
Abstract
Elucidating the content of a DNA sequence is critical to deeper understand and decode the genetic information for any biological system. As next generation sequencing (NGS) techniques have become cheaper and more advanced in throughput over time, great innovations and breakthrough conclusions have been generated in various biological areas. Few of these areas, which get shaped by the new technological advances, involve evolution of species, microbial mapping, population genetics, genome-wide association studies (GWAs), comparative genomics, variant analysis, gene expression, gene regulation, epigenetics and personalized medicine. While NGS techniques stand as key players in modern biological research, the analysis and the interpretation of the vast amount of data that gets produced is a not an easy or a trivial task and still remains a great challenge in the field of bioinformatics. Therefore, efficient tools to cope with information overload, tackle the high complexity and provide meaningful visualizations to make the knowledge extraction easier are essential. In this article, we briefly refer to the sequencing methodologies and the available equipment to serve these analyses and we describe the data formats of the files which get produced by them. We conclude with a thorough review of tools developed to efficiently store, analyze and visualize such data with emphasis in structural variation analysis and comparative genomics. We finally comment on their functionality, strengths and weaknesses and we discuss how future applications could further develop in this field.
Similar content being viewed by others
Introduction
High throughput sequencing (NGS) techniques have brought a remarkable revolution in the field of biology and other closely related fields and have shaped a new trend of how modern biological research can be done at a large scale level. With the advances of these techniques, it is feasible nowadays to scan and sequence a whole genome or exome at a base pair level at a low error rate, in an acceptable time frame and at a lower cost.
Based on the first Sanger sequencing technique, the Human Genome Project (1990–2003), allowed the release of the first human reference genome by determining the sequence of ~3 billion base pairs and identifying the approximately ~25,000 human genes [1–3]. That stood as a great breakthrough in the field of comparative genomics and genetics as one could in theory directly compare any healthy or non-healthy sample against a golden standard reference and detect genetic polymorphisms or variants that occur in a genome. Few years later, as sequencing techniques became more advanced, more accurate and less expensive, the 1000 Human Genome Project[4] was launched (January 2008). The main scope of this consortium is to sequence, ~1000 anonymous participants of different nationalities and concurrently compare these sequences to each other in order to better understand human genetic variation. Recently, as a result of the project, 1092 such human genomes were sequenced and published [5]. The International HapMap Project (short for “haplotype map”) [6–10] aims to identify common genetic variations among people and is currently making use of data from six different countries.
Shortly after the 1000 Human Genome Project, the 1000 Plant Genome Project (http://www.onekp.com) was launched, aiming to sequence and define the transcriptome of ~1000 plant species from different populations around the world. Notably, out of the 370,000 green plants that are known today, only ~125,000 species have recorded gene entries in GenBank and many others still remain unclassified [11]. While the 1000 Plant Genome Project was focused on comparing different plant species around the world, within the 1001 Genomes Project[12], 1000 whole genomes of A. Thaliana plants across different places of the planet were sequenced.
Similar to other consortiums, the 10,000 Genome Project[13] aims to create a collection of tissue and DNA specimens for 10,000 vertebrate species specifically designated for whole-genome sequencing. In addition, the overarching goal of the 1000 Fungal Genome Project (F1000 - http://1000.fungalgenomes.org) is to explore all areas of fungal biology by providing broad, genomic coverage of Kingdom Fungi. Notably, sequencing advances have paved the way to metagenome sequencing, which is defined as an approach for the study of microbial populations in a sample representing a community by analysing the nucleotide sequence content. Moreover, NGS will allow for the accurate detection of pan-genomes which describe the full complement of a superset of all the genes in all the strains of a species, typically applied to bacteria and archaea [14].
In the near future, sequencing techniques are expected to become even less time-consuming and more cost-effective in order to screen whole genomes within a few hours or even minutes. While sequencing techniques improve and develop overtime, the amount of data produced increases exponentially and therefore the implementation of efficient platforms to analyze and visualize such large amounts of data in fast and efficient ways has become a necessity. Following a top-down approach, the current review starts with an overview of generic visualization and analysis tools and file formats that can be used in any next generation sequencing analysis. While such tools are of a broad usage, the current review progressively focuses on their application in structural variation detection and representation and in parallel, commenting on their strengths and weaknesses, giving insights on how they could further develop to handle the overload of information and cope with the data complexity. It is not the scope of this article to describe in depth the existing sequencing techniques, but readers are strongly encouraged to follow a more detailed description about the widely used sequencing technologies in [15, 16]. Thorough explanations of how hundreds of thousands or even millions of sequences can be generated by such high-throughput techniques is presented in [17, 18] while sequence assembly strategies are extensively explained in [19]. The advantages and the limitations of the aforementioned techniques are discussed in [20, 21].
Sequencing technologies
First, second and third generation
Sequencing techniques are chronologically divided into 3 generations: the first, the second and the third. The key principle of the first generation (Sanger or dideoxy) sequencing techniques, which was discovered in 1977, was the use of dideoxy nucleotide triphosphates (ddNTPs) as DNA chain terminators so that the labeled fragments could be separated by size using gel electrophoresis. Dye-terminator sequencing discovered in the late 90s, utilizes labeling in a single reaction, instead of four reactions (A,T,C,G). In dye-terminator sequencing, each of the four ddNTPs is labeled with fluorescent dyes, each of which emits light at different wavelengths. Dye-terminator sequencing combined with capillary electrophoresis succeeded in speeding up performance and became one of the most standardized and widely used techniques.
Second generation high-throughput sequencing techniques generate thousands or millions of short sequences (reads) at higher speed and better accuracy. Such sequencing approaches can immediately be applied in relevant medical areas where previous Sanger-based trials fell short in capturing the desired sequencing depth in a manageable time-scale [22]. High-throughput second generation commercial technologies have already been developed by Illumina [23, 24], Roche 454 [25] and Biosystems/SOLiD. Today Illumina is the most widely used platform despite its lower multiplexing capability of samples allowed [26]. Recent HiSeq Illumina systems make it possible for researchers to perform large and complex sequencing studies at a lower cost. Cutting-edge innovations can dramatically increase the number of reads, sequence output and data generation rate. Thus, researchers are now able to sequence more than five human genomes at ~30x coverage simultaneously or ~100 exome samples in a single run.
Helicos BioSciences (http://www.helicosbio.com/), Pacific Biosciences (http://www.pacificbiosciences.com/), Oxford Nanopore (http://www.nanoporetech.com/) and Complete Genomics (http://www.completegenomics.com/) belong to the third generation of sequencing techniques, each of which have their pros and cons [16, 27, 28]. These techniques are promising to sequence a human genome at a very low cost within a matter of hours.
