
Abstract
Metagenomics, also known as environmental genomics, is the study of the genomic content of a sample of organisms (microbes) obtained from a common habitat. Metagenomics and other “omics” disciplines have captured the attention of researchers for several decades. The effect of microbes in our body is a relevant concern for health studies. There are plenty of studies using metagenomics which examine microorganisms that inhabit niches in the human body, sometimes causing disease, and are often correlated with multiple treatment conditions. No matter from which environment it comes, the analyses are often aimed at determining either the presence or absence of specific species of interest in a given metagenome or comparing the biological diversity and the functional activity of a wider range of microorganisms within their communities. The importance increases for comparison within different environments such as multiple patients with different conditions, multiple drugs, and multiple time points of same treatment or same patient. Thus, no matter how many hypotheses we have, we need a good understanding of genomics, bioinformatics, and statistics to work together to analyze and interpret these datasets in a meaningful way. This chapter provides an overview of different data analyses and statistical approaches (with example scenarios) to analyze metagenomics samples from different medical projects or clinical trials.
You have full access to this open access chapter, Download protocol PDF
Similar content being viewed by others


Key words
1 Introduction
The diversity of species on earth is high, and most of them are microorganisms. Their ubiquitous presence makes it extremely difficult to identify and classify all microbes in a laboratory environment. Standard genomics tries to enrich pure cultures and study them: for example, the taxonomy, the genome, the genes, and the pathways. However, only a miniscule fraction of all microbes can be cultured because of their complex symbiosis and nutrient requirements in other organisms. The scientific community is now equipped with the development of new sequencing techniques and high-throughput analysis. The study of the genomic content of a sample of microorganisms obtained from a common habitat is made possible with the field of metagenomics, also known as environmental genomics [1]. Instead of taking the DNA for sequencing from isolated cultures it is obtained directly from the environment. Therefore, the analysis of microbes that are deemed unculturable (which means current laboratory culturing techniques are unable to grow them) with standard laboratory techniques becomes possible. Two main approaches commonly used in metagenomic studies: marker gene-based metagenomics (e.g., 16S amplicon sequencing) and metagenomic shotgun sequencing. In the first approach, DNA is used as the template for PCR to amplify a segment of the conserved 16S ribosomal RNA (rRNA) gene sequence. Universal primers complementary to conserved regions are used so that the region can be amplified from any bacteria. After purification of PCR products, sequencing of the 16S rRNA gene is performed [2]. In the second approach, shotgun sequencing, DNA is broken up randomly into multiple small segments, which are sequenced using the chain termination method to obtain reads. Multiple overlapping reads for the target DNA are obtained by performing several rounds of this fragmentation and sequencing. Computer programs then use the overlapping ends of different reads to assemble them into a continuous sequence [3].There are several publications discussing the differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing, for example see [4]. In a recent study using water samples from Brazil’s major river floodplain systems, authors showed shotgun sequencing outdone by amplicon [5]. Here, the authors ascribed the poor performance of shotgun sequencing mainly to the weakness of the database used in the study, as compared to databases for the 16S rRNA gene. This study can be used as a caution for people working with rare environments (See article by Catherine Offord in The Scientist Footnote 1). Comparisons of the two methods in well-studied systems such as the gut microbiome have generally found that shotgun sequencing identifies more microbial diversity [6].
Further recent advancement of culturomics approach is shedding light on multiple high-throughput culture conditions [7, 8]. As the samples used in metagenomics do not contain the genome of just one but many different microorganisms, the possibility of analyzing their functional and metabolic interplay arises. Next-generation sequencing technology (NGS) has effectively transformed infectious disease research throughout the last decade, fuelling the growth in genetic data and providing huge number of DNA reads at an affordable cost. Many studies use these techniques, which examine microorganisms that inhabit niches in the human body, sometimes causing disease, and researchers often try to correlate these microorganisms and their change with multiple treatment conditions (e.g., see [9]). Gene annotations in these studies support the association of specific genes or metabolic pathways with health and with specific diseases. In a recent article authors discussed how host gene–microbial interactions are major determinants for the development of multifactorial chronic disorders and thus for the relationship between genotype and phenotype [10]. There are many other reports based on the application of metagenomics in understanding oral health and disease [11,12,13]. As recently described by Forbes et al., metagenomics and other “omics” disciplines could provide the solution to a cultureless future in clinical microbiology, food safety, and public health [14].
No matter from which environment it comes, the analysis of datasets from such studies are similar to some extent. Most projects aim at determining either the presence or absence of specific species of interest, or to obtain an overview of the taxa represented in a given metagenome and comparing the biological diversity and the functional activity of a wider range of microorganisms within their communities. The importance increases for comparison of different datasets, as researchers will need to determine and understand the similarities and dissimilarities within the metagenomes of different environments. These environments can be multiple patients with different conditions, multiple drugs, or multiple time points of same treatment or same patient. Further, sometimes researchers also may compare different environments for example to study antibiotic resistance genes (ARG) and understand which environments are more prone to such ARGs. Thus, no matter how many hypotheses we have, we need a good understanding of genomics, bioinformatics, and statistics to work together to analyze and interpret these datasets in a meaningful way.
This chapter provides an overview of different data analyses and statistical approaches to analyze metagenomics samples from a number of clinically derived datasets. The methodological description of this chapter will be guided by three main scenarios. The first one is a published data set from human atherosclerotic plaque samples (Scenario 1) [15]; the second one is a clinical trial example comparing the effects of two omega-3 polyunsaturated fatty acids (PUFAs) supplements on healthy volunteers (Scenario 2) [16]; and the third one is another clinical trial example comparing the efficacy of two drugs for an infectious disease (Scenario 3).
The Scenarios 3 came from an ongoing unpublished project; therefore, the real datasets are not provided. This chapter is mainly focused on multiple data analyses/annotation and statistical approaches that can be used in similar situations, but any biological finding of the example scenarios is not explained here. Although all of these scenarios are derived from medical projects, the analyses approach can be adapted to environmental samples as well. On this occasion, I must emphasize the importance to have good metadata, that is, a detailed description of each parameter like health status or sampling site or age or any similar information relating to specific samples that may be important for the analyses. Good metadata are key to good analyses and noise reduction in data analysis processes.
2 Description of Example Studies
2.1 Scenario 1: Metagenomic Analyses of Human Atherosclerotic Plaque Samples
To investigate microbiome diversity within human atherosclerotic tissue samples high-throughput metagenomic analysis was employed on (1) atherosclerotic plaques obtained from a group of patients who underwent endarterectomy due to recent transient cerebral ischemia or stroke and (2) presumed stabile atherosclerotic plaques obtained from autopsy from a control group of patients who all died from causes not related to cardiovascular disease. Our data provides evidence that suggest a wide range of microbial agents in atherosclerotic plaques, and an intriguing new observation that shows this microbiota displayed differences between symptomatic and asymptomatic plaques, as judged from the taxonomic profiles in these two groups of patients. Additionally, functional annotations reveal significant differences in basic metabolic and disease pathway signatures between these groups.
