
Abstract
Chimeric antigen receptor (CAR)-T cell therapy has shown promising results in patients with hematological malignancies. However, many patients still have poor prognoses or even fatal outcomes due to the life-threatening toxicities associated with the therapy. Moreover, even after improving the known influencing factors (such as number or type of CAR-T infusion) related to CAR-T cell infusion, the results remain unsatisfactory. In recent years, it has been found that endothelial cells (ECs), which are key components of the organization, play a crucial role in various aspects of immune system activation and inflammatory response. The levels of typical markers of endothelial activation positively correlated with the severity of cytokine release syndrome (CRS) and immune effector cell-associated neurotoxic syndrome (ICANS), suggesting that ECs are important targets for intervention and toxicity prevention. This review focuses on the critical role of ECs in CRS and ICANS and the intervention strategies adopted.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Chimeric antigen receptor (CAR)-T cell therapy is a rapidly advancing tumor immunotherapy approach that has been widely used and shown to be effective in the treatment of hematologic malignancies, including acute lymphoblastic leukemia [1, 2], non-Hodgkin’s lymphoma [3,4,5], and multiple myeloma [6, 7]. A certain amount of cytokine release is a marker of efficacy; however, excessive cytokine release leads to treatment-related toxicity. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxic syndrome (ICANS), which most treatment-related deaths are attributed to, are the most common and severe toxicities that require early intervention and standardized management [8,9,10].
The development and severity of CRS cannot be effectively predicted and attenuated by adjusting the relevant factors affecting its growth and severity, such as the number and type of CAR-T infusions [11, 12]. Simultaneously, treatment with interleukin (IL)-6 inhibitors is effective only during the early stages of CRS. This may cause severe ICANS due to binding to IL-6R, which allows high levels of serum IL-6 to cross the blood-brain barrier (BBB). CAR-T cell therapy releases cytokines during endothelial cell (EC) stimulation, suggesting that the patient’s factors is as essential as those factors of CAR-T infusion for the associated toxicity. As the first line of defense against inflammatory stress, EC dysfunction and activity can affect disease severity and progression. The elevated biomarkers associated with endothelial activation, hemodynamic instability, capillary leakage, and coagulopathy observed in severe CRS further corroborated that CRS and ICANS may be mediated by endothelial activation and malfunction to a certain extent [13,14,15,16,17,18].
In this review, we aimed to highlight the role ECs play in intervening CAR-T cell-related toxicity and promoting the broader application of CAR-T cell therapy.
Cytokine release syndrome and immune effector cell-associated neurotoxic syndrome
Studies using autologous anti-CD19 CAR-T cells have shown that CRS occurs in 42%–93% of patients and ICANS in 30%–67% [3, 19]. CRS is a systemic inflammatory response caused by CAR-T cell activation and proliferation and significant concomitant elevations of multiple serum cytokines [20], with the most pronounced peaks being IL-6, IL-1, interferon (IFN)-γ, and tumor necrosis factor-α (TNF-α), followed by IL-8, IL-10, monocyte chemotactic protein-1, and granulocyte-macrophage colony-stimulating factor [18, 21,22,23], leading to capillary leakage, elevated transaminases, and coagulation disorders [24,25,26]. Some of the clinical manifestations include fever, malaise, hypotension, shock, multiorgan dysfunction, and death [27, 28]. It can also result in severe CRS (grade ≥ 3) in up to 46% of patients [27,28,29].
CAR-T cell therapy–associated neurotoxicity, on the other hand, is due to high levels of systemic inflammatory cytokines (IL-6, TNF-γ, and TNF-β), leading to EC activation, BBB destruction, and infiltration of peripheral cytokines and immune cells into the central nervous system (CNS) [15, 16, 27, 29,30,31,32,33,34,35], which subsequently initiates a feedback loop that continuously activates the endothelium, making toxic events irreversible. The most distinct risk factors for ICANS include systemic cytokine release and CRS severity [36]. The American Society for Transplantation and Cellular Therapy Consensus Panel on Toxicity [37] named this novel neurological syndrome “immune effector cell-associated neurotoxic syndrome,” and it can occur after or alone with CRS or can develop concurrently with CRS [28] and manifests as delirium, somnolence, coma, cognitive impairment, dysphagia, tremor, ataxia, myoclonus, sensory deficits, seizures, and cerebral edema [26] (Fig. 1).

The clinical manifestations of CRS and ICANS
Biological behaviors of endothelial activation
Vascular ECs are essential for the activation of the coagulation system and the maintenance of vascular homeostasis. Healthy ECs naturally express substances that cause vasodilation, improve blood flow, decrease platelet (PLT) aggregation, and enhance fibrinolysis, whereas dysfunctional ECs lead to vasoconstriction and thrombosis. EC activation results in sequential activation of intracellular angiopoietin (Ang), TIE-2, JAK-STAT, PI3K-AKT, NF-kappa B, and other pathways, leading to the initiation of self-DNA damage apoptosis and the large-scale release of cytokines, such as IL-6 and IFN-γ, to participate in the “cytokine storm” while significantly upregulating a variety of adhesion molecules; promoting lymphocyte adhesion and infiltration into the organ interior [38], recruiting macrophages, neutrophils, and NK cells and promoting their activation; and releasing inflammatory mediators to join the inflammatory response [39].
