
Abstract
Introduction
Plasma circulating tumor DNA (ctDNA) is an ideal approach to detecting the epidermal growth factor receptor (EGFR) T790M mutation, which is a major mechanism of resistance to first-generation EGFR-tyrosine kinase inhibitor (TKI) therapy. The present study aimed to explore the association of ctDNA-identified T790M mutation with disease failure sites and clinical prognosis in non-small cell lung cancer (NSCLC) patients.
Methods
Patients who progressed on first-generation TKIs were categorized into failure site groups of chest limited (CF), brain limited (BF) and other (OF). Amplification refractory mutation system (ARMS) and droplet digital PCR (ddPCR) were used to identify the T790M mutation in ctDNA. Prognosis was analyzed with Kaplan–Meier methods.
Results
Overall concordance between the two methods was 78.3%. According to both ARMS and ddPCR, patients in the OF group had a significantly higher rate of T790M mutation than did patients in the BF and CF groups (P < 0.001), and a significantly higher T790M mutation rate was also observed in OF-group patients than in those in the CF and BF groups (P < 0.001). AZD9291 was found to be an excellent treatment option and yielded the longest survival for T790M+ patients in all groups who had progressed on EGFR-TKIs; for other treatments, the prognosis of T790M− patient subgroups varied.
Conclusions
The present study demonstrates that T790M mutation in ctDNA is associated with failure sites for NSCLC patients after EGFR-TKI therapy and indicates that both failure site and T790M mutational status greatly influence treatment selection and prognosis.
Similar content being viewed by others


Introduction
Approximately 85% of lung cancers are classified as non-small cell lung cancer (NSCLC). Among these patients, approximately 10% in the US and 35% in East Asia harbor epidermal growth factor receptor (EGFR) mutations in the tyrosine kinase domain (exons 18–21). EGFR mutations are usually heterozygous with amplification in the mutant allele [1]. Approximately 90% of these mutations are the exon 19 deletion (19del) or exon 21 L858R point mutation [2], which can increase the activity of EGFR kinase and activate downstream pro-survival signaling pathways [3]. EGFR-tyrosine kinase inhibitors (EGFR-TKIs) are highly effective clinical agents for NSCLC patients with EGFR mutations and can result in an objective response rate (ORR) of approximately 70% and prolong progression-free survival (PFS) to 8–13 months [4,5,6,7,8,9]. Although the initial response to EGFR-TKI therapy is rapid, prolonging ORR and PFS, the favorable therapeutic effects are not maintained due to acquisition of resistance to inhibitors after a median response duration of 9–13 months [10]. The most common mutation associated with acquired resistance is T790M, a secondary point mutation located in exon 20 that results in the substitution of methionine for threonine at position 790. The EGFR T790M mutation is present in over 50% of NSCLC patients with EGFR-TKI resistance [10]. Other molecular mechanisms for EGFR-TKI resistance include hepatocyte growth factor receptor (c-MET) amplification [11], erbb2 receptor tyrosine kinase 2 (HER2) and phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA) mutation [12], BCL2-like 11 (BIM) polymorphism [13], and transformation to small cell lung cancer [14].
Recurrence or lesion growth in advanced NSCLC patients can be found in the lung, the mediastinum, distant organs such as the liver and bone, or the central nervous system (CNS), requiring different treatment. For example, isolated or metastatic lesions in the lung, mediastinum and CNS may benefit from radiation or other local therapies, whereas distant lesions need to be treated with chemotherapy or third-generation EGFR-TKIs (AZD9291). Compared to those with other resistance mechanisms, patients with T790M mutation after EGFR-TKI treatment may present distinct modes of recurrence or progression. A previous study showed that the presence of T790M mutation in patients with acquired resistance to EGFR-TKIs was associated with a favorable prognosis and that these patients had longer PFS and overall survival (OS) than did those who acquired resistance via other mechanisms [15]. A preclinical model also revealed indolent growth for cells with acquired T790M mutation [16,17,18,19,20]. Another study reported that T790M mutation is more readily detected in the plasma of patients with extra-thoracic metastatic disease (M1b) than in the plasma of patients with intra-thoracic lesions (M1a/M0); thus, patients with T790M mutation in circulating tumor DNA (ctDNA) have a high likelihood of developing distant metastases [21]. In addition, T790M mutation in ctDNA is associated with a significantly shorter OS than is ctDNA negative for the mutation [22]. Therefore, T790M mutation identified in ctDNA may serve as a marker for clinical outcomes and failure after EGFR-TKI therapy.
Modes of clinical failure for EGFR-TKI therapy are typically based on the duration of disease control and evaluation of the tumor burden and clinical symptoms [23]. However, the relationship between failure sites for EGFR-TKIs and the T790M mutational status have remained unclear, and this issue needs to be resolved. Although Carrera et al. [12] reported no significant difference in the distribution of EGFR T790M mutation within various failure sites after TKI therapy, the recent study referred to above showed that T790M mutation was more readily detected in the plasma of M1b patients than in that of M1a/M0 patients [21]. Moreover, ctDNA-identified T790M mutation is more frequently observed in patients with new lesions or distant metastasis than in those with local lesions, indicating the prognostic value of T790M mutation with regard to tumor progression and metastasis [24]. Nevertheless, the relationship between failure sites of TKI treatment and T790M mutation in ctDNA has yet to be clarified. Therefore, it is necessary to investigate the potential mechanisms and roles of the T790M mutation in NSCLC patients who exhibit different failure sites after treatment with EGFR-TKIs.
Detection of ctDNA-based mutations is very promising due to several notable advantages, including the non-invasive nature of the assay, the accessibility of samples and the potential for repeated sampling, especially following progression after first-line TKI therapy. The detection rate of T790M mutation in ctDNA from NSCLC patients with acquired resistance to TKIs ranges from 30–50% via qualitative assays such as BEAMing (beads, emulsion, amplification, and magnetics) digital PCR [25], droplet digital PCR (ddPCR) [26], and next-generation sequencing (NGS)-based methods [26]. Although several studies have assessed the prognostic value of T790M mutation identified in ctDNA [20, 22, 27], associations of failure sites with TKI treatment and T790M mutation in ctDNA have not been explored. Hence, the present study aimed to determine whether the frequency and abundance of T790M mutation in ctDNA indicates failure sites and enables analysis of the prognostic value of these mutations in patients with disease failure sites after the acquisition of resistance to first-generation EGFR-TKI treatment.
