
Abstract
The therapeutic potential of mesenchymal stem cell (MSC) transplantation for the treatment of ischemic conditions such as coronary artery disease, peripheral arterial disease, and stroke has been explored in animal models and early-phase clinical trials. A substantial database documents the safety profile of MSC administration to humans in a large number of disease states. The mechanism of the therapeutic effect of MSC transplantation in ischemic disease has been postulated to be due to paracrine, immunomodulatory, and differentiation effects. This review provides an overview of the potential role of MSC-based therapy for critical limb ischemia (CLI), the comparison of MSC cellular therapy with angiogenesis gene therapy in CLI, and the proposed mechanism of action of MSC therapy. Preclinical efficacy data in animal models of hindlimb ischemia, current early-phase human trial data, and considerations for future MSC-based therapy in CLI will also be discussed.
Similar content being viewed by others
Introduction
Mesenchymal stem cell (MSC) transplantation has been proposed as a novel treatment approach for tissue engineering and regenerative medicine for various disease states. MSC-based therapy has been explored in pre-clinical animal models and recently has been used in early clinical trials for ischemic disorders, including stroke, coronary artery disease and peripheral arterial disease (PAD). In this review, we discuss the comparison of MSC cellular therapy with angiogenesis gene therapy in critical limb ischemia (CLI), the possible mechanism of action and safety profile of MSC therapy. We also highlight the potential role of MSC for the management of patients with CLI by describing the relevant preclinical and early clinical trial results. We conclude by discussing the several practical considerations for future clinical trials.
Peripheral arterial disease: unmet clinical need
Up to 10% of the population in the Western world suffers from PAD and this represents a major health problem [1]. The prevalence of PAD has increased exponentially due to the increase in the prevalence of diabetes mellitus (DM) and an aging population. The increasing prevalence of PAD has resulted in a substantial increase in the consumption of health-care costs [2]. DM is prevalent in patients with PAD. In fact, DM itself increases the risk of lower-extremity PAD by two- to fourfold [3]. Poor glycemic control is associated with an increased risk of PAD independently of other known cardiovascular risk factors [4]. Individuals with poor glycemic control (A1c >7.5%) are five times more likely to develop intermittent claudication and be hospitalized for PAD as compared with those with better glycemic control (A1c <6%) [4]. In fact, 1% increment in hemoglobin A1c in patients with Type 2 DM correlates with a 28% increase in the risk of PAD [5].
Patients with PAD may be asymptomatic or suffer from intermittent claudication, ischemic ulceration, rest pain or limb loss. CLI is the most advanced clinical stage of PAD. It is defined as rest pain or impending limb loss secondary to an objectively proven arterial occlusive disease for more than two weeks. The current treatment options aim at improving distal arterial perfusion by endovascular or surgical approaches or a combination of the two [1]. However, amputation is often inevitable in the majority of patients because of co-morbidities or unsuitable vasculature, and since these patients have no alternative therapeutic options, they have been termed no-option patients. These patients also had a 20% mortality within six months [1]. Hence, this condition represents an unmet clinical need. Cell transplantation has been suggested as a possible approach for the treatment of CLI. A variety of cell types have been proposed. Currently, clinical trials using cells from both the autologous and allogeneic sources for the treatment of CLI either have been completed or are under way. There is a particular interest in the use of both fractionated and unfractionated bone marrow cells as well as MSCs derived from various sources. Table 1 shows the current registered clinical trials at ClinicalTrials.gov [6] for various stem cell therapies for CLI.
Prior to the era of cellular therapy, gene therapy was proposed as a therapeutic option for CLI. Several phase 2 gene therapy clinical trials - with vascular endothelial growth factor (VEGF), del1, hypoxia-inducible factor 1a (HIF1a)/VP16, hepatocyte growth factor, and fibroblast growth factor 1 (FGF1) - have been completed and demonstrated their safety and feasibility in patients with PAD [7]. A phase 3 trial with FGF1 was recently completed but did not reach the combined primary outcome of reduction in major amputation or death [8]. The comparison between MSC cellular therapy and angiogenesis gene therapy for patients with CLI is summarized in Table 2.
Mesenchymal stem cells
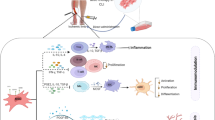
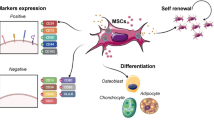
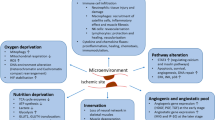
MSCs are multipotent non-hematopoetic, fibroblast-like plastic adherent cells that can be isolated from various tissue sources, including the bone marrow, adipose tissue, placenta, and umbilical cord blood [9]. They are capable of differentiating into different cell types such as bone, fat, cartilage, and muscle and demonstrate specific surface antigen expression [9]. In addition to possibly undergoing cell differentiation, MSC transplantation may exert its therapeutic effects via secretion of paracrine factors that may have anti-inflammatory and immunomodulatory effects [10, 11]. MSCs are relatively resistant to apoptosis induced by conditions such as serum starvation and hypoxia [12]. In fact, MSCs exposed to hypoxia produce more VEGF in vitro, and transplantation of hypoxic preconditioned MSCs into murine ischemic limbs has been reported to lead to an increase in vessel density [13]. MSCs can also home to hypoxic muscle following intravenous administration [14]. Even though MSCs represent a very small fraction of the total population of nucleated cells in the bone marrow (0.001% to 0.01%), they can be culture-expanded readily to yield a large number of cells. These cells can be stored for both autologous and allogeneic use. The latter may be possible due to the immune-privileged status of these cells. This property offers substantial practical advantage in the clinical setting, especially when autologous cell transplantation would be ineffective because of disease-induced cell dysfunction [15]. Furthermore, MSCs can be programmed to become a specific differentiated cell type prior to transplantation, thereby decreasing the likelihood of aberrant differentiation of MSCs after transplantation.
Mechanism of action of mesenchymal stem cells
A. Paracrine effects
MSCs can be isolated from a variety of sources, including bone marrow, adipose tissue, and induced pluripotent stem cells (iPSCs) [16, 17]. The conditioned media from the undifferentiated MSCs promote in vitro angiogenesis and migration [18, 19]. As compared with bone marrow-derived MSCs, adipose tissue-derived MSCs secrete more VEGF, hepatocyte growth factor, and transforming growth factor-beta [20], and the iPSC-derived MSCs secrete more stromal-derived factor 1 alpha (SDF-1-alpha), hepatocyte growth factor, stem cell factor, basic nerve growth factor, basic fibroblast growth factor, and VEGF [16]. The conditioned media from adipose tissue-derived MSCs also have higher matrix metalloproteinase 3 (MMP3) and MMP9 and showed enhanced in vitro tube formation in comparison with that from bone marrow-derived MSCs [17]. The MSC conditioned media can augment in vivo angiogenesis [21]. Under hypoxic conditions, bone marrow-derived MSCs produced more VEGF in comparison with normoxic conditions, and transplantation of these MSCs into murine ischemic limbs led to an increase in vessel density [13]. The MSC conditioned media can also enhance proliferation of endothelial cells and smooth muscle cells in a dose-dependent manner [22]. MSCs stimulate capillary morphogenesis via distinct proteolytic mechanisms [23].