EC activation has two outcomes: adaptive changes, which may be beneficial, and tissue-specific expression of vascular and secretory factors, which can facilitate organ repair and regeneration [40]. By contrast, excessive activation can lead to 1) damaged barrier function, which could allow soluble effectors to pass (such as TNF-α) and cause edema, damaging the endothelium; 2) an increased risk of thrombosis, which could lead to vascular thrombotic events (such as ischemia or stroke), and increased shear effects, which may result in erythrocyte fragmentation, anemia, thrombocytopenia, and neurological dysfunction; 3) increased expression of cell adhesion molecules (such as endothelial intercellular adhesion molecule-1 [ICAM-1], selectins, and integrins), leading to leukocyte migration and vascular adhesion, thus facilitating an inflammatory response and further exacerbating edema; and 4) upregulation of proinflammatory mediators. The upregulation of proinflammatory mediators is triggered by NF-κB translocation and Toll-like receptors. Inflammatory mediators enhance the expression of procoagulant tissue factors while inhibiting the anticoagulant system and indirectly activating the renin-angiotensin system [15, 40, 41]. In conclusion, inflammatory mediators created during EC activation continuously boost the activation process, resulting in a positive feedback loop and cytokine storms.
Role of EC in CRS and ICANS
ECs are the first line of defense against inflammatory stress and are central regulators of cytokine storms [42]. In capillary leak syndromes (such as acute respiratory distress syndrome and severe burns), alternative markers of endothelial injury, such as IL-6, IL-8, and fibrinogen activator inhibitor-1, are elevated [43], suggesting a link between inflammation and endothelial dysfunction. Many prevalent and serious infectious diseases can be distinguished based on EC activation caused by inflammation. The degree of EC activation and subsequent dysfunction affect the severity and progression of the disease [44] .
CRS and ICANS have similar characteristics to the endothelial injury syndrome that occurs after hematopoietic cell transplantation (HCT) [26]. Vascular endothelial activation due to high levels of inflammatory cytokines is believed to favor the development of CRS and ICANS after CAR-T treatment. Cytokines such as IL-1 and IL-6 inhibit the natural anticoagulant pathway [45] and lead to the activation of ECs’ MAPK/NF-κB pathway [46,47,48], producing and releasing procoagulant granules, such as Weibel-Palade vesicles, while the cytoskeleton of ECs is recombined [49] and tight junctions are lost [50]. Brain microvascular ECs are critical regulators of systemic inflammatory signaling into the CNS; brain ECs mediate central febrile responses; express IL-1β, IL-6, and TNF receptors; and locally produce cytokines that enhance pro-inflammatory responses, altering endothelial transporter protein function [16, 40]. Clinical evidence supporting strong endothelial activation and enhanced BBB permeability has been found in patients with CRS and ICANS, including inflammation, impaired coagulation, and enhanced vascular permeability [15, 16, 18, 26]. Correlations between EC activation biomarkers, the development and severity of CRS, and the degree of liver, renal, and hematopoietic dysfunction also confirm that endothelial activation is one of the mechanisms for CAR-T cell immune-mediated toxicity after CAR-T cell therapy [51, 52].
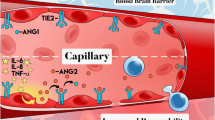
The Ang-TIE-2 system may explain the association between endothelial activation, systemic cytokine release, and microvascular dysfunction [16]. TIE-2 is expressed on the EC surface [53], and Ang-1, produced mainly by perivascular cells and PLTs [16], binds to TIE-2 on the EC surface and activates downstream pathways to maintain EC stability. Ang-2, stored in endothelial Weibel-Palade vesicles, is released from EC upon EC activation, displacing Ang-1 and inhibiting TIE-2 signaling [54,55,56,57], thereby impairing EC-cell junctions, inducing expression of pro-inflammatory adhesion molecules (including ICAM-1 and vascular cell adhesion molecule-1 [VCAM-1]) and epithelial procoagulant protein levels [58,59,60,61]. The combination of high serum cytokine levels and endothelial activation leads to a cascade response that progressively amplifies endothelial activation and injury. Biomarkers of endothelial activation, such as Ang-2, Ang-2/Ang-1 ratio, and von Willebrand factor, are elevated in patients with severe CRS and severe neurotoxicity (grades 3–4) [16, 18, 30], and the Ang-2/Ang-1 ratio is elevated before lymphatic depletion in patients with grade 4 and 5 ICANS [16]. Patients with endothelial activation before CAR-T cell infusion are more prone to develop CRS and ICANS [18]. Furthermore, as PLTs produce Ang-1, changes in the Ang-2/Ang-1 ratio may be driven by thrombocytopenia alone, and patients with severe thrombocytopenia before or immediately following CAR-T cell infusion may be more susceptible to developing CRS and ICANS-related endothelial activation and damage. This is because PLTs are one of the limited sources of endothelium-stabilizing cytokines (Ang-1) [54] (Fig. 2).

Simple schematic diagram of Ang-TIE-2 system. Ang, angiopoietin
EC activation is closely associated with immune-mediated inflammatory responses and organ damage; however, because the degree of EC activation varies among states, endothelial activation, a necessary step after initiation, is of great importance in intervening or mitigating the degree of endothelial activation in response to toxicity associated with CAR-T cell therapy.
How to predict CRS and ICANS by endothelial related indexes
Lactate dehydrogenase (LDH), a surrogate marker of tumor infiltration, and the general inflammatory markers C-reactive protein (CRP) and ferritin all shared a connection with severe CRS and/or ICANS [18, 19, 62,63,64,65,66]. They could potentially be applied to predict CRS and ICANS risks in patients; however, the reliability of a single index is low. The endothelial activation and stress index (EASIX) score (LDH [U/L] × creatinine [mg/dL]/PLTs [109 cells/L]) is a surrogate indicator of systemic endothelial activation that has been validated in the allogeneic hematopoietic stem cell transplantation [51, 67,68,69,70,71,72,73]; however, it is also applicable to other complex endothelial activation or injury situations, such as coronavirus disease 2019 [67,68,69,70,71,72,73,74]. It is significantly correlated with various endothelial dysfunction and complement activation biomarkers [75]. Higher EASIX score before CAR-T cell infusion parallels a significant elevation of multiple endothelial serum markers [17]. A study demonstrated that EASIX correlates with the incidence rate and seriousness of CRS and ICANS in axi-cel-treated large B-cell lymphoma patients [51]. This suggested that EASIX is also a useful prognostic marker of endothelial dysfunction in CAR-T cell therapy, with lateral evidence of EC involvement in the pathogenesis of CRS/ICANS. Modification of EASIX to remove creatinine (s-EASIX) or to replace creatinine with CRP (m-EASIX) was found to improve the predictive power [76, 77], and combining EASIX with inflammatory markers (CRP and ferritin) enhanced the prediction effect [51].
