
Abstract
It becomes more and more important to develop appropriate models for the efficient design of the cell factory for microbial biofuels and biochemical productions, since the appropriate model can predict the effect of culture environment and/or the specific pathway genes knockout on the growth characteristics. Among various modeling approaches, kinetic modeling is promising in the sense of realizing the essential feature of metabolic regulation. A brief overview is given for the current status of the kinetic modeling of the cell metabolism from the point of view of metabolic regulation focusing on Escherichia coli (but not limited to E. coli). For the proper modeling, it is important to realize the systems behavior by integrating different levels of information to understand and unravel the underlying principles of the living organisms, namely, it is important to properly understand how the environmental stimuli are detected by the cell, how those are transduced, and how the cell metabolism is regulated, and to express these into the model. In particular, it is important to incorporate the enzymatic regulations of Pyk, Pfk, and Ppc by fructose-1,6-bisphosphate (FBP), phosphoenol pyruvate (PEP), and acetyl-coenzyme A (AcCoA) to realize the flux-sensing and homeostatic behavior. The proper modeling for phosphotransferase system (PTS) and the transcriptional regulation by cAMP-Crp and Cra is also important to simulate the main metabolism in relation to catabolite regulation. The coordinated regulation between catabolic and anabolic (nitrogen source-assimilation) metabolisms may be simulated by the behavior of keto acid such as α-ketoglutarate (αKG). The metabolism under micro-aerobic conditions may be made by incorporating the global regulators such as ArcA/B and Fnr. It is quite important to develop quantitative kinetic models, which incorporate enzyme level and gene level regulations from the biological science and metabolic engineering points of view.
Similar content being viewed by others


Introduction
Microbial production of biofuels and biochemicals from renewable resources or biomass has been paid recent attention from global sustainability and environmental protection points of view, and many attempts have been made for the cell design by metabolic engineering approach. However, the practical application is limited in many cases, and more innovative design of cell factories is desired [1]. On the other hand, significant progress has been made on molecular biology from the reductionist point of view. However, the molecular knowledge alone is in many cases not sufficient to understand the cell system's behavior, where the system's behavior emerges from the interactions between the characterized molecules [2]. Thus, the systems biology approach has been paid recent attention in the post genome era. The ultimate goal of systems biology is to reconstruct a cell system into the computer which can predict observable phenotypes. If this could be attained, the effects of culture environment and/or the specific genetic mutation on the metabolism can be predicted without conducting many exhaustive experiments, and metabolic engineering may be made more efficient with verification by the experiment for the selected mutants in the optimized condition based on the computer simulation. Thus, the appropriate model can contribute for the efficient design of cell factories from the practical application point of view.
It is quite important to quantitatively understand the complex and highly interrelated cellular behavior from biological science and metabolic engineering points of view. This may be attained with the help of informatics and systems biology by integrating different levels of ever increasing data with deep insight into the available data by biological knowledge [2-4].
In living organisms, metabolic network, defined as the set and topology of metabolic biochemical reactions in a cell, plays an essential role for the cell to survive, where it is under well-organized control. Thousands of different biochemical reactions as well as transport processes are linked together to break down organic compounds to generate energy (catabolism) and to synthesize macromolecular compounds (anabolism) for the cell synthesis. Similarly, complex signaling networks interconvert signals or stimuli that are important for the cellular function and interactions with the environment. This implies the importance of the transfer of information in signal transduction pathways and cascades designed to maximize the efficiency for cellular responses to environmental perturbations.
In order to understand the cell system in response to culture environment, the coupling between the recognition or sensing of the environmental condition and adjustment of the metabolic system must be properly incorporated into the model. In particular, it is important to incorporate the coupling between enzymatic reactions and the transcriptional regulation [5]. Moreover, although local regulation mechanisms are known to exist, it is not clear how those local regulation systems are coordinated on the systems level, where this may be made by ‘distributed sensing of the intracellular metabolic fluxes’ [5].
In the present article, current status of kinetic modeling is overviewed from the point of view of proper modeling with incorporation of metabolic regulation mechanism. Metabolic regulation analysis is critical for the proper modeling and has to be made in evaluating the performance of the designed cell as well as for reengineering the cell factories [6]. In bacterial adaptation to the culture environment, the global regulators detect the change in culture environment and control the metabolic pathway genes [7-9]. Here, the modeling of the metabolic regulation is considered focusing on Escherichia coli (but not limited to E. coli) based on the kinetic modeling approach with consideration of metabolic regulation.