Cross-talk occurs between MSCs and endothelial cells. MSCs attenuate myocardial ischemic reperfusion injury by secreting exosomes [24]. Under hypoxic conditions, bone marrow-derived MSCs secreted higher amounts of VEGF with enhanced proliferative capacity as compared with fibroblasts [13]. Co-culturing of MSCs with endothelial cells upregulate gene expression of extra-cellular proteases such as MMP2, MMP9, and MT1-MMP in endothelial cells [23, 25]. Bone marrow-derived MSCs have also been shown to be attracted to in vitro formed vascular structures [26]. These studies supported the notion that MSCs exert the angiogenic properties via paracrine and autocrine effects, and the intensity is dependent on the MSC source.
B. Differentiation effect
There is considerable doubt that the mode of action of MSCs occurs via in vivo differentiation given that only small numbers of cells engraft at the site of ischemia following intravenous administration and those that do are rapidly lost from the site. In the mouse models of myocardial infarction, the majority (83%) of the xenogeneic MSCs were found in the lung at one hour after intravenous administration [27]. Another study showed that, after intravenous administration, allogeneic MSCs were detected for up to 20 days and completely undetectable at 40 days because of graft rejection but that syngeneic MSCs survived up to 40 days [28]. This result suggests that an allogeneic immune response may occur after MSC transplantation. On the other hand, xenogeneic MSCs administered intramuscularly to non-ischemic thigh muscle remain confined to the site of injection; the highest level was detected after one day, and cells were detectable for up to three weeks [29]. These results suggested that small numbers of MSCs engraft in the ischemic site following intravenous administration, that the beneficial effect of allogeneic transplantation may be attenuated by graft rejection, and that local administration may be the optimal approach for MSC-based therapy for CLI. However, there is evidence that MSCs can acquire myogenic and endothelial properties.
i. Myogenic differentiation
MSCs derived from adipose tissue, bone marrow, and synovial membrane are capable of inducing skeletal muscle regeneration, and adipose tissue-derived MSCs are the most efficient [30]. Adipose tissue-derived MSCs can enhance muscle regeneration even in dystrophin-deficient mice following intramuscular administration [31]. Hypoxia preconditioned MSCs can further enhance skeletal muscle regeneration [32]. MSCs can also differentiate into cardiomyocytes and these predifferentiated MSCs further augment cardiac regeneration more efficiently than undifferentiated cells [33].
MSCs derived from bone marrow, iPSCs, and adipose tissue were also capable of differentiating into smooth muscle cells [16, 34, 35]. However, iPSC-derived MSCs differentiate into smooth muscle more efficiently than bone marrow-derived MSCs [16]. MSC-derived smooth muscle cells have been used to engineer small-diameter vessel wall grafts [35]. In a dog model of peripheral and coronary artery bypass, canine grafts were able to maintain their patency and prevent dilatation, calcification, and intimal hyperplasia [36]. Furthermore, human engineered grafts were successfully made and tested in a baboon model of arteriovenous access for hemodialysis [36]. Currently, a functional urinary bladder tissue is being engineered by using a combination of smooth muscle cells and urothelium-like cells derived from human bone marrow-derived MSCs [37].
ii. Endothelial differentiation
MSCs derived from bone marrow, iPSCs, dental pulp, amniotic fluid, and adipose tissue have been reported to undergo endothelial differentiation [16, 34, 38, 39]. Direct comparison of umbilical cord-derived MSCs and bone marrow-derived MSCs showed that both cell types expressed MSC-specific markers and demonstrated trilineage differentiation ability with the ability to take up low-density lipoprotein following endothelial differentiation. However, umbilical cord-derived MSCs had higher proliferative potential and higher expression of the endothelial-specific factors and were able to form more capillary networks than bone marrow-derived MSCs [40].
Endothelial differentiation of MSCs can be stimulated by growth factors, including VEGF, and shear force [38, 39, 41]. We have shown that overexpression of Ephrin-B2 in MSCs resulted in an earlier endothelial differentiation with simultaneous reduction of osteogenic potential [42]. Endothelial differentiated MSCs have diminished capacity to differentiate into adipocytes, and subcutaneous implantation of these cells in collagen plugs in immuno-deficient mice resulted in the formation of functional blood vessels incorporating these cells [43]. In response to hypoxia, these differentiated cells secrete angiogenic factors (VEGF, placental growth factor, and hepatocyte growth factor) [38].
There is a complex cross-talk between MSCs and endothelial cells. MSCs increase endothelial cell proliferation and migration, promoting early events of angiogenesis and decrease endothelial cell monolayer permeability. In direct co-culture with endothelial cells, MSCs increase the persistence of pre-existing vessels in a time- and dose-dependent manner, and complex vessels remain stable for more than 10 days [44]. The conditioned media from MSCs also stimulate the proliferation of the local endothelial cells [19]. MSCs exposed to epidermal growth factor enhance adhesion and migration on cultured endothelial cells [45]. Co-culturing of bone marrow-derived endothelial progenitor cells (EPCs) and MSCs upregulate angiogenesis-related transcripts and result in the formation of elongated structures after three days, even with serum starvation and the absence of growth factors [46]. Besides exhibiting direct contact, these cells exhibited vesicle transport phenomena [46]. Both MSCs and EPCs contributed to these tubule structures [46]. Co-culture of MSCs with macro-vascular endothelial cells led to an increase in expression of both endothelial and smooth muscle cell markers. On the other hand, co-culture with micro-vascular endothelial cells increases the expression of endothelial cell markers only [47]. Secreted frizzled-related protein-1 enhances MSC function in angiogenesis and contributes to the maturation of new vessels [48].
C. Immunomodulatory effect of mesenchymal stem cells
A large body of literature supports the immune-modulatory properties of MSCs. It is outside the scope of the present article to review this in detail, but the topic was recently reviewed [49]. In a rat model of acute myocarditis, allogeneic administration of fetal membrane-derived MSCs attenuated the host cell-mediated immune response [50]. In a rat model of intra-cerebral hemorrhage, intra-cerebral administration of umbilical cord-derived MSCs attenuated inflammation and promoted angiogenesis, leading to earlier neurological function recovery [10]. In a mouse model of hind-limb ischemia, xenogeneic intramuscular administration of MSCs attenuated the local oxidative stress and endothelial inflammation [51]. Furthermore, intramuscular injection of adipose tissue-derived MSCs can reduce local inflammation in the dystrophin-deficient mice [31]. Intravenous administration of MSCs to brain-injured rodents reduces injury-induced enhanced blood-brain barrier permeability, thereby reducing the associated inflammatory response [52]. The immunosuppressive role of MSCs is promoted by CD14+ monocytes [53].
Despite the wealth of evidence that these cells have immune-modulatory properties, a recent publication demonstrated that allogeneic MSC administration was not completely immune-privileged as compared with syngeneic MSCs [28]. Of note, the optimal time for functional benefit of MSC transplantation after myocardial infarction was one week given that the absence of scar formation and the reduction in inflammation at this time point facilitate integration of transplanted cells, leading to functional recovery [54]. This result is supported by another study, in which MSC transplantation improved cardiac function, reduced the apoptosis of cardiomyocytes, and increased vessel density much better when administered at one week but not within one hour or after two weeks [55]. In a mouse model of hind-limb ischemia, administration of syngeneic MSCs or conditioned media immediately after induction of hind-limb ischemia did not improve revascularization but did do so when administered one day after induction of ischemia [22]. These studies suggest that local inflammatory processes can impede the therapeutic efficacy of MSC transplantation and that the optimal timing of administration is crucial.