Intervention strategies targeting the endothelium
Intervention strategies to reduce endothelial activation and endothelial injury include statins, defibrillated polynucleotides, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers, and a new type of p38/MAPK inhibitors [78] (Fig. 3).

Intervention strategies to reduce endothelial activation and endothelial injury. ox-LDL, oxidized low-density lipoprotein; NOX, nicotinamide adenine dinucleotide phosphate oxidase; ROS, reactive oxygen species; DF, defibrillated polynucleotides; NOS, nitric oxide synthase; MAC, membrane attack complex; CART, chimeric antigen receptor (CAR)-T cell ; TNF-α, tumor necrosis factor-α; FAK, focal adhesion kinase
Statins
Statins regulate immunological responses at various levels, including immune cell adhesion and migration, antigen presentation, and cytokine production [79], and exert endothelial protection against inflammation and oxidative stress. Antagonizing Ang2 through the angiopoietin/Tie-2 signaling axis [80] exerts its endothelial protective function and maintains endothelial cell quiescence, which antagonizes the pathological process characteristic of endothelial dysfunction associated with hematopoietic stem cell transplantation and CAR-T cell therapy [81], and there is a clinical trial investigating the use of simvastatin and intrathecal dexamethasone for the prevention of neurotoxicity after treatment with axicabtagene-ciloleucel (NCT04514029) [81]. In addition, statins may help reduce oxidized low-density lipoprotein levels and nicotinamide adenine dinucleotide phosphate oxidase activity, thereby decreasing reactive oxygen species (ROS), impacting either directly or indirectly NF-κB transcription, or strengthening endothelial nitric oxide synthase coupling [41]; statins should be studied further as potential targets for the prevention and therapy of endothelial damage.
Defibrillated polynucleotides
Defibrillated polynucleotides (DF) is a unique, naturally derived EC protector with potential effects on angiogenesis, EC activation, and endothelial inflammation, reducing EC activation through antithrombosis, anti-inflammatory, antioxidant, and antiadhesion activities [82], and restoring thrombolysis-fibrinolysis balance, with broad potential in a range of serious diseases based on EC injury and inflammation. The reduced release of inflammatory mediators manifests the anti-inflammatory effects, reduced production of ROS and nitric oxide synthase levels during oxidative stress [83, 84], and significant inhibition of heparanase expression and activity [85], promoting repair of endothelial integrity and function. DF significantly improved survival in patients with hepatic sinusoidal obstruction syndrome (SOS) and small hepatic vein occlusive disease (VOD) by stabilizing the endothelium [86, 87]. It has also been authorized to treat VOD/SOS in both adults and kids [88, 89], demonstrating effectiveness as well as safety. It is well tolerated and is reasonably presumed to be used to prevent or treat CAR-T cell immune-related toxicity. An in vitro study using endothelial cell lines revealed that defibrillating peptides inhibited endothelial proliferation in a dose-dependent manner and suppressed serum-induced endothelial activation and angiogenesis in graft-versus-host disease (GVHD) patients, and that DF had a significant positive effect on endothelial biological properties during aGVHD [83]. Another study found that adding defibrillating peptides reduced the alterations associated with endothelial dysfunction [90]. The administration of DF regulates EC injury in models of acute respiratory distress syndrome and idiopathic pneumonia syndrome [91]. A clinical trial showed that DF therapy slightly reduced the rate of CAR-T-associated neurotoxicity/high-grade event duration compared to previous data [92]. More research is required in the future to validate its efficacy.
Complement inhibitors
The complement system is linked to EC activation, inflammation, leukocyte recruitment, PLT activation, and coagulation. All three pathways (classical, alternative, and lectin pathways) trigger proximal complement activation, leading to C3 activation and C3 convertase formation. Activation of C3 via the alternative complement route also amplifies this effect, ultimately leading to the deposition of C3 fragments on target cells. When sufficient C3b is deposited, the terminal cleavage pathway is triggered, leading to the formation of a membrane attack complex (MAC) on the surface of the target cells [26]. CAR-T cell-related toxicity can also lead to these changes, and endothelial damage continues to occur through the activation of a variety of complement pathways and interactions between complements and interferons.
Complement inhibition is safe and effective in patients with endothelial dysfunction syndromes (such as transplant-associated thrombotic microangiopathy) [26]. Complement inhibitors are currently approved by the Food and Drug Administration for the treatment of paroxysmal nocturnal hemoglobinuria, blocking terminal complement activation by binding to C5 so that the process of producing pro-inflammatory C5a molecules and MAC scleral complex formation is stopped. Complement inhibitors at the C3 and C5 levels reduced whole blood-induced endothelial cell activation by 89% and eliminated TNF release [93]; complement inhibition may be a viable new strategy to control the systemic complement-mediated inflammatory response.
Adalimumab in combination with anti-IL-1β antibodies
During CAR-T cell treatment, vascular ECs are exposed to several stimuli in the bloodstream. Central cytokines that cause endothelial activation include IL-1β, which is released by activated myeloid cells, and TNF-α, which is produced by CAR-T cells when they recognize tumors. These cytokines highly activate EC by upregulating the expression of adhesion molecules (E-selectin, VCAM-1, and ICAM-1), while focal adhesion kinase (FAK) process, NF-кB process, and MAPK process are activated.
