
Abstract
Background
Liver fibrosis results from chronic liver injury and is characterized by excessive deposition of extracellular matrix proteins including collagen. It can progress to cirrhosis and liver failure.
Main body of the abstract
Multiple cellular signaling pathways drive hepatic stellate cell activation and fibrogenesis. Advances in biomarkers, imaging modalities, and omics platforms enable noninvasive diagnosis and staging of liver fibrosis. Emerging antifibrotic approaches include medications like pirfenidone, obeticholic acid, and monoclonal antibodies targeting pro-fibrotic mediators. Cell therapies using mesenchymal stem cells demonstrate antifibrotic potential through paracrine immunosuppression. Tissue-engineered liver grafts and biomaterial carriers for localized drug delivery are promising technologies. Microfluidic liver-on-a-chip platforms with patient-derived cells provide unprecedented models to study human liver fibrosis and test drug candidates.
Short conclusion
Significant progress has elucidated mechanisms underlying liver fibrogenesis and uncovered novel therapeutic targets. Ongoing challenges include translating preclinical findings, improving antifibrotic efficacy, and enabling personalized precision medicine approaches. Further research into combinatorial therapies, biomarkers, and tissue engineering technologies will advance the treatment of liver fibrosis from all causes.
Similar content being viewed by others


Background
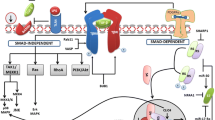
Liver fibrosis involves excessive deposition of extracellular matrix proteins like collagen. It occurs due to chronic liver injury and is an active wound-healing response that can progress to cirrhosis [1]. Fibrosis develops from sustained inflammation caused by chronic viral hepatitis, fatty liver disease, alcohol, autoimmune disorders, metabolic disorders, and cholestatic diseases [2]. During the initiation of liver injury, the activation of Kupffer cells and recruitment of inflammatory immune cells result in the release of cytokines, chemokines, and growth factors that trigger a response from hepatic stellate cells. Hepatic stellate cells are the major effector cells that become activated, proliferate, and transform into myofibroblast-like cells that deposit excessive amounts of extracellular matrix [3]. Key cytokines and growth factors involved in this process include transforming growth factor beta, platelet-derived growth factor, connective tissue growth factor, and endothelin-1. Activated stellate cells increase their expression of alpha-smooth muscle actin and exhibit contractile, proinflammatory, and fibrogenic properties [4]. The pathological accumulation of extracellular matrix in liver fibrosis occurs primarily from increased synthesis and deposition of collagen type I and type III. This is mediated by activated stellate cells which upregulate tissue inhibitors of metalloproteinases, causing an imbalance in extracellular matrix turnover favoring excessive accumulation. In addition to activated stellate cells, other cell types contribute to the fibrogenic response including portal fibroblasts, bone marrow-derived fibroblasts, and hepatocytes undergoing epithelial to mesenchymal transition. Chronic inflammation also perpetuates liver fibrosis through paracrine signaling between macrophages, stellate cells, and damaged hepatocytes. Figure 1 illustrates the role of hepatic stellate cells (HSCs) in the progression and regression of liver fibrosis. In a healthy liver, HSCs are quiescent (qHSCs). Liver injury triggers qHSC activation mediated by factors like hepatocytes, liver sinusoidal endothelial cells (LSECs), macrophages, transforming growth factor-beta (TGF-β), and platelet-derived growth factor (PDGF). Activated HSCs (aHSCs) proliferate, secrete excessive extracellular matrix (ECM) proteins, and promote fibrosis. Fibrosis can be reversed by inhibiting aHSC proliferation, inducing aHSC death, or inactivating aHSCs (iHSCs) [5]. Liver fibrosis results from chronic damage from etiologies like viral hepatitis and can progress to cirrhosis with distortion of hepatic architecture and regenerative nodules. Cirrhosis has complications like portal hypertension and liver failure. Accurate fibrosis staging is crucial for prognosis, guiding treatment, and monitoring progression. Liver fibrosis advances through stages of increasing collagen deposition and architectural distortion. Scoring systems categorize fibrosis stages based on patterns of fibrotic areas [6, 7]. Collagen first appears around vessels, then bridges portal and central areas causing distortion and nodules-defining cirrhosis [8]. Cirrhosis disrupts liver function and blood flow, increasing complications like cancer, bleeding, ascites, infections, and organ failure [9,10,11,12,13]. Genetic and epigenetic factors also influence fibrosis progression [14]. Biomarkers and imaging techniques are being developed for noninvasive diagnosis [15]. This review comprehensively summarizes recent advances across the spectrum of liver fibrosis research, from elucidating molecular pathways to emerging diagnostics, biomarker technologies, and novel therapeutic candidates. Notably, we highlight promising areas including stem cell, tissue engineering, nanotechnology, and microfluidic model approaches offering hope for personalized, precision antifibrotic therapies based on rapidly expanding knowledge of pathways underlying fibrogenesis. The background explains the focus is providing a comprehensive review of the multifaceted progress in understanding and treating liver fibrosis. It emphasizes emerging tools enabling precision, and personalized approaches as a key aspect differentiating this review.

Role of hepatic stellate cells in liver fibrosis progression and reversal [5]
Cellular and molecular mechanisms
Liver fibrosis results from a complex interplay between multiple cell types and signaling pathways that together stimulate hepatic stellate cell activation and excessive extracellular matrix (ECM) production and remodeling. Key events include hepatic stellate cell (HSC) activation into proliferative, fibrogenic myofibroblasts under paracrine stimuli from damaged hepatocytes and cells like Kupffer cells releasing inflammatory cytokines like tumor necrosis factor-alpha (TNF-α) and interleukin-6 (IL-6) [16]. Activated HSCs upregulate ECM components like fibrillar collagens and tissue inhibitors of metalloproteinases (TIMPs). Chronic inflammation further drives fibrosis progression through recruited leukocytes secreting profibrogenic cytokines. Epithelial-mesenchymal transition (EMT) of hepatocytes into migratory, invasive fibroblast-like cells is triggered by stimuli like transforming growth factor-beta (TGF-β) activating transcription factors like Snail and Zeb [17]. Strategies targeting HSC activation, inflammation, EMT, oxidative stress, and extracellular matrix balance show promise for antifibrotic therapies.
Genetic and epigenetic regulation
Liver fibrosis progression and severity exhibit interindividual variability influenced by genetic and epigenetic factors. Polymorphisms in genes encoding pro- and antifibrogenic mediators can regulate an individual’s fibrotic response to injury. Epigenetic changes including DNA methylation, histone modifications, and non-coding RNAs control hepatic stellate cell activation and profibrogenic gene expression. Functional polymorphisms affecting cytokines, the renin-angiotensin system, collagen remodeling enzymes like matrix metalloproteinases (MMPs), and genes regulating oxidative stress and immunity modify fibrosis progression [18]. Genome-wide association studies have uncovered additional genetic loci correlated with fibrosis severity, like toll-like receptor 4 (TLR4), neurocan (NCAN), and patatin-like phospholipase domain containing 3 (PNPLA3) variants [19]. Key epigenetic mechanisms include DNA methylation, post-translational histone modifications, and non-coding RNAs such as microRNAs (miRNAs). Hypermethylation of antifibrotic genes promotes stellate cell activation. MiR-29 downregulation correlates with progression [20]. Histone modifications alter chromatin structure to promote profibrogenic gene transcription. Manipulating these changes using histone deacetylase inhibitors or miR-29 restoration reduced fibrosis in models [21]. Hepatic stellate cells exhibit major DNA methylation reprogramming during activation warranting further epigenome analysis [22].