Current safety profile of mesenchymal stem cells
A substantial amount of evidence supports the safety of administration of human MSCs in a variety of disease states, and this has been reviewed by Ankrum and Karp [56]. Selected examples demonstrating safety of MSC administration will be reviewed here. The study with the longest period of follow-up assessed the autologous intra-articular bone marrow-derived MSC transplantation for cartilage repair for up to 11 years and 5 months [57]. No tumors or infections were reported in this cohort of 41 patients with MSC administration to 45 joints [57].
Yamout and colleagues [15] used intrathecal administration of ex vivo-expanded autologous bone marrow-derived MSCs for the treatment of multiple sclerosis. This phase 1 trial showed that intrathecal administration of a mean dose of 3 to 5 × 107 MSCs per patient was safe and feasible. Concomitant intrathecal and intravenous administration of MSCs with a mean dose of 63.2 × 106 in patients with amyotrophic lateral sclerosis with a follow-up period of up to 25 months was also safe and feasible [58].
A five-year follow-up study of intravenous autologous administration of two doses of 5 × 107 culture-expanded autologous bone marrow-derived MSCs into 16 patients with severe middle cerebral artery territory infarction has been completed. A significant clinical improvement was demonstrated in the MSC-treated group and this improvement correlated with serum SDF-1 levels and the extent of the stroke. No serious adverse effects or increase in the incidence of seizures or recurrent vascular events was observed [59].
Intravenous administration of two doses of culture-expanded autologous bone marrow-derived MSCs at a dose of 1 to 2 × 106 MSCs/kg seven days apart in patients with refractory Crohn's disease appeared to be safe and feasible [60]. In addition, intravenous administration of allogeneic MSCs with a dose escalation of 0.5, 1.6, and 5.0 × 106 bone marrow-derived MSCs/kg body weight was shown to be safe and feasible in patients with acute myocardial infarction [61]. Interestingly, MSC therapy also led to an improvement in pulmonary function and a lower incidence of arrhythmias in this cohort [61].
Preclinical data
Autologous, allogeneic, and xenogeneic administration of MSCs derived from various sources such as bone marrow, umbilical cord blood, fetal membrane, and adipose tissue has demonstrated significant improvement in mouse/rat models of hind-limb ischemia (Table 2). Each paper will not be reviewed in detail, but the accumulated evidence suggests that MSCs represent an attractive target to advance to clinical trials in humans. Although these studies demonstrate the efficacy of MSCs, there is a suggestion that MSCs derived from different sources may have variable in vivo therapeutic effects. iPSC- and adipose tissue-derived MSCs were more efficacious in therapeutic revascularization than bone marrow-derived MSCs [16, 17]. On the other hand, the fetal membrane-derived and bone marrow-derived MSCs demonstrated comparable efficacy for improvement in blood perfusion and capillary density [11].
In addition to the efficacy of transplantation of unmodified MSCs described above, the effect of modified MSCs, such as the exposure to the cells to hypoxia prior to transplantation, has been explored. Rosova and colleagues [62] demonstrated that inter-ventricular administration of normoxic and hypoxic preconditioned human bone marrow-derived MSCs restored blood flow following induction of hind-limb ischemia and that earlier improvement was observed in the hypoxic preconditioned group. However, in a subsequent study using intramuscular administration to a similar model of hind-limb ischemia, hypoxic preconditioned MSCs were superior, and no difference between the MSCs cultured in normoxia and controls was detected, suggesting that non-preconditioned MSCs do not improve blood flow [63]. Various approaches used to augment the therapeutic efficacy of MSCs are listed in Table 3[11, 12, 16, 17, 20, 21, 32, 34, 45, 51, 62–83].
A critical factor in the design of human trials is that of the cell dose to be administered. To date, various doses of MSCs derived from different sources were administered to rodent models of hind-limb ischemia and resulted in significant therapeutic revascularization. A dose of one million MSCs has been the most commonly used in these preclinical studies. Another key consideration for human translation is the route of administration. Most pre-clinical studies reported to date have transplanted the cells via the intramuscular route.
The timing of administration in relation to the induction of ischemia and the number and site (or sites) of administration also varied in different studies. The cells were administered most commonly 24 hours after induction of hind-limb ischemia. The sites of intra-muscular injections vary from the medial thigh alone or a combination of different sites such as the gastrocnemius, tibialis anterior, hamstring, and the adductor muscle groups (Table 3).
In addition to cell dose, cell type, and timing and route of administration, the endpoints to be assessed are crucial. Most of these studies used a combination of laser Doppler perfusion imaging, in vivo functional assessment (which included the ambulatory score and necrotic score), and histological assessment. However, in aggregate, these preclinical studies have demonstrated the proof of principle that MSCs derived from various sources are therapeutically effective in models of hind-limb ischemia in many different rodent species and strains. In addition, there appears to be a substantial preclinical safety profile. This provides a substantial impetus for the progression of MSC-based therapy into clinical trials for patients with CLI.
Clinical data
The preclinical efficacy and toxicology data reviewed above have provided a platform for the initiation of clinical trials of MSCs in CLI. The first reported human study using intramuscular administration of allogeneic human umbilical cord-derived MSCs was conducted in four patients with Buerger's disease. Allogeneic umbilical cord-derived MSCs improved ulcer recovery time, enhanced limb perfusion, and relieved the symptoms of rest pain [82]. Dash and colleagues [84] have shown that intramuscular administration of autologous bone marrow-derived MSCs to patients with non-healing ulcers accelerated ulcer healing and improved pain-free walking distance. In this cohort, nine patients with Buerger's disease and three with diabetic foot ulcers were included.
Whereas the two reports above used intramuscular cell delivery, the first study using intravenous administration of autologous bone marrow-derived MSCs was recently published. That report was of a single case of a patient with systemic sclerosis who developed acute gangrene of the upper and lower limbs, and three intravenous pulses of autologous bone marrow-derived MSCs were administered. The areas of necrotic skin were reduced following the first infusion. Following the third infusion, the revascularization of the patient's extremities was confirmed by angiography. The angiogenic role of MSC therapy was confirmed microscopically by using histological analysis of the skin section, which showed cell clusters with tube-like structures with high expression of multiple angiogenic factors [85].
Lasala and colleagues [86, 87] assessed 40 intramuscular injections per patient of a combination of up to 30 × 106 bone marrow-derived MSCs and 30 × 108 bone marrow-derived mononuclear cells. Walking time, ankle brachial pressure, and quality of life were improved with no reported adverse events after a mean follow-up period of 10 months. The angiogenic effect of MSCs was confirmed by both the angiographic and 99mTc-TF perfusion scintigraphy scores. These patients had diabetes mellitus with moderate to severe PAD (Fontaine class IIb to IV).
Lu and colleagues [88] compared the therapeutic effect of autologous intramuscular administration of bone marrow-derived MSCs with bone marrow-derived mononuclear cells in 20 patients with diabetes and severe PAD (Fontaine class IV). The authors showed that the ulcer healing rate was significantly higher in the bone marrow-derived MSC group than the bone marrow-derived mononuclear cell group at six weeks. Furthermore, the bone marrow-derived MSC group achieved complete ulcer healing four weeks earlier than the bone marrow-derived mononuclear cell group. In addition, the bone marrow-derived MSC group demonstrated a significant improvement in pain-free walking time, ankle brachial index, transcutaneous oxygen pressure, and magnetic resonance angiography analysis after 24 weeks of follow-up. It is important to point out that there was no significant difference among the groups in terms of pain relief and amputation. Of note, neither cell type resulted in any adverse effects.