While today, first generation sequencing is not used due to its forbidden cost and time consumption, second generation sequencing technologies are widely used due to their lower cost and time efficiency. Such techniques have led to a plethora of applications such as DNA-seq and assembly to determine an unknown genome from scratch or look for variations among genome samples, RNA-seq [29, 30] to analyse gene expression or ChIP-seq [31] to mainly identify DNA regions that are binding sites for proteins, such as transcription factors. It is not the scope of this review to describe the aforementioned techniques into depth but we give a short description of DNA sequencing and assembly and we explain below how this can be used to discover structural variations.
DNA sequencing and assembly
DNA sequencing can be applied to very long pieces of DNA such as whole chromosomes or whole genomes, but also on targeted regions such as the exome or a selection of genes pulled-down from assays or in solution. There are two different scenarios under which DNA sequencing is carried out. In the first case a reference genome for the organism of interest already exists, whereas in the second case of de novo sequencing, there is no reference sequence available. The main idea behind the reference genome approach consists of 3 general steps: Firstly, DNA molecules are broken down into smaller fragments at random positions by using restriction enzymes or mechanical forces. Secondly, a sequencing library consisting of such fragments of known insert size is created, while during a third step, these fragments are sequenced and finally mapped back to an already known reference sequence. The general methodology is widely known as shotgun sequencing. The aforementioned process is depicted in Figure 1. In the case of de novo sequencing, where there is no a priory catalogued reference sequence for the given organism, the small sequenced fragments are assembled into contigs (groups of overlapping, contiguous fragments) and the consensus sequence is finally established from these contigs. This process is often compared to putting together the pieces of a jigsaw puzzle. Thus, the short DNA fragments produced are assembled electronically into one long and contiguous sequence. No prior knowledge about the original sequence is needed. While short read technologies produce higher coverage, longer reads are easier to process computationally and interpret analytically, as they are faster to align compared to short reads because they have higher significant probabilities to align to unique locations on a genome. Notable tools for sequence assembly are the: Celera [32], Atlas [33], Arachne [34], JAZZ [35], PCAP [36], ABySS [37], Velvet [38] and Phusion [39]. The accuracy of this approach increases when comparing larger sized fragments (resulting in larger overlaps) of less repetitive DNA molecules. For larger genomes, this method has many limitations mainly due to the smaller size of reads and its high cost. The aforementioned process is displayed in Figure 2.

DNA sequencing. DNA sequencing: 1st step: The DNA of interest is purified and extracted. 2nd step: Creation of multiple copies of DNA. 3nd step: DNA is shattered into smaller pieces. 4rd step: DNA fragment sequencing. 5th step: A computer maps the small pieces to an already known reference genome.

DNA assembly. DNA assembly: 1st step: The DNA is purified and extracted. 2nd step: DNA is fragmented into smaller pieces. 3rd step: DNA fragment sequencing. 4th step: A computer matches the overlapping parts of the fragments to get a continuous sequence. 5th step: The whole sequence is reassembled. No prior knowledge about the DNA sequence is necessary.
The structural variome
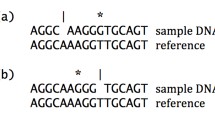
A single nucleotide polymorphism (SNP), or equally a single nucleotide variation (SNV), refers to a single nucleotide change (adenine-A, thymine-T, guanine-G, and cytosine-C) in genomic DNA which is observed between members of the same biological species or paired chromosomes in a single individual. A SNP example is shown in Figure 3. SNPs are single nucleotide substitutions, which are mainly divided into two types: transitions (interchanges of two purines or two pyrimidines such as A-G or C-T) and transversions (interchanges between purines and pyrimidines A-T, A-C, G-T and G-C). There are multiple public databases which store information about SNPs. The National Center for Biotechnology Information (NCBI) has released dbSNP [40], a public archive for genetic variation within and across different species. The Human Gene Mutation Database (HGMD) [41] holds information about gene mutations associated with human inherited diseases and functional SNPs. The International HapMap Project (short for “haplotype map”) [6–10] holds information about genetic variations among people, so far from containing data from six countries. The data includes haplotypes (several SNPs that cluster together on a chromosome), their locations in the genome and their frequencies in different populations throughout the world. Other databases to be mentioned are the HGBASE [42], HGVbase [43], GWAS Central [44] and SNPedia [45]. A great variety of tools to detect SNVs and predict their impact is analytically reviewed in [46].

SNP example. A difference in a single nucleotide between two DNA fragments from different individuals. In this case we say that there are two alleles: C and T.
Recently, the focus has been shifted to understanding genetic differences in the form of short sequence fragments or structural rearrangements (rather than variation at the single nucleotide level). This type of variation is known as the structural variome. The structural variome refers to the set of structural genetic variations in populations of a single species that have been acquired in a relatively short time on an evolutionary scale. Structural variations are mainly separated in two categories; namely the balanced and the unbalanced variations. The basic variations include insertions, deletions, duplications, translocations and inversions. Balanced variations refer to genome rearrangements, which do not change the total content of the DNA. These are mainly inversions or intra/inter-chromosomal translocations. Unbalanced variations on the other hand, refer to rearrangements that change the total DNA content. These are insertions and deletions. Unbalanced variations are also called copy number variations (CNVs). Figure 4 shows a schematic representation of such intra/inter-chromosomal balanced and unbalanced structural variations.

Structural Variations. This figure illustrates the basic structural variations. A) Inversion. B) Translocation within the same chromosome. C) Translocation across different chromosomes. D) Duplication. E) Deletion.
Methods to detect structural variations
During the past years, a great effort has been made towards the development of several techniques [47] and software applications [46] to detect structural variations in genomes. In the case of SNP detection, the differences are extracted from local alignments whereas for the detection of structural variations approaches, such as read-pair (RP), read-depth (RD) and split-reads can be used.