Molecular pathogenesis of liver fibrosis in major liver diseases
Liver fibrosis can occur as a consequence of virtually all chronic liver diseases. However, the molecular triggers for hepatic stellate cell activation and fibrogenesis show some variations between different disease etiologies. In chronic hepatitis C virus (HCV) infection, fibrosis results from hepatic inflammation triggered by the immune response against the virus. HCV proteins directly activate stellate cells through oxidative stress. Loss of antifibrotic miRNAs like miR-29 contributes to fibrosis. Hepatitis B virus (HBV) similarly elicits fibrosis indirectly through immune-mediated inflammation and directly through profibrogenic viral factors altering the epigenetic regulation of genes in stellate cells. Effective antiviral suppression can normalize fibrosis. In nonalcoholic fatty liver disease (NAFLD), fibrosis is driven by inflammation from fat accumulation, insulin resistance, oxidative stress, and lipotoxicity activating Kupffer cells and recruiting macrophages. Hepatocyte apoptosis from lipotoxicity provokes stellate cell matrix deposition [23]. Oxidative stress is a key driver in NAFLD and alcoholic liver disease, with alcohol metabolism generating reactive oxygen species inducing inflammatory and fibrogenic genes. In cholestatic diseases like primary biliary cholangitis, fibrosis occurs from bile acid-induced cholangiocyte and hepatocyte apoptosis recruiting immune cells to stimulate stellate cells. While shared pathways potentiate fibrosis across diseases, elucidating molecular nuances related to the initial injury is imperative for targeted therapies, like suppressing inflammation, inhibiting lipotoxicity, and oxidative stress, blocking stellate activation, and normalizing dysregulated epigenetic/metabolic pathways [24].
Emerging biomarkers for liver fibrosis assessment
The development of non-invasive biomarkers to diagnose and stage liver fibrosis is an area of intense research. Beyond traditional protein and metabolic biomarkers, newer classes include microRNAs (miRNAs), long non-coding RNAs (lncRNAs) and extracellular vesicles (EVs), and their molecular cargos. Multiple miRNA families show altered expression during fibrogenesis and directly target extracellular matrix component mRNAs for repression. Declining miR-29 levels correlate with the advancing fibrosis stage. A 5-miRNA panel accurately differentiated significant fibrosis in nonalcoholic fatty liver disease. LncRNAs (long non-coding RNAs) are RNA transcripts longer than 200 nucleotides that regulate gene expression but do not encode proteins. In fibrotic diseases affecting the liver, lungs, kidneys, and heart, the expression levels of many lncRNAs become dysregulated. One prominent example is HOTAIR (HOX transcript antisense RNA), which shows increased expression across multiple fibrotic diseases. Higher circulating blood levels of HOTAIR correlate with more severe fibrosis and worse clinical outcomes, suggesting it could potentially serve as a biomarker for fibrosis severity. Mechanistically, HOTAIR interacts with chromatin remodeling complexes and can silence antifibrotic genes. Overexpression of HOTAIR promotes collagen deposition and myofibroblast differentiation. Other pro-fibrotic lncRNAs include MEG3, PVT1, and FENDRR, while GAS5 and lncRNA-ATB have antifibrotic effects. Modulating the levels of dysregulated lncRNAs impacts fibrosis progression in preclinical disease models. The functional roles and dysregulation of lncRNAs like HOTAIR in fibrosis point to their potential as biomarkers and therapeutic targets in fibrotic diseases across different organs [25]. LncRNA panels may enhance diagnostic accuracy. EVs provide an enriched liquid biopsy source of miRNAs, proteins, mRNAs, and lipids reflective of liver status. Exosome surface proteins and miRNAs distinguished mild from significant fibrosis in chronic hepatitis C. Better characterization of liver-specific EVs is still needed [26, 27].
Advanced imaging techniques for assessing liver fibrosis and cirrhosis
Liver fibrosis and cirrhosis represent advanced stages of chronic liver disease characterized by excessive extracellular matrix protein accumulation, including collagen. Liver biopsy has limitations including invasiveness, sampling errors, and qualitative scoring. Noninvasive quantitative methods for assessing fibrosis are increasingly of interest for safe, dynamic monitoring. Advanced imaging techniques show promise, including elastography methods, MRI-based techniques, and second harmonic generation microscopy [28, 29].
Elastography methods
Elastography methods assess the mechanical properties of tissues by measuring their response to applied stress. Tissues stiffen with fibrosis since collagen deposition increases elastic modulus. Methods used to assess liver fibrosis include transient elastography, shear wave elastography, and magnetic resonance elastography (MRE). Transient elastography involves inducing a shear wave using a vibrating probe on the skin to measure its velocity, which correlates with tissue stiffness. Higher velocity indicates more advanced fibrosis [30]. It is rapid and validated but can be limited by obesity. Shear wave elastography methods include point shear wave elastography and 2D shear wave elastography. These induce localized shear waves in the liver to quantify stiffness. 2D methods generate stiffness maps across larger regions. Shear wave elastography accurately stages fibrosis and avoids limitations of ribs/vessels. MRE involves inducing shear waves using a driver on the body wall and imaging propagation using MRI. This generates quantitative maps of tissue stiffness. MRE is highly accurate for staging fibrosis due to sampling larger regions and quantitative measurements. However, MRE requires specialized equipment and expertise. Still, MRE is increasingly used in clinical trials as a noninvasive reference standard [31].
MRI-based techniques
Beyond magnetic resonance elastography, other MRI-based imaging techniques are being developed to noninvasively assess liver fibrosis, including gadoxetate MRI which provides functional information about liver function and fibrosis. Gadoxetate is a liver-specific contrast agent taken up by hepatocytes. Delayed phase gadoxetate MRI identifies alterations in liver function and contrast uptake kinetics associated with chronic liver disease that correlate with increased fibrosis stages [32,33,34].
Machine learning applied to multiphasic gadoxetate MRI can accurately stage fibrosis by detecting complex hepatic function loss patterns. Gadoxetate MRI also calculates the relative enhancement ratio that reflects hepatocyte uptake function, correlates with worsening fibrosis, and predicts clinical outcomes. Gadoxetate MRI provides concurrent quantitative functional information related to liver fibrosis severity [35].
Second harmonic generation microscopy
Second harmonic generation (SHG) microscopy allows visualization of tissue collagen fibers without exogenous stains by generating optical signals specifically from fibrillar collagen. SHG microscopy of unstained liver tissue can visualize collagen architecture, quantify collagen content, and determine the collagen-to-parenchyma ratio. Second harmonic generation microscopy distinguishes significant fibrosis from mild disease and normal livers. Since collagen remodeling is a key event during fibrogenesis, the second harmonic generation provides direct quantitative information on progression [36,37,38]. A major advantage is the ability to perform in vivo longitudinal monitoring of liver fibrosis to assess response to therapies. In animal models, SHG microscopy detected early regression of fibrosis reflected by decreasing collagen signals following injury resolution. Potential clinical uses include SHG endomicroscopy to quantify and monitor fibrosis. Limitations include limited depth penetration and sampling variability. Overall, second harmonic generation microscopy enables direct noninvasive visualization of liver collagen alterations occurring with fibrogenesis and fibrosis resolution [39]. Table 1 summarizes key parameters, strengths, and limitations of leading noninvasive technologies spanning imaging modalities, circulating biomarkers, and multi-omics data integration for assessing liver fibrosis stage. It provides an overview of evaluating these emerging tools.