Basic modeling approach
A variety of models have been proposed in the past, where they are discriminated from others depending on the underlying assumptions for the modeling, the data they require, and the accuracy of the model prediction [10]. The types of modeling formalism depend on such characteristics [11].
The model development may start with considering the network structure with kinetic rate expressions, model structure, parameter identification, and model validation, which may differ depending on the purpose of using a model [12]. It must be careful that the determination of kinetic rate expression is not straightforward due to the difficulty in identifying the mechanisms of enzymes and transporters [13], and therefore, some appropriate model simplification may be considered. Although parameter identification, sensitivity analysis, identifiability, experimental design, and optimization are important for the modeling in practice [12,14,15], here, we rather focus on the kinetic modeling with consideration of metabolic regulation.
Metabolic flux analysis
Among different levels of information, the metabolic fluxes are located on top of those, and it is the most important information from the phenotypic fermentation point of view [16-18], and it can be used for the analysis of the specific pathway gene knockout on the metabolism [19,20]. 13C-metabolic flux analysis (13C-MFA) has been shown to be useful for the metabolic regulation analysis [16,19-23]. However, this is essentially the analysis method for the physiological state of the organism based on mass balance together with isotopomer balance, and it does not have the predictability. It is highly desirable and useful to be able to predict the cell growth characteristics and the metabolic changes in response to the change in culture environment and/or the specific pathway mutation.
Flux balance analysis and its extensions
Flux balance analysis (FBA) and its extension to genome scale has made significant progress as it requires only basic knowledge of the metabolic reaction stoichiometry and has a reasonably accurate predictability. Significant efforts have also been made to integrate gene level regulation and metabolic networks to reveal the regulation mechanism [24,25]. In such approach, however, some appropriate objective functions such as the maximization of the cell growth rate, the specific substrate consumption rate, and/or the metabolite production rate must be introduced due to excess degrees of freedom. It was, however, shown that no single objective function can accurately represent the flux data for the different culture condition [26]. Rather, a vector-valued objective function or multiple objective functions must be considered, resulting in the Pareto optimal set to represent the metabolic fluxes [27], where the influential objective function may be the maximum ATP yield, maximum biomass yield, and minimum sum of absolute fluxes (which corresponds to minimum enzyme investment).
The formulation may be as follows:
Subject to:
where j i is the ith objective function, S ij is the stoichiometric coefficient, and v j is the flux. However, it is not easy to determine the optimal point on the Pareto optimal set in practice depending on the variety of culture conditions.
FBA approach together with MFA information may be considered for the metabolic engineering purpose such as OptKnock [28], a bi-level programming framework for identifying gene knockout for the strain improvement. This has been extended as OptReg to consider not only knockouts but also overexpression and downregulations of various reactions in the network [29]. Another extension has also been made as OptForce [30], OptFlux [31], and differential bees FBA (DFFBA) with OptKnock to identify the optimal gene knockout strategies for maximizing the yield of the desired phenotypes while sustaining the growth rate [32]. Further extension has been made as OptStrain aiming at guiding pathway modifications, through reaction additions and deletions, of microbial networks for the overproduction of targeted compounds based on stoichiometrically balanced approach imposing maximum product yield requirements, pinpointing the optimal substrates, and evaluating the different microbial hosts such as Helicobacter pylori, E. coli, S. cerevisiae, and other microorganisms [33].
Stoichiometry-based strain design algorithms are often formulated as bi-level mixed integer linear programming problems [28-30,34,35], where outer level optimizes the objective function(s), while the inner level optimizes the cellular system that counteract any externally imposed genetic or environmental perturbations [36,37]. Different fitness functions may be considered [38,39].
The linear property of stoichiometric equations underlying FBA is the computational advantage and allows for genome-scale extension. However, it is not easy to confirm the designed cell metabolism in view of enzymatic reactions with intracellular metabolite concentrations.
