lower site with low N load, where the highest at.% 15N excess of emitted N2O occurred already after 4 h (Fig. 6b). In general, the at.% 15N excess of emitted N2O peaked faster at the lower site than the upper site (Fig. 6b). Application of 1 g N m−1 (high N load) resulted in relatively stable at.% 15N excess for N2O throughout the experiment at both sites, whereas a decreasing trend was seen at both sites with low N load (Fig. 6b).
During the 147 h of observation, in total 2.6, 6.0, 6.5 and 13.2 mg 15N–N2O m−2 was emitted from the soil at upper site low and high N load, and lower site low and high N load, respectively. These fluxes represented 1.3, 0.6, 3.2 and 1.3 %, respectively, of the total applied 15N–NO3 − (Table 2). From about 22 h after label application onwards, 71–100 % of the emitted N2O was due to denitrification, with one exception for the low N load at the upper site at 22 h, which was 61 % (Fig. 7). There was no significant difference in N2O source partitioning between N loads or between the upper and lower site, despite the clear differences in WFPS (Fig. 3b). The sharp increase in 15N values of N2O emitted during the initial 10 h after label addition reflects the time needed for the labeled NO3 − to mix with the soil’s NO3 − pool and to diffuse to the sites in which denitrification occurred. The percentage of N2O apportioned to denitrification was positively related with N2O emission rate but showed no correlation with either soil temperature or WFPS.

%N2O from denitrification in soils O/A horizon of each treatment at upper (T1/T2) and lower (T5) sites during the experiment. Values are means of three replicates. The insert indicates the standard errors of the means
Discussion
The addition of labeled KNO3 caused an immediate increase in the concentration of NO3 − and its 15N abundance in both pore water and soil extracts, reaching peak values within 4 h after application (Figs. 4a, 4b, 5a and 5b). The N2O flux at the lower site responded almost as fast with peak values occurring after 7.5 h (Fig. 6a). The N2O flux at the drier upper site reached peak values about 33 h after application, likely because soil O2 needed to be depleted at the previously aerobic micro sites before denitrification increased. The increase in N2O flux at both sites was associated with increased WFPS (Fig. 3b) in response to the addition of 10.5 mm labeling solution. Overall, the N2O emission rate at both sites and at both loadings appeared to be positively related to WFPS, albeit with a delay of about 1 day, illustrating the importance of WFPS for N2O emission in this soil. The WFPS was significantly smaller at the upper (48–60 %) than the lower site (55–70 %), resulting in significantly larger N2O fluxes at the lower site (p = 0.002; Fig. 6a). This supports our hypothesis that denitrification is an important source of N2O in this subtropical forested catchment with high N2O emission (4.1–5.0 kg N ha−1 year−1), as proposed previously by Zhu et al (2013a), who studied the temporal and spatial variability of N2O flux in this watershed. Higher cumulative N2O fluxes at the lower site than at the upper site were also found in summer of 2009, but not in summer of 2010.
The peak in 15N abundance in emitted N2O occurred after about 30 h in all cases, except for the lower N addition at the lower site, where 15N–N2O already peaked after 7.5 h (Fig. 6b). The timing of the 15N peak in N2O was closely linked to the 15N peak in NO3 −–N in both pore water and soil extracts (Fig. 5a, 5b), likely reflecting the gradual replacement and mixing of native NO3 − with labeled NO3 − at micro-sites. The N2O source partitioning estimated by comparing at.% of 15N–N2O with that of 15N–NO3 − in soil extracts from O/A horizon suggests that after an unstable phase on average 71–100 % of the N2O was produced from denitrification (Fig. 7). This finding is supported by high denitrification potentials and impeded N2O reduction reported previously from a laboratory study with soils from the same sites (Zhu et al. 2013b).
Changes in WFPS (Fig. 3b) did not affect N2O source partitioning appreciably. It appeared that nitrification contributed very little to N2O production within the observed WFPS range. This was in contrast to several other studies (Bateman and Baggs 2005; Mathieu et al. 2006; Zhang et al. 2011a; Khalil et al. 2004; Ambus et al. 2006; Mørkved et al. 2006; Russow et al. 1994; Wolf and Brumme 2002; Wolf and Russow 2000; Eickenscheidt et al. 2011) who found that significant contributions of nitrification to N2O production occurred at intermediate WFPS ranges whereas denitrification generally occurred at high soil moisture content. Comparing soil types in the above cited studies, it appears that also soil pH could be an important factor for N2O source partitioning, as it interacts with the soil moisture relationship; in neutral to alkaline soils the contribution of nitrification to N2O emission seems to be high and less sensitive to WFPS than the contribution of denitrification (Khalil et al. 2004; Mathieu et al. 2006; Bateman and Baggs 2005). Vice versa, in acid soils, the contribution of denitrification seems to be more prominent in a wider range of WFPSs (Ambus et al. 2006; Wolf and Brumme 2002; Zhang et al. 2011a; Eickenscheidt et al. 2011).