Patients and methods
Study population
This prospective, observational, multi-institutional study was performed between March 2015 and March 2016. The protocol was approved by the Institutional Review Board of Hangzhou First People’s Hospital (No. HZFH CA15-02). All patients signed informed consent. The present study was registered in clinicaltrials.gov (NCT02418234). All experiments were performed in accordance with relevant guidelines and regulations. Patients were considered eligible and enrolled in the present study if they met the following criteria: (1) presence of histologically confirmed stage III/IV NSCLC; (2) presence of activating EGFR mutations (G719A/C/S/X; exon 19 insertion (Ins)/Del; L858R; L861Q; or 20Ins) or response to treatment or durable stable disease (≥ 6 months) to EGFR-TKIs (erlotinib, gefitinib and icotinib) followed by progression during TKI treatment; (3) presence of tumors clinically resistant to first-generation EGFR-TKIs according to Jackman’s criteria [28]; (4) samples collected before all treatment; and (5) available data corresponding to clinical features, best TKI response, site(s) of failure, and duration of response. Follow-up was performed every 3 months via telephone calls.
Definition of disease failure sites
To analyze failure sites, two senior thoracic radiologists reviewed radiological images to evaluate disease progression at the original (primary and metastatic) or new site(s). Patients were categorized into three groups according to the following failure sites: (1) chest limited (CF), defined as progressive disease (PD) that was limited to the chest in lung/pleural tissues and lymph nodes, with no evidence of progression beyond the chest; (2) brain limited (BF), defined as PD in an previously existing site or a new site of metastatic disease in the brain, with no evidence of extracranial progression; and (3) other (OF), defined as PD at other distant sites or multiple sites including the chest and intracranial region.
Sample collection and DNA extraction
Blood samples were collected within 14 days after the development of TKI resistance, as assessed by the physician according to the Jackman criteria [28]. Approximately 10–15 mL of peripheral blood was collected into a cell-free DNA protection vacuum tube (AmoyDx, Xiamen, Fujian, China) containing a cell-free DNA protection reagent that promotes DNA stability for 7 days at 4–25 °C. Blood samples were transported to the Center for Translational Medicine of Hangzhou First People’s Hospital within 36 h for further processing. For DNA extraction, the blood samples were centrifuged at 2500×g for 10 min at 4 °C. The supernatant was transferred to a new tube and centrifuged at 15,800×g for 15 min at 4 °C. The supernatant (plasma) was stored at −80 °C. Cell-free DNA from 1.5 mL plasma was extracted with a QIAamp Circulating Nucleic Acid kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany).
Detection of EGFR mutations in plasma ctDNA by ARMS
EGFR mutations in plasma ctDNA were determined by using the ADx-ARMS kit (AmoyDx), and all experiments and genotype calling were performed according to the manufacturer’s instructions [29].
ddPCR assays for EGFR T790M mutation determination
Recently, ddPCR has become a well-developed method for rapidly and quantitatively assessing EGFR mutations with high specificity and sensitivity [22, 26, 30,31,32,33]. EGFR T790M mutational status was determined by ddPCR with AmoyDx EGFR Exon 20 T790M Mutation Detection Kit. The principle of ddPCR for T790M mutation testing is shown in Additional file 1: Figure S1. Briefly, two probes with one nucleotide difference that target the mutated region were labeled with carboxyfluorescein (FAM) and green fluorescent protein (VIC) dyes to detect mutant and wild-type EGFR alleles, respectively. The customized primers and probes were synthesized by Life Technologies (Thermo Fisher Scientific Inc., Boston, MA, USA). To evaluate the sensitivity of the T790M detection assay, DNA from NCI-H1975 cells, which harbor the T790M mutation, was serially diluted with reference human genomic DNA to achieve decreasing ratios from 1:1 to 1:2500 of the T790M mutant allele versus the wild-type allele. The final 20 μL of the TaqMan PCR mixture contained 1× ddPCR Master Mix (Bio-Rad Laboratories, Hercules, CA, USA), 900 nM of each primer, 450 nM of each probe, and 50 ng of DNA template. A maximum of 20,000 droplets could be generated from each sample for PCR. The thermal cycling conditions for the T790M detection assay comprised a 10-min incubation at 95 °C followed by 45 cycles at 95 °C for 15 s and 60 °C for 1 min, followed by a hold step at 4 °C. Analysis of ddPCR data for allele calling was performed with Quanta Soft software version 1.3.2.0 (Bio-Rad). Reference human genomic DNA (Catalog No. G1471, Promega, Wisconsin, USA) was routinely included as a negative control and used to determine the cut-off value for allele calling. Considering that single, non-specific droplets were occasionally found in the positive area, the presence of at least two droplets with the FAM signal was defined as a positive signal for the mutation. After the establishment of the ddPCR protocol, 7.3 µL of plasma cell-free DNA was added to the reaction mixture described above. The number of positive droplets and sample input followed the Poisson distribution. Plasma DNA input per reaction (I) was calculated with the following equation: \(I\left( {{\text{copies}}/{\text{reaction}}} \right) = - { \ln }\left( { 1- {\text{p}}} \right)/{\text{V}}\; \times \; 1000\; \times \; 20\), where p represents the fraction of positive droplets, V represents the volume of each droplet (0.91 nL), and I represents the total number of copies of EGFR-mutant and wild-type DNA templates (corresponding to FAM and VIC signals, respectively).
The protocol for PCR was the same as that mentioned above. Four reference human genomic DNA samples were used as negative controls. Two positive controls with a 1:2500 ratio of mutant allele to wild-type allele and two non-template controls (NTC) were included in each run. The samples were considered positive for target mutations when they contained at least 2 droplets positive for the FAM signal. The fraction of EGFR T790M mutation (F1) was calculated as follows:\({\text{F1}} = {\text{I}}\left( {\text{FAM}} \right)/\left[ {{\text{I}}\left( {\text{FAM}} \right) \, + {\text{ I}}\left( {\text{VIC}} \right)} \right]\). We defined a sample as ddPCRT790M+ when it contained at least 2 droplets positive for the FAM signal.