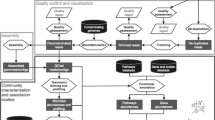
In this project, we demonstrate the feasibility of novel high-resolution techniques aimed at identification and characterization of microbial genomes in human atherosclerotic tissue samples. Our analysis suggests that distinct groups of microbial agents might play different roles during the development of atherosclerotic plaques. These findings may serve as a reference point for future studies in this area of research. The workflow in Fig. 1 provides a brief description of the sample processing and analyses pipeline for the study described in Scenario 1. If readers want to know more details of the methodology, please refer to (15). This scenario is an example of analyzing host-associated metagenome samples.

Analysis pipeline for the study of human atherosclerotic plaque samples. Interested readers may refer to the full study here [15]
2.1.1 Methodology Details
For this study, we used atherosclerotic tissue samples from a group of 15 patients that underwent elective carotid endarterectomy following repeated transient ischemic attacks or minor strokes (samples from symptomatic atherosclerotic plaques as cases).Footnote 2 Further, we have asymptomatic atherosclerotic plaques from seven persons who died from causes not related to atherosclerotic disease (samples from stable plaques as controls).Footnote 3
All 22 arterial plaque samples resulted in 2,610,268,774 shotgun sequencing reads. After mapping these reads against Hg19 using bowtie 2 [17] with “very-sensitive” parameters to filter all human-like sequences from our samples. The average amount of non-Hg19 reads is 884,727,044 (average 33.89% per sample, Table 1). These non-Hg19 reads were extracted and aligned against nonredundant (nr) protein database (version 30.07.2012) [18] using BLASTX (ncbi-blast-2.2.25+; Max e-value 10e−3) [19]. After performing the BLASTX alignment, all output files of paired read sequences were imported and analyzed using the paired-end protocol of MEGAN5 [20]. For all non-Hg19 annotated reads, 2–16% (mean 4.6%) were assigned as bacteria in different samples. The rest of reads were assigned to Eukaryota. Table 1 provides details of sequencing read statistics and assignments of reads after different stages of data processing. R statistical programming language [21] was used for multivariate statistics. Later in Subheading 3, we will describe few of the analysis approaches revisiting this study.
In this study our data provided evidence that suggest a wide range of microbial agents (some pathogens) in atherosclerotic plaques, and these microbes displayed differences between symptomatic and asymptomatic plaques as judged from the taxonomic profiles in these two groups of patients. Further, fluorescence in situ hybridization (FISH) was performed to validate the presence of biofilm-like structures of few pathogens (which have been previously predicted from taxonomic analyses) in the symptomatic atherosclerotic plague samples. FISH staining demonstrates the presence of live bacteria; thus, this is a very good approach for cross-validation of any computational finding in the lab.
There are also potentials of using this data for not only taxonomic annotation but also to reveal the functional profiles through partial assembly of specific members and their functional annotations. Functional annotations reveal significant differences in basic metabolic and disease pathway signatures between these groups. Here, we will not provide details of the whole study, but interested readers may refer to [15].
On this occasion, it is necessary to mention that in any similar project in future, for alignment purpose, we would have used DIAMOND [22] which uses improved algorithms and additional heuristics and works much faster compared to available other aligners. Scenario 1 is an example of analyzing shotgun sequence datasets obtained from tissue samples or host-associated metagenome. In case readers have shotgun sequence datasets from environmental samples or from fecal samples, they do not need to perform alignment step to get rid of the host-associated sequences, unless there is any doubt of contamination. Normally we suggest to have control or blank samples in two wells per 96-well plate to address any issue with contaminations.
2.2 Scenario 2: The Effect of Omega-3 Polyunsaturated Fatty Acid Supplements on the Human Intestinal Microbiota
2.2.1 Study Design
A randomized, open-label, crossover trial of 8 weeks’ treatment with 4 g mixed eicosapentaenoic acid (EPA)/docosahexaenoic acid (DHA) in two formulations (soft-gel capsules and drinks) with a 12-week “washout” period [16] is chosen. Healthy volunteers aged greater than 50 years of both genders were included in this study. Participants were randomized to take two types of EPA and DHA compositions (Fig. 2):
-
1.
Two 200 mL drinks per day (providing approximately as the triglyceride daily) at any suitable time of day, or
-
2.
Four soft-gel capsules (each containing 250 mg EPA and 250 mg DHA as the ethyl ester) twice daily with meals (providing 2000 mg EPA and 2000 mg DHA per day), both for 8 weeks.

Schedule of visits for the study to understand the effect of omega-3 polyunsaturated fatty acid supplements on the human intestinal microbiota
After a 12-week “washout” period, participants took the second intervention for 8 weeks. We also included a final study visit after a second 12-week “washout” period (V5; Fig. 2). Fecal samples were collected at five time-points for microbiome analysis by 16S rRNA PCR and Illumina MiSeq sequencing. Parallel red blood cell (RBC) fatty acid analysis was performed by liquid chromatography–tandem mass spectrometry.
2.2.2 Sample Preparation and Sequencing
Microbial DNA extractions were performed based on the method of Yu and Morrison, [23] with slight modifications. DNA was extracted from approximately 250 mg feces using the QIAamp DNA Stool Mini Kit (Qiagen, Germany) with bead beating. DNA Library Prep Kit for Illumina, NEBNext Singleplex Oligos for Illumina (New England Biolabs, UK), and unique in-house-designed index primers (Integrated DNA Technologies, UK) were used to allow for multiplexing of samples. Twelve cycles of enrichment PCR were performed, and final libraries were cleaned with AMPure Beads (Beckman Coulter, UK). Successful libraries were confirmed by DNA 1000 bioanalyzer chips or DNA Analysis screen tapes (Agilent, UK). Quantification was performed with the Quant-iT dsDNA Assay Kit, broad range. A total of 30 ng of each library was pooled and sequenced on an Illumina MiSeq (2 × 250 bp) [24]. The variable region (V4) of the 16S rRNA gene was sequenced for these samples.
2.2.3 Data Analyses
Demultiplexed FASTQ files were trimmed of adapter sequences using cutadapt [25]. Paired reads were merged using fastq-join [26] under default settings and then converted to FASTA format. Consensus sequences were removed if they contained any ambiguous base calls, two contiguous bases with a PHRED quality score lower than 33, or a length more than 2 bp different from the expected length of 240 bp. Further analysis was performed using QIIME [27]. Operational taxonomy units (OTUs) were picked using usearch [28] and aligned to the Greengenes reference database using PyNAST [29]. Taxonomy was assigned using the RDP 2.2 classifier [30]. The resulting OTU BIOM files from the above analyses were imported in MEGAN for detailed group-specific analyses, annotations, and plots [31]. R statistical programming language [21] was used for multivariate statistics and other plots.