Artificial intelligence and big data applications in clinical research
The advent of artificial intelligence (AI) and big data has opened new frontiers in clinical research. In chronic liver disease, AI and big data analytics are being applied in areas including image analysis, biomarker discovery, disease subgroups, and drug discovery [40,41,42,43]. A major application of AI is the automated analysis of medical images to detect liver pathology. AI image analysis algorithms can rapidly quantify specific features in a standardized, objective manner. For example, algorithms can evaluate liver collagen as a marker of the fibrosis stage from histopathology slides stained to enhance collagen visualization. Studies have shown AI collagen quantitation strongly correlates with stage and outcomes, outperforming qualitative scoring [44, 45]. Algorithms can also assess additional histological features like cell ballooning and inflammation. Integrating analysis of multiple domain-specific features allows AI assessment of biopsy samples rivaling or exceeding pathologists. Radiomic analysis extracts quantitative data from medical images by evaluating relationships between voxels. Algorithms can discern texture and shape patterns in MRI associated with fibrosis stages. Models identified imaging biomarkers related to the stage with high accuracy. Radiomics has the potential to identify prognostic imaging biomarkers for monitoring. Integrating multidimensional molecular data is another promising application. Machine learning can integrate genomics, transcriptomics, proteomics, metabolomics, and microbiome data from patients. Analysis can identify molecular signatures differentiating disease subclasses and predict trajectories. For example, a meta-analysis combining serum metabolomics data from over 1000 nonalcoholic fatty liver disease patients revealed distinct subgroups, including increased severe fibrosis risk, and prognostic biomarkers [46]. Harmonizing big data uncovers insights not discernible when analyzing individual datasets. Big data also aids drug discovery through high-throughput screening and machine learning to identify antifibrotic candidates, refine hits in models, and facilitate in silico modeling of drug interactions. AI-enabled discovery expedites the search for promising new liver disease therapeutics [47].
Molecular targets for antifibrotic therapies in liver fibrosis
Extensive research into the cellular and molecular mechanisms driving liver fibrosis has uncovered numerous potential therapeutic targets to halt or reverse fibrosis progression. Key strategies include directly targeting activated hepatic stellate cells, inhibiting proinflammatory and profibrogenic signaling pathways, blocking epithelial-mesenchymal transition, and normalizing extracellular matrix remodeling. Hepatic stellate cells are the predominant collagen-producing myofibroblast-like cells in the fibrotic liver. Selectively inducing stellate cell apoptosis or reverting their activation state represents a direct antifibrotic approach [48]. Inflammation is a major driver of liver fibrosis, making inflammatory signaling pathways like TNF-α, TGF-β, IL-6, toll-like receptor 4, and JAK/STAT key targets. Anti-TNF biologics like infliximab reduce hepatitis and fibrosis in animal models. Blocking TGF-β signaling with neutralizing antibodies or TGF-β receptor inhibitors effectively inhibits collagen deposition [49]. Oxidative stress generated during chronic liver injury directly activates stellate cells and increases collagen deposition. Antioxidants like vitamin E, superoxide dismutase (SOD) mimetics, and nuclear factor erythroid 2-related factor 2 (NRF2) activators inhibit stellate cell activation in preclinical studies. Imbalances between matrix metalloproteinases (MMPs) that degrade extracellular matrix and their inhibitors TIMPs drive net collagen accumulation in fibrosis. Recombinant MMPs like MMP-8 and MMP-9 degrade fibrotic matrix in preclinical models [50, 51].
Novel interventions for liver fibrosis
As understanding of the molecular mechanisms of liver fibrosis has advanced, numerous therapeutic approaches have emerged to target different aspects of the pathogenesis. These novel antifibrotic interventions include medications, stem cell therapies, RNA therapeutics, artificial liver technology, transplantation, and early-stage interventions undergoing preclinical investigation. Several antifibrotic medications are under clinical investigation after showing promise in animal models. Pirfenidone is an oral agent with anti-inflammatory, antioxidant, and antifibrotic properties. By inhibiting TGF-β and TNF-α, pirfenidone reduced fibrosis progression in idiopathic pulmonary fibrosis (IPF) patients and is undergoing trials for liver fibrosis. The farnesoid X nuclear receptor agonist obeticholic acid improved fibrosis in NASH clinical trials, likely by suppressing stellate cell activation and inflammation. Pentoxifylline, a TNF-α inhibitor, also exhibited antifibrotic effects in patients by reducing inflammation [52]. Cell therapies aim to repopulate the liver with healthy cells or deliver antifibrotic factors. Mesenchymal stem cells (MSCs) are administered intravenously home to injured tissues and secrete immunomodulatory and regenerative factors. MSC therapy alleviated fibrosis in animal models through paracrine effects on stellate cells. Early-stage trials found that MSCs were safe and improved liver function in cirrhosis patients. Co-administration with MSCs may enhance hepatocyte engraftment, while engineered MSCs overexpressing MMPs could boost collagen degradation. Hematopoietic stem cell therapy also improved fibrosis in early clinical studies potentially by promoting hepatic regeneration [53]. RNA interference therapeutics using small interfering RNAs (siRNAs) allow specific silencing of disease-causing genes. SiRNAs targeting collagen, Timp1, integrin β1, and heat shock proteins have shown antifibrotic effects in rodent models. CRISPR/Cas9 gene editing could correct disease-causing mutations or delete pro-fibrogenic genes. Induced pluripotent stem cell-derived hepatocytes offer another cell-based approach, although challenges exist in maintaining function and integration [54]. Artificial liver support systems aim to bridge patients with acute liver failure to transplant or recovery. The molecular adsorbents recirculating system (MARS) removes circulating albumin-bound toxins using dialysis and adsorption columns. Bioartificial liver devices contain hepatocytes or stem cell-derived hepatocyte-like cells to provide metabolic and synthetic support. While promising, the clinical efficacy of these technologies requires further evaluation [55]. Liver transplantation remains the only curative option for end-stage cirrhosis. However, organ shortages and the need for lifelong immunosuppression limit its applicability. Emerging approaches like hepatocyte transplantation, bioengineered liver grafts, and 3D bio-printed liver tissues could eventually expand curative options [56]. Looking ahead, several preclinical stage interventions show promise for translating into antifibrotic therapies. These include engineered exosomes or nanoparticles for targeted siRNA/microRNA delivery, compounds targeting epigenetic modifiers, and modulating gut-liver axes implicated in fibrosis. Integrating cutting-edge techniques like nanotechnology, induced pluripotent stem cells, and gene editing could enable personalized cell and gene therapies. While significant challenges remain, the expanding antifibrotic pipeline offers hope for treating this major unmet need [57].
Preclinical studies targeting molecular pathways implicated in liver fibrosis
Preclinical research in cell and animal models has enabled extensive investigation into targeting the major molecular pathways that drive liver fibrosis progression. Strategies undergoing active investigation at the preclinical stage include blocking the profibrogenic cytokine TGF-β, suppressing inflammation via cytokines like TNF-α and IL-6, inhibiting oxidative stress through antioxidant activation, and preventing epithelial-mesenchymal transition. The TGF-β signaling pathway is a predominant target for antifibrotic drug development due to its critical role in stellate cell activation and fibrosis. Neutralizing antibodies against all three TGF-β isoforms (TGF-β 1, 2, and 3) demonstrate antifibrotic effects in animal models through reducing inflammation, matrix deposition, and stellate cell activation [58]. Multiple TGF-β receptor inhibitors such as galunisertib, SB525334, and SM305 are in preclinical testing. These compounds blunt liver, lung, kidney, and cardiac fibrosis by blocking receptor-mediated Smad signaling [5, 59,60,61,62,63,64,65]. Anti-TNF biologics like infliximab demonstrate antifibrotic efficacy in rodent models by suppressing inflammatory cell infiltration and stellate cell activation. IL-6 neutralization with monoclonal antibodies attenuates stellate cell activation and collagen production in mice. Oxidative stress is another key driver of fibrogenesis [66, 67]. Antioxidants like vitamin E, SOD mimetics, and mitochondrial-targeted antioxidants reduce ROS production and associated inflammation and matrix deposition in animal studies. Inhibiting epithelial-mesenchymal transition represents another antifibrotic approach. Transcription factor inhibitors targeting Zeb, Snail, and Twist suppress epithelial-mesenchymal transition in vitro, reducing ECM production. Given the key role of inflammation in driving hepatic stellate cell activation and liver fibrosis progression, prophylactic vaccination approaches targeting high-risk inflammatory mediators may hold promise for halting or reversing fibrogenesis in susceptible individuals before significant matrix deposition and architectural damage ensues [68]. Overall, Table 2 concisely summarizes key details on preclinical studies targeting 5 major pro-fibrogenic pathways using various animal models and intervention strategies. It aims to provide a snapshot of cutting-edge antifibrotic approaches under active investigation at the preclinical stage.