Statistical analysis
Continuous data with a normal distribution were compared between two groups using Student’s t tests. Wilcoxon two-sample tests were performed when continuous data did not follow a normal distribution. Categorical data between two groups were compared using the Chi square or Fisher’s exact test. Concordance of T790M mutational status in ctDNA detected by ddPCR and ARMS assays was compared by McNemar’s test. PFS curves were constructed using the Kaplan–Meier method and compared using the log-rank test. Unconditional multiple logistic regression was performed to estimate risk factors of radical metastasis, and variables included EGFR mutation, age, smoking history, Eastern Cooperative Oncology Group (ECOG) score, therapy before TKI administration, sex and tumor pathology. OS was calculated from the time of development of TKI resistance to the time of death for any reason or last follow-up. Significance in all analyses was assessed at P < 0.05. All tests were two tailed. All analyses were performed using SAS software (version 9.3, SAS Institute, Cary, NC, USA).
Results
Patient characteristics
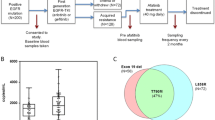
In total, 307 patients (44.0% males and 56.0% females) with advanced or recurrent NSCLC who had progressed during EGFR-TKI treatment with gefitinib, erlotinib or icotinib between March 2015 and March 2016 were consecutively enrolled in the present study. The median age of the patients was 63 years (range 32–89 years). Approximately 74.6% of the patients did not have a history of smoking. The type of NSCLC was predominantly adenocarcinoma (97.1%), and 76.5% of patients were diagnosed with stage IV disease. Moreover, 53.1% of the patients had EGFR 19del; 39.4% had the L858R mutation. Response to EGFR-TKI treatment was as follows: 32.9% stable disease (SD), 58.6% partial response (PR) and 8.5% complete response (CR) (Table 1). Figure 1 shows the scheme of the clinical trial design. The median follow-up time was 11 months (range 2–22 months).

The scheme of the clinical trial design. TKI tyrosine kinase inhibitor, ctDNA circulating tumor DNA, EGFR epidermal growth factor receptor, ARMS amplification refractory mutation system, ddPCR droplet digital PCR
EGFR mutation profiles in TKI-resistant ctDNA samples by ARMS
A common reason for disease progression during EGFR-TKI treatment is the acquisition of secondary mutations such as T790M. To gain insight into T790M mutation in patients with failure at different sites, we used ARMS and ddPCR to detect possible EGFR mutations, including T790M, in ctDNA samples isolated from TKI-resistant patients. ARMS detected T790M mutation (ARMST790M+) in 95 (30.9%) samples (Fig. 2a). In total, 84 (27.4%) samples had original EGFR activating mutations (19del, L858R and other rare mutations) that had been detected in primary tumor specimens before TKI administration, whereas 128 (41.7%) samples exhibited wild-type EGFR (Fig. 2a). Among the 95 samples with T790M mutation, 52 (54.7%) had 19del and T790M mutation, and 39 (41.1%) had L858R and T790M mutation; single T790M mutation was detected in only 4 (4.2%) samples (Fig. 2b). For T790M− samples, 128 (60.4%), 38 (17.9%), and 37 (17.5%) samples had wild-type EGFR, single 19del, and single L858R mutation, respectively (Fig. 2b).

EGFR mutations in ctDNA samples from patients with TKI resistance. a EGFR mutation profiles determined by ARMS. b EGFR mutation profiles in T790M+ and T790M− groups assessed by ARMS. c T790M mutation determined by ddPCR. d Scatter plot of the T790M abundance in the groups according to the T790M status by ARMS (top) and the number of patients with different abundance of T790M (bottom). TKI tyrosine kinase inhibitor, ctDNA circulating tumor DNA, EGFR epidermal growth factor receptor, ARMS amplification refractory mutation system, ddPCR droplet digital PCR
Development of a ddPCR assay for T790M mutation in TKI-resistant ctDNA samples
ddPCR was used in the present study to specifically detect and quantify ctDNA with EGFR T790M mutation. In total, 145 (47.2%) samples had a detectable T790M mutation (termed ddPCRT790M+); 162 (52.8%) samples were negative for T790M (termed ddPCRT790M−) (Fig. 2c). The median abundance of T790M was 1.20% (range 0.04%–70.30%) for 145 ddPCRT790M+ samples, with 0.44% (range 0.04%–10.77%) for ddPCR T790M+ ARMST790M− samples and 3.73% (range 0.04%–70.30%) for ddPCRT790M+ ARMST790M+ samples (Fig. 2d). Of note, ddPCR detected a significantly greater abundance of T790M mutation in the ARMST790M+ group than in the ARMST790M− group. In addition, among 145 ddPCRT790M+ patients, T790M abundance for 11 patients (7.6%) was within the range of 0–0.1%, 56 (38.6%) within 0.1–1%, 37 (25.5%) within 1–5%, 20 (13.8%) within 5–10%, and 21 (14.5%) above 10% (Fig. 2d).
The overall concordance of ARMS and ddPCR was 78.5% (241/307) (with a kappa value of 0.561), indicating that both methods were highly consistent. Relative to the corresponding sensitivity and specificity of ARMS, ddPCR showed a sensitivity of 91.6% (95% confidence interval (CI) 86–96.7%) and a specificity of 72.6% (95% CI 67.0–78.4%) in detecting T790M mutation (Table 2).
Association between T790M mutation and disease failure site
To assess correlations between T790M mutation and failure site, the 307 patients were categorized into three groups: 192 (62.5%) into the CF group, 32 (10.4%) into the BF group, and 83 (27.0%) into the OF group. The clinical characteristics of these three groups are shown in Table 1. The median ages were 64, 62, and 63 years old in patients with CF, BF, and OF groups, respectively. There was no significant difference in age, smoking history, pathology or initial EGFR mutation type among the three groups. However, the BF group was predominantly male and the OF group predominantly female (P = 0.01). TKI response also differed, with patients in the BF group having the highest percentage of SD but a lower rate of PR and CR than did patients in the CF and OF groups (P = 0.023) (Table 1).