This dataset and method pipeline are purely described as an example for similar analyses; thus, we will not explain the results here, but interested, readers may see [16]. Scenario 2 is a typical example of analyzing 16S sequence data. In Subheading 3, we will describe few of the analysis approaches using data from this study.
2.3 Scenario 3: Comparing Effects of Two Drug Treatments for an Infectious Disease
In a given situation suppose we need to compare treatment effect of two drugs (e.g., X and Y) or more, where we have time series data, that is, patient samples from multiple time points of the treatment course for both drugs. This time series data can be either collected every day of the treatment period or in intervals. Furthermore, for practical reasons we might not be able to obtain data at a desired day but ±1/2 days. It is important to select an error threshold and be consistent with that throughout the project. For example, we need to have a similar depth of sequencing reads or need to follow subsample comparison as detailed later, and, also, we need to discard samples with very low number of reads. Further during alignment to reference database and during mapping to taxonomy similar scores and thresholds should be used for all samples (please check best parameter selections in individual websites while using specific tools). Additionally, there can be multiple fundamental factors in patient samples such as age, gender, and geography that may not contribute in a similar manner to resiliency. Figure 3 shows a schematic of the metadata structure, which may help to understand the complexity of a typical clinical trial.

Schematic diagram of multiple factors in a clinical study
2.3.1 Sample Preparation and Sequencing and Data Analyses
In a clinically relevant setting this type of study wants to know which drug works better for a similar group of patients. Patients are randomized between drug arms to control any selection bias. Usually in this type of projects as we want to compare several factors, we need many samples to start with. Readers are advised to seek statistics help to do power calculation to obtain the preferred sample size. In general, as we end up having hundreds of samples, we usually go for 16S sequencing as a cost-effective solution. However, some projects can also use shotgun sequencing. Similar to previous examples, we assume that we have sequenced (either 16S or shotgun sequencing) our samples and performed further analysis process as outlined earlier to obtain taxonomic profile (following data analyses methods as described in previous scenarios) for each patient at each time point. Besides analyzing time series of each individual separately, we have also grouped them in certain time points such as baseline, mid-treatment, end of treatment, and follow-up. Besides treatment groups, patients are also compared based on multiple factors such as age, gender, and geography.
3 General Methods for Annotation and Statistical Analyses
Broadening our focus beyond these studies, additional analysis techniques are explained below which are used in these studies and also can be used in similar projects.
3.1 Taxonomic and Functional Annotation
Taxonomic annotation addresses the question, ‘Who is out there?’ or in other words tries to obtain information regarding the species composition of a given metagenome. On the other hand, functional annotation attempts to answer the question, ‘What are they doing?’ There are different approaches for metagenome analyses, among which one type of approach is to use phylogenetic markers to distinguish between different species in a sample. The most widely used marker is the small subunit ribosomal ribonucleic acid (SSU rRNA) gene (16S or 18S) and a second type of method is based on analyzing the nucleotide composition of reads. In a supervised approach the nucleotide composition of a collection of reference genomes is used to train a classifier, which is then used to place a given set of reads into taxonomic bins. In an unsupervised approach, reads are clustered by composition similarity and then the resulting clusters are analyzed in an attempt to place the reads. Subheading 4 of this chapter provide details of multiple approaches and available different tools which readers can use according to their preferences.
In general, for annotating 16S rRNA sequences we use QIIME [27] and for shotgun sequencing we use MEGAN [31] which can also be used for 16S. MEGAN is a highly efficient program for interactive analysis and comparison of microbiome data, allowing one to explore hundreds of samples and billions of reads. While taxonomic profiling is performed based on the NCBI taxonomy, MEGAN also provides a number of different functional profiling approaches. MEGAN Community Edition also supports the use of metadata in the context of principal coordinate analysis and clustering analysis [31]. In all the three scenarios explained in this chapter, MEGAN is used as primary tool for annotations. For more details on MEGAN tool, see Chapter 23 .
If we have shotgun sequencing then we have good option for functional annotation, but with 16S sequences we can only perform taxonomic analyses with confidence although there are few tools which might predict metagenome functional content from marker genes [32, 33]. Most shotgun annotation pipelines (such as MEGAN [31], MG-RAST [34], IMG/MER [35], EBI Metagenomics [36]) support functional annotations and they often use databases such as KEGG [37], SEED [38], eggNOG [39], and COG/KOG [40], as well as protein domain databases such as TIGRFAM [41] and PFAM [42].
3.2 Metagenome Assembly
Similar in nature to the genomic assembly, which is the reconstruction of genomes from the sequenced DNA segments (or reads), metagenome assembly is more complex. The main goal is to stitch together the fragments of the reads that could be from the same genome. Here the reads consist of mixture of DNA from different organisms and also may have widely different levels of abundance. Few recent reviews discussed new challenges and opportunities as well as assessed the most common and freely available metagenome assembly tools with respect to their output statistics, their sensitivity for low-abundance community members and variability in resulting community profiles as well as their ease of use. Interested readers please refer to reviews [43, 44].
3.3 Rarefaction Curves
Rarefaction curves represent a powerful method for comparing species richness among habitats on an equal-effort basis based on the construction of the so-called rarefaction curves [45]. This is a very useful tool for statistical data analyses that helps us to Correct for bias in species number due to unequal sample sizes by standardization to the number of species expected in a sample if it had the same total size as the smallest sample. As an example, we have two sample groups, first having 50 individuals and second 30 individuals with multiple number of species obtained from their taxonomic analyses. Rarefaction helps us to compare the situation, if we would have same number of individuals in two sample groups. Rarefaction curves are used differently in case of 16S and shotgun metagenomics. Ni and colleagues have described methods for estimating a reasonable and practical amount for SSU rRNA gene sequencing and explained how much metagenomic sequencing is enough to achieve a given goal [46]. In metagenomic shotgun sequencing, the fraction of the metagenome represented in the data set is termed coverage, which can be assessed through rarefaction curve. Interested readers may refer to a recent publication which has advocated for the estimation of the average coverage obtained in metagenomic studies, and briefly presented the advantages of different approaches [47].
In Scenario 1, for comparing case and control groups from human atherosclerotic plaque samples, we computed rarefaction curves from the normalized profile of 22 samples using the bacterial reads, showing the number of nodes that would be present in the analysis if based from 10% to 90% of the reads (Fig. 4). From sequence statistics (Table 1) and the rarefaction curve (Fig. 4), it is apparent that 2 (sample 233 and 238) of the 22 samples had much higher sequencing depth than the other samples. Later in the study we therefore omitted these two samples from merged case vs. control analyses.

Rarefaction. Rarefaction plot using annotated species profile for all 22 (unstable and stable) atherosclerotic plaque samples. These curves show the number of nodes that would be present if based on 10%, 20%, and up to 90% of the reads
Similarly, in Scenario 2 also, rarefaction was performed at various levels to compare diversity for different sample groupings. All groups were rarefied to the lowest read number, and the diversity calculated using weighted and unweighted UniFrac as well as the non-phylogenetic Bray–Curtis dissimilarity measure.