Pair-end mapping (PEM)
According to this approach, the DNA is initially fragmented into smaller pieces. The two ends of each DNA fragment (paired end reads or mate pairs) are then sequenced and finally get mapped back to the reference sequence. Notably, the two ends of each read are long enough to allow for unique mapping back to the reference genome. The idea behind this strategy is that the ends of the reads, which align back to the reference genome, map back to specific positions of an expected distance according to information from stored DNA libraries. For certain cases, the mapping distance appears to be different from the expected length, or mapping displays an alternative orientation from that anticipated. These observations can be considered as strong indicators for the occurrence of a possible structural variation. Thus, if the mapped distance is smaller than the expected one, it could indicate a deletion or vice versa an insertion. The main difference between the terms paired end reads and mate pairs, is that while pair-end reads provide tighter insert sizes, the mate pairs give the advantage of larger insert sizes [47]. Differences and structural variations among genomes can be tracked by observing PEM signatures. While PEM signatures together with approaches to detect them are analytically described elsewhere [47], some common signatures are shown in Figure 5.

PEM signatures. Basic PEM signatures. A) Insertion. B) Deletion. C) Inversion. More PEM signatures are visually presented in [47].
Single-end
According to this methodology, multiple copies of a DNA molecule get produced and randomly chopped into smaller fragments (reads). These reads are eventually aligned and mapped back to a reference genome. The reasoning behind this approach is that various reads will map back to various positions across the genome, and exhibit significant overlap of read mapping. By measuring the frequency of nucleotides mapped by the reads across the depth of coverage (DOC), it is possible to obtain an evaluation of the number of reads that have been mapped to a specific genomic position (see Figure 6). The Depth of coverage (DOC) is a significant way to detect insertions or deletions, gains or losses in a donor sample comparing to the reference genome. Thus, a region that has been deleted will have less reads mapped to it, and vice versa in cases of insertions. Similarly to PEM, the aforementioned methodology is an alternative way to extract information about possible structural variations described by DOC signatures. While read-depth has a higher resolution, it gives no information about the location of the variation and it can only detect unbalanced variations. DOC signatures, compared to PEM signatures are more suitable to detect larger events, since the stronger the event, the stronger the signal of the signature. On the other hand, PEM signatures are more suitable to detect smaller events, even with low coverage, but are far less efficient in localizing breakpoints. Available tools to detect structural variations and cluster them according to different methodologies are presented below.

Read depth. Read depth: A) Fragments of DNA (Reads) are mapped to the original reference genome. B) Plotting the frequency of each nucleotide that was mapped at the reference genome.
Split-reads
According to this approach, a read is mapped to two separate locations because of possible structural variation. The prefix and the suffix of a match may be interrupted by a longer gap. This split read mapping strategy is useful for small to medium-sized rearrangements in a base pair level resolution. It is suitable for mRNA sequencing, where absent intronic arrangements can cause junction reads that span exon-exon boundaries. Often, local assembly is used to detect regions of micro-homology or non-template sequences around a breakpoint. This is done to detect the actual sequence around the break points.
File formats
Sequencing techniques generate vast amounts of data that need to be efficiently stored, parsed and analyzed. A typical sequencing experiment might produce files ranging from few gigabytes to terabytes in size, containing thousands or millions of reads together with additional information such as read identifiers, descriptions, annotations, other meta-data, etc. Therefore, file formats such as FASTQ [48], SAM/BAM [49] or VCF [50] have been introduced to efficiently store such information.
FASTQ
It comes as a simple extension of the FASTA format and it is widely used in DNA sequencing mainly due to its simplicity. Its main strength is its ability to store a numeric quality score (PHRED [51]) for every nucleotide in a sequence. FASTQ mainly consists of four lines. The first line starts with the symbol ‘@’ which is followed by the sequence identifier. The second line contains the whole sequence as a series of nucleotides in uppercase. Tabs or spaces are not permitted. The third line starts with the ‘+’ symbol which indicates the end of the sequence and the start of the quality string which follows in the 4th line. Often, the third line contains a repetition of the same identifier like in line 1. The quality string, which is shown in the 4th line, uses a subset of the ASCII printable character representation. Each character of the quality string corresponds to one nucleotide of the sequence; thus the two strings should have the same length. Encoding quality scores in ASCII format, makes FASTQ format easier to be edited. The range of printed ASCII characters to represent quality scores varies between different technologies. Sanger format accepts a PHRED quality score from 0 to 93 using ASCII 33 to 126. Illumina 1.0 encodes a Illumina quality score from −5 to 62 using ASCII 59 to 126. Illumina 1.3+ format can encode a PHRED quality score from 0 to 62 using ASCII 64 to 126. Using different ranges for every technology is often confusing, and therefore the Sanger version of the FASTQ format has found the broadest acceptance. Quality scores and how they are calculated per platform is described in [52]. A typical FASTQ file is shown in Figure 7. Compression algorithms such as [53] and [54] succeed in storing FASTQ using lower disk space. In order to interconvert files between Sanger, Illumina 1.3+ platforms, Biopython [55], EMBOSS, BioPerl [56] and BioRuby [57] come with file conversion modules.

FASTQ file. 1st line always starts with the symbol ‘@’ followed by the sequence identifier. 2nd line contains the sequence. 3rd line starts with the symbol ‘+’ symbol which is optionally followed by the same sequence identifier and any description. It indicates the end of the sequence and the beginning of the quality score string. 4th line contains the quality score (QS) in ASCII format. The current example shows an Illumina representation.
Sequence alignment/Map (SAM) format
It describes a flexible and a generic way to store information about alignments against a reference sequence. It supports both short and long reads produced by different sequencing platforms. It is compact in size, efficient in random access and represents the format, which was mostly used by the 1000 Genomes Project to release alignments. It mainly supports 11 mandatory and many other optional fields. For better performance, store efficiency and intensive data processing, the BAM file, a binary representation of SAM, was implemented. BAM files are compressed in the BGZF format and hold the same information as SAM, while they require less disk space. SAM can be indexed and processed by specific tools. While Figure 8 shows an example of a SAM file, a very detailed description of the SAM and BAM files is presented in [58].

BAM/SAM files. Example of an alignment to the reference sequence (pileup). A) Read r001/1 and r001/2 constitute a read pair; r003 is a chimeric read; r004 represents a split alignment. B) The corresponding SAM file and their tags for each field.
Variant call format (VCF)
This specific file type was initially introduced by the 1000 Genomes Project to store the most prevalent types of sequence variation, such as SNPs and small indels (inserions/deletions) enriched by annotations. VCFtools [50] are equipped with numerous functionalities to process VCF files. Such functionalities include validations, merges and comparisons. An example of a VCF file is shown in Figure 9.

VCF file. This figure demonstrates an example of a CVF file. A) Different types of variations and polymorphisms that can be stored in CVF format. B) Example of a CVF format and its fields.