Clinical trials of antifibrotic drugs for liver fibrosis
As the complex cellular and molecular mechanisms of liver fibrosis have been unraveled, a growing number of targeted antifibrotic compounds have progressed into clinical testing. Key drug classes reaching clinical trials include pirfenidone, obeticholic acid, and monoclonal antibodies against profibrotic factors and pathways. Pirfenidone is a small-molecule drug with anti-inflammatory, antioxidant, and antifibrotic properties. In cell and animal models, pirfenidone reduced fibrosis by decreasing TNF-α, TGF-β1, collagen I, and other pro-fibrogenic mediators. In a phase 2 trial for liver fibrosis, pirfenidone treatment for 1 year significantly reduced liver stiffness compared to placebo in chronic hepatitis C patients. Phase 3 trials FAILED-B and REGENERATE evaluating pirfenidone in non-alcoholic steatohepatitis (NASH) patients are ongoing [69]. Obeticholic acid is a synthetic bile acid agonist for the farnesoid X nuclear receptor (FXR). FXR activation is antifibrotic by inhibiting stellate cell activation and reducing inflammatory cytokine production. In the phase 2 FLINT trial for NASH treatment, obeticholic acid improved histological features including fibrosis. However, adverse effects included pruritis and elevated LDL cholesterol. The phase 3 REGENERATE study confirmed its fibrosis improvement, leading to recent FDA approval for NASH fibrosis [70]. Monoclonal antibody therapies are also emerging as antifibrotics by targeting specific mediators. Simtuzumab is a humanized monoclonal antibody against lysyl oxidase homolog 2 (LOXL2), an enzyme involved in collagen crosslinking. In phase 2 studies, simtuzumab treatment yielded significant hepatic collagen reductions in primary sclerosing cholangitis and NASH fibrosis patients. Another recently evaluated monoclonal antibody is BMS-986036, which blocks lysophosphatidic acid 1 receptor (LPA1) signaling, implicated in myofibroblast activation. Treatment for 15 weeks showed antifibrotic activity by reducing hepatic collagen, TGF-β1, and αSMA in a phase 2 study of patients with NASH and liver fibrosis [71]. Ongoing and planned trials are evaluating other compounds like emricasan, an apoptosis signal-regulating kinase 1 inhibitor, galectin inhibitors, and cytokine inhibitors. Despite the promise, challenges exist in replicating preclinical efficacy in humans and optimizing delivery specifically to the liver. Combination treatment regimens may augment the modest efficacy of single agents. Further research on bioactive phytoconstituents, targeted delivery, and clinical translation is needed to realize the full potential of nano-herbal antifibrotics [16, 72,73,74,75,76].
Tissue engineering and biomaterials for treating liver fibrosis
Tissue engineering and biomaterials offer new approaches to recreating functional liver tissue and deliver targeted antifibrotic therapies. Key technologies include decellularized liver scaffolds, biomaterial implants for localized drug delivery, and 3D bioprinting of liver constructs. Decellularized liver scaffolds leverage the innate extracellular matrix environment of the liver to promote implanted cell survival, differentiation, and tissue remodeling. In rodent models with liver injury, recellularized liver scaffolds improved survival compared to hepatocyte-only transplantation. Implanted cells proliferated, displayed metabolic activity, reduced fibrosis, and integrated with the host tissue. Challenges remain in recapitulating the intricate liver cell populations, perfusable vasculature, and biliary drainage system [77]. Controlled delivery of antifibrotics using biomaterial carriers is another promising approach. Both natural polymers like alginate, chitosan, and collagen and synthetic polymers like PLA, PLGA, and PEG have been formulated into nanoparticles, hydrogels, and implants for sustained drug release in the liver. Targeted delivery using vessels or molecules that are home to liver myofibroblasts can increase bioavailability and reduce off-target effects. Three-dimensional bioprinting of liver constructs offers precise control over the microarchitecture. Bioprinted liver mimics aim to replicate the lobule structure, hepatic zones, and vascular channels. However, challenges exist in printing multiple cell types, vascular integration, and scalable tissue generation. Further advances in bioprinting technology, bioinks, and tissue maturation will help realize implantable engineered livers [78].
Cell therapies for liver fibrosis
Cell-based therapies aim to repair or regenerate damaged tissue and provide beneficial paracrine signals that modulate liver inflammation and fibrosis. Key cell types under investigation include mesenchymal stem cells, fibrocytes, and hepatocytes. Mesenchymal stem cells (MSCs) exert antifibrotic effects through immunomodulation, inflammation resolution, and matrix remodeling. MSCs secrete hepatocyte growth factor, interleukin-10, and other anti-inflammatory mediators that reduce liver injury. In rodent models, MSC transplantation attenuated liver fibrosis by downregulating collagen, smoothening reticular fiber density, and inhibiting TGF-β1 [58]. Several clinical trials have evaluated autologous MSC infusion in cirrhosis patients. A study found umbilical cord-derived MSC infusion was safe and improved liver function and model for end-stage liver disease (MELD) scores. While MSCs suppress fibrosis, fibrocytes originating from bone marrow appear to contribute to fibrosis progression. Fibrocytes infiltrate injured livers and deposit type I collagen. In mice, antibody-mediated fibrocyte depletion or chemokine receptor blockade ameliorated injury and fibrosis in the liver, pancreas, and other organs. Hepatocyte transplantation aims to restore metabolic and synthetic functions in liver failure as a bridge to recovery or transplantation. Limitations include the availability of donor cells and poor long-term engraftment. Co-transplanting mesenchymal stem cells improves hepatocyte engraftment in animal models. Bioengineered liver grafts and primary human hepatocyte 3D spheroids show improved engraftment over isolated hepatocytes. Challenges remain in scaling up technologies for widespread application [79].
Liver-on-a-chip platforms for modeling liver disease
Liver-on-a-chip platforms are emerging as advanced in vitro models that integrate microfluidics, tissue engineering, and microfabrication to create biochips that recapitulate functional units of the human liver. These miniaturized devices allow precise control of the cellular microenvironment and application of physiologically relevant mechanical forces. Liver-on-a-chip technology enables researchers to investigate liver biology and model liver diseases such as fibrosis with unprecedented ability compared to conventional 2D culture systems [80].
Primary human hepatocytes are considered the gold standard cell type for liver-on-a-chip platforms. Isolation of hepatocytes from donor livers that are unsuitable for transplantation provides a source of highly functional human liver cells. Recent advances have enabled the use of stem cell-derived hepatocytes cultured on chips. Human induced pluripotent stem cells (iPSCs) can be differentiated into hepatocyte-like cells and integrated into liver-on-a-chip devices. Additionally, self-organizing 3D hepatic organoids generated from stem cells represent an exciting option for cellularizing liver chips. These new cell sources provide a scalable, renewable supply of human liver cells [81].