The 15N abundance of NH4 + was not determined in our experiment, since we believe that DNRA, a strictly anaerobic process (Tiedje 1988), was negligible in our soil. This is supported by the low NH4 + concentrations in soil pore water on the HS at TSP (Fig. 4c), where subsurface water is quickly drained and accumulation of 15N–NH4 + via DNRA is likely to be small. Earlier, Wolf and Brumme (2002) reported that 15N–NH4 + did not increase in a 15N–NO3 − labeling experiment in a temperate forest with acidic soil.
With the assumption that only the soil NO3 − pool was labeled with 15N, strictly speaking we can only address the percentage of N2O derived from the labeled soil NO3 − pool. Potentially other processes such as DNRA and immobilization and re-mineralization of NO3 − may also contribute to the production of labeled N2O. However, we believe these processes to be insignificant for N2O production in this study, since small NH4 + accumulation and stable 15N signal of NO3 − suggest small production rates of NH4 + and NO3 − in the experiment.
The small gross nitrification rates determined by 15N–NO3 − pool-dilution in the low-N addition treatments also suggest a minor contribution of nitrification to N2O emission. By contrast to the 15N–NO3 − pool in soil water and soil extracts from the low N load treatment, those of the high load did not show a clear dilution pattern. Apparently, the nitrification rate was too low to show a detectable decline in 15N signal of NO3 − due to dilution at the high N load. Even though the in situ nitrification rate was greater at the lower than at the upper site, values were generally small (0.2–0.6 μg N g−1 dw soil day−1 in the O/A horizon). Although, this estimate of in situ gross nitrification is crude (no 15N–NH4 + treatment was performed and probably homogeneous distribution of the label and ideal mixing with indigenous soil NO3 − was not achieved), these values are similar to the nitrification potential (including both autotrophic and heterotrophic nitrification) found in the laboratory (1.45 μg N g−1 dw soil day−1 for O/A horizon at T3 on the hill slope; Zhu et al. unpublished). Both observations suggest low nitrification activity in TSP soils, with rates at the lower end of a large range of values (from 0.1 to 12 μg N g−1 day−1) reported for terrestrial ecosystems with various soil pH and in different climate zones (Booth et al. 2005). In temperate spruce forests in Germany with soil pH below 4, Corre et al. (2007) observed gross nitrification rates of similar magnitude as we found for TSP. By contrast in a temperate mixed deciduous forest in Belgium, Vervaet et al. (2004) observed gross nitrification rates which were one order of magnitude greater. Low nitrification rates in acid soils are commonly explained by low NH3 availability, limiting autotrophic nitrification (Mayer et al. 2001; Zhang et al. 2011b; De Boer and Kowalchuk 2001). Heterotrophic nitrification and archaeal ammonia oxidation are believed to be more tolerant to low pH (Prosser 2007). However, the low gross nitrification rates in our study suggest that these processes did not play an important role in NO3 − production in our study. The small in situ gross nitrification rates at TSP do not explain the observed rapid disappearance of atmogenic NH4 + in soil water. Follow up 15N–NH4 + labeling experiments to study the rapid disappearance of NH4 + are underway.
Although the NO3 − concentration in pore water at plots with high N application (1 g m−2) were 1.5–2 times greater than those at plots with low N application (0.2 g m−2), this did not result in greater N2O fluxes. This result confirms in situ measurements of N2O flux and ancillary variables at TSP, which suggested that NO3 − was not a limiting substrate for N2O production on the HS (Zhu et al. 2013a). By contrast, several other studies of forest soils report a significant response of N2O emission to elevated NO3 − availability. However, in most of these studies N availability was artificially increased from a very low level in soil by N fertilization (e.g. Kim et al. 2012; Hall and Matson 1999; Nobre et al. 2001). In the present study, 70–100 % increase in available NO3 − only had a moderate effect on N2O emission (11 and 25 % greater at high N load than at low N load for the upper and lower site, respectively), suggesting that denitrification was not N-limited after adding 0.2 g N m−2 (equivalent to 4 % of the annual N input in the N-saturated TSP catchment). A similar observation was made in an experiment with NH4NO3–N addition in the forested Dinghushan catchment in south China (Zhang et al. 2008). Their results showed little increase of N2O emission in disturbed or rehabilitated forests along with control, low, medium and high N addition (0, 5, 10, 15 g N m−2 year−1, respectively; sprayed once every month).