3.4 Subsample Comparison
In situations like Fig. 3, where two samples have much higher sequencing depth, another option can be subsample comparison. In this process without excluding high-depth samples from further study, another approach is to simulate subsample of lowest sample size (of other samples in the study) for sufficient number of times. And then take a median of the subsamples to generate a pseudo profile, which can serve as a good comparable sample for the group. For example, if in a study for most of the samples sequence reads are in a range of 200,000–300,000. However, only few samples have approx. 1 million reads, in those cases we simulate subsample of 200,000 reads from them for large number of times (say 1000) and we take median of the profiles, which we can then compare with other samples.
3.5 Comparative Visualization
Comparative visualization includes different types of plots and charts (pie charts, histograms, and many other kinds of plots) which can help us to draw basic conclusions regarding our data. For example, Fig. 5 depicts basic comparison of patients in two drug treatment groups for certain time points such as baseline, mid-treatment, end of treatment and follow up (from Scenario 3). Form this figure we can easily see that the microbiome pattern in drug X over treatment period is more consistent (or more stable over the time) than in drug Y. Here with visual comparison we are not making any conclusion, but with these types of plots we can start to see if there is any trend in our data, which can later be investigated with appropriate statistical tests.

Genus level taxonomic comparison of patients’ microbiome (median of each time point group) in two drug treatment groups for certain time points such as baseline, mid-treatment, end of treatment and follow up. Here different colors indicate different genera and the size of each color in the pie reflects the percentage of those genus in median microbiome for each time point group and for each drug
Further as metagenomic data are often hierarchical in nature, besides doing basic plots which can be done only at certain taxonomic levels (e.g., family/genus), often it is helpful to display the whole data as comparative tree view. For example in Scenario 1, samples from cases and controls have grouped closely (as can be seen later in Subheading 3.9), we can explore their broad differences by comparing total biome from cases and controls using comparative tree view (Fig. 6). This kind of tree view also help us to assess multiple time point samples from single patient or grouped data comparison for multiple factors (e.g., in Scenario 3).

Tree view at “family” level taxonomy comparing merged data from cases and control samples using data from Scenario 1
3.6 Diversity Analyses
Diversity analyses is one of the prominent statistical analysis approaches that address some of the downstream analysis steps associated with metagenomic studies. Species abundance estimates in the community are used to make inference about diversity on the whole community. The terms alpha, beta, and gamma diversity were all introduced by R. H. Whittaker to describe the spatial component of biodiversity [48]. Alpha diversity is just the diversity of each site (samples in each group). Beta diversity represents the differences in species composition among sites. Gamma diversity is the diversity of the entire landscape of different sites (all species pool from multiple samples). A diversity index measures how many different types (such as species) are there in a dataset (a community) and simultaneously takes into account how evenly the basic entities (such as individuals) are distributed among these types. Three commonly used measures of diversity, Simpson’s index, Shannon’s entropy, and the total number of species, are related to Renyi’s definition of a generalized entropy, and are well explained and compared by Hill [49]. Interested readers may also refer to [50] for consistent terminology for quantifying species diversity. Many other publications also explain this topic very well.
3.7 Comparison Using Distance Matrices
Another common technique to compare metagenomic datasets is using distance matrices. First, a taxonomic profile is computed for each data set. Second, a matrix of pairwise distances is determined using one of several possible ecological indices. Finally, the distances are represented using an appropriate visualization technique. Mitra et al. [51] explained multiple distance matrices (such as Bray–Curtis, Kulczynski, χ2, Hellinger, and Goodall) in the context of multiple metagenome comparison. In addition to these UniFrac is another distance metric used for comparing biological communities. It differs from dissimilarity measures such as Bray–Curtis by incorporating information on the relative relatedness of community members by incorporating phylogenetic distances between observed organisms in the computation [52,53,54]. Both weighted (quantitative) and unweighted (qualitative) variants of UniFrac are often used in microbial ecology, where the former accounts for abundance of observed organisms, while the latter only considers their presence or absence.
3.8 Boxplots
In descriptive statistics, “boxplot” or alternatively called “box and whisker plot,” is an important and one of the most informative tools that is used for graphically depicting groups of numerical data through their quartiles [55]. The boxplot is a quick way of examining multiple groups of data graphically, which easily provides information regarding quartiles, range, variation, and even outliers and enables us to compare within and between group samples. For example, Fig. 7 shows distribution of samples in multiple time point for both drugs (example data in Scenario 3). From this plot we can clearly gather the idea that diversity with drug X is consistently higher than that with drug Y. Further in Fig. 5 we have already seen that microbiome pattern in drug X showed less disruption, thus from these two figures we can hypothesize that drug Y being more disruptive to the microbiome. Such hypotheses can help us in further statistical analyses.

Boxplot showing Simpson diversity indices for samples from each time point and for both the drugs X and Y
3.9 Hierarchical Clustering
Cluster analysis, especially hierarchical clustering [56, 57], is an important tool for the exploratory and unsupervised analysis (where we do not need a training dataset to feed the programme) of high dimensional datasets and often used in genomics and other fields for their ability to simultaneously uncover multiple layers of clustering structure. In our example, Fig. 8 depicts a hierarchical clustering result of family level taxonomic comparison data for all 22 samples. Interestingly, samples 238 and P0613 were mostly different, and among the other samples, all unstable plaques clustered together, apart from all stable plaque controls that clustered separately.

Taxonomic comparison of all DNA samples. Hierarchical clustering result of “family” level taxonomic comparisons of data from Scenario 1: unstable atherosclerotic plaques from 15 patients with symptomatic atherosclerotic disease (unstable plaques) and stable plaques from a control group of seven patients that died from other causes than atherosclerosis (controls). Red indicates downregulation, green indicates upregulation, and black indicates no change in read abundance level comparing to all samples. Hierarchical clustering was computed with average linkage, whereas Pearson correlation was used for clustering the families (rows) and Spearman correlation was used for clustering the datasets (columns), respectively
Interestingly, the asymptomatic atherosclerotic plaques have more abundance of host microbiome-associated microbial families such as Porphyromonadaceae, Bacteroidaceae, Micrococcaceae, and Streptococcaceae than the symptomatic atherosclerotic plaques. In contrast, the symptomatic atherosclerotic plaques have more abundance of pathogenic microbial families such as Helicobacteraceae, Neisseriaceae, and sulfur-consuming families such as sulfur-oxidizing symbionts and Thiotrichaceae than the asymptomatic atherosclerotic plaques (Fig. 8). For P0613, the species profile appeared very different from all other samples. Thus, this sample also treated as an outlier in further analyses (see [15] if interested in actual study).