A major application of liver-on-a-chip platforms is modeling the development and progression of chronic liver diseases. The stepwise accumulation of extracellular matrix proteins and scar formation that occurs during liver fibrosis and cirrhosis can be recapitulated on-chip. Researchers developed a fibrosis-on-a-chip platform consisting of iPSC-derived hepatocytes cultured in a collagen hydrogel. By modulating cell–matrix interactions and culture conditions, they induced activation of stellate cells, focal matrix deposition, and progressive scarring reminiscent of early fibrotic remodeling. The biomimetic fibrosis model responded to profibrotic stimuli and known antifibrotic drugs similar to human liver. Such micro-engineered fibrotic tissues open new avenues for investigating mechanisms of fibrogenesis and testing potential treatments [82]. The schematic in Fig. 2A illustrates NAFLD progression mediated by free fatty acids (FFAs) through the gut-liver axis (GLA). The photograph in Fig. 2B shows an integrated gut-liver chip (iGLC) platform with separate perfusion and control layers colored pink and blue. Figure 2C depicts the iGLC system used to model NAFLD with Caco-2 gut cells and HepG2 hepatocytes in separate chambers connected by a microfluidic channel and micropump for closed FFA circulation. Integrated microvalves within the chip allow individual cell chamber access without cross-contamination. This iGLC platform enables the investigation of inter-tissue interactions between the gut and liver. Cross-section A-A' shows the Caco-2 and HepG2 cell culture chambers. Cross-section B-B' illustrates open and closed microvalves using an elastic PDMS membrane and differential pressure control [83].

Microfluidic iGLC platform design to model NAFLD [83]
In the future, personalized liver-on-a-chip models derived from patient-specific iPSCs could be used for therapeutic testing to identify optimal treatments for that individual. Integrated sensors could enable continuous monitoring of drug responses. Along with clinical information, data from patient-specific organ chip models could provide an invaluable tool for precision medicine. However, significant research is still required to fully realize the potential of liver-on-a-chip systems. Current challenges include recapitulating the full complexity of human liver physiology, the inclusion of non-parenchymal cells, vascularization, reproducibility, and validation of observations against the human liver [84].
Conclusions
Recent advances have uncovered critical pathways driving liver fibrosis, revealing numerous therapeutic targets. Approaches like pirfenidone, obeticholic acid, and monoclonal antibodies have shown promise in early clinical trials after demonstrating preclinical efficacy. Mesenchymal stem cell therapies also exhibit antifibrotic potential. Non-invasive biomarkers and advanced imaging enable accurate fibrosis diagnosis, staging, and monitoring. Omics data integration facilitates disease subclassification and prognosis. Liver tissue engineering platforms provide unprecedented human models to study fibrosis and screen therapies. Looking ahead, combination therapies targeting multiple pathways may yield greater efficacy. Further research into biomarkers, tissue engineering, and AI will help translate discoveries into effective antifibrotics. A multifaceted approach harnessing expanding knowledge offers hope for managing fibrosis from all causes.
Recommendations
(1) Evaluate combination regimens in clinical trials; (2) develop additional non-invasive biomarkers; (3) leverage AI for data integration and analysis; (4) engineer biomimetic liver tissues for drug screening; (5) elucidate genetics and epigenetics of fibrosis subtypes. Implementation of these recommendations, alongside continuing research, will accelerate the development of safe, efficacious antifibrotic therapies.
Availability of data and materials
All data are available and sharing is available as well as publication.
Abbreviations
- ECM:
-
Extracellular matrix
- TGF-β:
-
Transforming growth factor beta
- PDGF:
-
Platelet-derived growth factor
- CTGF:
-
Connective tissue growth factor
- α-SMA:
-
Alpha smooth muscle actin
- HOTAIR:
-
HOX transcript antisense RNA
- TIMPs:
-
Tissue inhibitors of metalloproteinases
- MMPs:
-
Matrix metalloproteinases
- TNF-α :
-
Tumor necrosis factor alpha
- IL-6:
-
Interleukin 6
- SOD:
-
Superoxide dismutase
- NRF2:
-
Nuclear factor erythroid 2-related factor 2
- EMT:
-
Epithelial-mesenchymal transition
- miRNAs:
-
MicroRNAs
- lncRNAs:
-
Long non-coding RNAs
- EVs:
-
Extracellular vesicles
- MRE:
-
Magnetic resonance elastography
- CPA:
-
Collagen proportional area
- NAFLD:
-
Nonalcoholic fatty liver disease
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- FXR:
-
Farnesoid X nuclear receptor
- NASH:
-
Non-alcoholic steatohepatitis
- LOXL2:
-
Lysyl oxidase homolog 2
- LPA1:
-
Lysophosphatidic acid 1 receptor
- MSCs:
-
Mesenchymal stem cells
- IPF:
-
Idiopathic pulmonary fibrosis
References
Lee MJ (2023) A Review of liver fibrosis and cirrhosis regression. Journal of pathology and translational medicine 57(4):189–195. https://doi.org/10.4132/jptm.2023.05.24
Addissouky TA, Wang Y, Megahed FAK et al (2021) Novel biomarkers assist in detection of liver fibrosis in HCV patients. Egypt Liver Journal 11:86. https://doi.org/10.1186/s43066-021-00156-x
Addissouky TA, Ali MMA, El Tantawy El Sayed I, Wang Y, El Baz A, Elarabany N et al (2023) Preclinical promise and clinical challenges for innovative therapies targeting liver fibrogenesis. Arch Gastroenterol Res 4(1):14–23. https://doi.org/10.33696/Gastroenterology.4.044
Park S-J, Garcia Diaz J, Um E, Hahn YS (2023) Major Roles of Kupffer Cells and Macrophages in NAFLD Development. Front Endocrinol 14. https://doi.org/10.3389/fendo.2023.1150118
Pei Q, Qian Y, Tang L (2023) Liver fibrosis resolution: from molecular mechanisms to therapeutic opportunities. Int J Mol Sci 24(11):9671–9671. https://doi.org/10.3390/ijms24119671
Matsuzaki S, Hase E, Takanari H et al (2023) Quantification of collagen fiber properties in alcoholic liver fibrosis using polarization-resolved second harmonic generation microscopy. Sci Rep 13:22100. https://doi.org/10.1038/s41598-023-48887-8
Addissouky TA, Sayed IETE, Ali MMA et al (2024) Latest advances in hepatocellular carcinoma management and prevention through advanced technologies. Egypt Liver Journal 14:2. https://doi.org/10.1186/s43066-023-00306-3
Lingas EC (2023) Hematological abnormalities in cirrhosis: a narrative review. Cureus. https://doi.org/10.7759/cureus.39239
Abbas N et al (2023) Guidance document: risk assessment of patients with cirrhosis prior to elective non-hepatic surgery. Frontline Gastroenterology 14(5):359–370. https://doi.org/10.1136/flgastro-2023-102381
Addissouky, T.A., Ayman E. El-Agroudy, Abdel Moneim A.K. El-Torgoman and 1Ibrahim E. El-Sayed, Efficacy of biomarkers in detecting fibrosis levels of liver diseases. IDOSI Publications, World Journal of Medical Sciences, Volume 16, Issue (1): PP. 11–18, March 2019, ISSN 1817–3055, DOI: https://doi.org/10.5829/idosi.wjms.2019.11.18
Addissouky, T.A., Detecting liver fibrosis by recent reliable biomarkers in viral hepatitis patients, American Journal of Clinical Pathology, Volume 152, Issue Supplement_1, October 2019, Page S85, https://doi.org/10.1093/ajcp/aqz117.000, published: 11 September 2019
El Agroudy AE, Elghareb MS, Addissouky TA, Elshahat EH, Hafez EH (2016) Serum hyaluronic acid as a non-invasive biomarker to predict liver fibrosis in viral hepatitis patients. Journal of Biochemical and Analytical Research 43(2):108377. https://doi.org/10.21608/JBAAR.2016.108377
El Agroudy AE, Elghareb MS, Addissouky TA, Elshahat EH, Hafez EH (2016) Biochemical study of some non-invasive markers in liver fibrosis patients. Journal of Biochemical and Analytical Research 43(2):108375. https://doi.org/10.21608/JBAAR.2016.108375
Deyamira Matuz-Mares; Héctor Vázquez-Meza; M. Magdalena Vilchis-Landeros. NOX as a therapeutic target in liver disease. Antioxidants 2022, 11 (10), 2038–2038. https://doi.org/10.3390/antiox11102038.