At TSP, the N2O emission flux was significantly related to soil temperature and soil moisture, which is in accordance with findings from an earlier N2O field study at TSP (Zhu et al. 2013a).
At low N load, the recovery of applied 15N in N2O was on average 2.3 % (Table 2) within 6 days (when only 15N–N2O is taken into account). Eickenscheidt et al. (2011) reported annual values of 0.1 and 0.6 % for N2O loss in a German spruce and beech stand, respectively. Even though the latter was a long-term study with tracer applied 18-times during one year with low N application rate each time, the big difference to our values for only 6-days suggests a very high percentage of N2O loss at TSP. At low N addition, the total amount of N2O emitted (labeled and unlabeled) in 6 days amounted to 8 and 15 % of the N addition at the upper and lower site, respectively, illustrating the large potential of these soils to quickly convert NO3 − to N2O. Our emission factors (the percentage of applied N emitted as N2O), albeit based on a 6-days period only, are well above the value of 1 % recommended for both direct N2O emissions from mineral N application to managed soils as well as for indirect N2O emissions from re-deposited N to uncultivated soils (IPCC 2006). In a recent field study at TSP, we found that about 8–10% of the atmogenically derived N was emitted as N2O annually (Zhu et al. 2013a). This study clearly revealed that high N deposition in subtropical forests in south China with wide-spread acidic soils can result in high N2O/N2 product ratios during denitrification.
References
Ambus P, Zechmeister-Boltenstern S, Butterbach-Bahl K (2006) Sources of nitrous oxide emitted from European forest soils. Biogeosciences 3(2):135–145
Baggs EM (2011) Soil microbial sources of nitrous oxide: recent advances in knowledge, emerging challenges and future direction. Curr Opin Environ Sustain 3(5):321–327
Baggs EM, Smales CL, Bateman EJ (2010) Changing pH shifts the microbial sourceas well as the magnitude of N(2)O emission from soil. Biol Fertil Soils 46(8):793–805
Barraclough D (1995) N-15 isotope dilution techniques to study soil nitrogen transformations and plant uptake. Fertil Res 42(1–3):185–192
Bateman EJ, Baggs EM (2005) Contributions of nitrification and denitrification to N2O emissions from soils at different water-filled pore space. Biol Fertil Soils 41(6):379–388
Bergaust L, Mao YJ, Bakken LR, Frostegard A (2010) Denitrification response patterns during the transition to anoxic respiration and posttranscriptional effects of suboptimal pH on nitrogen oxide reductase in Paracoccus denitrificans. Appl Environ Microbiol 76(19):6387–6396
Booth MS, Stark JM, Rastetter E (2005) Controls on nitrogen cycling in terrestrial ecosystems: a synthetic analysis of literature data. Ecol Monogr 75(2):139–157
Casciotti KL, Sigman DM, Hastings MG, Bohlke JK, Hilkert A (2002) Measurement of the oxygen isotopic composition of nitrate in seawater and freshwater using the denitrifier method. Anal Chem 74(19):4905–4912
Chen XY, Mulder J (2007a) Indicators for nitrogen status and leaching in subtropical forest ecosystems. South China. Biogeochem 82(2):165–180
Chen XY, Mulder J (2007b) Atmospheric deposition of nitrogen at five subtropical forested sites in South China. Sci Total Environ 378(3):317–330
Corre MD, Brumme R, Veldkamp E, Beese FO (2007) Changes in nitrogen cycling and retention processes in soils under spruce forests along a nitrogen enrichment gradient in Germany. Glob Change Biol 13(7):1509–1527
Dalal RC, Allen DE (2008) Greenhouse gas fluxes from natural ecosystems. Aust J Bot 56(5):369–407
De Boer W, Kowalchuk GA (2001) Nitrification in acid soils: micro-organisms and mechanisms. Soil Biol Biochem 33(7–8):853–866
Eickenscheidt N, Brumme R, Veldkamp E (2011) Direct contribution of nitrogen deposition to nitrous oxide emissions in a temperate beech and spruce forest: a 15N tracer study. Biogeosciences 8(3):621–635