TNF receptor 1 (TNFR1), the primary TNF-α receptor on the endothelial membrane, is involved in the inflammatory process. By deleting TNFR1 in human ECs, the degree of CAR-T cell-induced endothelial activation was reduced. Endothelial activation was also prevented by selective small-molecule inhibitors of the TNFR1, NF-кB, and MAPK signaling pathways [94]. Adalimumab, anti-IL-1β, and FAK inhibitors effectively blocked TNF-α, IL-1β, and reduced FAK activity; improved endothelial dysfunction caused by CAR-T cells, malignant cells, and myeloid cells in CAR-T cells treatment; and reduced endothelial leakage caused by CAR-T and other cells. Moreover, the combination of adalimumab and anti-IL-1β antibodies showed synergistic effects [94]. As a result, the above drugs may have therapeutic potential for immunotoxicity associated with CAR-T therapy.
Others
Corticosteroids are frequently used to regulate proinflammatory states linked to endothelium-related HCT complications, such as interstitial pneumonia syndrome, because they have anti-inflammatory properties that can reduce endothelial damage. However, they are only anti-inflammatory and not endothelial-specific. In order to combat endothelial activation and coagulation impairment, other strategies including increasing Ang-1 or PLT transfusions can be included [16, 55]. Symptomatic treatment with ACE inhibitors, angiotensin receptor blockers (ARBs), and cytokine inhibitors, such as IL-6 receptor antibodies, may be used for the different steps of the cascade response. ACE inhibitors [95, 96] and ARBs [97] are important strategies with endothelial protective potential because they have anti-inflammatory effects and improve endothelial dysfunction. As antagonists of the Ang-2 pathway, ACE inhibitors and ARB inhibit the p38/MAPK activation signal cascade reaction of endothelial cells, which is triggered by a high level of Ang-2 [78]; therefore, direct p38/MAPK inhibitors [98] might provide immediate and long-term protection of the endothelium by affecting the Ang-2 and p38/MAPK signaling axes, thus further improving patient outcomes.
Conclusions and future directions
CAR-T cell therapy has achieved excellent results in patients with hematological malignancies. Its use has also been extended to other fields such as solid tumors; however, its response varies significantly among patients after infusion. ECs, an essential link in the role of CAR-T cells, are expected to be a target for intervening in CAR-T cell-related toxicity and promoting the broader application of CAR-T cell therapy. In the future, the relationship between EC dysfunction and the efficacy of CAR T-cell therapy should be investigated further.
References
Park JH, Riviere I, Gonen M et al (2018) Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia [J]. N Engl J Med 378(5):449–59. https://doi.org/10.1056/NEJMoa1709919
Maude SL, Laetsch TW, Buechner J et al (2018) Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia [J]. N Engl J Med 378(5):439–48. https://doi.org/10.1056/NEJMoa1709866
Neelapu SS, Locke FL, Bartlett NL et al (2017) Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-Cell lymphoma [J]. N Engl J Med 377(26):2531–44. https://doi.org/10.1056/NEJMoa1707447
Schuster SJ, Bishop MR, Tam CS et al (2019) Tisagenlecleucel in adult relapsed or refractory diffuse large B-cell lymphoma [J]. N Engl J Med 380(1):45–56. https://doi.org/10.1056/NEJMoa1804980
Wang M, Munoz J, Goy A et al (2020) KTE-X19 CAR T-cell therapy in relapsed or refractory mantle-cell lymphoma [J]. N Engl J Med 382(14):1331–42. https://doi.org/10.1056/NEJMoa1914347
Wu C, Zhang L, Brockman QR et al (2019) Chimeric antigen receptor T cell therapies for multiple myeloma [J]. J Hematol Oncol 12(1):120. https://doi.org/10.1186/s13045-019-0823-5
Munshi NC, Anderson LD, Jr Shah N et al (2021) Idecabtagene vicleucel in relapsed and refractory multiple myeloma [J]. N Engl J Med 384(8):705–16. https://doi.org/10.1056/NEJMoa2024850
Li W, Wu L, Huang C et al (2020) Challenges and strategies of clinical application of CAR-T therapy in the treatment of tumors-a narrative review [J]. Ann Transl Med 8(17):1093. https://doi.org/10.21037/atm-20-4502
Greenbaum U, Kebriaei P, Srour SA et al (2021) Chimeric antigen receptor T-cell therapy toxicities [J]. Br J Clin Pharmacol 87(6):2414–24. https://doi.org/10.1111/bcp.14403
Neelapu SS, Tummala S, Kebriaei P et al (2018) Chimeric antigen receptor T-cell therapy - assessment and management of toxicities [J]. Nat Rev Clin Oncol 15(1):47–62. https://doi.org/10.1038/nrclinonc.2017.148
Schubert ML, Schmitt A, Sellner L et al (2019) Treatment of patients with relapsed or refractory CD19+ lymphoid disease with T lymphocytes transduced by RV-SFG.CD19.CD28.4-1BBzeta retroviral vector: a unicentre phase I/II clinical trial protocol [J]. BMJ Open 9(5):e026644. https://doi.org/10.1136/bmjopen-2018-026644
Bachmann M (2019) The UniCAR system: a modular CAR T cell approach to improve the safety of CAR T cells [J]. Immunol Lett 211:13–22. https://doi.org/10.1016/j.imlet.2019.05.003