3.10 Principal Component Analysis (PCA) and Principal Coordinates Analysis (PCoA)
PCA and PCoA are tools for multivariate analysis. PCA uses an orthogonal transformation to convert a set of observations of possibly correlated variables into a set of values of linearly uncorrelated variables called principal components [58]. This is often used for quantitative variables, so the axes in graphic have a quantitative weight, and the positions of the samples are in relation with those weight. On the other hand, PCoA or multidimensional scaling (MDS) is a means of visualizing the level of similarity of individual cases of a dataset [59]. PCoA is similar to Polar ordination (PO; [60]) arranges samples between endpoints or ‘poles’ according to the distance matrix maximizing the linear correlation between the distances in the distance matrix. If further interested in these methods please see [61].
For multiple sample comparison we often use PCoA and PCA, these are among the best tools available for multivariate analysis. These can give us powerful information of similarities and dissimilarities within samples. When coupled with phenotypic data or metadata (using colors and symbols etc.), these can be very helpful tools to understand within group variations. As an example, we have used PCoA on 22 plaque samples from Scenario 1 (Fig. 9). Here we can see that sample 238 and 238 being very different possibly due to high sequence depth (as also seen in Fig. 4).

principal coordinate analyses (PCoA) of “family” level taxonomic comparisons of data from Scenario 1: unstable atherosclerotic plaques from 15 patients with symptomatic atherosclerotic disease (cases: cyan) and stable plaques from a control group of seven patients that died from other causes than atherosclerosis (controls: magenta)
Biplots: In addition to PCA or PCoA, variables can also be plotted on the same diagram (this is called a biplot). The biplot provides a useful tool of data analysis and allows the visual appraisal of the structure of large data matrices [62]. In our examples, where taxa are variables, biplot can show important taxa which helps in determining relatedness represented as arrows. For example, in Scenario 2, β diversity was compared using principal coordinate analysis (PCoA) on all samples from all visits, where biplots are displayed with green arrows (Fig. 10). From this PCoA with biplot, we interpret that samples from volunteers 8, 13, and 16 are different than the other volunteers and that they have higher abundance of Succinivibrionaceae, Gammaproteobacteria, Aeromonadales, etc.

principal coordinate analyses (PCoA) of level taxonomic comparisons of data from Scenario 2: all samples (V1–V5) for all participants, where biplots are displayed with green arrows. Each visit is denoted by a different color
3.11 Canonical-Correlation Analysis (CCA) and Canonical-Correspondence Analysis (CCA)
CCA (correlation) seeks to find the linear combination of the X i and Y j that have the greatest correlation with each other where X = (X 1, …, X n) and Y = (Y 1, …, Y m) of random variables thus it is often used as a dimension–reduction method. The method was first introduced by Harold Hotelling [63]. On the other hand, CCA (correspondence) is a multivariate method to elucidate the relationships between biological assemblages of species and their environment. This method by Cajo J. F. ter Braak involves a canonical correlation analysis and a direct gradient analysis [64]. By environment we mean any kind of metadata, such as some physicochemical parameters obtained from same group where the species data is obtained. The idea is to relate the prevalence of a set of species to a collection of environmental variables. Biplots are often used in CCA (correspondence) for visualization purpose. For example, in our Scenario 2, a typical illustration of correlation and correspondence analyses between the microbiome and RBC fatty acid data is displayed in Fig. 11.

(a) Pearson correlation between genus level microbiome and RBC fatty acid data. (b) Canonical correspondence analysis of microbiome (genus level taxonomy) distribution in relation to blood parameters (biplot: represented by blue arrows). Red crosses represent taxa and black circles represents individual samples
In this occasion it is important to note that CCA does not perform variable selection. Further, when the number of variables exceeds the number of observations (or sample size), CCA cannot be applied directly due to singularity of the covariance matrix. In a recent study [65] the authors have discussed this problem and a few existing solutions. Additionally, they developed a method for structure-constrained sparse canonical correlation analysis (ssCCA) in a high-dimensional setting. ssCCA takes into account the phylogenetic relationships among bacteria, which provides important prior knowledge on evolutionary relationships among bacterial taxa (see [65] if interested).
3.12 Multivariate Analyses
Multivariate data analysis refers to any statistical approach used to analyze data with more than one variable. For example, as described in Scenario 3 we have multiple factors. The key to identifying important microbial taxa associated with two treatments is that the large datasets from each patient are compared within groups, and then the metadata from the patients’ groups are compared against each other. Analysis of multivariate data in response to factors, groups, or treatments in an experimental design needs sophisticated methods.
To achieve this, we can use PERMANOVA (permutational multivariate analysis of variance) [66] to test the homogeneity of multivariate dispersions within groups, on the basis of any resemblance measure. PERMANOVA is a better approach than ANOVA (Analysis of variance)/MANOVA (Multivariate analysis of variance) for our study as PERMANOVA works with any distance measure that is appropriate to the data, and uses permutations to make it distribution free, unlike assuming normal distributions. Finally, in addition to the above multiple comparisons, we can examine if there is consistency of microbiota changes and patterns across the geographical locales of treatment subjects; as our samples are from different countries. We are not showing the details of multivariate analyses, but there are multiple available packages for such analyses with good tutorials. Interested readers may visit these packages and websites as detailed below.
The Primer-E package [67] is commonly used by microbial ecologists and allows for multiple multivariate statistical analyses. We often use R statistical programming language [21] for multivariate statistics. Moreover R is used for several types of graphical representations. Particular packages provide in-built functions and libraries (within R environment) specially for metagenomic datasets such as Bioconductor [68], vegan [69], and phyloseq [70].
4 Tools and Packages Commonly Used in Metagenomic Studies
A list of multiple tools is provided below for analyzing metagenomic data from raw sequence reads to final comparisons and statistical analyses. Discussion of all these tools are beyond the scope of this chapter, but interested readers can see recent review articles [71,72,73,74] and it must be noted that there can be other tools as well outside this list.
-
1.
Processing of raw sequence reads and quality control (QC):
-
(a)
FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/).
-
(b)
Fastx_toolkit (http://hannonlab.cshl.edu/fastx_toolkit/).
-
(c)
Cut-adapt (both adapter trimming and quality trim) [25].
-
(d)
BBTools (http://jgi.doe.gov/data-and-tools/bbtools/).
-
(e)
Condetri (Read trimmer for Illumina data) [75].
-
(f)
Trimmomatic (allows multiple threads) [76].
-
(g)
SolexaQA [77].
-
(h)
PRINSEQ [78].
-
(a)
-
2.
Alignment tool:
-
3.
Analyses for 16S projects: OTU clustering, picking, and taxonomic assignment.
-
4.
Assembly of shotgun metagenomics data.
-
(a)
Reference-based assembly.
-
MIRA 4 [84].
-
MetaAMOS (https://www.cbcb.umd.edu/software/metamos).
-
-
(b)
De novo assembly.
-
(c)
The next generation of assembly tools.