Overall, 49 (25.5%) of 192 patients in the CF group and 45 (54.2%) of 83 patients in the OF group were ARMST790M+, whereas only 1 (3.1%) of 32 patients in the BF group was ARMST790M+ (P < 0.001) (Fig. 3a, left). According to ddPCR, a similar distribution of T790M mutation was observed among the three groups (Fig. 3b): the proportion of ddPCRT790M+ patients was 40.6% (78 of 192) for the CF group, 21.9% (7 of 32) for the BF group, and 72.3% (60 of 83) for the OF group (P < 0.001) (Fig. 3b, left).

Distribution of EGFR mutations between different failure sites. a The proportion of ARMST790M+ and ARMST790M− in different failure sites (left). The proportion of ARMS19del+T790M, ARMSL858R+T790M, and ARMST790M+ in different failure sites (up-right). The distribution of ARMSwild-type, ARMS19del, and ARMSL858R in different failure sites (down-right). b The proportion of ddPCRT790M+ and ddPCRT790M− (left), as well as the abundance of T790M in patients with detectable T790M mutation (up-right) and in total patients (down-right). ARMS amplification refractory mutation system, ddPCR droplet digital PCR
We next explored the distribution of different subtypes of T790M mutation (T790M, 19del + T790M, L858R + T790M) in the three groups (Fig. 3a, right). However, no significant difference in the distribution of the different mutational subtypes was observed for either ARMST790M+ (P = 0.460) or ARMST790M− (P = 0.156) patients (Fig. 3a, right).
Furthermore, the median abundance of T790M mutation in ddPCRT790M+ patients was 0.77, 0.59, and 3% for the CF, BF, and OF groups, respectively (P = 0.065) (Fig. 3b, right). Of note, pair-wise comparison showed that the difference between the OF and CF groups was significant (P = 0.047) (Fig. 3b, right). Nonetheless, when all patients were included in the analysis, the median abundance of T790M mutation was significantly different among the groups, with 0, 0 and 0.5% for the CF, BF, and OF groups, respectively (P < 0.001) (Fig. 3b, right).
Effect of T790M mutation in plasma on clinical prognosis after the development of resistance to first-generation EGFR-TKI
To assess the effect of the presence of T790M mutation in plasma samples on prognosis after first-generation TKI resistance development, we followed up the patients every 3 months via telephone until April 2017; we recorded data for 278 patients, as 29 were lost to follow-up. As shown in Table 3, most patients received subsequent treatment. The T790M− patients in all three groups were likely to continue receiving the original TKI, whereas the T790M+ patients in the OF group were most likely to receive AZD9291.
The median survival time after progression on TKIs was 17.5 months (95% CI 15–20 months) (Fig. 4a). Patients with/without T790M mutation had similar survival durations (Fig. 4b). Although patients in the CF and OF groups had longer survival times than did patients in the BF group, there was no significant difference in survival among the three groups according to T790M mutational status (Fig. 5). In the CF group, T790M+ patients undergoing AZD9291 treatment had the best survival duration (median survival time not reached), followed by patients treated with chemotherapy ± radiotherapy (chemo ± RT) and TKI + chemo/RT (17.8 and 11.0 months, respectively). Continuation of TKIs and best supportive care (BSC) yielded the shortest survival duration (9.7 and 6.1 months, respectively) (Fig. 6a). In T790M− patients in the CF group, TKI + chemo/RT conferred the longest survival duration (median survival time not reached), and the continuation of TKIs yielded the shortest survival duration (14.6 months) (Fig. 6a). Due to the small sample size, we did not observe a significant difference among T790M+ patients in the BF group (Fig. 6b). In the T790M− patients in the BF group, AZD9291 and TKI + chemo/RT resulted in better survival durations than did the other treatments (Fig. 6b). In T790M+ patients in the OF group, AZD9291 was the best option, whereas the median survival duration for the chemo ± RT group and BSC was merely 4.0 and 1.3 months, respectively (Fig. 6c). In the T790M− patients in the OF group, TKI + chemo/RT and chemo ± RT resulted in the longest survival durations (Fig. 6c). In total, 85 patients (51 with T790M mutation) received AZD9291 after progressing on first-generation EGFR-TKIs. Two patients were lost to follow-up. Furthermore, 49 T790M+ patients had response records. The disease control rates of T790M+ patients after AZD9291 administration were 92.3% (24/26), 100% (1/1), and 77.3% (17/22) for the CF, BF, and OF groups, respectively.

Survival curves of patients with different T790M mutations. a Survival curves after TKI failure in 307 patients. b OS curves for patients with different T790M status, as assessed by ARMS and ddPCR. MST median survival time, ARMS amplification refractory mutation system, ddPCR droplet digital PCR

Survival curves of patients with different failure sites. a Survival curves categorized by failure site. b Survival curves of T790M+ and T790M− CF patients. c Survival curves of T790M+ and T790M− BF patients. d Survival curves of T790M+ and T790M− OF patients. CF, progressive disease limited to the chest in lung/pleural tissues and lymph nodes, with no evidence of progression beyond the chest; BF, progressive disease in a previously existing site or a new site of metastatic disease in the brain, with no evidence of extracranial progression; OF, progressive disease in other distant sites or multiple sites including the chest and intracranial region. T790M mutation was detected by ddPCR. MST median survival time, ARMS amplification refractory mutation system, ddPCR droplet digital PCR

Survival curves of patients receiving different subsequent treatments. a Curve for T790M+ and T790M− CF patients. b Curve for T790M+ and T790M− BF patients. c Curve for T790M+ and T790M− OF patients. CF, progressive disease limited to the chest in lung/pleural tissues and lymph nodes, with no evidence of progression beyond the chest; BF, progressive disease in a previously existing site or a new site of metastatic disease in the brain, with no evidence of extracranial progression; OF, progressive disease in other distant sites or multiple sites including the chest and intracranial region. T790M mutation was detected by ddPCR
Discussion
In the present study, we used ARMS and ddPCR to detect possible T790M mutation in plasma ctDNA collected from patients after the failure of EGFR-TKIs. The results demonstrate a strong concordance between ARMS and ddPCR for the detection of T790M mutation in plasma ctDNA. Because ddPCR also possesses high sensitivity and specificity, we suggest that ddPCR is a useful tool for determining T790M mutation and for quantifying both the frequency and abundance of T790M mutation in plasma ctDNA. Based on our ddPCR results, 47.2% of patients had detectable T790M mutation, similar to the findings of previous studies [22, 34, 35]. The relatively low abundance (median abundance of 1.2%) indicates the complexity of detecting T790M mutation in plasma.