Variant calling pipelines
Variant discovery still remains a major challenge for sequencing experiments. Bioinformatics approaches that aim to detect variations across different human genomes, have identified 3–5 million variations for each individual compared to the reference. It is noticeable that most of the current comparative sequencing-based studies are mainly targeting the exome and not the whole genome, initially due to the lower cost. It is believed that variations in the exome can have a higher chance of having a functional impact in human diseases [59]. However, recent studies show that non-coding regions contain equally important disease related information [60]. Sophisticated tools that can cope with the large data size, efficiently analyze a whole genome or an exome and accurately detect genomic variations such as deletions, insertions, inversions or inter/intra chromosomal translocation are currently necessary. Today, only few of such tools exist and are summarized in Table 1. Many of the tools are error sensitive, as false negatives in base calling may lead to the identification of non-existent variants or to missing true variants in the sample, something that still remains a bottleneck in the field.
Variant annotation
As genetic diseases can be caused by a variety of different possible mutations in DNA sequences, the detection of genetic variations that are associated to a specific disease of interest is very important. Even though most of the variations detected by variant callers are found to be functionally neutral [74] and do not contribute to the phenotype of a disease [75], many of them have concluded to important results. In order to better identify the causative variations for genetic disorders and characterize them, the implementation of efficient variant annotation tools emerges and is one of the most challenging aspects of the field. Table 2 summarizes the available software which serves this purpose by highlighting the strengths and the weaknesses of each application.
Visualization of structural variation
Visualization of high throughput data to provide meaningful views and make pattern extraction easier still remains a bottleneck in systems biology. More than ever, such applications represent a precious tool for biologists in order to allow them to directly visualize large scale data generated by sequencing. The vast amounts of data produced by deep sequencing can be impossible to analyze and visualize due to high storage, memory and screen size requirements. Therefore, the field of biological data visualization is an ever-expanding field that is required to address new tasks in order to cope with the increasing complexity of information. While a recent review [87] discusses the perspectives and the challenges of visualization approaches in sequencing, the tables below emphasize on the strengths and the weaknesses of the available tools respectively.
Alignment tools
Aligning sequences of long length is not a trivial task. Therefore, efficient tools able to handle this load of data and provide intuitive layouts using linear or alternative representations i.e. circular are of importance. Table 3 shows a list of the widely used applications while also providing an overview of the strengths and weaknesses of each tool.
Genome browsers
Genome browsers are mainly developed to display sequencing data and genome annotations from various data sources in one common graphical interface. Initially genome browsers were mainly developed to display assemblies of smaller genomes of specific organisms, but with the latest rapid technological innovations and sequencing improvements, it is essential today to be able to navigate through sequences of huge length, and simultaneously browse for genomic annotations and other known sources of information available for these sequences. While recent studies [94–96] try to review the overlaps and comment on the future of genome browsers, we focus on the most widely used ones and we comment on their usability and their strengths as shown in Table 4.
Visualization for comparative genomics
Comparative genomics is expected to be one of the main challenges of the next decade in bioinformatics research, mainly due to sequencing innovations that currently allow sequencing of whole genomes at a lower cost and a reasonable timeframe. Microbial studies, evolutionary studies and medical approaches already take advantage of such methods to compare sequences of patients against controls, newly discovered species with other closely related species and identifying the presence of specific species in a population. Therefore, a great deal of effort has been made to develop algorithms that are able to cope with multiple, pairwise and local alignments of complete genomes. Alignment of unfinished genomes, intra/inter chromosome rearrangements and identification of functional elements are some important tasks that are amenable to analysis by comparative genomics approaches. Visualization of such information is essential to obtain new knowledge and reveal patterns that can only be perceived by the human eye. In this section we present a list of lately developed software applications that aim to address all of the aforementioned tasks and we emphasize on their main functionality, their strengths and their weaknesses (see Table 5).
Discussion
Advances in high throughput next generation sequencing techniques allow the production of vast amounts of data in different formats that currently cannot be analyzed in a non-automated way. Visualization approaches are today called upon to handle huge amounts of data, efficiently analyze them and deliver the knowledge to the user in a visual way that is concise, coherent and easy to comprehend and interpret. User friendliness, pattern recognition and knowledge extraction are the main goals that an optimal visualization tool should achieve. Therefore, tasks like handling the overload of information, displaying data at different resolutions, fast searching or smoother scaling and navigation are not trivial when the information to be visualized consists of millions of elements and often reaches an enormous level of complexity. Modern libraries, able to visually scale millions of data points at different resolutions are nowadays essential.
Current tools lack dynamic data structure and dynamic indexing support for better processing performance. Multi-threaded programming or parallel processing support would also be a very intuitive approach to reduce the processing time, when applications run in multicore machines with many CPUs. Efficient architecture setup, that would decentralize data and distribute the work between the servers and the clients, is also a step towards the reduction of processing time.
While knowledge is currently stored in various databases, distributed across the world and analyzed by various workflows, the need of integration among available tools is becoming a necessity. Next-generation visualization tools should be able to extract, combine and analyze knowledge and deliver it in a meaningful and sensible way. For this to happen, international standards should be defined to describe how next generation sequencing techniques should store their results and exchange them through the web. Unfortunately today, many visualization and analysis approaches are being developed independently. Many of the new methods come with their own convenient file format to store and present the information, something that will become a problem in the future when hundreds of methods will become available. Such cases are widely discussed in biological network analysis approaches [124, 125].
Visual analytics in the future will play an important role to visually allow parameterizations of various workflows. So far it may be confusing and misleading to the user, when various software packages often produce significantly different results just by slightly changing the value of a single parameter. Furthermore, different approaches can come up with completely different results despite the fact that they try to answer the same question. This can be attributed to the fact that they follow a completely different methodology, therefore highlighting the need for enforcing a more general output format. Future visualization tools should offer the flexibility to easily integrate and perform fine-tuning of parameters in such a way that it allows the end users to readily adjust their research to their needs.
Finally, data integration at different levels varying from tools to concepts is a necessity. Combining functions from diverse sources varying from annotations to microarrays, RNA-Seq and ChIP-Seq data emerges towards a better understanding of the information hidden in a genome. Similarly, visual representations well established in other scientific areas, such as economics or social studies, should be shared and applied to the current field of sequencing.
References
Finishing the euchromatic sequence of the human genome. Nature. 2004, 431 (7011): 931-945. 10.1038/nature03001. PMID: 15496913
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W: Initial sequencing and analysis of the human genome. Nature. 2001, 409 (6822): 860-921. 10.1038/35057062.