Lee and colleagues [89] later demonstrated that autologous adipose tissue-derived MSC transplantation in patients with Buerger's disease and diabetic foot (a total of 3 × 108 cells) was feasible and safe. It improved claudication walking distance, collateral vessel formation, wound healing, and clinical symptoms, especially pain relief. There was a trend toward an improvement in maximal walking distance. However, there was no change in ankle brachial index [89]. The details of these published human studies are summarized in Table 4[82, 84–89].
From the commercial perspective, Stempeutics Research Pvt. Ltd. (Bangalore, India) has completed a phase 1/2 clinical trial using intramuscular administration of off-the-shelf allogeneic bone marrow-derived MSCs into patients with CLI. The company has reported that the MSCs were well tolerated with no adverse events or rejection. A positive efficacy trend toward improvement in ankle brachial pressure index and a reduction in the number of ulcers were demonstrated. No significant increase in amputation rate was observed. The efficacy of allogeneic bone marrow-derived MSCs is currently being assessed in phase 2/3 clinical trials [90].
In parallel to the trial by Stempeutics Research Pvt. Ltd., two phase 1 trials using intramuscular administration of allogeneic placenta-derived MSCs have been conducted by Pluristem Therapeutics Inc. (Haifa, Israel) since 2010. Their six-month follow-up interim analysis demonstrated that these cells were safe with no adverse effects. No specific anti-MSC HLA class I or II antibodies were detected. Strikingly, only one out of 27 patients (3.7%) had a major amputation within six months. This therapy significantly improved blood flow and quality of life and reduced pain score. Phase 2/3 clinical trials for CLI and Buerger's disease and a phase 2 clinical trial for intermittent claudication will be under way by the end of this year [91].
Considerations for future clinical trials using mesenchymal stem cell therapy
Although using MSCs to treat CLI is a rather novel therapeutic concept, abundant data are available from the preclinical studies and recent early clinical trials to draw conclusions on the beneficial effect of MSC therapy, beyond the recurring safety profile and feasibility evaluation. However, there is a general lack of consensus on several crucial issues on the recent early clinical trials, rendering the direct comparison among these studies impossible. These issues include the patient type, cell dosing, relevant clinical endpoints, and long-term follow-up (Table 4).
a. Patient type
Whereas atherosclerosis is the commonest cause of peripheral vascular disease, Buerger's disease or thromboangiitis obliterans is a less common but important cause. The latter is an inflammatory disorder, a distinct form of vascular occlusive disease that afflicts the peripheral arteries of young smokers. It is often characterized by an inexorable downhill course, even in patients who discontinue smoking, once a stage of CLI associated with ulceration or gangrene is reached. Regardless of these two causes of PAD, the early clinical trials that included both group of patients have demonstrated the safety and feasibility of the therapy with suggestion of efficacy. Currently, there is no evidence that either condition may respond better to MSC-based therapy. This needs to be confirmed in larger trials.
b. Cell dosing
As in preclinical studies, the site of administration and the total cell number appeared not to affect efficacy, and the current regimes (either single or multiple doses and either intramuscularly or intravenously administered) used in early clinical trials were safe and seemed to be efficacious (Table 4). However, these parameters need to be standardized to allow direct comparison with other trials.
c. Relevant clinical endpoint
Future clinical trials should include relevant clinical endpoints beyond measurement of ankle brachial index, walking time, and quality of life. Specific objective performance goals (OPGs) have been developed by the Society for Vascular Surgery to define the therapeutic benchmarks for revascularization therapies in CLI [92]. These OPGs for both the safety and efficacy endpoints are: 1) major adverse limb event (MALE), 2) MALE and peri-operative death (POD), 3) major adverse cardiovascular events (MACEs), 4) above-ankle amputation of the index limb, 5) amputation-free survival (AFS), 6) any re-intervention of above-ankle amputation of the index limb (RAO), 7) any re-intervention, above-ankle amputation of the index limb or stenosis (RAS), and 8) all-cause mortality [92]. These OPGs are crucial, particularly to allow direct comparison with other trials.
d. Assessment of long-term effects
Table 1 showed that the duration for post-administration follow-up ranged from three months to one year. According to the Society for Vascular Surgery, the assessment of safety endpoints, including the MACE, MALE, and amputation within 30 days was considered the standard duration for post-procedural events for new devices [92]. On the other hand, the minimal exposure time for relevant clinical efficacy is one year: MALE and POD are the primary efficacy endpoints and amputation-free survival is the secondary efficacy endpoint [92]. These OPGs should be adopted in clinical trials involving patients with CLI to allow direct comparison among trials.
Conclusions
MSCs have been shown to be effective in multiple reports in preclinical models of CLI. In addition, there is a substantial amount of evidence on the safety of MSC administration to humans. So far, there has been no evidence of toxicity in terms of either aberrant differentiation or tumorigenesis noted in human studies. Larger studies with longer follow-up will be required to confirm the safety demonstrated by recent studies. The published human data reviewed in this article have enrolled small numbers of patients with relatively short follow-up periods. Since CLI represents the most severe form of PAD, it may also reduce the likelihood of demonstrating efficacy given the severity of the disorder. This is, however, the easiest regulatory pathway to the clinic. Once additional safety data are collected, it may be reasonable to progress to studies to patients with intermittent claudication who represent the majority of patients with PAD and in whom therapeutic efficacy may be easier to demonstrate. This review did not focus on good manufacturing practice (GMP) production of cells or issues surrounding the need to scale up manufacture to generate therapeutic product with predicted efficacy. The challenge for the field remains to undertake clinical trials that progress from phase 1 to 3 while using cells manufactured under GMP conditions. The issue of whether to use autologous or allogeneic 'off the shelf' cells will also need to be addressed.
Abbreviations
- CLI:
-
critical limb ischemia
- DM:
-
diabetes mellitus
- EPC:
-
endothelial progenitor cell
- FGF:
-
fibroblast growth factor
- GMP:
-
good manufacturing practice
- iPSC:
-
induced pluripotent stem cell
- MACE:
-
major adverse cardiovascular event
- MALE:
-
major adverse limb event
- MMP:
-
matrix metalloproteinase
- MSC:
-
mesenchymal stem cell
- OPG:
-
objective performance goal
- PAD:
-
peripheral arterial disease
- POD:
-
peri-operative death
- SDF-1:
-
stromal-derived factor 1
- VEGF:
-
vascular endothelial growth factor.
References
Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG: Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg. 2007, 45 (Suppl S): S5-67.
Mahoney EM, Wang K, Keo HH, Duval S, Smolderen KG, Cohen DJ, Steg G, Bhatt DL, Hirsch AT: Vascular hospitalization rates and costs in patients with peripheral artery disease in the United States. Circ Cardiovasc Qual Outcomes. 2010, 3: 642-651. 10.1161/CIRCOUTCOMES.109.930735.
Hirsch AT, Haskal ZJ, Hertzer NR, Bakal CW, Creager MA, Halperin JL, Hiratzka LF, Murphy WR, Olin JW, Puschett JB, Rosenfield KA, Sacks D, Stanley JC, Taylor LM, White CJ, White J, White RA, Antman EM, Smith SC, Adams CD, Anderson JL, Faxon DP, Fuster V, Gibbons RJ, Hunt SA, Jacobs AK, Nishimura R, Ornato JP, Page RL, Riegel B: ACC/AHA 2005 Practice Guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation. 2006, 113: e463-654.