The problem in FBA and its extension to genome-scale is the difficulty for the dynamic analysis as compared to kinetic modeling approach. Some extension has been made by incorporating kinetic expressions of multiple carbon sources and other nutrients into the quasi steady-state [40-42]. The dynamic multi-species metabolic modeling (DMMM) approach has been considered by incorporating the metabolite uptake kinetics into stoichiometric models of a microbial consortium [43,44]. On the other hand, steady-state flux distributions obtained from FBA and stoichiometric information have been used to parameterize genome-scale kinetic models applicable for small perturbations [45-48]. Lin-log kinetic expression and thermodynamics may be incorporated to constrain FBA simulation [49].
Although some attempts have been made by the hybrid type of stoichiometric/kinetics-based modeling approach [45,50,51], its potential may not be fully investigated. The dynamic flux balance analysis (dFBA) may be considered for diauxic growth of E. coli consuming glucose and acetate by taking into account the constraints that govern the cell growth at different phases in the batch culture [52]. Moreover, dFBA may be used for the co-culture with multiple sugars for the cellulosic biofuels production [53-55]. Recently, OptForce formalism has been extended as k-OptForce by bridging the gap between stoichiometric approach and kinetics-based approach, where the procedure seamlessly integrates the mechanistic detail given by kinetic models within a constraint-optimization framework tractable for genome-scale models [56].
The proper formulation for the interaction between the metabolism and gene expression by applying the principle of growth optimization enables the accurate prediction of multi-scale phenotypes [25], where constitutively expressed genes show growth-rate-dependent expression trends [57,58]. This implies the economic ways of the cell system that is regulated in response to global change in metabolic efficiency [59]. Moreover, such optimality model may be used for the adaptive laboratory evolution [60].
The construction of a virtual microbe will be an ambitious but realistic target that builds a novel resource that can provide significant benefits in the variety of practical applications. As an extension of the constraint-based genome-scale models [61], a whole cell computational model was developed for Mycoplasma genitalium, a urogenital parasite adored by synthetic biologists for its reduced genome [62]. This model constitutes 28 processes of the cell's operation, where these include processes that track exchanges with the extracellular medium, all the metabolic fluxes, the state of the supercoiled chromosome, transcription of all active genes, processing of all mRNAs, translation of all proteins, formation of all macromolecular complexes including RNA polymerases and ribosomes, and progresses of cytokinesis and FtsZ polymerization [63]. This may be the dawn of virtual cell biology [64], and this might even go beyond the previous attempt of the so-called ‘a grand challenge of the twenty-first century’ [65].
Once again, although powerful and attractive for the possible extension to the whole cell modeling or the so-called virtual microbes, the main drawback of the above approach is the difficulty in incorporating ‘explicitly’ the metabolic regulation mechanism.
Kinetic modeling and incorporation of metabolic regulation
Kinetic models for the metabolism require quantitative expressions that connect fluxes and metabolite concentrations. Those can be obtained with respect to time by solving a set of ordinary differential equations (ODEs) such as:
where the typical mechanistic expression may be the Michaelis-Menten type such as:
or Hill type expression may be considered, where v j max and K s are the model parameters, and S is the substrate concentration for the corresponding pathway reaction. These expressions require detailed enzyme reaction mechanism and characterization [66,67]. In order to reduce the computational burden in association with kinetic modeling, various approximate kinetic forms such as lin-log [68-70] and log-lin [71] kinetics, power-law kinetic expressions such as S-system [72], generalized mass action [45], and others [73-75] have been proposed.
As mentioned in the previous section, although the stoichiometric constraint-based genome-scale metabolic models have been developed for a variety of organisms [76], it is not easy to incorporate or express the effects of intracellular metabolites and enzyme activities appropriately with such approach. Although some attempts have been made for incorporating transcriptional regulation into FBA framework in the form of Boolean rules [77-80], the regulatory rules are not based on the metabolic regulation mechanisms, but on the basis of the available data which may be the manifestation of part or snapshot of the real regulation mechanism [81].
In contrast to the stoichiometric models, the kinetic modeling approach is attractive in the sense that such mechanism can be incorporated into the model appropriately. The primary attempts of incorporating the regulation mechanisms into kinetic models have been made by cybernetic modeling approach, where the organisms are considered to utilize the available nutrient sources with the maximum efficiency by the optimal strategy [82]. This approach has been extended to more structural models that contain detailed pathways [83]. More recently, this approach has been considered for the potential applications to metabolic engineering [84,85]. In such modeling approach, an elementary mode was considered as a metabolic subunit to model cellular regulatory processes, where the elementary modes are a set of metabolic pathways by which the cellular metabolic routes can be completely described, and any feasible fluxes can be represented by their combinations at steady state [86]. The elementary modes consist of a minimal set of reactions that function at steady state, which implies that the elementary mode cannot be a functional unit if any reaction is removed [86]. The hybrid type modeling has also been developed by assuming quasi-steady-state for the intracellular metabolites [87,88], where several applications were made for E. coli [88] and for yeast [81].