-
(a)
-
5.
Removing near-exact matches by maping to specific genomes.
-
(a)
Bowtie 2 [17].
-
(a)
-
6.
Binning tools for metagenomes.
-
(a)
Composition-based binning algorithms.
-
(b)
Similarity-based binning software include tools.
-
(c)
Unsupervised binning.
-
(a)
-
7.
Binning of metagenome contigs for reconstructing single genomes.
-
8.
Identification of genes within the reads/assembled contigs or “gene calling”.
-
9.
Predict for clustered regularly interspaced short palindromic repeats (CRISPRs).
-
10.
Annotation pipelines.
-
11.
Prediction of functional content from metagenomics.
-
12.
Statistical computing.
-
(a)
R [21].
-
(b)
Many other tools can be used for statistical analyses.
-
(a)
-
13.
Web service for the analysis of metagenomic data.
5 Concluding Remarks
This chapter has illustrated multiple data analyses and annotation techniques in metagenomic studies with three case studies. This is not a chapter about any new method development but a description of optimized pipelines using various available tools. With these example scenarios, the use of multiple pipelines has been demonstrated to analyze and interpret the data starting from very raw sequence to the final statistical outputs. Example scenarios describe some of the tools that we have used for analyzing the projects selected for demonstration, but besides these there are plenty of other available tools for metagenomics, most of which are listed in Subheading 4. This chapter does not provide the details of the tools or describe their pros and cons but this can be a good starting point for the readers to explore available options to analyze and interpret their datasets. From this chapter readers shall get an idea of current research projects in medical studies and multiple approaches used to analyze the data originating from these projects, although readers should keep in mind that this is not an exclusive list of possible pipelines for analyzing metagenomic samples. There might be other approaches as well. While step-by-step instructions of all the tools is beyond the scope of this chapter, the methods outline here might be useful to researchers to plan, analyze, and interpret their research projects successfully.
Notes
- 1.
- 2.
All methods and experimental manuals were approved by The National Committee on Health Research Ethics (Danish) and was granted by the Ethical Committee of the region of Copenhagen (H-3-2011-013).
- 3.
These samples originated from the tissue bank at the Department of Forensic Medicine (Approval No. 1501230).
References
Riesenfeld CS, Schloss PD, Handelsman J (2004) Metagenomics: genomic analysis of microbial communities. Annu Rev Genet 38:525–552
Clarridge JE (2004) Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev 17(4):840–862
Staden R (1979) A strategy of DNA sequencing employing computer programs. Nucleic Acids Res 6(7):2601–2610
Ranjan R, Rani A, Metwally A, McGee HS, Perkins DL (2016) Analysis of the microbiome: advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem Biophys Res Commun 469(4):967–977
Tessler M, Neumann JS, Afshinnekoo E, Pineda M, Hersch R, Velho LFM et al (2017) Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci Rep 7(1):6589
Jovel J, Patterson J, Wang W, Hotte N, O’Keefe S, Mitchel T et al (2016) Characterization of the gut microbiome using 16S or shotgun metagenomics. Front Microbiol 7:459
Greub G (2012) Culturomics: a new approach to study the human microbiome. Clin Microbiol Infect 18(12):1157–1159
Lagier JC, Khelaifia S, Alou MT, Ndongo S, Dione N, Hugon P et al (2016) Culture of previously uncultured members of the human gut microbiota by culturomics. Nat Microbiol 1(12):8
Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA et al (2009) The NIH human microbiome project. Genome Res 19(12):2317–2323
Virgin HW, Todd JA (2011) Metagenomics and personalized medicine. Cell 147(1):44–56
Wang J, Qi J, Zhao H, He S, Zhang Y, Wei S et al (2013) Metagenomic sequencing reveals microbiota and its functional potential associated with periodontal disease. Sci Rep 3:1843
Xu P, Gunsolley J (2014) Application of metagenomics in understanding oral health and disease. Virulence 5(3):424–432
Ai D, Huang R, Wen J, Li C, Zhu J, Xia LC (2017) Integrated metagenomic data analysis demonstrates that a loss of diversity in oral microbiota is associated with periodontitis. BMC Genomics 18(Suppl 1):1041
Forbes JD, Knox NC, Ronholm J, Pagotto F, Reimer A (2017) Metagenomics: the next culture-independent game changer. Front Microbiol 8:1069
Mitra S, Drautz-Moses DI, Alhede M, Maw MT, Liu Y, Purbojati RW et al (2015) In silico analyses of metagenomes from human atherosclerotic plaque samples. Microbiome 3:14
Watson H, Mitra S, Croden FC, Taylor M, Wood HM, Perry SL et al (2018) A randomised trial of the effect of omega-3 polyunsaturated fatty acid supplements on the human intestinal microbiota. Gut 67(11):1974–1983
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9(4):357–U54
Benson DA, Karsch-Mizrachi I, Clark K, Lipman DJ, Ostell J, Sayers EW (2012) GenBank. Nucleic Acids Res 40(D1):D48–D53
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215(3):403–410
Huson DH, Mitra S, Ruscheweyh HJ, Weber N, Schuster SC (2011) Integrative analysis of environmental sequences using MEGAN4. Genome Res 21(9):1552–1560
R Development Core Team (2008) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Buchfink B, Xie C, Huson DH (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12(1):59–60
Yu ZT, Morrison M (2004) Improved extraction of PCR-quality community DNA from digesta and fecal samples. BioTechniques 36(5):808–812
Taylor M, Wood HM, Halloran SP, Quirke P (2017) Examining the potential use and long-term stability of guaiac faecal occult blood test cards for microbial DNA 16S rRNA sequencing. J Clin Pathol 70(7):600–606
Martin M (2011) Cutadapt removes adapter sequences from high-throughput sequencing reads. Genomics 98(1):152–153
Aronesty E (2011) ea-utils: “Command-line tools for processing biological sequencing data”. https://github.com/ExpressionAnalysis/ea-utils
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK et al (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7(5):335–336
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26(19):2460–2461
Caporaso JG, Bittinger K, Bushman FD, DeSantis TZ, Andersen GL, Knight R (2010) PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26(2):266–267
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73(16):5261–5267
Huson DH, Beier S, Flade I, Gorska A, El-Hadidi M, Mitra S et al (2016) MEGAN community edition—interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput Biol 12(6):12
Aßhauer KP, Wemheuer B, Daniel R, Meinicke P (2015) Tax4Fun: predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 31(17):2882–2884