Obstfeld AE, Frey NV, Mansfield K et al (2017) Cytokine release syndrome associated with chimeric-antigen receptor T-cell therapy: clinicopathological insights [J]. Blood 130(23):2569–72. https://doi.org/10.1182/blood-2017-08-802413
Maude SL (2017) CAR emissions: cytokines tell the story [J]. Blood 130(21):2238–40. https://doi.org/10.1182/blood-2017-10-808592
Mackall CL, Miklos DB (2017) CNS endothelial cell activation emerges as a driver of CAR T cell-associated neurotoxicity [J]. Cancer Discov 7(12):1371–3. https://doi.org/10.1158/2159-8290.CD-17-1084
Gust J, Hay KA, Hanafi LA et al (2017) Endothelial activation and blood-brain barrier disruption in neurotoxicity after adoptive immunotherapy with CD19 CAR-T cells [J]. Cancer Discov 7(12):1404–19. https://doi.org/10.1158/2159-8290.CD-17-0698
Korell F, Penack O, Mattie M et al (2022) EASIX and severe endothelial complications after CD19-directed CAR-T cell therapy-a cohort study [J]. Front Immunol 13:877477. https://doi.org/10.3389/fimmu.2022.877477
Hay KA, Hanafi LA, Li D et al (2017) Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy [J]. Blood 130(21):2295–306. https://doi.org/10.1182/blood-2017-06-793141
Abramson JS, Palomba ML, Gordon LI et al (2020) Lisocabtagene maraleucel for patients with relapsed or refractory large B-cell lymphomas (TRANSCEND NHL 001): a multicentre seamless design study [J]. Lancet 396(10254):839–52. https://doi.org/10.1016/S0140-6736(20)31366-0
Santomasso B, Bachier C, Westin J et al (2019) The other side of CAR T-cell therapy: cytokine release syndrome, neurologic toxicity, and financial burden [J]. Am Soc Clin Oncol Educ Book 39:433–44. https://doi.org/10.1200/EDBK_238691
Norelli M, Camisa B, Barbiera G et al (2018) Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells [J]. Nat Med 24(6):739–48. https://doi.org/10.1038/s41591-018-0036-4
Wang Z, Han W (2018) Biomarkers of cytokine release syndrome and neurotoxicity related to CAR-T cell therapy [J]. Biomark Res 6:4. https://doi.org/10.1186/s40364-018-0116-0
Lee DW, Gardner R, Porter DL et al (2014) Current concepts in the diagnosis and management of cytokine release syndrome [J]. Blood 124(2):188–95. https://doi.org/10.1182/blood-2014-05-552729
Wang J, Doran J (2021) The many faces of cytokine release syndrome-related coagulopathy [J]. Clin Hematol Int 3(1):3–12. https://doi.org/10.2991/chi.k.210117.001
van der Poll T, van de Veerdonk FL, Scicluna BP et al (2017) The immunopathology of sepsis and potential therapeutic targets [J]. Nat Rev Immunol 17(7):407–20. https://doi.org/10.1038/nri.2017.36
Gavriilaki E, Sakellari I, Gavriilaki M, Anagnostopoulos A (2020) A new era in endothelial injury syndromes: toxicity of CAR-T cells and the role of immunity. Int J Mol Sci 21(11):3886. https://doi.org/10.3390/ijms21113886
Neelapu SS (2019) Managing the toxicities of CAR T-cell therapy [J]. Hematol Oncol 37(Suppl 1):48–52. https://doi.org/10.1002/hon.2595
Schubert ML, Schmitt M, Wang L et al (2021) Side-effect management of chimeric antigen receptor (CAR) T-cell therapy [J]. Ann Oncol 32(1):34–48. https://doi.org/10.1016/j.annonc.2020.10.478
Zahid A, Siegler EL, Kenderian SS (2020) CART Cell toxicities: new insight into mechanisms and management [J]. Clin Hematol Int 2(4):149–55. https://doi.org/10.2991/chi.k.201108.001
Santomasso BD, Park JH, Salloum D et al (2018) Clinical and biological correlates of neurotoxicity associated with CAR T-cell therapy in patients with B-cell acute lymphoblastic leukemia [J]. Cancer Discov 8(8):958–71. https://doi.org/10.1158/2159-8290.CD-17-1319
Gust J, Taraseviciute A, Turtle CJ (2018) Neurotoxicity associated with CD19-targeted CAR-T cell therapies [J]. CNS Drugs 32(12):1091–101. https://doi.org/10.1007/s40263-018-0582-9
Siegler EL, Kenderian SS (1973) Neurotoxicity and cytokine release syndrome after chimeric antigen receptor T cell therapy: insights into mechanisms and novel therapies [J]. Front Immunol 2020:11. https://doi.org/10.3389/fimmu.2020.01973
Morris EC, Neelapu SS, Giavridis T et al (2022) Cytokine release syndrome and associated neurotoxicity in cancer immunotherapy [J]. Nat Rev Immunol 22(2):85–96. https://doi.org/10.1038/s41577-021-00547-6
Akbari P, Katsarou A, Daghighian R et al (2022) Directing CAR T cells towards the tumor vasculature for the treatment of solid tumors [J]. Biochim Biophys Acta Rev Cancer 1877(3):188701. https://doi.org/10.1016/j.bbcan.2022.188701
Jung S, Greiner J, von Harsdorf S et al (2021) Fatal late-onset CAR T-cell-mediated encephalitis after axicabtagene-ciloleucel in a patient with large B-cell lymphoma [J]. Blood Adv 5(19):3789–93. https://doi.org/10.1182/bloodadvances.2021004889
Gust J, Finney OC, Li D et al (2019) Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy [J]. Ann Neurol 86(1):42–54. https://doi.org/10.1002/ana.25502
Lee DW, Santomasso BD, Locke FL et al (2019) ASTCT consensus grading for cytokine release syndrome and neurologic toxicity associated with immune effector cells [J]. Biol Blood Marrow Transplant 25(4):625–38. https://doi.org/10.1016/j.bbmt.2018.12.758