Selvin E, Wattanakit K, Steffes MW, Coresh J, Sharrett AR: HbA1c and peripheral arterial disease in diabetes: the Atherosclerosis Risk in Communities study. Diabetes Care. 2006, 29: 877-882. 10.2337/diacare.29.04.06.dc05-2018.
Selvin E, Marinopoulos S, Berkenblit G, Rami T, Brancati FL, Powe NR, Golden SH: Meta-analysis: glycosylated hemoglobin and cardiovascular disease in diabetes mellitus. Ann Intern Med. 2004, 141: 421-431.
ClinicalTrials.gov homepage. [http://www.clinicaltrials.gov]
Sedighiani F, Nikol S: Gene therapy in vascular disease. Surgeon. 2011, 9: 326-335. 10.1016/j.surge.2011.05.003.
Belch J, Hiatt WR, Baumgartner I, Driver IV, Nikol S, Norgren L, Van Belle E: Effect of fibroblast growth factor NV1FGF on amputation and death: a randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet. 2011, 377: 1929-1937. 10.1016/S0140-6736(11)60394-2.
Barry FP, Murphy JM: Mesenchymal stem cells: clinical applications and biological characterization. Int J Biochem Cell Biol. 2004, 36: 568-584. 10.1016/j.biocel.2003.11.001.
Liao W, Zhong J, Yu J, Xie J, Liu Y, Du L, Yang S, Liu P, Xu J, Wang J, Han Z, Han ZC: Therapeutic benefit of human umbilical cord derived mesenchymal stromal cells in intracerebral hemorrhage rat: implications of anti-inflammation and angiogenesis. Cell Physiol Biochem. 2009, 24: 307-316. 10.1159/000233255.
Ishikane S, Ohnishi S, Yamahara K, Sada M, Harada K, Mishima K, Iwasaki K, Fujiwara M, Kitamura S, Nagaya N, Ikeda T: Allogeneic injection of fetal membrane-derived mesenchymal stem cells induces therapeutic angiogenesis in a rat model of hind limb ischemia. Stem Cells. 2008, 26: 2625-2633. 10.1634/stemcells.2008-0236.
Iwase T, Nagaya N, Fujii T, Itoh T, Murakami S, Matsumoto T, Kangawa K, Kitamura S: Comparison of angiogenic potency between mesenchymal stem cells and mononuclear cells in a rat model of hindlimb ischemia. Cardiovasc Res. 2005, 66: 543-551. 10.1016/j.cardiores.2005.02.006.
Hoffmann J, Glassford AJ, Doyle TC, Robbins RC, Schrepfer S, Pelletier MP: Angiogenic effects despite limited cell survival of bone marrow-derived mesenchymal stem cells under ischemia. Thorac Cardiovasc Surg. 2010, 58: 136-142. 10.1055/s-0029-1240758.
Gruenloh W, Kambal A, Sondergaard C, McGee J, Nacey C, Kalomoiris S, Pepper K, Olson S, Fierro F, Nolta JA: Characterization and in vivo testing of mesenchymal stem cells derived from human embryonic stem cells. Tissue Eng Part A. 2011, 17: 1517-1525. 10.1089/ten.tea.2010.0460.
Yamout B, Hourani R, Salti H, Barada W, El-Hajj T, Al-Kutoubi A, Herlopian A, Baz EK, Mahfouz R, Khalil-Hamdan R, Kreidieh NM, El-Sabban M, Bazarbachi A: Bone marrow mesenchymal stem cell transplantation in patients with multiple sclerosis: a pilot study. J Neuroimmunol. 2010, 227: 185-189. 10.1016/j.jneuroim.2010.07.013.
Lian Q, Zhang Y, Zhang J, Zhang HK, Wu X, Zhang Y, Lam FF, Kang S, Xia JC, Lai WH, Au KW, Chow YY, Siu CW, Lee CN, Tse HF: Functional mesenchymal stem cells derived from human induced pluripotent stem cells attenuate limb ischemia in mice. Circulation. 2010, 121: 1113-1123. 10.1161/CIRCULATIONAHA.109.898312.
Kim Y, Kim H, Cho H, Bae Y, Suh K, Jung J: Direct comparison of human mesenchymal stem cells derived from adipose tissues and bone marrow in mediating neovascularization in response to vascular ischemia. Cell Physiol Biochem. 2007, 20: 867-876. 10.1159/000110447.
Gruber R, Kandler B, Holzmann P, Vogele-Kadletz M, Losert U, Fischer MB, Watzek G: Bone marrow stromal cells can provide a local environment that favors migration and formation of tubular structures of endothelial cells. Tissue Eng. 2005, 11: 896-903. 10.1089/ten.2005.11.896.
Wang CY, Yang HB, Hsu HS, Chen LL, Tsai CC, Tsai KS, Yew TL, Kao YH, Hung SC: Mesenchymal stem cell-conditioned medium facilitates angiogenesis and fracture healing in diabetic rats. J Tissue Eng Regen Med. 2012, 6: 559-569. 10.1002/term.461.
Rehman J, Traktuev D, Li J, Merfeld-Clauss S, Temm-Grove CJ, Bovenkerk JE, Pell CL, Johnstone BH, Considine RV, March KL: Secretion of angiogenic and antiapoptotic factors by human adipose stromal cells. Circulation. 2004, 109: 1292-1298. 10.1161/01.CIR.0000121425.42966.F1.
Kinnaird T, Stabile E, Burnett MS, Lee CW, Barr S, Fuchs S, Epstein SE: Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ Res. 2004, 94: 678-685. 10.1161/01.RES.0000118601.37875.AC.
Kinnaird T, Stabile E, Burnett MS, Shou M, Lee CW, Barr S, Fuchs S, Epstein SE: Local delivery of marrow-derived stromal cells augments collateral perfusion through paracrine mechanisms. Circulation. 2004, 109: 1543-1549. 10.1161/01.CIR.0000124062.31102.57.
Ghajar CM, Kachgal S, Kniazeva E, Mori H, Costes SV, George SC, Putnam AJ: Mesenchymal cells stimulate capillary morphogenesis via distinct proteolytic mechanisms. Exp Cell Res. 2010, 316: 813-825. 10.1016/j.yexcr.2010.01.013.
Lai RC, Arslan F, Lee MM, Sze NS, Choo A, Chen TS, Salto-Tellez M, Timmers L, Lee CN, El Oakley RM, Pasterkamp G, de Kleijn DP, Lim SK: Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4: 214-222. 10.1016/j.scr.2009.12.003.
Ghajar CM, Blevins KS, Hughes CC, George SC, Putnam AJ: Mesenchymal stem cells enhance angiogenesis in mechanically viable prevascularized tissues via early matrix metalloproteinase upregulation. Tissue Eng. 2006, 12: 2875-2888. 10.1089/ten.2006.12.2875.
Sorrell JM, Baber MA, Caplan AI: Influence of adult mesenchymal stem cells on in vitro vascular formation. Tissue Eng Part A. 2009, 15: 1751-1761. 10.1089/ten.tea.2008.0254.
Lee RH, Pulin AA, Seo MJ, Kota DJ, Ylostalo J, Larson BL, Semprun-Prieto L, Delafontaine P, Prockop DJ: Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell. 2009, 5: 54-63. 10.1016/j.stem.2009.05.003.