Addissouky, T. A.; Ayman E. El Agroudy, Abdel Moneim A.K. El-Torgoman, Ibrahim El Tantawy El Sayed, Eman Mohamad Ibrahim. Efficiency of alternative markers to assess liver fibrosis levels in viral hepatitis B patients. 2019, 30 (2). PP. 351–356, https://doi.org/10.35841/biomedicalresearch.30-19-107
Zhang C, Liu S, Yang M (2023) Treatment of liver fibrosis: past, current, and future. World J Hepatol 15(6):755–774. https://doi.org/10.4254/wjh.v15.i6.755
Zhang J, Hu Z, Horta CA, Yang J (2023) Regulation of epithelial-mesenchymal transition by tumor microenvironmental signals and its implication in cancer therapeutics. Semin Cancer Biol 88:46–66. https://doi.org/10.1016/j.semcancer.2022.12.002
VPS Punia; Agrawal, N.; Akash Bharti; Mittal, S.; Chaudhary, D.; Mathur, A.; Anwar, S.; Chakravorty, A. Association of TGF-β1 polymorphism and TGF-β1 levels with chronic hepatitis c and cirrhosis: a systematic review and meta-analysis. Cureus 2023. https://doi.org/10.7759/cureus.41157.
Siti Aishah Sulaiman; Vicneswarry Dorairaj; Muhammad (2022) Genetic Polymorphisms and Diversity in Nonalcoholic Fatty Liver Disease (NAFLD): A Mini Review. Biomedicines 11(1):106–106. https://doi.org/10.3390/biomedicines11010106
Liu, Y.; Yang, Z.; Yang, Y.; Liu, K.; Wu, J.-Y.; Gao, P.; Zhang, C. Exosomes in liver fibrosis: the role of modulating hepatic stellate cells and immune cells, and prospects for clinical applications. Frontiers in Immunology 2023, 14. https://doi.org/10.3389/fimmu.2023.1133297.
Liu Y, Wen D, Ho C, et al (2023) Epigenetics as a versatile regulator of fibrosis. J Transl Med 21:164. https://doi.org/10.1186/s12967-023-04018-5
Harsh Vardhan Charan, Durgesh Kumar Dwivedi, Khan S, Jena G (2023) Mechanisms of NLRP3 inflammasome-mediated hepatic stellate cell activation: therapeutic potential for liver fibrosis. Genes and Diseases 10(2):480–494. https://doi.org/10.1016/j.gendis.2021.12.006
Yang X, Li; Liu, W, Zong C, Wei L, Shi Y, Han Z (2023) Mesenchymal stromal cells in hepatic fibrosis/cirrhosis: from pathogenesis to treatment. Cellular & Molecular Immunology 20(6):583–599. https://doi.org/10.1038/s41423-023-00983-5.
Zhang D, Zhang Y et al (2022) The molecular mechanisms of liver fibrosis and its potential therapy in application. Int J Mol Sci 23(20):12572–12572. https://doi.org/10.3390/ijms232012572
Ahmad M, Weiswald LB, Poulain L, Denoyelle C, Meryet-Figuiere M (2023) Involvement of lncRNAs in cancer cells migration, invasion and metastasis: cytoskeleton and ECM crosstalk. J Exp Clin Cancer Res 42(1):173. https://doi.org/10.1186/s13046-023-02741-x.PMID:37464436;PMCID:PMC10353155
Eun JW, Cheong JY, Jeong J-Y, Kim HS (2023) A new understanding of long non-coding RNA in hepatocellular carcinoma—from M6A modification to blood biomarkers. Cells 12(18):2272. https://doi.org/10.3390/cells12182272
Jianhao Jiang, Ilgiz Gareev, Tatiana Ilyasova, Alina Shumadalova, Weijie Du, Baofeng Yang, The role of lncRNA-mediated ceRNA regulatory networks in liver fibrosis, Non-coding RNA Research, 2024, ISSN 2468–0540, https://doi.org/10.1016/j.ncrna.2024.01.001.
Bachir Taouli, Ehman R. L, Reeder S. B (2009) Advanced MRI methods for assessment of chronic liver disease. American Journal of Roentgenology 193(1):14–27. https://doi.org/10.2214/ajr.09.2601.
Wegrzyniak, O., Zhang, B., Rokka, J. et al. Imaging of fibrogenesis in the liver by [18F]TZ-Z09591, an Affibody molecule targeting platelet derived growth factor receptor β. EJNMMI radiopharm. chem. 8, 23 (2023). https://doi.org/10.1186/s41181-023-00210-6
Lemine AS, Ahmad Z, Al-Thani NJ et al (2023) Mechanical properties of human hepatic tissues to develop liver-mimicking phantoms for medical applications. Biomech Model Mechanobiol. https://doi.org/10.1007/s10237-023-01785-4
Villani R, Lupo P, Moris Sangineto, Antonino Davide Romano, Serviddio Gaetano (2023) Liver ultrasound elastography in non-alcoholic fatty liver disease: a state-of-the-art summary. Diagnostics 13(7):1236–1236. https://doi.org/10.3390/diagnostics13071236
Petitclerc, Léonie; et al. Liver fibrosis quantification by magnetic resonance imaging. topics in magnetic resonance imaging 26(6):p 229–241, December 2017. | DOI: https://doi.org/10.1097/RMR.0000000000000149
Ringe KI, Yoon JH. Strategies and techniques for liver magnetic resonance imaging: new and pending applications for routine clinical practice. Korean J Radiol. 2023 Mar;24(3):180–189. doi: https://doi.org/10.3348/kjr.2022.0838. Epub 2023 Feb 6. Erratum in: Korean J Radiol. 2023 Apr;24(4):372–373. PMID: 36788770; PMCID: PMC9971842.
Guglielmo FF et al (2023) Liver fibrosis, fat, and iron evaluation with MRI and fibrosis and fat evaluation with US: a practical guide for radiologists. Radiographics 43(6):e220181. https://doi.org/10.1148/rg.220181. (PMID: 37227944)
Jung Hwan Yu; Yong Sang Lee; Seung Up Kim. Noninvasive imaging biomarkers for liver fibrosis in nonalcoholic fatty liver disease: current and future. Clinical and molecular hepatology 2023, 29 (Suppl), S136–S149. https://doi.org/10.3350/cmh.2022.0436.
Ng N, Tai D, Ren Y, Chng E, Seneshaw M, Mirshahi F, Idowu M, Asgharpour A, Sanyal AJ (2023) Second-harmonic generated quantifiable fibrosis parameters provide signatures for disease progression and regression in nonalcoholic fatty liver disease. Clinical Pathology. https://doi.org/10.1177/2632010X231162317
Park H, Li B, Liu Y, Nelson MS, Wilson HM, Sifakis E, Eliceiri KW (2023) Collagen fiber centerline tracking in fibrotic tissue via deep neural networks with variational autoencoder-based synthetic training data generation. Med Image Anal 90:102961. https://doi.org/10.1016/j.media.2023.102961
Rodimova S et al (2023) Optical biomedical imaging reveals criteria for violated liver regenerative potential. Cells 12(3):479. https://doi.org/10.3390/cells12030479.PMID:36766821;PMCID:PMC9914457
Nicole Ng, Dean Tai, Yayun Ren, Elaine Chng, Mulugeta Seneshaw, Faridoddin Mirshahi, et al. Second-harmonic generated quantifiable fibrosis parameters provide signatures for disease progression and regression in nonalcoholic fatty liver disease - 2023. Clinical Pathology.https://journals.sagepub.com/doi/https://doi.org/10.1177/2632010X231162317.