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA et al (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31(9):814–821
Wilke A, Bischof J, Gerlach W, Glass E, Harrison T, Keegan KP et al (2016) The MG-RAST metagenomics database and portal in 2015. Nucleic Acids Res 44(Database issue):D590–D5D4
Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Pillay M et al (2014) IMG 4 version of the integrated microbial genomes comparative analysis system. Nucleic Acids Res 42(Database issue):D560–D5D7
Hunter S, Corbett M, Denise H, Fraser M, Gonzalez-Beltran A, Hunter C et al (2014) EBI metagenomics—a new resource for the analysis and archiving of metagenomic data. Nucleic Acids Res 42(Database issue):D600–D6D6
Du JL, Yuan ZF, Ma ZW, Song JZ, Xie XL, Chen YL (2014) KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol BioSyst 10(9):2441–2447
Overbeek R, Begley T, Butler RM, Choudhuri JV, Chuang HY, Cohoon M et al (2005) The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res 33(17):5691–5702
Powell S, Forslund K, Szklarczyk D, Trachana K, Roth A, Huerta-Cepas J et al (2014) eggNOG v4.0: nested orthology inference across 3686 organisms. Nucleic Acids Res 42(D1):D231–D2D9
Tatusov RL, Galperin MY, Natale DA, Koonin EV (2000) The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res 28(1):33–36
Haft DH, Selengut JD, White O (2003) The TIGRFAMs database of protein families. Nucleic Acids Res 31(1):371–373
Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, Mitchell AL et al (2016) The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44(D1):D279–DD85
Vollmers J, Wiegand S, Kaster AK (2017) Comparing and evaluating Metagenome assembly tools from a microbiologist’s perspective—not only size matters! PLoS One 12(1):e0169662
Ghurye JS, Cepeda-Espinoza V, Pop M (2016) Metagenomic assembly: overview, challenges and applications. Yale J Biol Med 89(3):353–362
Bacaro G, Rocchini D, Ghisla A, Marcantonio M, Neteler M, Chiarucci A (2012) The spatial domain matters: spatially constrained species rarefaction in a free and open source environment. Ecol Complex 12:63–69
Ni J, Yan Q, Yu Y (2013) How much metagenomic sequencing is enough to achieve a given goal? Sci Rep 3:1968
Rodriguez RL, Konstantinidis KT (2014) Estimating coverage in metagenomic data sets and why it matters. ISME J 8(11):2349–2351
Whittaker RH (1972) Evolution and measurement of species diversity. Taxon 21(2/3):213–251
Hill MO (1973) Diversity and evenness: a unifying notation and its consequences. Ecology 54(2):427–432
Tuomisto H (2010) A consistent terminology for quantifying species diversity? Yes, it does exist. Oecologia 164(4):853–860
Mitra S, Gilbert JA, Field D, Huson DH (2010) Comparison of multiple metagenomes using phylogenetic networks based on ecological indices. ISME J 4(10):1236–1242
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71(12):8228–8235
Hamady M, Lozupone C, Knight R (2010) Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J 4(1):17–27
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R (2011) UniFrac: an effective distance metric for microbial community comparison. ISME J 5(2):169–172
McGill R, Tukey JW, Larsen WA (1978) Variations of box plots. Am Stat 32(1):12–16
Kaufman L, Rousseeuw PJ (1990) Finding groups in data: an introduction to cluster analysis, 1st edn. John Wiley, New York
Hastie T, Tibshirani R, Friedman J (2009) Hierarchical clustering. In: The elements of statistical learning, 2nd edn. Springer, New York
Pearson K (1901) On lines and planes of closest fit to systems of points in space. Philos Mag 2(7–12):559–572
Borg I, Groenen PJ (2005) Modern multidimensional scaling: theory and applications. Springer Science & Business Media, New York
Bray JR, Curtis JT (1957) An ordination of the upland forest communities of southern Wisconsin. Ecol Monogr 27(4):326–349
Michael PW Ordination methods—an overview. http://ordination.okstate.edu/overview.htm
Gabriel KR (1971) Biplot graphic display of matrices with application to principal component analysis. Biometrika 58(3):453–467
Hotelling H (1936) Relations between two sets of variates. Biometrika 28:321–377
Terbraak CJF (1986) Canonical correspondence-analysis—a new eigenvector technique for multivariate direct gradient analysis. Ecology 67(5):1167–1179
Chen J, Bushman FD, Lewis JD, Wu GD, Li H (2013) Structure-constrained sparse canonical correlation analysis with an application to microbiome data analysis. Biostatistics 14(2):244–258
Anderson MJ (2001) A new method for non-parametric multivariate analysis of variance. Austral Ecol 26(1):32–46
Clarke KG, Gorley RN (2006) PRIMER v6: user manual/tutorial. PRIMER-E, Plymouth
Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S et al (2004) Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5(10):16
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D, et al (2008) The vegan package. https://cran.r-project.org/web/packages/vegan/vegan.pdf
McMurdie PJ, Holmes S (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8(4):11
Teeling H, Glockner FO (2012) Current opportunities and challenges in microbial metagenome analysis-a bioinformatic perspective. Brief Bioinform 13(6):728–742
Oulas A, Pavloudi C, Polymenakou P, Pavlopoulos GA, Papanikolaou N, Kotoulas G et al (2015) Metagenomics: tools and insights for analyzing next-generation sequencing data derived from biodiversity studies. Bioinform Biol Insights 9:75–88
Pavlopoulos GA, Oulas A, Iacucci E, Sifrim A, Moreau Y, Schneider R et al (2013) Unraveling genomic variation from next generation sequencing data. BioData Min 6:13
Lindgreen S, Adair KL, Gardner PP (2016) An evaluation of the accuracy and speed of metagenome analysis tools. Sci Rep 6:19233
Smeds L, Künstner A (2011) ConDeTri—a content dependent read trimmer for Illumina data. PLoS One 6(10):e26314
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for illumina sequence data. Bioinformatics 30(15):2114–2120
Cox MP, Peterson DA, Biggs PJ (2010) SolexaQA: at-a-glance quality assessment of illumina second-generation sequencing data. BMC Bioinformatics 11:485
Schmieder R, Edwards R (2011) Quality control and preprocessing of metagenomic datasets. Bioinformatics 27(6):863–864
Ye Y, Choi J-H, Tang H (2011) RAPSearch: a fast protein similarity search tool for short reads. BMC Bioinformatics 12:159
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P et al (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41(D1):D590–D5D6
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB et al (2009) Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75(23):7537–7541
Quince C, Lanzen A, Davenport RJ, Turnbaugh PJ (2011) Removing noise from Pyrosequenced amplicons. BMC Bioinformatics 12:38
Schloss PD, Handelsman J (2008) A statistical toolbox for metagenomics: assessing functional diversity in microbial communities. BMC Bioinformatics 9:34