Zangi L, Margalit R, Reich-Zeliger S, Bachar-Lustig E, Beilhack A, Negrin R, Reisner Y: Direct imaging of immune rejection and memory induction by allogeneic mesenchymal stromal cells. Stem Cells. 2009, 27: 2865-2874. 10.1002/stem.217.
Ramot Y, Meiron M, Toren A, Steiner M, Nyska A: Safety and biodistribution profile of placental-derived mesenchymal stromal cells (PLX-PAD) following intramuscular delivery. Toxicol Pathol. 2009, 37: 606-616. 10.1177/0192623309338383.
de la Garza-Rodea AS, van der Velde-van Dijke L, Boersma H, Goncalves MA, van Bekkum DW, de Vries AA, Knaan-Shanzer S: Myogenic properties of human mesenchymal stem cells derived from three different sources. Cell Transplant. 2012, 21: 153-173.
da Justa Pinheiro CH, de Queiroz JC, Guimaraes-Ferreira L, Vitzel KF, Nachbar RT, de Sousa LG, de Souza-Jr AL, Nunes MT, Curi R: Local injections of adipose-derived mesenchymal stem cells modulate inflammation and increase angiogenesis ameliorating the dystrophic phenotype in dystrophin-deficient skeletal muscle. Stem Cell Rev. 2012, 8: 363-374. 10.1007/s12015-011-9304-0.
Leroux L, Descamps B, Tojais NF, Séguy B, Oses P, Moreau C, Daret D, Ivanovic Z, Boiron JM, Lamazière JM, Dufourcq P, Couffinhal T, Duplàa C: Hypoxia preconditioned mesenchymal stem cells improve vascular and skeletal muscle fiber regeneration after ischemia through a Wnt4-dependent pathway. Mol Ther. 2010, 18: 1545-1552. 10.1038/mt.2010.108.
Guan J, Wang F, Li Z, Chen J, Guo X, Liao J, Moldovan NI: The stimulation of the cardiac differentiation of mesenchymal stem cells in tissue constructs that mimic myocardium structure and biomechanics. Biomaterials. 2011, 32: 5568-5580. 10.1016/j.biomaterials.2011.04.038.
Kang Y, Park C, Kim D, Seong CM, Kwon K, Choi C: Unsorted human adipose tissue-derived stem cells promote angiogenesis and myogenesis in murine ischemic hindlimb model. Microvasc Res. 2010, 80: 310-316. 10.1016/j.mvr.2010.05.006.
Gong Z, Niklason LE: Small-diameter human vessel wall engineered from bone marrow-derived mesenchymal stem cells (hMSCs). FASEB J. 2008, 22: 1635-1648. 10.1096/fj.07-087924.
Dahl SL, Kypson AP, Lawson JH, Blum JL, Strader JT, Li Y, Manson RJ, Tente WE, DiBernardo L, Hensley MT, Carter R, Williams TP, Prichard HL, Dey MS, Begelman KG, Niklason LE: Readily available tissue-engineered vascular grafts. Sci Transl Med. 2011, 3: 68ra9-10.1126/scitranslmed.3001426.
Tian H, Bharadwaj S, Liu Y, Ma PX, Atala A, Zhang Y: Differentiation of human bone marrow mesenchymal stem cells into bladder cells: potential for urological tissue engineering. Tissue Eng Part A. 2010, 16: 1769-1779. 10.1089/ten.tea.2009.0625.
Zhang P, Baxter J, Vinod K, Tulenko TN, Di Muzio PJ: Endothelial differentiation of amniotic fluid-derived stem cells: synergism of biochemical and shear force stimuli. Stem Cells Dev. 2009, 18: 1299-1308. 10.1089/scd.2008.0331.
Marchionni C, Bonsi L, Alviano F, Lanzoni G, Di Tullio A, Costa R, Montanari M, Tazzari PL, Ricci F, Pasquinelli G, Orrico C, Grossi A, Prati C, Bagnara GP: Angiogenic potential of human dental pulp stromal (stem) cells. Int J Immunopathol Pharmacol. 2009, 22: 699-706.
Chen MY, Lie PC, Li ZL, Wei X: Endothelial differentiation of Wharton's jelly derived mesenchymal stem cells in comparison with bone marrow-derived mesenchymal stem cells. Exp Hematol. 2009, 37: 629-640. 10.1016/j.exphem.2009.02.003.
Wang H, Riha GM, Yan S, Li M, Chai H, Yang H, Yao Q, Chen C: Shear stress induces endothelial differentiation from a murine embryonic mesenchymal progenitor cell line. Arterioscler Thromb Vasc Biol. 2005, 25: 1817-1823. 10.1161/01.ATV.0000175840.90510.a8.
Duffy GP, D'Arcy S, Ahsan T, Nerem RM, O'Brien T, Barry F: Mesenchymal stem cells overexpressing ephrin-B2 rapidly adopt an early endothelial phenotype with simultaneous reduction of osteogenic potential. Tissue Eng Part A. 2010, 16: 2755-2768. 10.1089/ten.tea.2009.0623.
Liu JW, Dunoyer-Geindre S, Serre-Beinier V, Mai G, Lambert JF, Fish RJ, Pernod G, Buehler L, Bounameaux H, Kruithof EK: Characterization of endothelial-like cells derived from human mesenchymal stem cells. J Thromb Haemost. 2007, 5: 826-834. 10.1111/j.1538-7836.2007.02381.x.
Duffy GP, Ahsan T, O'Brien T, Barry F, Nerem RM: Bone marrow-derived mesenchymal stem cells promote angiogenic processes in a time- and dose-dependent manner in vitro. Tissue Eng Part A. 2009, 15: 2459-2470. 10.1089/ten.tea.2008.0341.
Amin AH, Abd Elmageed ZY, Nair D, Partyka MI, Kadowitz PJ, Belmadani S, Matrougui K: Modified multipotent stromal cells with epidermal growth factor restore vasculogenesis and blood flow in ischemic hind-limb of type II diabetic mice. Lab Invest. 2010, 90: 985-996. 10.1038/labinvest.2010.86.
Aguirre A, Planell JA, Engel E: Dynamics of bone marrow-derived endothelial progenitor cell/mesenchymal stem cell interaction in co-culture and its implications in angiogenesis. Biochem Biophys Res Commun. 2010, 400: 284-291. 10.1016/j.bbrc.2010.08.073.
Lozito TP, Kuo CK, Taboas JM, Tuan RS: Human mesenchymal stem cells express vascular cell phenotypes upon interaction with endothelial cell matrix. J Cell Biochem. 2009, 107: 714-722. 10.1002/jcb.22167.
Dufourcq P, Descamps B, Tojais NF, Leroux L, Oses P, Daret D, Moreau C, Lamaziere JM, Couffinhal T, Duplaa C: Secreted frizzled-related protein-1 enhances mesenchymal stem cell function in angiogenesis and contributes to neovessel maturation. Stem Cells. 2008, 26: 2991-3001. 10.1634/stemcells.2008-0372.
Griffin MD, Ritter T, Mahon BP: Immunological aspects of allogeneic mesenchymal stem cell therapies. Hum Gene Ther. 2010, 21: 1641-1655. 10.1089/hum.2010.156.