Addissouky T, Ali M, El Tantawy El Sayed I, Wang Y. Revolutionary innovations in diabetes research: from biomarkers to genomic medicine. IJDO 2023; 15 (4) :228–242 https://doi.org/10.18502/ijdo.v15i4.14556
Allaume, Pierre, Noémie Rabilloud, Bruno Turlin, Edouard Bardou-Jacquet, Olivier Loréal, Julien Calderaro, Zine-Eddine Khene, Oscar Acosta, Renaud De Crevoisier, Nathalie Rioux-Leclercq, and et al. 2023. Artificial intelligence-based opportunities in liver pathology—a systematic review Diagnostics 13, no. 10: 1799. https://doi.org/10.3390/diagnostics13101799
Addissouky TA, Wang Y, El Sayed IE et al (2023) Recent trends in Helicobacter pylori management: harnessing the power of AI and other advanced approaches. Beni-Suef Univ J Basic Appl Sci 12:80. https://doi.org/10.1186/s43088-023-00417-1
Balsano C, Burra P, Duvoux C, Alisi A, Piscaglia F, Gerussi A (2023) Artificial Intelligence and Liver: Opportunities and Barriers. Dig Liver Dis 55(11):1455–1461. https://doi.org/10.1016/j.dld.2023.08.048
Addissouky TA, El Tantawy El Sayed I, Ali MMA, Wang Y, El Baz A, Elarabany N et al (2024) Shaping the future of cardiac wellness: exploring revolutionary approaches in disease management and prevention. J Clin Cardiol 5(1):6–29. https://doi.org/10.33696/cardiology.5.048
Xiong, M.; Xu, Y.; Zhao, Y.; He, S.; Zhu, Q.; Wu, Y.; Hu, X.; Liu, L. Quantitative analysis of artificial intelligence on liver cancer: a bibliometric analysis. Frontiers in Oncology 2023, 13. https://doi.org/10.3389/fonc.2023.990306.
Addissouky TA, El Sayed IET, Ali MMA et al (2024) Oxidative stress and inflammation: elucidating mechanisms of smoking-attributable pathology for therapeutic targeting. Bull Natl Res Cent 48:16. https://doi.org/10.1186/s42269-024-01174-6
Popa S.-L, Abdulrahman Ismaiel, Ludovico Abenavoli, Alexandru Marius Padureanu, Miruna Oana Dita, Bolchis R, Munteanu M et al (2023) Diagnosis of liver fibrosis using artificial intelligence: a systematic review. Medicina-lithuania 59(5):992–992. https://doi.org/10.3390/medicina59050992
Brennan, P.; Elsharkawy, A. M.; Kendall, T. J.; Rohit Loomba; Mann, D. A.; Fallowfield, J. Antifibrotic therapy in nonalcoholic steatohepatitis: time for a human-centric approach. Nature Reviews Gastroenterology & Hepatology 2023. https://doi.org/10.1038/s41575-023-00796-x.
Dong, Q.; Bao, H.; Wang, J.; Shi, W.; Zou, X.; Sheng, J.; et al. Liver fibrosis and MAFLD: the exploration of multi-drug combination therapy strategies. Frontiers in Medicine 2023, 10. https://doi.org/10.3389/fmed.2023.1120621.
Park, Jeong-Su, Nodir Rustamov, and Yoon-Seok Roh. 2023. The roles of NFR2-regulated oxidative stress and mitochondrial quality control in chronic liver diseases Antioxidants 12, no. 11: 1928. https://doi.org/10.3390/antiox12111928
Vyas K, Patel MM (2023) Insights on drug and gene delivery systems in liver fibrosis. Asian J Pharm Sci 18(2):100779–100779. https://doi.org/10.1016/j.ajps.2023.100779
Blas-García, A.; Nadezda Apostolova. Novel therapeutic approaches to liver fibrosis based on targeting oxidative stress. Antioxidants 2023, 12 (8), 1567–1567. https://doi.org/10.3390/antiox12081567.
AbouSamra, M. M.; Rania Elgohary; Mansy, S. S. Innovated pirfenidone loaded lecithin nanocapsules for targeting liver fibrosis: formulation, characterization and in vivo study. International Journal of Pharmaceutics 2023, 631, 122539–122539. https://doi.org/10.1016/j.ijpharm.2022.122539.
Hu, X.; Chen, L.; Wu, H.; Tang, Y.-B.; Zheng, Q.; et al. Cell therapy in end-stage liver disease: replace and remodel. Stem Cell Research & Therapy 2023, 14 (1). https://doi.org/10.1186/s13287-023-03370-z.
In Sook Ahn; Kang, C.; Han, J. Where should SiRNAs Go: applicable organs for SiRNA drugs. Experimental and Molecular Medicine 2023, 55 (7), 1283–1292. https://doi.org/10.1038/s12276-023-00998-y.
Maurine, O.; Akhmadu Muradi; Radiana Dhewayani Antarianto. Bio-artificial liver support system: a prospective future therapy. Livers 2023, 3 (1), 65–75. https://doi.org/10.3390/livers3010006.
François Villeret; Sébastien Dharancy; Erard, D.; Abergel, A.; Barbier, L.; Besch, C.; et al. Inevitability of disease recurrence after liver transplantation for NAFLD cirrhosis. JHEP reports 2023, 5 (3), 100668–100668. https://doi.org/10.1016/j.jhepr.2022.100668.
Li X, Yu M, Zhao Q, Ye Y (2023) Prospective therapeutics for intestinal and hepatic fibrosis. Bioengineering & translational medicine. https://doi.org/10.1002/btm2.10579
Shakour, Neda, Shima Karami, Mehrdad Iranshahi, Alexandra E. Butler, and Amirhossein Sahebkar. Antifibrotic effects of sodium-glucose cotransporter-2 inhibitors: a comprehensive review. Diabetes & Metabolic Syndrome: Clinical Research & Reviews 18, no. 1 (2023): 102934. Accessed January 25, 2024. https://doi.org/10.1016/j.dsx.2023.102934.
Liu Y, Deng S, Song Z, Zhang Q, Guo Y, Yu Y, Wang Y, Li T, Megahed FAK, Addissouky TA, Mao J, Zhang Y (2021) MLIF modulates microglia polarization in ischemic stroke by targeting EEF1A1. Front Pharmacol 12. https://doi.org/10.3389/fphar.2021.725268
T A Addissouky, A A Khalil, Detecting lung cancer stages earlier by appropriate markers rather than biopsy and other techniques, American Journal of Clinical Pathology, Volume 154, Issue Supplement_1, October 2020, Pages S146–S147, https://doi.org/10.1093/ajcp/aqaa161.320
Lowe, Kirstin O., Constantin E. Tanase, Susan Maghami, Leanne E. Fisher, and Amir M. Ghaemmaghami. 2023. Inflammatory network of liver fibrosis and how it can be targeted therapeutically Immuno 3, no. 4: 375–408. https://doi.org/10.3390/immuno3040023
T A Addissouky, A E El Agroudy, A A Khalil, Developing a novel non-invasive serum-based diagnostic test for early detection of colorectal cancer, American Journal of Clinical Pathology, Volume 160, Issue Supplement_1, November 2023, Page S17, https://doi.org/10.1093/ajcp/aqad150.037
Wang S, Friedman SL. Found in translation-Fibrosis in metabolic dysfunction-associated steatohepatitis (MASH). Sci Transl Med. 2023 Oct 4;15(716):eadi0759. doi: https://doi.org/10.1126/scitranslmed.adi0759. Epub 2023 Oct 4. PMID: 37792957; PMCID: PMC10671253.