Interestingly, we identified a significant association between the plasma T790M mutational status and failure sites in the present study. We found that patients in the OF group had a higher frequency of T790M mutation and a higher mutational abundance than did patients in the CF and BF groups, which was consistent with the results reported by Thress et al. [21]. Theoretically, ctDNA levels are generally correlated with the tumor burden; more metastatic tumors shed more DNA into the bloodstream, resulting in higher levels of tumor-derived DNA. Consistent with this hypothesis, a high rate of T790M mutation was detected in patients in the OF group. However, tumor burden alone was not sufficient to explain the higher T790M abundance in patients in the OF group compared to those in the CF and BF groups because T790M abundance is calculated from the ratio of T790M mutation to total EGFR copy number. A higher capacity for invasion and migration in cancer cells with T790M mutation may have led to the higher abundance of these mutations in patients in the OF group.
Brain metastasis (BM) accounts for approximately 50% of NSCLC recurrence after TKI treatment [36]. First-generation EGFR-TKIs commonly have a low capacity to penetrate the blood brain barrier (BBB) (0.6–1.3% for gefitinib and 2.8–4.4% for erlotinib). TKIs are also a substrate of P-glycoprotein, an efflux pump located on the membrane of endothelial cells [37, 38]. Therefore, intracranial failure of EGFR-TKIs is mainly considered a pharmacokinetic limitation rather than emergence of T790M mutation [39,40,41,42]. Zhao et al. [43] analyzed paired plasma and cerebrospinal fluid (CSF) samples from seven NSCLC patients with CNS metastases after failure with first-generation EGFR-TKIs and found that all harbored EGFR-TKI-sensitive mutations in CSF but that only one harbored T790M mutation in CSF. Exposure of gefitinib for all patients was significantly low in CSF, with a mean CSF/plasma mutational rate of 1.8%. Another study identified T790M mutation in 7 of 9 patients with extracranial lesions using circulating tumor cells (CTCs) from CSF but in only 1 of 14 patients using CSF samples [44]. In the present study, the lowest frequency and abundance of T790M mutation in plasma ctDNA was detected in patients in the BF group. In addition, several other studies have reported a low incidence of T790M mutation in CSF of a small number of BM patients [45, 46]. Therefore, we suggest that T790M mutation, one of the predominant resistance mechanisms, might not contribute substantially to BM. Future studies using brain tumor tissues might provide a better understanding of the role of T790M mutation in patients developing BM during EGFR-TKI therapy.
Several retrospective studies have reported controversial results regarding the prognostic value of acquired T790M mutation. Studies using tumor re-biopsies showed that NSCLC patients with acquired T790M mutation had prolonged survival durations; however, several studies using ctDNA-based mutational analysis demonstrated that ctDNA T790M+ patients had significantly short OS [15, 18,19,20]. Zheng et al. [22] reported that among patients receiving TKI treatment as a 2nd-line or later treatment, those with detectable T790M mutation in ctDNA had poor survival. In our study, ddPCRT790M− patients had a longer median PFS on first-generation EGFR-TKIs than did ddPCRT790M+ patients; a similar trend was also observed by ARMS, indicating that the tumors that had progressed in patients with plasma ctDNA-detectable T790M mutation were highly aggressive. We also observed longer survival in T790M− patients than in T790M+ patients based on the time of initial TKI administration (data not shown). However, when we calculated survival from the initial development of resistance to TKIs, we failed to observe any difference between; these findings were similar to the results of a study by Zhang et al. [47].
Furthermore, by following up the patients every 3 months, we assessed the effect of T790M mutation on subsequent clinical prognosis after progression on first-generation TKI treatment. Different lesions from patients with advanced NSCLC including recurrent or enlarged tumors or metastases requiring different treatments. The median survival time after progression on TKIs was 17.5 months, and the five types of treatments used in patients were continuation of the original TKI, AZD9291, chemo ± radiation, TKI + chemo/RT and BSC. For each subgroup of T790M+ patients in the CF, BF and OF groups, AZD9291 treatment appeared to be the best option, conferring the longest survival duration. These data are consistent with the findings of AURA trials, reporting a higher response rate to AZD9291 (61%) among patients with T790M mutation than among patients without this mutation (21%), with a median PFS of 9.6 months in T790M+ patients and 2.8 months in T790M− patients [48]. Moreover, in our study, the survival of patients treated with chemo ± RT and TKI + chemo/RT was acceptable in the T790M + CF group but not in the T790M + OF group (4.0 to 9.1 months). Patients receiving concurrent chemoradiation or TKI + RT had long survival durations, even those in the T790M− CF group. These data indicate that local treatment may have contributed to clinical benefit when the disease was localized, though this may not have been the case for patients with extensive metastasis. TKI + chemo/RT yielded the best survival in T790M− patients of the CF and OF groups. In the IMPRESS study, concurrent chemotherapy combined with TKIs after PD did not confer any benefit in the total population, whereas TKIs in combination with chemotherapy did provide a clinical benefit for patients with T790M− plasma ctDNA [49]. Nevertheless, further investigation is required to determine whether TKI + chemo can benefit T790M− patients after resistance to first-generation EGFR-TKIs. In the present study, patients, both T790M+ and T790M− in the BF group, treated with AZD9291 had the best survival. Considering the better penetration of AZD9291, as reported in the BLOOM study, and its great potency for targeting both sensitive and resistant mutant NSCLC cells [50], the reason for the excellent efficacy of AZD9291 in both T790M+ and T790M− BF groups is rather clear. Thus, both T790M mutational status and failure site strongly influence treatment selection and therapeutic effect.