Ishikane S, Yamahara K, Sada M, Harada K, Kodama M, Ishibashi-Ueda H, Hayakawa K, Mishima K, Iwasaki K, Fujiwara M, Kangawa K, Ikeda T: Allogeneic administration of fetal membrane-derived mesenchymal stem cells attenuates acute myocarditis in rats. J Mol Cell Cardiol. 2010, 49: 753-761. 10.1016/j.yjmcc.2010.07.019.
Prather WR, Toren A, Meiron M, Ofir R, Tschope C, Horwitz EM: The role of placental-derived adherent stromal cell (PLX-PAD) in the treatment of critical limb ischemia. Cytotherapy. 2009, 11: 427-434. 10.1080/14653240902849762.
Pati S, Khakoo AY, Zhao J, Jimenez F, Gerber MH, Harting M, Redell JB, Grill R, Matsuo Y, Guha S, Cox CS, Reitz MS, Holcomb JB, Dash PK: Human mesenchymal stem cells inhibit vascular permeability by modulating vascular endothelial cadherin/beta-catenin signaling. Stem Cells Dev. 2011, 20: 89-101. 10.1089/scd.2010.0013.
Wang D, Chen K, Du WT, Han ZB, Ren H, Chi Y, Yang SG, Bayard F, Zhu D, Han ZC: CD14+ monocytes promote the immunosuppressive effect of human umbilical cord matrix stem cells. Exp Cell Res. 2010, 316: 2414-2423. 10.1016/j.yexcr.2010.04.018.
Hu X, Wang J, Chen J, Luo R, He A, Xie X, Li J: Optimal temporal delivery of bone marrow mesenchymal stem cells in rats with myocardial infarction. Eur J Cardiothorac Surg. 2007, 31: 438-443. 10.1016/j.ejcts.2006.11.057.
Jiang CY, Gui C, He AN, Hu XY, Chen J, Jiang Y, Wang JA: Optimal time for mesenchymal stem cell transplantation in rats with myocardial infarction. J Zhejiang Univ Sci B. 2008, 9: 630-637. 10.1631/jzus.B0820004.
Ankrum J, Karp JM: Mesenchymal stem cell therapy: two steps forward, one step back. Trends Mol Med. 2010, 16: 203-209. 10.1016/j.molmed.2010.02.005.
Wakitani S, Okabe T, Horibe S, Mitsuoka T, Saito M, Koyama T, Nawata M, Tensho K, Kato H, Uematsu K, Kuroda R, Kurosaka M, Yoshiya S, Hattori K, Ohgushi H: Safety of autologous bone marrow-derived mesenchymal stem cell transplantation for cartilage repair in 41 patients with 45 joints followed for up to 11 years and 5 months. J Tissue Eng Regen Med. 2011, 5: 146-150. 10.1002/term.299.
Karussis D, Karageorgiou C, Vaknin-Dembinsky A, Gowda-Kurkalli B, Gomori JM, Kassis I, Bulte JW, Petrou P, Ben-Hur T, Abramsky O, Slavin S: Safety and immunological effects of mesenchymal stem cell transplantation in patients with multiple sclerosis and amyotrophic lateral sclerosis. Arch Neurol. 2010, 67: 1187-1194. 10.1001/archneurol.2010.248.
Lee JS, Hong JM, Moon GJ, Lee PH, Ahn YH, Bang OY: A long-term follow-up study of intravenous autologous mesenchymal stem cell transplantation in patients with ischemic stroke. Stem Cells. 2010, 28: 1099-1106. 10.1002/stem.430.
Duijvestein M, Vos AC, Roelofs H, Wildenberg ME, Wendrich BB, Verspaget HW, Kooy-Winkelaar EM, Koning F, Zwaginga JJ, Fidder HH, Verhaar AP, Fibbe WE, van den Brink GR, Hommes DW: Autologous bone marrow-derived mesenchymal stromal cell treatment for refractory luminal Crohn's disease: results of a phase I study. Gut. 2010, 59: 1662-1669. 10.1136/gut.2010.215152.
Hare JM, Traverse JH, Henry TD, Dib N, Strumpf RK, Schulman SP, Gerstenblith G, DeMaria AN, Denktas AE, Gammon RS, Hermiller JB, Reisman MA, Schaer GL, Sherman W: A randomized, double-blind, placebo-controlled, dose-escalation study of intravenous adult human mesenchymal stem cells (prochymal) after acute myocardial infarction. J Am Coll Cardiol. 2009, 54: 2277-2286. 10.1016/j.jacc.2009.06.055.
Rosova I, Dao M, Capoccia B, Link D, Nolta JA: Hypoxic preconditioning results in increased motility and improved therapeutic potential of human mesenchymal stem cells. Stem Cells. 2008, 26: 2173-2182. 10.1634/stemcells.2007-1104.
Rosova I, Link D, Nolta JA: shRNA-mediated decreases in c-Met levels affect the differentiation potential of human mesenchymal stem cells and reduce their capacity for tissue repair. Tissue Eng Part A. 2010, 16: 2627-2639. 10.1089/ten.tea.2009.0363.
Nakagami H, Morishita R, Maeda K, Kikuchi Y, Ogihara T, Kaneda Y: Adipose tissue-derived stromal cells as a novel option for regenerative cell therapy. J Atheroscler Thromb. 2006, 13: 77-81. 10.5551/jat.13.77.
Zhang D, Li Y, Zhu T, Zhang F, Yang Z, Miao D: Zinc supplementation results in improved therapeutic potential of bone marrow-derived mesenchymal stromal cells in a mouse ischemic limb model. Cytotherapy. 2011, 13: 156-164. 10.3109/14653249.2010.512633.
Zhang Y, Zhang R, Li Y, He G, Zhang D, Zhang F: Simvastatin augments the efficacy of therapeutic angiogenesis induced by bone marrow-derived mesenchymal stem cells in a murine model of hindlimb ischemia. Mol Biol Rep. 2012, 39: 285-293. 10.1007/s11033-011-0737-y.
Li Y, Zhang D, Zhang Y, He G, Zhang F: Augmentation of neovascularization in murine hindlimb ischemia by combined therapy with simvastatin and bone marrow-derived mesenchymal stem cells transplantation. J Biomed Sci. 2010, 17: 75-10.1186/1423-0127-17-75.
Ishii M, Numaguchi Y, Okumura K, Kubota R, Ma X, Murakami R, Naruse K, Murohara T: Mesenchymal stem cell-based gene therapy with prostacyclin synthase enhanced neovascularization in hindlimb ischemia. Atherosclerosis. 2009, 206: 109-118. 10.1016/j.atherosclerosis.2009.02.023.
Murphy MP, Wang H, Patel AN, Kambhampati S, Angle N, Chan K, Marleau AM, Pyszniak A, Carrier E, Ichim TE, Riordan NH: Allogeneic endometrial regenerative cells: an "Off the shelf solution" for critical limb ischemia?. J Transl Med. 2008, 6: 45-10.1186/1479-5876-6-45.
Al-Khaldi A, Al-Sabti H, Galipeau J, Lachapelle K: Therapeutic angiogenesis using autologous bone marrow stromal cells: improved blood flow in a chronic limb ischemia model. Ann Thorac Surg. 2003, 75: 204-209. 10.1016/S0003-4975(02)04291-1.
Piao W, Wang H, Inoue M, Hasegawa M, Hamada H, Huang J: Transplantation of sendai viral angiopoietin-1-modified mesenchymal stem cells for ischemic limb disease. Angiogenesis. 2010, 13: 203-210. 10.1007/s10456-010-9169-x.