Levy S, Sutton G, Ng PC, Feuk L, Halpern AL, Walenz BP, Axelrod N, Huang J, Kirkness EF, Denisov G: The diploid genome sequence of an individual human. PLoS Biol. 2007, 5 (10): e254-10.1371/journal.pbio.0050254.
Abecasis GR, Altshuler D, Auton A, Brooks LD, Durbin RM, Gibbs RA, Hurles ME, McVean GA: A map of human genome variation from population-scale sequencing. Nature. 2010, 467 (7319): 1061-1073. 10.1038/nature09534.
Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA: An integrated map of genetic variation from 1,092 human genomes. Nature. 2012, 491 (7422): 56-65. 10.1038/nature11632.
Buchanan CC, Torstenson ES, Bush WS, Ritchie MD: A comparison of cataloged variation between international HapMap consortium and 1000 genomes project data. J Am Med Inform Assoc. 2012, 19 (2): 289-294. 10.1136/amiajnl-2011-000652.
Tanaka T: [International HapMap project]. Nihon Rinsho. 2005, 12 (63 Suppl): 29-34.
Thorisson GA, Smith AV, Krishnan L, Stein LD: The international HapMap project Web site. Genome Res. 2005, 15 (11): 1592-1593. 10.1101/gr.4413105.
Integrating ethics and science in the international HapMap project. Nat Rev Genet. 2004, 5 (6): 467-475. 10.1038/nrg1351. PMID: 15153999
The international HapMap project. Nature. 2003, 426 (6968): 789-796. 10.1038/nature02168. PMID: 14685227
Pitman NC, Jorgensen PM: Estimating the size of the world's threatened flora. Science. 2002, 298 (5595): 989-10.1126/science.298.5595.989.
Weigel D, Mott R: The 1001 genomes project for arabidopsis thaliana. Genome Biol. 2009, 10 (5): 107-10.1186/gb-2009-10-5-107.
Genome 10K: a proposal to obtain whole-genome sequence for 10,000 vertebrate species. J Hered. 2009, 100 (6): 659-674. PMID: 19892720
Medini D, Donati C, Tettelin H, Masignani V, Rappuoli R: The microbial pan-genome. Curr Opin Genet Dev. 2005, 15 (6): 589-594. 10.1016/j.gde.2005.09.006.
Cullum R, Alder O, Hoodless PA: The next generation: using new sequencing technologies to analyse gene regulation. Respirology. 2011, 16 (2): 210-222. 10.1111/j.1440-1843.2010.01899.x.
Metzker ML: Sequencing technologies - the next generation. Nat Rev Genet. 2010, 11 (1): 31-46. 10.1038/nrg2626.
Church M: Genomes for All. Sci Am. 2006, 294: 46-54.
Hall N: Advanced sequencing technologies and their wider impact in microbiology. J Exp Biol. 2007, 210 (Pt 9): 1518-1525.
Nagarajan N, Pop M: Sequencing and genome assembly using next-generation technologies. Methods Mol Biol. 2010, 673: 1-17. 10.1007/978-1-60761-842-3_1.
Git A, Dvinge H, Salmon-Divon M, Osborne M, Kutter C, Hadfield J, Bertone P, Caldas C: Systematic comparison of microarray profiling, real-time PCR, and next-generation sequencing technologies for measuring differential microRNA expression. RNA. 2010, 16 (5): 991-1006. 10.1261/rna.1947110.
Hert DG, Fredlake CP, Barron AE: Advantages and limitations of next-generation sequencing technologies: a comparison of electrophoresis and non-electrophoresis methods. Electrophoresis. 2008, 29 (23): 4618-4626. 10.1002/elps.200800456.
Thomas RK, Baker AC, Debiasi RM, Winckler W, Laframboise T, Lin WM, Wang M, Feng W, Zander T, MacConaill L: High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007, 39 (3): 347-351. 10.1038/ng1975.
Bennett S: Solexa Ltd. Pharmacogenomics. 2004, 5 (4): 433-438. 10.1517/14622416.5.4.433.
Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, Hall KP, Evers DJ, Barnes CL, Bignell HR: Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008, 456 (7218): 53-59. 10.1038/nature07517.
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z: Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005, 437 (7057): 376-380.
Luo C, Tsementzi D, Kyrpides N, Read T, Konstantinidis KT: Direct comparisons of illumina vs. Roche 454 sequencing technologies on the same microbial community DNA sample. PLoS One. 2012, 7 (2): e30087-10.1371/journal.pone.0030087.
Liu L, Li Y, Li S, Hu N, He Y, Pong R, Lin D, Lu L, Law M: Comparison of next-generation sequencing systems. J Biomed Biotechnol. 2012, 2012: 251364-
Xu M, Fujita D, Hanagata N: Perspectives and challenges of emerging single-molecule DNA sequencing technologies. Small. 2009, 5 (23): 2638-2649. 10.1002/smll.200900976.
Wang Z, Gerstein M, Snyder M: RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet. 2009, 10 (1): 57-63. 10.1038/nrg2484.
Morin R, Bainbridge M, Fejes A, Hirst M, Krzywinski M, Pugh T, McDonald H, Varhol R, Jones S, Marra M: Profiling the HeLa S3 transcriptome using randomly primed cDNA and massively parallel short-read sequencing. Biotechniques. 2008, 45 (1): 81-94. 10.2144/000112900.
Furey TS: ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 2012, 13 (12): 840-852. 10.1038/nrg3306.
Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA: A whole-genome assembly of drosophila. Science. 2000, 287 (5461): 2196-2204. 10.1126/science.287.5461.2196.
Havlak P, Chen R, Durbin KJ, Egan A, Ren Y, Song XZ, Weinstock GM, Gibbs RA: The atlas genome assembly system. Genome Res. 2004, 14 (4): 721-732. 10.1101/gr.2264004.
Batzoglou S, Jaffe DB, Stanley K, Butler J, Gnerre S, Mauceli E, Berger B, Mesirov JP, Lander ES: ARACHNE: a whole-genome shotgun assembler. Genome Res. 2002, 12 (1): 177-189. 10.1101/gr.208902.
Aparicio S, Chapman J, Stupka E, Putnam N, Chia JM, Dehal P, Christoffels A, Rash S, Hoon S, Smit A: Whole-genome shotgun assembly and analysis of the genome of fugu rubripes. Science. 2002, 297 (5585): 1301-1310. 10.1126/science.1072104.
Huang X, Wang J, Aluru S, Yang SP, Hillier L: PCAP: a whole-genome assembly program. Genome Res. 2003, 13 (9): 2164-2170. 10.1101/gr.1390403.
Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I: ABySS: a parallel assembler for short read sequence data. Genome Res. 2009, 19 (6): 1117-1123. 10.1101/gr.089532.108.
Zerbino DR, Birney E: Velvet: algorithms for de novo short read assembly using de bruijn graphs. Genome Res. 2008, 18 (5): 821-829. 10.1101/gr.074492.107.
Mullikin JC, Ning Z: The phusion assembler. Genome Res. 2003, 13 (1): 81-90. 10.1101/gr.731003.
Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, DiCuccio M, Edgar R, Federhen S: Database resources of the national center for biotechnology information. Nucleic Acids Res. 2007, 35 (Database issue): D5-12.
Stenson PD, Ball EV, Mort M, Phillips AD, Shaw K, Cooper DN: The human gene mutation database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Current protocols in bioinformatics. Edited by: Baxevanis AD. 2012, Chapter 1:Unit1 13. PMID:22948725
Brookes AJ, Lehvaslaiho H, Siegfried M, Boehm JG, Yuan YP, Sarkar CM, Bork P, Ortigao F: HGBASE: a database of SNPs and other variations in and around human genes. Nucleic Acids Res. 2000, 28 (1): 356-360. 10.1093/nar/28.1.356.
Fredman D, Siegfried M, Yuan YP, Bork P, Lehvaslaiho H, Brookes AJ: HGVbase: a human sequence variation database emphasizing data quality and a broad spectrum of data sources. Nucleic Acids Res. 2002, 30 (1): 387-391. 10.1093/nar/30.1.387.
The GWAS central.http://www.gwascentral.org,
The SNPedia.http://www.snpedia.com/index.php/SNPedia,
Karchin R: Next generation tools for the annotation of human SNPs. Brief Bioinform. 2009, 10 (1): 35-52.
Medvedev P, Stanciu M, Brudno M: Computational methods for discovering structural variation with next-generation sequencing. Nat Methods. 2009, 6 (11 Suppl): S13-20.
Cock PJ, Fields CJ, Goto N, Heuer ML, Rice PM: The sanger FASTQ file format for sequences with quality scores, and the solexa/illumina FASTQ variants. Nucleic Acids Res. 2010, 38 (6): 1767-1771. 10.1093/nar/gkp1137.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: Genome project data processing S: the sequence alignment/Map format and SAMtools. Bioinformatics. 2009, 25 (16): 2078-2079. 10.1093/bioinformatics/btp352.
Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST: The variant call format and VCFtools. Bioinformatics. 2011, 27 (15): 2156-2158. 10.1093/bioinformatics/btr330.
Ewing B, Hillier L, Wendl MC, Green P: Base-calling of automated sequencer traces using phred. I. Accuracy assessment. Genome Res. 1998, 8 (3): 175-185. 10.1101/gr.8.3.175.
Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin ML, Ordonez GR, Bignell GR: A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010, 463 (7278): 191-196. 10.1038/nature08658.
Deorowicz S, Grabowski S: Compression of genomic sequences in FASTQ format. Bioinformatics. 2011, PMID: 21252073
Tembe W, Lowey J, Suh E: G-SQZ: compact encoding of genomic sequence and quality data. Bioinformatics. 2010, 26 (17): 2192-2194. 10.1093/bioinformatics/btq346.
Cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B: Biopython: freely available python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009, 25 (11): 1422-1423. 10.1093/bioinformatics/btp163.
Stajich JE, Block D, Boulez K, Brenner SE, Chervitz SA, Dagdigian C, Fuellen G, Gilbert JG, Korf I, Lapp H: The bioperl toolkit: perl modules for the life sciences. Genome Res. 2002, 12 (10): 1611-1618. 10.1101/gr.361602.
Goto N, Prins P, Nakao M, Bonnal R, Aerts J, Katayama T: BioRuby: bioinformatics software for the ruby programming language. Bioinformatics. 2010, 26 (20): 2617-2619. 10.1093/bioinformatics/btq475.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R: The sequence alignment/Map format and SAMtools. Bioinformatics. 2009, 25 (16): 2078-2079. 10.1093/bioinformatics/btp352.
Botstein D, Risch N: Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat Genet. 2003, 33 (Suppl): 228-237.
Altshuler D, Daly MJ, Lander ES: Genetic mapping in human disease. Science. 2008, 322 (5903): 881-888. 10.1126/science.1156409.
Chen K, Wallis JW, McLellan MD, Larson DE, Kalicki JM, Pohl CS, McGrath SD, Wendl MC, Zhang Q, Locke DP: BreakDancer: an algorithm for high-resolution mapping of genomic structural variation. Nat Methods. 2009, 6 (9): 677-681. 10.1038/nmeth.1363.
Xie C, Tammi MT: CNV-seq, a new method to detect copy number variation using high-throughput sequencing. BMC Bioinforma. 2009, 10: 80-10.1186/1471-2105-10-80.
Sindi S, Helman E, Bashir A, Raphael BJ: A geometric approach for classification and comparison of structural variants. Bioinformatics. 2009, 25 (12): i222-230. 10.1093/bioinformatics/btp208.
Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, Mell JC, Hall IM: Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2010, 20 (5): 623-635. 10.1101/gr.102970.109.
Lee S, Hormozdiari F, Alkan C, Brudno M: MoDIL: detecting small indels from clone-end sequencing with mixtures of distributions. Nat Methods. 2009, 6 (7): 473-474. 10.1038/nmeth.f.256.
Hach F, Hormozdiari F, Alkan C, Hormozdiari F, Birol I, Eichler EE, Sahinalp SC: mrsFAST: a cache-oblivious algorithm for short-read mapping. Nat Methods. 2010, 7 (8): 576-577. 10.1038/nmeth0810-576.
Hajirasouliha I, Hormozdiari F, Alkan C, Kidd JM, Birol I, Eichler EE, Sahinalp SC: Detection and characterization of novel sequence insertions using paired-end next-generation sequencing. Bioinformatics. 2010, 26 (10): 1277-1283. 10.1093/bioinformatics/btq152.
Korbel JO, Abyzov A, Mu XJ, Carriero N, Cayting P, Zhang Z, Snyder M, Gerstein MB: PEMer: a computational framework with simulation-based error models for inferring genomic structural variants from massive paired-end sequencing data. Genome Biol. 2009, 10 (2): R23-10.1186/gb-2009-10-2-r23.
Ye K, Schulz MH, Long Q, Apweiler R, Ning Z: Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. 2009, 25 (21): 2865-2871. 10.1093/bioinformatics/btp394.