Fang YT, Gundersen P, Zhang W, Zhou GY, Christiansen JR, Mo JM, Dong SF, Zhang T (2009) Soil-atmosphere exchange of N2O, CO2 and CH4 along a slope of an evergreen broad-leaved forest in southern China. Plant Soil 319(1–2):37–48
Hall SJ, Matson PA (1999) Nitrogen oxide emissions after nitrogen additions in tropical forests. Nature 400(6740):152–155
Hirsch AI, Michalak AM, Bruhwiler LM, Peters W, Dlugokencky EJ, Tans PP (2006) Inverse modeling estimates of the global nitrous oxide surface flux from 1998–2001. Glob Biogeochem Cycles. doi:10.1029/2004gb002443
Hutchinson GL, Mosier AR (1981) Improved soil cover method for field measurement of nitrous oxide fluxes. Soil Sci Soc Am J 45(2):311–316
IPCC (2006) Agriculture, forestry and other land use. In: Eggleston HS, Buendia L, Miwa K, Ngara T, Tanabe K (eds) Guidelines for national greenhouse gas inventories. IGES, Japan
IPCC (2007) Summary for policy makers. In: Solomon S, Qin D, Manning M, Chen Z, Marquis M, Averyt KB, Tignor M, Miller HL (eds) Climate change 2007: the physical science basis. contribution of working group i to the fourth assessment report of the intergovernmental panel on climate change. IPCC, Cambridge, New York
Khalil K, Mary B, Renault P (2004) Nitrous oxide production by nitrification and denitrification in soil aggregates as affected by O2 concentration. Soil Biol Biochem 36(4):687–699
Kim YS, Imori M, Watanabe M, Hatano R, Yi MJ, Koike T (2012) Simulated nitrogen inputs influence methane and nitrous oxide fluxes from a young larch plantation in northern Japan. Atmos Environ 46:36–44
Kort EA, Patra PK, Ishijima K, Daube BC, Jimenez R, Elkins J, Hurst D, Moore FL, Sweeney C, Wofsy SC (2011) Tropospheric distribution and variability of N2O: evidence for strong tropical emissions. Geophys Res Lett. doi:10.1029/2011gl047612
Larssen T, Tang D, He Y (eds) (2004) Integrated monitoring program on acidification of Chinese terrestrial systems—IMPACTS—Annual report results 2003.NIVA Report 4905-2004., vol NIVA Report 4905-2004. NIVA, Oslo
Larssen T, Duan L, Muder J (2011) Deposition and leaching of sulfur, nitrogen and calcium in four forested catchments in China: implications for acidification. Environ Sci Technol 45(4):1192–1198
Liu BB, Mørkved PT, Frostegard A, Bakken LR (2010) Denitrification gene pools, transcription and kinetics of NO, N2O and N2 production as affected by soil pH. FEMS Microbiol Ecol 72(3):407–417
Liu XJ, Duan L, Mo JM, Du EZ, Shen JL, Lu XK, Zhang Y, Zhou XB, He CN, Zhang FS (2011) Nitrogen deposition and its ecological impact in China: an overview. Environ Pollut 159(10):2251–2264
Mathieu O, Henault C, Leveque J, Baujard E, Milloux MJ, Andreux F (2006) Quantifying the contribution of nitrification and denitrification to the nitrous oxide flux using N-15 tracers. Environ Pollut 144(3):933–940
Mayer B, Bollwerk SM, Mansfeldt T, Hutter B, Veizer J (2001) The oxygen isotope composition of nitrate generated by nitrification in acid forest floors. Geochim Cosmochim Acta 65(16):2743–2756
Mørkved PT, Dörsch P, Henriksen TM, Bakken LR (2006) N2O emissions and product ratios of nitrification and denitrification as affected by freezing and thawing. Soil Biol Biochem 38(12):3411–3420
Mørkved PT, Dörsch P, Bakken LR (2007) The N2O product ratio of nitrification and its dependence on long-term changes in soil pH. Soil Biol Biochem 39(8):2048–2057
Nobre AD, Keller M, Crill PM, Harriss RC (2001) Short-term nitrous oxide profile dynamics and emissions response to water, nitrogen and carbon additions in two tropical soils. Biol Fertil Soils 34(5):363–373
Phillips DL (2001) Mixing models in analyses of diet using multiple stable isotopes: a critique. Oecologia 127:166–170
Prosser JI (2007) The ecology of nitrifying bacteria. In: Bothe H, Ferguson SJ, Newton WE (eds) Biology of the nitrogen cycle. Elsevier, Amsterdam, pp 223–244
Raich JW, Schlesinger WH (1992) The global carbon dioxide flux in soil respiration and its relationship to vegetation and climate. Tellus B 44(2):81–99