Li Q, Yao D, Ma J, Zhu J, Xu X, Ren Y, Ding X, Mao X: Transplantation of MSCs in combination with netrin-1 improves neoangiogenesis in a rat model of hind limb ischemia. J Surg Res. 2011, 166: 162-169. 10.1016/j.jss.2009.08.031.
Iwashima S, Ozaki T, Maruyama S, Saka Y, Kobori M, Omae K, Yamaguchi H, Niimi T, Toriyama K, Kamei Y, Torii S, Murohara T, Yuzawa Y, Kitagawa Y, Matsuo S: Novel culture system of mesenchymal stromal cells from human subcutaneous adipose tissue. Stem Cells Dev. 2009, 18: 533-543. 10.1089/scd.2008.0358.
Moon MH, Kim SY, Kim YJ, Kim SJ, Lee JB, Bae YC, Sung SM, Jung JS: Human adipose tissue-derived mesenchymal stem cells improve postnatal neovascularization in a mouse model of hindlimb ischemia. Cell Physiol Biochem. 2006, 17: 279-290. 10.1159/000094140.
Bhang SH, Cho SW, La WG, Lee TJ, Yang HS, Sun AY, Baek SH, Rhie JW, Kim BS: Angiogenesis in ischemic tissue produced by spheroid grafting of human adipose-derived stromal cells. Biomaterials. 2011, 32: 2734-2747. 10.1016/j.biomaterials.2010.12.035.
Bhang SH, Cho SW, Lim JM, Kang JM, Lee TJ, Yang HS, Song YS, Park MH, Kim HS, Yoo KJ, Jang Y, Langer R, Anderson DG, Kim BS: Locally delivered growth factor enhances the angiogenic efficacy of adipose-derived stromal cells transplanted to ischemic limbs. Stem Cells. 2009, 27: 1976-1986. 10.1002/stem.115.
Cho HH, Kim YJ, Kim JT, Song JS, Shin KK, Bae YC, Jung JS: The role of chemokines in proangiogenic action induced by human adipose tissue-derived mesenchymal stem cells in the murine model of hindlimb ischemia. Cell Physiol Biochem. 2009, 24: 511-518. 10.1159/000257495.
Laurila JP, Laatikainen L, Castellone MD, Trivedi P, Heikkila J, Hinkkanen A, Hematti P, Laukkanen MO: Human embryonic stem cell-derived mesenchymal stromal cell transplantation in a rat hind limb injury model. Cytotherapy. 2009, 11: 726-737. 10.3109/14653240903067299.
Kim HG, Choi OH: Neovascularization in a mouse model via stem cells derived from human fetal amniotic membranes. Heart Vessels. 2011, 26: 196-205. 10.1007/s00380-010-0064-6.
Nishishita T, Ouchi K, Zhang X, Inoue M, Inazawa T, Yoshiura K, Kuwabara K, Nakaoka T, Watanabe N, Igura K, Takahashi TA, Yamashita N: A potential pro-angiogenic cell therapy with human placenta-derived mesenchymal cells. Biochem Biophys Res Commun. 2004, 325: 24-31. 10.1016/j.bbrc.2004.10.003.
Bhang SH, Lee TJ, La WG, Kim DI, Kim BS: Delivery of fibroblast growth factor 2 enhances the viability of cord blood-derived mesenchymal stem cells transplanted to ischemic limbs. J Biosci Bioeng. 2011, 111: 584-589. 10.1016/j.jbiosc.2011.01.003.
Kim SW, Han H, Chae GT, Lee SH, Bo S, Yoon JH, Lee YS, Lee KS, Park HK, Kang KS: Successful stem cell therapy using umbilical cord blood-derived multipotent stem cells for Buerger's disease and ischemic limb disease animal model. Stem Cells. 2006, 24: 1620-1626. 10.1634/stemcells.2005-0365.
Koponen JK, Kekarainen T, S EH, Laitinen A, Nystedt J, Laine J, Yla-Herttuala S: Umbilical cord blood-derived progenitor cells enhance muscle regeneration in mouse hindlimb ischemia model. Mol Ther. 2007, 15: 2172-2177. 10.1038/sj.mt.6300302.
Dash NR, Dash SN, Routray P, Mohapatra S, Mohapatra PC: Targeting nonhealing ulcers of lower extremity in human through autologous bone marrow-derived mesenchymal stem cells. Rejuvenation Res. 2009, 12: 359-366. 10.1089/rej.2009.0872.
Guiducci S, Porta F, Saccardi R, Guidi S, Ibba-Manneschi L, Manetti M, Mazzanti B, Dal Pozzo S, Milia AF, Bellando-Randone S, Miniati I, Fiori G, Fontana R, Amanzi L, Braschi F, Bosi A, Matucci-Cerinic M: Autologous mesenchymal stem cells foster revascularization of ischemic limbs in systemic sclerosis: a case report. Ann Intern Med. 2010, 153: 650-654.
Lasala GP, Silva JA, Gardner PA, Minguell JJ: Combination stem cell therapy for the treatment of severe limb ischemia: safety and efficacy analysis. Angiology. 2010, 61: 551-556. 10.1177/0003319710364213.
Lasala GP, Silva JA, Minguell JJ: Therapeutic angiogenesis in patients with severe limb ischemia by transplantation of a combination stem cell product. J Thorac Cardiovasc Surg. 2012, 144: 377-382. 10.1016/j.jtcvs.2011.08.053.
Lu D, Chen B, Liang Z, Deng W, Jiang Y, Li S, Xu J, Wu Q, Zhang Z, Xie B, Chen S: Comparison of bone marrow mesenchymal stem cells with bone marrow-derived mononuclear cells for treatment of diabetic critical limb ischemia and foot ulcer: a double-blind, randomized, controlled trial. Diabetes Res Clin Pract. 2011, 92: 26-36. 10.1016/j.diabres.2010.12.010.
Lee HC, An SG, Lee HW, Park JS, Cha KS, Hong TJ, Park JH, Lee SY, Kim SP, Kim YD, Chung SW, Bae YC, Shin YB, Kim JI, Jung JS: Safety and effect of adipose tissue-derived stem cell implantation in patients with critical limb ischemia. Circ J. 2012, 76: 1750-1760.
Stempeutics announces clinical trial outcome of India's first stem cell product stempeucel. [http://www.stempeutics.com/html/Article%201.pdf]
Pluristem Therapeutics, Inc. (PSTI)-Buy. [http://www.pluristem.com/CPY155053[1].pdf]
Conte MS: Understanding objective performance goals for critical limb ischemia trials. Semin Vasc Surg. 2010, 23: 129-137. 10.1053/j.semvascsurg.2010.06.001.
Acknowledgements
This work was supported by Science Foundation Ireland (SFI), Strategic Research Cluster (SRC), Grant No. SFI: 09/SRC B1794 and the European Regional Development Fund.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
TO is a founder and director of and equity holder in Orbsen Therapeutics Ltd. (Galway, Ireland). AL declares that he has no competing interests.
Rights and permissions
About this article
Cite this article
Liew, A., O'Brien, T. Therapeutic potential for mesenchymal stem cell transplantation in critical limb ischemia. Stem Cell Res Ther 3, 28 (2012). https://doi.org/10.1186/scrt119
Published:
DOI: https://doi.org/10.1186/scrt119