Kim TM, Luquette LJ, Xi R, Park PJ: rSW-seq: algorithm for detection of copy number alterations in deep sequencing data. BMC Bioinforma. 2010, 11: 432-10.1186/1471-2105-11-432.
Hormozdiari F, Hajirasouliha I, Dao P, Hach F, Yorukoglu D, Alkan C, Eichler EE, Sahinalp SC: Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics. 2010, 26 (12): i350-357. 10.1093/bioinformatics/btq216.
Koboldt DC, Zhang Q, Larson DE, Shen D, McLellan MD, Lin L, Miller CA, Mardis ER, Ding L, Wilson RK: VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22 (3): 568-576. 10.1101/gr.129684.111.
Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L: VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009, 25 (17): 2283-2285. 10.1093/bioinformatics/btp373.
McClellan J, King MC: Genetic heterogeneity in human disease. Cell. 2010, 141 (2): 210-217. 10.1016/j.cell.2010.03.032.
Cantor RM, Lange K, Sinsheimer JS: Prioritizing GWAS results: a review of statistical methods and recommendations for their application. Am J Hum Genet. 2010, 86 (1): 6-22. 10.1016/j.ajhg.2009.11.017.
Sifrim A, Van Houdt JK, Tranchevent LC, Nowakowska B, Sakai R, Pavlopoulos GA, Devriendt K, Vermeesch JR, Moreau Y, Aerts J: Annotate-it: a swiss-knife approach to annotation, analysis and interpretation of single nucleotide variation in human disease. Genome Med. 2012, 4 (9): 73-10.1186/gm374.
Li MX, Gui HS, Kwan JS, Bao SY, Sham PC: A comprehensive framework for prioritizing variants in exome sequencing studies of mendelian diseases. Nucleic Acids Res. 2012, 40 (7): e53-10.1093/nar/gkr1257.
Wang K, Li M, Hakonarson H: ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38 (16): e164-10.1093/nar/gkq603.
Makarov V, O'Grady T, Cai G, Lihm J, Buxbaum JD, Yoon S: AnnTools: a comprehensive and versatile annotation toolkit for genomic variants. Bioinformatics. 2012, 28 (5): 724-725. 10.1093/bioinformatics/bts032.
Shetty AC, Athri P, Mondal K, Horner VL, Steinberg KM, Patel V, Caspary T, Cutler DJ, Zwick ME: SeqAnt: a web service to rapidly identify and annotate DNA sequence variations. BMC Bioinforma. 2010, 11: 471-10.1186/1471-2105-11-471.
Ge D, Ruzzo EK, Shianna KV, He M, Pelak K, Heinzen EL, Need AC, Cirulli ET, Maia JM, Dickson SP: SVA: software for annotating and visualizing sequenced human genomes. Bioinformatics. 2011, 27 (14): 1998-2000. 10.1093/bioinformatics/btr317.
Asmann YW, Middha S, Hossain A, Baheti S, Li Y, Chai HS, Sun Z, Duffy PH, Hadad AA, Nair A: TREAT: a bioinformatics tool for variant annotations and visualizations in targeted and exome sequencing data. Bioinformatics. 2012, 28 (2): 277-278. 10.1093/bioinformatics/btr612.
Yandell M, Huff C, Hu H, Singleton M, Moore B, Xing J, Jorde LB, Reese MG: A probabilistic disease-gene finder for personal genomes. Genome Res. 2011, 21 (9): 1529-1542. 10.1101/gr.123158.111.
Cheng YC, Hsiao FC, Yeh EC, Lin WJ, Tang CY, Tseng HC, Wu HT, Liu CK, Chen CC, Chen YT: VarioWatch: providing large-scale and comprehensive annotations on human genomic variants in the next generation sequencing era. Nucleic Acids Res. 2012, 40 (Web Server issue): W76-81.
Sincan M, Simeonov DR, Adams D, Markello TC, Pierson TM, Toro C, Gahl WA, Boerkoel CF: VAR-MD: a tool to analyze whole exome-genome variants in small human pedigrees with mendelian inheritance. Hum Mutat. 2012, 33 (4): 593-598. 10.1002/humu.22034.
Teer JK, Green ED, Mullikin JC, Biesecker LG: VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics. 2012, 28 (4): 599-600. 10.1093/bioinformatics/btr711.
O'Donoghue SI, Gavin AC, Gehlenborg N, Goodsell DS, Heriche JK, Nielsen CB, North C, Olson AJ, Procter JB, Shattuck DW: Visualizing biological data-now and in the future. Nat Methods. 2010, 7 (3 Suppl): S2-4.
Nielsen CB, Jackman SD, Birol I, Jones SJ: ABySS-explorer: visualizing genome sequence assemblies. IEEE Trans Vis Comput Graph. 2009, 15 (6): 881-888.
Huang W, Marth G: EagleView: a genome assembly viewer for next-generation sequencing technologies. Genome Res. 2008, 18 (9): 1538-1543. 10.1101/gr.076067.108.
Schatz MC, Phillippy AM, Sommer DD, Delcher AL, Puiu D, Narzisi G, Salzberg SL, Pop M: Hawkeye and AMOS: visualizing and assessing the quality of genome assemblies. Brief Bioinform. 2013, 14 (2): 213-224. 10.1093/bib/bbr074.
Manske HM, Kwiatkowski DP: LookSeq: a browser-based viewer for deep sequencing data. Genome Res. 2009, 19 (11): 2125-2132. 10.1101/gr.093443.109.
Hou H, Zhao F, Zhou L, Zhu E, Teng H, Li X, Bao Q, Wu J, Sun Z: MagicViewer: integrated solution for next-generation sequencing data visualization and genetic variation detection and annotation. Nucleic Acids Res. 2010, 38 (Web Server issue): W732-736.
Bao H, Guo H, Wang J, Zhou R, Lu X, Shi S: MapView: visualization of short reads alignment on a desktop computer. Bioinformatics. 2009, 25 (12): 1554-1555. 10.1093/bioinformatics/btp255.
Furey TS: Comparison of human (and other) genome browsers. Hum Genomics. 2006, 2 (4): 266-270. 10.1186/1479-7364-2-4-266.
Cline MS, Kent WJ: Understanding genome browsing. Nat Biotechnol. 2009, 27 (2): 153-155. 10.1038/nbt0209-153.
Nielsen CB, Cantor M, Dubchak I, Gordon D, Wang T: Visualizing genomes: techniques and challenges. Nat Methods. 2010, 7 (3 Suppl): S5-S15.