This is a large-scale multi-institutional study to demonstrate correlation between disease failure sites and clinical outcomes after TKI therapy with the frequency/abundance of T790M mutation in ctDNA. Nevertheless, several limitations exist. First, molecular profiling was not simultaneously performed on matched tissue biopsies and blood samples after disease progression, and such analysis may reveal more direct correlation and concordance between both sets of samples, thereby enabling a better understanding of their distinct clinical features. Second, dynamic changes in T790M mutation in ctDNA were not monitored during disease progression, which may have been helpful in following disease progression among the three groups based on failure site. Third, only a single EGFR mutation was investigated in the present study due to limitations in one-time ddPCR and ARMS. Further studies to overcome these limitations are needed in the future.
Conclusion
Our prospective study demonstrated an important clinical value of T790M mutation in the ctDNA of patients who progressed on EGFR-TKI therapy. We show that ddPCR has a higher sensitivity than does ARMS. Our study revealed that the T790M mutational frequency and abundance are correlated with disease failure sites. Patients in the OF group were more likely to harbor T790M mutation than were those in the CF and BF groups. In addition, the T790M mutational status in ctDNA has a substantial influence on clinical prognosis based on subsequent treatment in each subgroup stratified by different failure sites. AZD9291 was the most frequent choice for T790M+ patients and yielded the longest survival after resistance to first-generation EGFR-TKIs. These data suggest that different therapeutic strategies should be considered for TKI-resistant patients depending on their T790M mutational status and disease failure site.
Abbreviations
- ARMS:
-
amplification refractory mutation system
- CNS:
-
central nervous system
- CR:
-
complete disease
- ctDNA:
-
circulating tumor DNA
- ddPCR:
-
droplet digital PCR
- ECOG:
-
Eastern Cooperative Oncology Group
- EGFR:
-
epidermal growth factor receptor
- NSCLC:
-
non-small cell lung cancer
- TKI:
-
tyrosine kinase inhibitor
- ORR:
-
objective response rate
- OS:
-
overall survival
- PD:
-
progressive disease
- PFS:
-
progression-free survival
- PR:
-
partial disease
- SD:
-
stable disease
References
Soh J, Okumura N, Lockwood WW, Yamamoto H, Shigematsu H, Zhang W, et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS ONE. 2009;4:e7464. https://doi.org/10.1371/journal.pone.0007464.
Ladanyi M, Pao W. Lung adenocarcinoma: guiding EGFR-targeted therapy and beyond. Mod Pathol. 2008;21(Suppl 2):S16–22. https://doi.org/10.1038/modpathol.3801018.
Sordella R, Bell DW, Haber DA, Settleman J. Gefitinib-sensitizing EGFR mutations in lung cancer activate anti-apoptotic pathways. Science. 2004;305:1163–7. https://doi.org/10.1126/science.1101637.
Lee CK, Brown C, Gralla RJ, Hirsh V, Thongprasert S, Tsai CM, et al. Impact of EGFR inhibitor in non-small cell lung cancer on progression-free and overall survival: a meta-analysis. J Natl Cancer Inst. 2013;105:595–605. https://doi.org/10.1093/jnci/djt072.
Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–57. https://doi.org/10.1056/NEJMoa0810699.
Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–8. https://doi.org/10.1056/NEJMoa0909530.
Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–8. https://doi.org/10.1016/s1470-2045(09)70364-x.
Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–46. https://doi.org/10.1016/s1470-2045(11)70393-x.
Zhou C, Wu YL, Chen G, Feng J, Liu XQ, Wang C, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): a multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–42. https://doi.org/10.1016/s1470-2045(11)70184-x.
Pao W, Miller VA, Politi KA, Riely GJ, Somwar R, Zakowski MF, et al. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005;2:e73. https://doi.org/10.1371/journal.pmed.0020073.
Gou LY, Li AN, Yang JJ, Zhang XC, Su J, Yan HH, et al. The coexistence of MET over-expression and an EGFR T790M mutation is related to acquired resistance to EGFR tyrosine kinase inhibitors in advanced non-small cell lung cancer. Oncotarget. 2016. https://doi.org/10.18632/oncotarget.9697.
Carrera S, Buque A, Azkona E, Aresti U, Calvo B, Sancho A, et al. Epidermal growth factor receptor tyrosine-kinase inhibitor treatment resistance in non-small cell lung cancer: biological basis and therapeutic strategies. Clin Transl Oncol. 2014;16:339–50. https://doi.org/10.1007/s12094-013-1143-9.
Zhao M, Zhang Y, Cai W, Li J, Zhou F, Cheng N, et al. The Bim deletion polymorphism clinical profile and its relation with tyrosine kinase inhibitor resistance in Chinese patients with non-small cell lung cancer. Cancer. 2014;120:2299–307. https://doi.org/10.1002/cncr.28725.
Jakobsen KR, Demuth C, Sorensen BS, Nielsen AL. The role of epithelial to mesenchymal transition in resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Transl Lung Cancer Res. 2016;5:172–82. https://doi.org/10.21037/tlcr.2016.04.07.
Oxnard GR, Arcila ME, Sima CS, Riely GJ, Chmielecki J, Kris MG, et al. Acquired resistance to EGFR tyrosine kinase inhibitors in EGFR-mutant lung cancer: distinct natural history of patients with tumors harboring the T790M mutation. Clin Cancer Res. 2011;17:1616–22. https://doi.org/10.1158/1078-0432.ccr-10-2692.
Engelman JA, Mukohara T, Zejnullahu K, Lifshits E, Borras AM, Gale CM, et al. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J Clin Invest. 2006;116:2695–706. https://doi.org/10.1172/jci28656.
Ogino A, Kitao H, Hirano S, Uchida A, Ishiai M, Kozuki T, et al. Emergence of epidermal growth factor receptor T790M mutation during chronic exposure to gefitinib in a non small cell lung cancer cell line. Cancer Res. 2007;67:7807–14. https://doi.org/10.1158/0008-5472.can-07-0681.
Chaft JE, Oxnard GR, Sima CS, Kris MG, Miller VA, Riely GJ. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin Cancer Res. 2011;17:6298–303. https://doi.org/10.1158/1078-0432.ccr-11-1468.
Hata A, Katakami N, Yoshioka H, Takeshita J, Tanaka K, Nanjo S, et al. Rebiopsy of non-small cell lung cancer patients with acquired resistance to epidermal growth factor receptor-tyrosine kinase inhibitor: comparison between T790M mutation-positive and mutation-negative populations. Cancer. 2013;119:4325–32. https://doi.org/10.1002/cncr.28364.