Buser TA, Martinez M, Drexler B et al (2019) Biological markers of hemostasis and endothelial activation in patients with a hematological malignancy with or without stem cell transplants [J]. Eur J Haematol 103(5):472–7. https://doi.org/10.1111/ejh.13310
Furukawa M, Wang X, Ohkawara H et al (2019) A critical role of the Gas6-Mer axis in endothelial dysfunction contributing to TA-TMA associated with GVHD [J]. Blood Adv 3(14):2128–43. https://doi.org/10.1182/bloodadvances.2019000222
Hildebrandt GC, Chao N (2020) Endothelial cell function and endothelial-related disorders following haematopoietic cell transplantation [J]. Br J Haematol 190(4):508–19. https://doi.org/10.1111/bjh.16621
Nägele MP, Haubner B, Tanner FC et al (2020) Endothelial dysfunction in COVID-19: current findings and therapeutic implications [J]. Atherosclerosis 314:58–62. https://doi.org/10.1016/j.atherosclerosis.2020.10.014
Teijaro JR, Walsh KB, Cahalan S et al (2011) Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection [J]. Cell 146(6):980–91. https://doi.org/10.1016/j.cell.2011.08.015
Kang S, Tanaka T, Inoue H et al (2020) IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome [J]. Proc Natl Acad Sci U S A 117(36):22351–6. https://doi.org/10.1073/pnas.2010229117
Kim H, Higgins S, Liles WC et al (2011) Endothelial activation and dysregulation in malaria: a potential target for novel therapeutics [J]. Curr Opin Hematol 18(3):177–85. https://doi.org/10.1097/MOH.0b013e328345a4cf
Danese S, Vetrano S, Zhang L et al (2010) The protein C pathway in tissue inflammation and injury: pathogenic role and therapeutic implications [J]. Blood 115(6):1121–30. https://doi.org/10.1182/blood-2009-09-201616
Giddings JC, Shall L (1987) Enhanced release of von Willebrand factor by human endothelial cells in culture in the presence of phorbol myristate acetate and interleukin 1 [J]. Thromb Res 47(3):259–67. https://doi.org/10.1016/0049-3848(87)90139-3
Paleolog EM, Crossman DC, Mcvey JH et al (1990) Differential regulation by cytokines of constitutive and stimulated secretion of von Willebrand factor from endothelial cells [J]. Blood 75(3):688–95
Bernardo A, Ball C, Nolasco L et al (2004) Effects of inflammatory cytokines on the release and cleavage of the endothelial cell-derived ultralarge von Willebrand factor multimers under flow [J]. Blood 104(1):100–6. https://doi.org/10.1182/blood-2004-01-0107
Petrache I, Birukova A, Ramirez SI et al (2003) The role of the microtubules in tumor necrosis factor-alpha-induced endothelial cell permeability [J]. Am J Respir Cell Mol Biol 28(5):574–81. https://doi.org/10.1165/rcmb.2002-0075OC
Wang S, Le TQ, Kurihara N et al (2010) Influenza virus-cytokine-protease cycle in the pathogenesis of vascular hyperpermeability in severe influenza [J]. J Infect Dis 202(7):991–1001. https://doi.org/10.1086/656044
Greenbaum U, Strati P, Saliba RM et al (2021) CRP and ferritin in addition to the EASIX score predict CAR-T-related toxicity [J]. Blood Adv 5(14):2799–806. https://doi.org/10.1182/bloodadvances.2021004575
Hong F, Shi M, Cao J et al (2021) Predictive role of endothelial cell activation in cytokine release syndrome after chimeric antigen receptor T cell therapy for acute lymphoblastic leukaemia [J]. J Cell Mol Med 25(24):11063–74. https://doi.org/10.1111/jcmm.17029
Armulik A, Abramsson A, Betsholtz C (2005) Endothelial/pericyte interactions [J]. Circ Res 97(6):512–23. https://doi.org/10.1161/01.RES.0000182903.16652.d7
Page AV, Liles WC (2013) Biomarkers of endothelial activation/dysfunction in infectious diseases [J]. Virulence 4(6):507–16. https://doi.org/10.4161/viru.24530
Saharinen P, Eklund L, Alitalo K (2017) Therapeutic targeting of the angiopoietin-TIE pathway [J]. Nat Rev Drug Discov 16(9):635–61. https://doi.org/10.1038/nrd.2016.278
Milam KE, Parikh SM (2015) The angiopoietin-Tie2 signaling axis in the vascular leakage of systemic inflammation [J]. Tissue Barriers 3(1–2):e957508. https://doi.org/10.4161/21688362.2014.957508
Xing K, Murthy S, Liles WC et al (2012) Clinical utility of biomarkers of endothelial activation in sepsis–a systematic review [J]. Crit Care 16(1):R7. https://doi.org/10.1186/cc11145
Melrose J, Tsurushita N, Liu G et al (1998) IFN-gamma inhibits activation-induced expression of E- and P-selectin on endothelial cells [J]. J Immunol 161(5):2457–64
Maisonpierre PC, Suri C, Jones PF et al (1997) Angiopoietin-2, a natural antagonist for Tie2 that disrupts in vivo angiogenesis [J]. Science 277(5322):55–60. https://doi.org/10.1126/science.277.5322.55
Leong A, Kim M (2020) The Angiopoietin-2 and TIE Pathway as a Therapeutic Target for Enhancing Antiangiogenic Therapy and Immunotherapy in Patients with Advanced Cancer. Int J Mol Sci. 21(22):8689. https://doi.org/10.3390/ijms21228689
Klinger M, Zugmaier G, Nägele V et al (2020) Adhesion of T cells to endothelial cells facilitates blinatumomab-associated neurologic adverse events [J]. Cancer Res 80(1):91–101. https://doi.org/10.1158/0008-5472.CAN-19-1131