Chevreux B, Pfisterer T, Drescher B, Driesel AJ, Müller WEG, Wetter T et al (2004) Using the miraEST assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res 14(6):1147–1159
Zheng Y, Zhao LJ, Gao JP, Fei ZJ (2011) iAssembler: a package for de novo assembly of Roche-454/sanger transcriptome sequences. Bmc Bioinformatics 12:8
Pevzner PA, Tang H, Waterman MS (2001) An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci U S A 98(17):9748–9753
Zerbino DR, McEwen GK, Margulies EH, Birney E (2009) Pebble and rock band: heuristic resolution of repeats and scaffolding in the velvet short-read de novo assembler. PLoS One 4(12):9
Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J et al (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1:18
Jackman SD, Vandervalk BP, Mohamadi H, Chu J, Yeo S, Hammond SA et al (2017) ABySS 2.0: resource-efficient assembly of large genomes using a bloom filter. Genome Res 27(5):768–777
Afiahayati, Sato K, Sakakibara Y (2015) MetaVelvet-SL: an extension of the velvet assembler to a de novo metagenomic assembler utilizing supervised learning. DNA Res 22(1):69–77
Peng Y, Leung HCM, Yiu SM, Chin FYL (2011) Meta-IDBA: a de Novo assembler for metagenomic data. Bioinformatics 27(13):i94–i101
Lai B, Wang F, Wang X, Duan L, Zhu H (2015) InteMAP: integrated metagenomic assembly pipeline for NGS short reads. BMC Bioinformatics 16:244
Zhang Y, Sun Y, Cole JR (2014) A scalable and accurate targeted gene assembly tool (SAT-assembler) for next-generation sequencing data. PLoS Comput Biol 10(8):e1003737
Peng Y, Leung HCM, Yiu SM, Chin FYL (2012) IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28(11):1420–1428
Chan C-KK, Hsu AL, Tang S-L, Halgamuge SK (2008) Using growing self-Organising maps to improve the binning process in environmental whole-genome shotgun sequencing. J Biomed Biotechnol 2008:513701
Patil KR, Roune L, McHardy AC (2012) The PhyloPythiaS web server for taxonomic assignment of metagenome sequences. PLoS One 7(6):e38581
Diaz NN, Krause L, Goesmann A, Niehaus K, Nattkemper TW (2009) TACOA—taxonomic classification of environmental genomic fragments using a kernelized nearest neighbor approach. BMC Bioinformatics 10:56
Zheng H, Wu H (2010) Short prokaryotic DNA fragment binning using a hierarchical classifier based on linear discriminant analysis and principal component analysis. J Bioinforma Comput Biol 8(6):995–1011
Pati A, Heath LS, Kyrpides NC, Ivanova N (2011) ClaMS: a classifier for metagenomic sequences. Stand Genomic Sci 5(2):248–253
Krause L, Diaz NN, Goesmann A, Kelley S, Nattkemper TW, Rohwer F et al (2008) Phylogenetic classification of short environmental DNA fragments. Nucleic Acids Res 36(7):2230–2239
Liu B, Gibbons T, Ghodsi M, Treangen T, Pop M (2011) Accurate and fast estimation of taxonomic profiles from metagenomic shotgun sequences. BMC Genomics 12(2):S4
Gregor I, Dröge J, Schirmer M, Quince C, McHardy AC (2016) PhyloPythiaS+: a self-training method for the rapid reconstruction of low-ranking taxonomic bins from metagenomes. PeerJ 4:e1603
Brady A, Salzberg S (2011) PhymmBL expanded: confidence scores, custom databases, parallelization and more. Nat Methods 8(5):367
Dick GJ, Andersson AF, Baker BJ, Simmons SL, Thomas BC, Yelton AP et al (2009) Community-wide analysis of microbial genome sequence signatures. Genome Biol 10(8):R85
Laczny CC, Sternal T, Plugaru V, Gawron P, Atashpendar A, Margossian HH et al (2015) VizBin—an application for reference-independent visualization and human-augmented binning of metagenomic data. Microbiome 3:1
Liu Y, Hou T, Kang B, Liu F (2017) Unsupervised binning of metagenomic assembled Contigs using improved fuzzy C-means method. IEEE/ACM Trans Comput Biol Bioinform 14(6):1459–1467
Broeksema B, Calusinska M, McGee F, Winter K, Bongiovanni F, Goux X et al (2017) ICoVeR—an interactive visualization tool for verification and refinement of metagenomic bins. BMC Bioinformatics 18:233
Lin H-H, Liao Y-C (2016) Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci Rep 6:24175
Kang DD, Froula J, Egan R, Wang Z (2015) MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 3:e1165
Imelfort M, Parks D, Woodcroft BJ, Dennis P, Hugenholtz P, Tyson GW (2014) GroopM: an automated tool for the recovery of population genomes from related metagenomes. PeerJ 2:e603
Wu Y-W, Tang Y-H, Tringe SG, Simmons BA, Singer SW (2014) MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2:26
Alneberg J, Bjarnason BS, de Bruijn I, Schirmer M, Quick J, Ijaz UZ et al (2014) Binning metagenomic contigs by coverage and composition. Nat Methods 11(11):1144–1146
Zhu W, Lomsadze A, Borodovsky M (2010) Ab initio gene identification in metagenomic sequences. Nucleic Acids Res 38(12):e132
Hyatt D, Chen G-L, LoCascio PF, Land ML, Larimer FW, Hauser LJ (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119
Hoff KJ, Lingner T, Meinicke P, Tech M (2009) Orphelia: predicting genes in metagenomic sequencing reads. Nucleic Acids Res 37(Web Server issue):W101–W1W5
Rho M, Tang H, Ye Y (2010) FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res 38(20):e191
Bland C, Ramsey TL, Sabree F, Lowe M, Brown K, Kyrpides NC et al (2007) CRISPR recognition tool (CRT): a tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinformatics 8:209
Edgar RC (2007) PILER-CR: fast and accurate identification of CRISPR repeats. BMC Bioinformatics 8:18
Arndt D, Xia J, Liu Y, Zhou Y, Guo AC, Cruz JA et al (2012) METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res 40(Web Server issue):W88–W95
Laczny CC, Kiefer C, Galata V, Fehlmann T, Backes C, Keller A (2017) BusyBee web: metagenomic data analysis by bootstrapped supervised binning and annotation. Nucleic Acids Res 45(W1):W171–W1W9
Richardson EJ, Escalettes F, Fotheringham I, Wallace RJ, Watson M (2013) Meta4: a web application for sharing and annotating metagenomic gene predictions using web services. Front Genet 4:168
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Open Access This chapter is licensed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license and indicate if changes were made.
The images or other third party material in this chapter are included in the chapter's Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the chapter's Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
Copyright information
© 2019 The Author(s)
About this protocol

Cite this protocol
Mitra, S. (2019). Multiple Data Analyses and Statistical Approaches for Analyzing Data from Metagenomic Studies and Clinical Trials. In: Anisimova, M. (eds) Evolutionary Genomics. Methods in Molecular Biology, vol 1910. Humana, New York, NY. https://doi.org/10.1007/978-1-4939-9074-0_20
Download citation
DOI: https://doi.org/10.1007/978-1-4939-9074-0_20
Published:
Publisher Name: Humana, New York, NY
Print ISBN: 978-1-4939-9073-3
Online ISBN: 978-1-4939-9074-0
eBook Packages: Springer Protocols