Li W, Ren S, Li J, Li A, Fan L, Li X, et al. T790M mutation is associated with better efficacy of treatment beyond progression with EGFR-TKI in advanced NSCLC patients. Lung Cancer. 2014;84:295–300. https://doi.org/10.1016/j.lungcan.2014.03.011.
Thress KS, Brant R, Carr TH, Dearden S, Jenkins S, Brown H, et al. EGFR mutation detection in ctDNA from NSCLC patient plasma: across-platform comparison of leading technologies to support the clinical development of AZD9291. Lung Cancer. 2015;90:509–15. https://doi.org/10.1016/j.lungcan.2015.10.004.
Zheng D, Ye X, Zhang MZ, Sun Y, Wang JY, Ni J, et al. Plasma EGFR T790M ctDNA status is associated with clinical outcome in advanced NSCLC patients with acquired EGFR-TKI resistance. Sci Rep. 2016;6:20913. https://doi.org/10.1038/srep20913.
Yang JJ, Chen HJ, Yan HH, Zhang XC, Zhou Q, Su J, et al. Clinical modes of EGFR tyrosine kinase inhibitor failure and subsequent management in advanced non-small cell lung cancer. Lung Cancer. 2013;79:33–9. https://doi.org/10.1016/j.lungcan.2012.09.016.
Sakai K, Horiike A, Irwin DL, Kudo K, Fujita Y, Tanimoto A, et al. Detection of epidermal growth factor receptor T790M mutation in plasma DNA from patients refractory to epidermal growth factor receptor tyrosine kinase inhibitor. Cancer Sci. 2013;104:1198–204. https://doi.org/10.1111/cas.12211.
Taniguchi K, Uchida J, Nishino K, Kumagai T, Okuyama T, Okami J, et al. Quantitative detection of EGFR mutations in circulating tumor DNA derived from lung adenocarcinomas. Clin Cancer Res. 2011;17:7808–15. https://doi.org/10.1158/1078-0432.ccr-11-1712.
Oxnard GR, Paweletz CP, Kuang Y, Mach SL, O’Connell A, Messineo MM, et al. Noninvasive detection of response and resistance in EGFR-mutant lung cancer using quantitative next-generation genotyping of cell-free plasma DNA. Clin Cancer Res. 2014;20:1698–705. https://doi.org/10.1158/1078-0432.ccr-13-2482.
Wang Z, Chen R, Wang S, Zhong J, Wu M, Zhao J, et al. Quantification and dynamic monitoring of EGFR T790M in plasma cell-free DNA by digital PCR for prognosis of EGFR-TKI treatment in advanced NSCLC. PLoS ONE. 2014;9:e110780. https://doi.org/10.1371/journal.pone.0110780.
Jackman D, Pao W, Riely GJ, Engelman JA, Kris MG, Janne PA, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small-cell lung cancer. J Clin Oncol. 2010;28:357–60. https://doi.org/10.1200/jco.2009.24.7049.
Liu X, Lu Y, Zhu G, Lei Y, Zheng L, Qin H, et al. The diagnostic accuracy of pleural effusion and plasma samples versus tumour tissue for detection of EGFR mutation in patients with advanced non-small cell lung cancer: comparison of methodologies. J Clin Pathol. 2013;66:1065–9. https://doi.org/10.1136/jclinpath-2013-201728.
Sacher AG, Paweletz C, Dahlberg SE, Alden RS, O’Connell A, Feeney N, et al. Prospective validation of rapid plasma genotyping for the detection of EGFR and KRAS mutations in advanced lung cancer. JAMA Oncol. 2016;2:1014–22. https://doi.org/10.1001/jamaoncol.2016.0173.
Seki Y, Fujiwara Y, Kohno T, Takai E, Sunami K, Goto Y, et al. Picoliter-droplet digital polymerase chain reaction-based analysis of cell-free plasma DNA to assess EGFR mutations in lung adenocarcinoma that confer resistance to tyrosine-kinase inhibitors. Oncologist. 2016;21:156–64. https://doi.org/10.1634/theoncologist.2015-0288.
Xu Q, Zhu Y, Bai Y, Wei X, Zheng X, Mao M, et al. Detection of epidermal growth factor receptor mutation in lung cancer by droplet digital polymerase chain reaction. OncoTargets Ther. 2015;8:1533–41. https://doi.org/10.2147/OTT.S84938.
Watanabe M, Kawaguchi T, Isa S, Ando M, Tamiya A, Kubo A, et al. Ultra-sensitive detection of the pretreatment EGFR T790M mutation in non-small cell lung cancer patients with an EGFR-activating mutation using droplet digital PCR. Clin Cancer Res. 2015;21:3552–60. https://doi.org/10.1158/1078-0432.ccr-14-2151.
Balak MN, Gong Y, Riely GJ, Somwar R, Li AR, Zakowski MF, et al. Novel D761Y and common secondary T790M mutations in epidermal growth factor receptor-mutant lung adenocarcinomas with acquired resistance to kinase inhibitors. Clin Cancer Res. 2006;12:6494–501. https://doi.org/10.1158/1078-0432.ccr-06-1570.
Arcila ME, Oxnard GR, Nafa K, Riely GJ, Solomon SB, Zakowski MF, et al. Rebiopsy of lung cancer patients with acquired resistance to EGFR inhibitors and enhanced detection of the T790M mutation using a locked nucleic acid-based assay. Clin Cancer Res. 2011;17:1169–80. https://doi.org/10.1158/1078-0432.ccr-10-2277.
Dempke WC, Edvardsen K, Lu S, Reinmuth N, Reck M, Inoue A. Brain metastases in NSCLC—are TKIs changing the treatment strategy? Anticancer Res. 2015;35:5797–806.
Zhang J, Yu J, Sun X, Meng X. Epidermal growth factor receptor tyrosine kinase inhibitors in the treatment of central nerve system metastases from non-small cell lung cancer. Cancer Lett. 2014;351:6–12. https://doi.org/10.1016/j.canlet.2014.04.019.
Abbott NJ, Ronnback L, Hansson E. Astrocyte–endothelial interactions at the blood–brain barrier. Nat Rev Neurosci. 2006;7:41–53. https://doi.org/10.1038/nrn1824.