Russow R, Hofer M, Faust H (1994) N-15 tracer studies on the mechanism of the N2O formation in soils. Isotopenpraxis 30(2–3):157–164
Simek M, Cooper JE (2002) The influence of soil pH on denitrification: progress towards the understanding of this interaction over the last 50 years. Eur J Soil Sci 53(3):345–354
Sørbotten L (2011) Hill slope unsaturated flowpaths and soil moisture variability in a forested catchment in southwest China. Norwegian Univerisity of Life Sciences, Aas
Templer PH, Silver WL, Pett-Ridge J, DeAngelis KM, Firestone MK (2008) Plant and microbial controls on nitrogen retention and loss in a humid tropical froest. Ecology 89(11):3030–3040
Tiedje JM (1988) Ecology of denitrification and dissimilatory nitrate reduction to ammonium. In: Zehnder JBA (ed) Biology of anaerobic microorganisms. Wiley, New York, pp 197–244
VanCleemput O, Samater AH (1996) Nitrite in soils: accumulation and role in the formation of gaseous N compounds. Fertil Res 45(1):81–89
Vervaet H, Boeckx P, Boko AMC, Van Cleemput O, Hofman G (2004) The role of gross and net N transformation processes and NH4 + and NO3 − immobilization in controlling the mineral N pool of a temperate mixed deciduous forest soil. Plant Soil 264(1–2):349–357
Wolf I, Brumme R (2002) Contribution of nitrification and denitrification sources for seasonal N2O emissions in an acid German forest soil. Soil Biol Biochem 34(5):741–744
Wolf I, Russow R (2000) Different pathways of formation of N2O, N2 and NO in black earth soil. Soil Biol Biochem 32(2):229–239
WRB (2006) World reference base for soil resources, 2006. FAO, Rome
Yakir D, Sternberg LDL (2000) The use of stable isotopes to study ecosystem gas exchange. Oecologia 123(3):297–311
Zhang W, Mo JM, Yu GR, Fang YT, Li DJ, Lu XK, Wang H (2008) Emissions of nitrous oxide from three tropical forests in Southern China in response to simulated nitrogen deposition. Plant Soil 306(1–2):221–236
Zhang JB, Cai ZC, Zhu TB (2011a) N2O production pathways in the subtropical acid forest soils in China. Environ Res 111(5):643–649
Zhang JB, Muller C, Zhu TB, Cheng Y, Cai ZC (2011b) Heterotrophic nitrification is the predominant NO3 − production mechanism in coniferous but not broad-leaf acid forest soil in subtropical China. Biol Fertil Soils 47(5):533–542
Zhou GY, Guan LL, Wei XH, Tang XL, Liu SG, Liu JX, Zhang DQ, Yan JH (2008) Factors influencing leaf litter decomposition: an intersite decomposition experiment across China. Plant Soil 311(1–2):61–72
Zhu J, Mulder J, Bakken L, Wu LP, Meng XX, Wang YH, Dörsch P (2013a) Spatial and temporal variability of N2O emissions in a subtropical forest catchment in China. Biogeosciences 10:1309–1321. doi:10.5194/bg-10-1309-2013
Zhu J, Mulder J, Solheimslid S, Dörsch P (2013b) Functional traits of denitrification in a subtropical forest catchment in China with high atmogenic N deposition. Soil Biol Biochem 57:577–586
Acknowledgments
This study was supported by the Norwegian research council (Nordklima 193725/S30) and the Chinese Academy of Sciences (No. KZCX2-YW-GJ01 and GJHZ1205). We are grateful for technical support by Meng Xiaoxing, Wu Liping and Zhang Guanli at the Chongqing Academy of Environmental Sciences, Prof. Zhang Xiaoshan at the Research Center for Eco-environmental Sciences, Chinese Academy of Sciences and T. Fredriksen at the Norwegian University of Life Sciences. Many thanks to S. Solheimslid, H. Silvennoinen and L.E. Sørbotten at the Norwegian University of Life Sciences for assistance in the field. We also thank for the suggestions from H. Silvennoinen and P. T. Mørkved with respect to laboratory work.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Zhu, J., Mulder, J., Bakken, L. et al. The importance of denitrification for N2O emissions from an N-saturated forest in SW China: results from in situ 15N labeling experiments. Biogeochemistry 116, 103–117 (2013). https://doi.org/10.1007/s10533-013-9883-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10533-013-9883-8