AnnoJ.http://www.annoj.org,
Grant JR, Stothard P: The CGView server: a comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36 (Web Server issue): W181-184.
Engels R, Yu T, Burge C, Mesirov JP, DeCaprio D, Galagan JE: Combo: a whole genome comparative browser. Bioinformatics. 2006, 22 (14): 1782-1783. 10.1093/bioinformatics/btl193.
Flicek P, Amode MR, Barrell D, Beal K, Brent S, Carvalho-Silva D, Clapham P, Coates G, Fairley S, Fitzgerald S: Ensembl 2012. Nucleic Acids Res. 2012, 40 (Database issue): D84-90.
Hubbard T, Barker D, Birney E, Cameron G, Chen Y, Clark L, Cox T, Cuff J, Curwen V, Down T: The ensembl genome database project. Nucleic Acids Res. 2002, 30 (1): 38-41. 10.1093/nar/30.1.38.
Papanicolaou A, Heckel DG: The GMOD drupal bioinformatic server framework. Bioinformatics. 2010, 26 (24): 3119-3124. 10.1093/bioinformatics/btq599.
Wang H, Su Y, Mackey AJ, Kraemer ET, Kissinger JC: SynView: a GBrowse-compatible approach to visualizing comparative genome data. Bioinformatics. 2006, 22 (18): 2308-2309. 10.1093/bioinformatics/btl389.
Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A: The generic genome browser: a building block for a model organism system database. Genome Res. 2002, 12 (10): 1599-1610. 10.1101/gr.403602.
Arakawa K, Tamaki S, Kono N, Kido N, Ikegami K, Ogawa R, Tomita M: Genome projector: zoomable genome map with multiple views. BMC Bioinforma. 2009, 10: 31-10.1186/1471-2105-10-31.
Nicol JW, Helt GA, Blanchard SG, Raja A, Loraine AE: The integrated genome browser: free software for distribution and exploration of genome-scale datasets. Bioinformatics. 2009, 25 (20): 2730-2731. 10.1093/bioinformatics/btp472.
Thorvaldsdottir H, Robinson JT, Mesirov JP: Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013, 14 (2): 178-192. 10.1093/bib/bbs017.
Zhu J, Sanborn JZ, Benz S, Szeto C, Hsu F, Kuhn RM, Karolchik D, Archie J, Lenburg ME, Esserman LJ: The UCSC cancer genomics browser. Nat Methods. 2009, 6 (4): 239-240. 10.1038/nmeth0409-239.
Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D: The human genome browser at UCSC. Genome Res. 2002, 12 (6): 996-1006.
Yates T, Okoniewski MJ, Miller CJ: X:Map: annotation and visualization of genome structure for affymetrix exon array analysis. Nucleic Acids Res. 2008, 36 (Database issue): D780-786.
Sinha AU, Meller J: Cinteny: flexible analysis and visualization of synteny and genome rearrangements in multiple organisms. BMC Bioinforma. 2007, 8: 82-10.1186/1471-2105-8-82.
Yin T, Cook D, Lawrence M: Ggbio: an R package for extending the grammar of graphics for genomic data. Genome Biol. 2012, 13 (8): R77-10.1186/gb-2012-13-8-r77.
Yang J, Wang J, Yao ZJ, Jin Q, Shen Y, Chen R: GenomeComp: a visualization tool for microbial genome comparison. J Microbiol Methods. 2003, 54 (3): 423-426. 10.1016/S0167-7012(03)00094-0.
Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA: Circos: an information aesthetic for comparative genomics. Genome Res. 2009, 19 (9): 1639-1645. 10.1101/gr.092759.109.
Deng X, Rayner S, Liu X, Zhang Q, Yang Y, Li N: DHPC: a new tool to express genome structural features. Genomics. 2008, 91 (5): 476-483. 10.1016/j.ygeno.2008.01.003.
Anders S: Visualization of genomic data with the hilbert curve. Bioinformatics. 2009, 25 (10): 1231-1235. 10.1093/bioinformatics/btp152. PMID: 23605045
Qi J, Zhao F: InGAP-sv: a novel scheme to identify and visualize structural variation from paired end mapping data. Nucleic Acids Res. 2011, 39 (Web Server issue): W567-575.
Pavlopoulos GA, Kumar P, Sifrim A, Sakai R, Lin ML, Voet T, Moreau Y, Aerts J: Meander: visually exploring the structural variome using space-filling curves. Nucleic Acids Res. 2013
MEDEA: Comparative genomic visualization with adobe flash. [http://www.broadinstitute.org/annotation/medea/]
Meyer M, Munzner T, Pfister H: MizBee: a multiscale synteny browser. IEEE Trans Vis Comput Graph. 2009, 15 (6): 897-904.
Esteban-Marcos A, Darling AE, Ragan MA: Seevolution: visualizing chromosome evolution. Bioinformatics. 2009, 25 (7): 960-961. 10.1093/bioinformatics/btp096.
Crabtree J, Angiuoli SV, Wortman JR, White OR: Sybil: methods and software for multiple genome comparison and visualization. Methods Mol Biol. 2007, 408: 93-108. 10.1007/978-1-59745-547-3_6. Clifton, NJ
Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I: VISTA : visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000, 16 (11): 1046-1047. 10.1093/bioinformatics/16.11.1046.
Pavlopoulos GA, Soldatos TG, Barbosa-Silva A, Schneider R: A reference guide for tree analysis and visualization. BioData Min. 2010, 3 (1): 1-10.1186/1756-0381-3-1.
Pavlopoulos GA, Wegener AL, Schneider R: A survey of visualization tools for biological network analysis. BioData Min. 2008, 1: 12-10.1186/1756-0381-1-12.
Acknowledgements
This work was supported by iMinds [SBO 2012]; University of Leuven Research Council [SymBioSys PFV/10/016, GOA/10/009] and European Union Framework Programme 7 [HEALTH-F2-2008-223040 "CHeartED"]. GAP was financially supported by the European Commission FP7 programme 'Translational Potential' (TransPOT; EC contract number 285948). AO was funded by the European Union’s Seventh Framework Programme (FP7/2007-2013) under grant agreement no 264089 (MARBIGEN project). AS was supported by IWT Grant No. IWT-SB/ 093289.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
The authors wrote and revised the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pavlopoulos, G.A., Oulas, A., Iacucci, E. et al. Unraveling genomic variation from next generation sequencing data. BioData Mining 6, 13 (2013). https://doi.org/10.1186/1756-0381-6-13
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0381-6-13