Wang D, Zhou J, Liu X, Lu D, Shen C, Du Y, et al. Methylation of SUV39H1 by SET7/9 results in heterochromatin relaxation and genome instability. Proc Natl Acad Sci USA. 2013;110:5516–21. https://doi.org/10.1073/pnas.1216596110.
Chen Y, Wang M, Zhong W, Zhao J. Pharmacokinetic and pharmacodynamic study of gefitinib in a mouse model of non-small-cell lung carcinoma with brain metastasis. Lung Cancer. 2013;82:313–8. https://doi.org/10.1016/j.lungcan.2013.08.013.
Togashi Y, Masago K, Masuda S, Mizuno T, Fukudo M, Ikemi Y, et al. Cerebrospinal fluid concentration of gefitinib and erlotinib in patients with non-small cell lung cancer. Cancer Chemother Pharmacol. 2012;70:399–405. https://doi.org/10.1007/s00280-012-1929-4.
Deng Y, Feng W, Wu J, Chen Z, Tang Y, Zhang H, et al. The concentration of erlotinib in the cerebrospinal fluid of patients with brain metastasis from non-small-cell lung cancer. Mol Clin Oncol. 2014;2:116–20. https://doi.org/10.3892/mco.2013.190.
Zhao J, Ye X, Xu Y, Chen M, Zhong W, Sun Y, et al. EGFR mutation status of paired cerebrospinal fluid and plasma samples in EGFR mutant non-small cell lung cancer with leptomeningeal metastases. Cancer Chemother Pharmacol. 2016;78:1305–10. https://doi.org/10.1007/s00280-016-3155-y.
Jiang BY, Li YS, Guo WB, Zhang XC, Chen ZH, Su J, et al. Detection of Driver and resistance mutations in leptomeningeal metastases of NSCLC by next-generation sequencing of cerebrospinal fluid circulating tumor cells. Clin Cancer Res. 2017;23:5480–8. https://doi.org/10.1158/1078-0432.ccr-17-0047.
Hata A, Katakami N, Yoshioka H, Kaji R, Masago K, Fujita S, et al. Spatiotemporal T790M heterogeneity in individual patients with EGFR-mutant non-small-cell lung cancer after acquired resistance to EGFR-TKI. J Thorac Oncol. 2015;10:1553–9. https://doi.org/10.1097/jto.0000000000000647.
Hata A, Katakami N, Yoshioka H, Takeshita J, Tanaka K, Masago K, et al. Prognostic impact of central nervous system metastases after acquired resistance to EGFR-TKI: poorer prognosis associated with T790M-negative status and leptomeningeal metastases. Anticancer Res. 2015;35:1025–31.
Zhang Q, Ke E, Niu F, Deng W, Chen Z, Xu C, et al. The role of T790M mutation in EGFR-TKI re-challenge for patients with EGFR-mutant advanced lung adenocarcinoma. Oncotarget. 2017;8:4994–5002. https://doi.org/10.18632/oncotarget.14007.
Janne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, et al. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N Engl J Med. 2015;372:1689–99. https://doi.org/10.1056/NEJMoa1411817.
Soria JC, Wu YL, Nakagawa K, Kim SW, Yang JJ, Ahn MJ, et al. Gefitinib plus chemotherapy versus placebo plus chemotherapy in EGFR-mutation-positive non-small-cell lung cancer after progression on first-line gefitinib (IMPRESS): a phase 3 randomised trial. Lancet Oncol. 2015;16:990–8. https://doi.org/10.1016/s1470-2045(15)00121-7.
Yang JCH, Kim DW, Kim SW, Cho BC, Lee JS, Ye X, et al. Osimertinib activity in patients (pts) with leptomeningeal (LM) disease from non-small cell lung cancer (NSCLC): updated results from BLOOM, a phase I study. Am Soc Clin Oncol. 2016. https://doi.org/10.1200/JCO.2017.35.15_suppl.2020.
Authors’ contributions
SZ, LZ, and BX have been involved in drafting the article or revising it critically for important intellectual content. EC, QZ, XC, and XZ have made substantial contributions to acquisition of data. SZ, LZ, XZ and XC have made substantial contributions to analysis and interpretation of data. SM has made substantial contributions to conception and design of this manuscript and final approval of the version to be submitted. All authors read and approved the final manuscript.
Acknowledgements
We thank Xun Yan-Ping and Jiang Yan-Ping for their work in sample collection and performing EGFR test.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The main datasets supporting the conclusions of this article are included within the article and its additional files. Other clinical results of this trial have not been published. We cannot share a more detailed database.
Consent for publication
Not applicable.
Ethics approval and consent to participate
The protocol was approved by the Institutional Review Board of Hangzhou First People’s Hospital (No. HZFH CA15-02). All patients signed the informed consent.
Funding
The present study was supported by grants from Projects of Medical and Health Technology in Zhejiang Province (WKJ-2J-1532), the Zhejiang Provincial Natural Science Foundation (LY15H160010) and National Natural Science Foundation of China (Grant No. 81602671).
Author information
Authors and Affiliations
Corresponding author
Additional file
Additional file 1: Figure S1.
Design of the assay for detection of EGFR T790M mutations. (A) FAM- and VIC-labeled probes were designed to target mutant and wild-type EGFR alleles, respectively. (B) Sequence information of the primers and probes for the T790M ddPCR assay. (C) Selective sensitivity of the assay for T790M mutation. Up to 1:2,500 dilution of mutant to wild-type EGFR alleles; at least two positive droplets were stably detected by the ddPCR assay. The numbers shown in the positive area are the amounts of EGFR mutant allele-positive droplets by ddPCR. Mt, mutant allele; Wt, wild-type allele; ddPCR, droplet digital PCR.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhang, S., Zhu, L., Xia, B. et al. Epidermal growth factor receptor (EGFR) T790M mutation identified in plasma indicates failure sites and predicts clinical prognosis in non-small cell lung cancer progression during first-generation tyrosine kinase inhibitor therapy: a prospective observational study. Cancer Commun 38, 28 (2018). https://doi.org/10.1186/s40880-018-0303-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40880-018-0303-2