Strati P, Nastoupil LJ, Westin J et al (2020) Clinical and radiologic correlates of neurotoxicity after axicabtagene ciloleucel in large B-cell lymphoma [J]. Blood Adv 4(16):3943–51. https://doi.org/10.1182/bloodadvances.2020002228
Rubin DB, Al Jarrah A, Li K et al (2020) Clinical predictors of neurotoxicity after chimeric antigen receptor T-cell therapy [J]. JAMA Neurol 77(12):1536–42. https://doi.org/10.1001/jamaneurol.2020.2703
Nastoupil LJ, Jain MD, Feng L et al (2020) Standard-of-care axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma: results from the US lymphoma CAR T Consortium [J]. J Clin Oncol 38(27):3119–28. https://doi.org/10.1200/JCO.19.02104
Locke FL, Rossi JM, Neelapu SS et al (2020) Tumor burden, inflammation, and product attributes determine outcomes of axicabtagene ciloleucel in large B-cell lymphoma [J]. Blood Adv 4(19):4898–911. https://doi.org/10.1182/bloodadvances.2020002394
Gutgarts V, Jain T, Zheng J et al (2020) Acute kidney injury after CAR-T cell therapy: low incidence and rapid recovery [J]. Biol Blood Marrow Transplant 26(6):1071–6. https://doi.org/10.1016/j.bbmt.2020.02.012
Luft T, Benner A, Terzer T et al (2020) EASIX and mortality after allogeneic stem cell transplantation [J]. Bone Marrow Transplant 55(3):553–61. https://doi.org/10.1038/s41409-019-0703-1
Luft T, Benner A, Jodele S et al (2017) EASIX in patients with acute graft-versus-host disease: a retrospective cohort analysis [J]. Lancet Haematol 4(9):e414–e23. https://doi.org/10.1016/S2352-3026(17)30108-4
Varma A, Rondon G, Srour SA et al (2020) Endothelial activation and stress index (EASIX) at admission predicts fluid overload in recipients of allogeneic stem cell transplantation [J]. Biol Blood Marrow Transplant 26(5):1013–20. https://doi.org/10.1016/j.bbmt.2020.01.028
Shouval R, Fein JA, Shouval A et al (2019) External validation and comparison of multiple prognostic scores in allogeneic hematopoietic stem cell transplantation [J]. Blood Adv 3(12):1881–90. https://doi.org/10.1182/bloodadvances.2019032268
Jiang S, Penack O, Terzer T et al (2021) Predicting sinusoidal obstruction syndrome after allogeneic stem cell transplantation with the EASIX biomarker panel [J]. Haematologica 106(2):446–53. https://doi.org/10.3324/haematol.2019.238790
Merz A, Germing U, Kobbe G, Kaivers J, Jauch A, Radujkovic A, Hummel M, Benner A, Merz M, Dreger P, Luft T (2019) EASIX for prediction of survival in lower-risk myelodysplastic syndromes. Blood Cancer J 9(11):85. https://doi.org/10.1038/s41408-019-0247-z
Song GY, Jung SH, Kim K et al (2020) Endothelial activation and stress index (EASIX) is a reliable predictor for overall survival in patients with multiple myeloma [J]. BMC Cancer 20(1):803. https://doi.org/10.1186/s12885-020-07317-y
Luft T, Wendtner CM, Kosely F et al (2021) EASIX for prediction of outcome in hospitalized SARS-CoV-2 infected patients [J]. Front Immunol 12:634416. https://doi.org/10.3389/fimmu.2021.634416
Gavriilaki E, Sakellari I, Chatzikonstantinou T et al (2021) Endothelial and complement activation as predictors of survival in adult allogeneic hematopoietic cell transplantation [J]. Hemasphere 5(1):e487. https://doi.org/10.1097/HS9.0000000000000487
Pennisi M, Sanchez-Escamilla M, Flynn JR et al (2021) Modified EASIX predicts severe cytokine release syndrome and neurotoxicity after chimeric antigen receptor T cells [J]. Blood Adv 5(17):3397–406. https://doi.org/10.1182/bloodadvances.2020003885
Kadauke S, Myers RM, Li Y et al (2021) Risk-adapted preemptive tocilizumab to prevent severe cytokine release syndrome after CTL019 for pediatric B-cell acute lymphoblastic leukemia: a prospective clinical trial [J]. J Clin Oncol 39(8):920–30. https://doi.org/10.1200/JCO.20.02477
Castro P, Palomo M, Moreno-Castaño AB et al (2022) Is the endothelium the missing link in the pathophysiology and treatment of COVID-19 complications? [J]. Cardiovasc Drugs Ther 36(3):547–60. https://doi.org/10.1007/s10557-021-07207-w
Castiglione V, Chiriacò M, Emdin M et al (2020) Statin therapy in COVID-19 infection [J]. Eur Heart J Cardiovasc Pharmacother 6(4):258–9. https://doi.org/10.1093/ehjcvp/pvaa042
Fedson DS (2016) Treating the host response to emerging virus diseases: lessons learned from sepsis, pneumonia, influenza and Ebola [J]. Ann Transl Med 4(21):421. https://doi.org/10.21037/atm.2016.11.03
Sumransub N, El Jurdi N, Chiraphapphaiboon W et al (2022) Putting function back in dysfunction: endothelial diseases and current therapies in hematopoietic stem cell transplantation and cellular therapies [J]. Blood Rev 51:100883. https://doi.org/10.1016/j.blre.2021.100883
Richardson PG, Carreras E, Iacobelli M et al (2018) The use of defibrotide in blood and marrow transplantation [J]. Blood Adv 2(12):1495–509. https://doi.org/10.1182/bloodadvances.2017008375
Martinez-Sanchez J, Hamelmann H, Palomo M et al (2019) Acute graft-vs.-host disease-associated endothelial activation in vitro is prevented by defibrotide [J]. Front Immunol 10:2339. https://doi.org/10.3389/fimmu.2019.02339