T A Addissouky, A A Khalil, A E El Agroudy, Assessment of potential biomarkers for early detection and management of Glomerulonephritis patients with diabetic diseases, American Journal of Clinical Pathology, Volume 160, Issue Supplement_1, November 2023, Pages S18–S19, https://doi.org/10.1093/ajcp/aqad150.040
Vachliotis ID, Polyzos SA (2023) The role of tumor necrosis factor-alpha in the pathogenesis and treatment of nonalcoholic fatty liver disease. Curr Obes Rep 12:191–206. https://doi.org/10.1007/s13679-023-00519-y
Lopetuso LR, Cuomo C, Mignini I, Gasbarrini A, Papa A (2023) Focus on anti-tumour necrosis factor (TNF)-α-related autoimmune diseases. Int J Mol Sci 24(9):8187. https://doi.org/10.3390/ijms24098187.PMID:37175894;PMCID:PMC10179362
Addissouky TA, El Tantawy El Sayed I, Ali MMA, Wang Y, El Baz A, Khalil AA et al (2023) Can vaccines stop cancer before it starts? Assessing the promise of prophylactic immunization against high-risk preneoplastic lesions. J Cell Immunol 5(4):127–126. https://doi.org/10.33696/immunology.5.178
Antar SA, Saleh MA, Al-Karmalawy AA (2022) Investigating the possible mechanisms of pirfenidone to be targeted as a promising anti-inflammatory, anti-fibrotic, anti-oxidant, anti-apoptotic, anti-tumor, and/or anti-SARS-CoV-2. Life Sci 309:121048–121048. https://doi.org/10.1016/j.lfs.2022.121048
Caezaan Keshvani; Kopel, J.; Goyal, H. Obeticholic acid—a pharmacological and clinical review. Future Pharmacology 2023, 3 (1), 238–251. https://doi.org/10.3390/futurepharmacol3010017.
Radic, J.; Bojana Kožik; Nikolic, I.; Kolarov-Bjelobrk, I.; Tijana Vasiljevic; Bojana Vranjković; Sanja Despotović. Multiple roles of LOXL2 in the progression of hepatocellular carcinoma and its potential for therapeutic targeting. International Journal of Molecular Sciences 2023, 24 (14), 11745–11745. https://doi.org/10.3390/ijms241411745.
Singh S, Sharma N, Shukla S, Behl T, Gupta S, Khalid Anwer, De-La-Cruz Vargas C, Bungau S, Cristina Mihaela Brisc (2023) Understanding the potential role of nanotechnology in liver fibrosis: a paradigm in therapeutics. Molecules 28(6):2811–2811.https://doi.org/10.3390/molecules28062811.
Lei, Zhigang, Jiaojiao Yu, Yu Wu, Junyao Shen, Shibo Lin, Weijie Xue, Chenxu Mao et al. CD1d protects against hepatocyte apoptosis in non-alcoholic steatohepatitis. Journal of Hepatology 80, no. 2 (2024): 194–208. Accessed January 25, 2024. https://doi.org/10.1016/j.jhep.2023.10.025.
Addissouky TA, Ali MMA, El Tantawy El Sayed I, Wang Y. Recent advances in diagnosing and treating helicobacter pylori through botanical extracts and advanced technologies. Arch Pharmacol Ther. 2023;5(1):53–66. https://doi.org/10.33696/Pharmacol.4.045
Addissouky, T.A., et al. efficiency of mixture of olive oil and figs as an antiviral agents: a review and perspective, International Journal of Medical Science and Health Research, ISSN:2581–3366, Volume 4, Issue 4, Aug 2020, page 107–111,http://ijmshr.com/link/208
T A Addissouky, A A Khalil, A E El Agroudy, Assessing the efficacy of a modified triple drug regimen supplemented with mastic gum in the eradication of Helicobacter pylori infection, American Journal of Clinical Pathology, Volume 160, Issue Supplement_1, November 2023, Page S19, https://doi.org/10.1093/ajcp/aqad150.041
Qian Huai; Zhu, C.; Zhang, X.; Dai, H.; Li, X.; Wang, H. Mesenchymal stromal/stem cells and their extracellular vesicles in liver diseases: insights on their immunomodulatory roles and clinical applications. Cell & Bioscience 2023, 13 (1). https://doi.org/10.1186/s13578-023-01122-3.
Zhao P, Sun T, Lyu C, Liang K, Niu Y, Zhang Y, Cao C, Xiang C, Du Y (2022) Scar-degrading endothelial cells as a treatment for advanced liver fibrosis. Advanced Science 10(4):2203315–2203315. https://doi.org/10.1002/advs.202203315
Jiang C, Chen H, Kang Y, He XT, Huang J, Lu T et al (2023) Administration of AG490 decreases the senescence of umbilical cord-mesenchymal stem cells and promotes the cytotherapeutic effect in liver fibrosis. Cell Death Discov 9(1). https://doi.org/10.1038/s41420-023-01546-3
Li, W.; Liu, Z. A.; Tang, F.; Jiang, H.; Zhou, Z.; Hao, X.; Jia Ming Zhang. Application of 3D bioprinting in liver diseases. Micromachines 2023, 14 (8), 1648–1648. https://doi.org/10.3390/mi14081648.
Somnath Maji; Lee, M.; Lee, J.; Lee, J.; Lee, H. Development of lumen-based perfusable 3D liver in vitro model using single-step bioprinting with composite bioinks. Materials today bio 2023, 21, 100723–100723. https://doi.org/10.1016/j.mtbio.2023.100723.
Adiya Otumala; Hellen, D.; Cecilia Alessandra Luna; Delgado, P.; Anjana Dissanayaka; Chidozie Ugwumadu; et al. Opportunities and considerations for studying liver disease with microphysiological systems on a chip. Lab on a Chip 2023, 23 (13), 2877–2898. https://doi.org/10.1039/d2lc00940d.
Yang, J.; Hirai, Y.; Iida, K.; Ito, S.; Trumm, M.; Terada, S.; Sakai, R.; Tsuchiya, T.; Tabata, O.; Kamei, K. Integrated-gut-liver-on-a-chip platform as an in vitro human model of non-alcoholic fatty liver disease. Communications biology 2023, 6 (1). https://doi.org/10.1038/s42003-023-04710-8.
Qiu L, Kong B, Kong T, Wang H (2023) Recent Advances in Liver-on-chips: Design, Fabrication, and Applications. Smart Medicine 2(1):e20220010. https://doi.org/10.1002/SMMD.20220010
Acknowledgements
The authors thank all the researchers who have made great efforts in their studies. The authors would like to thank the Deanships of all the participating universities for supporting this work. Moreover, we are grateful to the editors, reviewers, and readers of this journal.
Funding
The corresponding author supplied all study materials. There was no further funding for this study.
Author information
Authors and Affiliations
Contributions
The authors completed the study protocol and were the primary organizers of data collection and the manuscript's draft and revision process. Tamer A. Addissouky wrote the article and ensured its accuracy. All authors contributed to the discussion, assisted in designing the study and protocol, and engaged in critical discussions of the draft manuscript. Lastly, the authors (TA, MA, IE, YW) reviewed and confirmed the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Addissouky, T.A., Ali, M.M.A., Sayed, I.E.T.E. et al. Emerging advanced approaches for diagnosis and inhibition of liver fibrogenesis. Egypt J Intern Med 36, 19 (2024). https://doi.org/10.1186/s43162-024-00283-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s43162-024-00283-y