García-Bernal D, Palomo M, Martínez CM et al (2020) Defibrotide inhibits donor leucocyte-endothelial interactions and protects against acute graft-versus-host disease [J]. J Cell Mol Med 24(14):8031–44. https://doi.org/10.1111/jcmm.15434
Mitsiades CS, Rouleau C, Echart C et al (2009) Preclinical studies in support of defibrotide for the treatment of multiple myeloma and other neoplasias [J]. Clin Cancer Res 15(4):1210–21. https://doi.org/10.1158/1078-0432.CCR-08-1270
Corbacioglu S, Cesaro S, Faraci M et al (2012) Defibrotide for prophylaxis of hepatic veno-occlusive disease in paediatric haemopoietic stem-cell transplantation: an open-label, phase 3, randomised controlled trial [J]. Lancet 379(9823):1301–9. https://doi.org/10.1016/S0140-6736(11)61938-7
Picod A, Bonnin A, Battipaglia G et al (2018) Defibrotide for sinusoidal obstruction syndrome/veno-occlusive disease prophylaxis in high-risk adult patients: a single-center experience study [J]. Biol Blood Marrow Transplant 24(7):1471–5. https://doi.org/10.1016/j.bbmt.2018.02.015
Richardson PG, Grupp SA, Pagliuca A et al (2017) Defibrotide for the treatment of hepatic veno-occlusive disease/sinusoidal obstruction syndrome with multiorgan failure [J]. Int J Hematol Oncol 6(3):75–93. https://doi.org/10.2217/ijh-2017-0015
Richardson PG, Riches ML, Kernan NA et al (2016) Phase 3 trial of defibrotide for the treatment of severe veno-occlusive disease and multi-organ failure [J]. Blood 127(13):1656–65. https://doi.org/10.1182/blood-2015-10-676924
Carmona A, Díaz-Ricart M, Palomo M et al (2013) Distinct deleterious effects of cyclosporine and tacrolimus and combined tacrolimus-sirolimus on endothelial cells: protective effect of defibrotide [J]. Biol Blood Marrow Transplant 19(10):1439–45. https://doi.org/10.1016/j.bbmt.2013.07.001
Klein OR, Ktena YP, Pierce E et al (2023) Defibrotide modulates pulmonary endothelial cell activation and protects against lung inflammation in pre-clinical models of LPS-induced lung injury and idiopathic pneumonia syndrome [J]. Front Immunol 14:1186422. https://doi.org/10.3389/fimmu.2023.1186422
Jacobson CA, Rosenthal AC, Arnason J et al (2023) A phase 2 trial of defibrotide for the prevention of chimeric antigen receptor T-cell-associated neurotoxicity syndrome [J]. Blood Adv 7(21):6790–9. https://doi.org/10.1182/bloodadvances.2023009961
Nymo S, Niyonzima N, Espevik T et al (2014) Cholesterol crystal-induced endothelial cell activation is complement-dependent and mediated by TNF [J]. Immunobiology 219(10):786–92. https://doi.org/10.1016/j.imbio.2014.06.006
Chen Y, Li R, Shang S et al (2021) Therapeutic potential of TNFα and IL1β blockade for CRS/ICANS in CAR-T therapy via ameliorating endothelial activation [J]. Front Immunol 12:623610. https://doi.org/10.3389/fimmu.2021.623610.10.3389/fimmu.2021.623610
Mukai Y, Shimokawa H, Higashi M et al (2002) Inhibition of renin-angiotensin system ameliorates endothelial dysfunction associated with aging in rats [J]. Arterioscler Thromb Vasc Biol 22(9):1445–50. https://doi.org/10.1161/01.atv.0000029121.63691.ce
Locatelli M, Rottoli D, Mahmoud R, Abbate M, Corna D, Cerullo D, Tomasoni S, Remuzzi G, Zoja C, Benigni A, Macconi D (2023) Endothelial glycocalyx of peritubular capillaries in experimental diabetic nephropathy: a target of ACE inhibitor-induced kidney microvascular protection. Int J Mol Sci 24(22):16543. https://doi.org/10.3390/ijms242216543
Garg N, Krishan P, Syngle A (2021) Angiotensin-receptor blockade improves inflammation and endothelial dysfunction in ankylosing spondylitis: ARB-AS study [J]. Int J Angiol 30(4):262–70. https://doi.org/10.1055/s-0040-1722738
Bühler S, Laufer SA (2014) p38 MAPK inhibitors: a patent review (2012–2013) [J]. Expert Opin Ther Pat 24(5):535–54. https://doi.org/10.1517/13543776.2014.894977
Funding
This research funded in part by the National Natural Science Foundation of China (No. 82070213 to YX) and the High value intellectual property cultivation project of Hubei Intellectual Property Office (No. 2022).
Author information
Authors and Affiliations
Contributions
Writing—original draft preparation, Q.Z.; writing—review and editing, X.Z. and Y.X.; funding acquisition, Y.X.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Key points
In this review, we highlight the critical role endothelial cells play in intervening CAR-T cell-related toxicity and promoting the broader application of CAR-T cell therapy. We believe that our study makes a significant contribution because it emphasizes the role endothelial cells play in cytokine release syndrome and immune effector cell-associated neurotoxic syndrome, which most treatment-related deaths are attributed to and are the most common and severe toxicities that require early intervention and standardized management.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, Q., Zhu, X. & Xiao, Y. The critical role of endothelial cell in the toxicity associated with chimeric antigen receptor T cell therapy and intervention strategies. Ann Hematol (2024). https://doi.org/10.1007/s00277-024-05640-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00277-024-05640-z