In the kinetic modeling approach, it is critical to identify kinetic parameter values and kinetic rate laws applicable to a variety of genetic and/or environmental perturbations. Moreover, the large-scale extension may be limited by considering unambiguous kinetic model parameterization [89]. Several attempts have been made towards postulating a generalized uniform kinetic expression such as approximate enzyme kinetic equations [68,73,90-93], S-system formalism [94,95], or a combination of in vitro-based lumped and approximate rate equations [96,97]. However, the predictability may not be the satisfied level [68,75].
Recently, the ensemble modeling (EM) has been considered to cope with large-scale kinetic modeling by successively reducing the size of parameter space based on the available experimental data such as fluxes and/or intracellular metabolite concentrations together with thermodynamic constraints for the direction of the net fluxes [98]. In EM approach, any type of pathway reaction mechanism can be considered as well as already known mechanism, where each reaction is decomposed into elementary reaction steps with mass action kinetics [99] such as:
where A and B are the metabolites and E is the enzyme.
The EM procedure starts with initially assumed kinetic models that predict the experimentally observed phenotypic characteristics, and the additional data such as those of the strain under environmental and/or genetic perturbations are used to screen the models until a minimal set of kinetic models are obtained [99]. This modeling approach has been successfully applied for lysine production [100], fatty acid production [101], aromatic production [98], robustness analysis for engineered non-native pathways [102], and modeling cancer cells [103]. Moreover, this approach has been applied for the modeling of E. coli that reasonably predicts the fluxes and intracellular metabolite concentrations of wild type and its single gene knockout mutants [104] based on the available multi-omics data [105].
Modeling of the main metabolism for catabolite regulation
The metabolic reactions of the central metabolism play important roles for energy generation and the production of the precursors for biosynthesis, and those form the hub on which all nearly catabolic and anabolic processes are built. Metabolic regulation of the central metabolism plays a key role in the adaptation of organisms to changes in their environment. The overall structure of central metabolic pathways is remarkably well conserved in the living organisms. Thus, the metabolic model of the central metabolism will provide a platform for further extension to peripheral metabolism and incorporation.
An attempt has been made for the modeling of the main metabolic pathways to simulate the dynamic behavior of Saccharomyces cerevisiae in response to the pulse addition of the carbon limited growth condition and measurement by fast sampling system [106,107]. The kinetic model equations for the glycolysis and pentose phosphate (PP) pathway have been developed for E. coli to simulate the transient data obtained by the fast sampling system [108]. The kinetic models for the tricarboxylic acid (TCA) cycle and anaplerotic pathways as well as glycolysis and PP pathway were also considered to simulate the typical batch and continuous cultures with some rule-based approach, where the cell growth rate was estimated based on the specific ATP production rate computed from the fluxes [109]. The kinetic modeling for the main metabolism of E. coli has also been made based on fluxomics and metabolomics data [110].
A wealth of information is available on genetic regulation, biochemistry, and physiology of cellular metabolism in response to culture environment, and some attempt has been made for the modeling and simulation, where it is important to make modeling based on the integrated information from gene level to flux level by incorporating the roles of transcription factors [5,111,112]. The important steps are how to incorporate (i) the effect of culture environment on global regulators, (ii) the effects of global regulators on the metabolic pathway genes, (iii) the effects of metabolic pathway genes on the corresponding enzyme activities, as well as (iv) the effects of enzyme level regulations (Figure 1).

Overall metabolic regulation scheme.
Importance of the modeling for the main metabolic pathways
Although the modeling of the restricted metabolic pathways such as glycolysis only or glycolysis plus PP pathway, etc. may be useful depending on the purpose of using the model such as short-time transient responses against pulse addition of substrate, it is by far important to model the whole main metabolic pathways such as glycolysis, TCA cycle, PP pathway, together with anaplerotic and gluconeogenic pathways. This enables us to simulate the typical batch culture, where the metabolic transition occurs from glucose-rich (glycolysis) condition to acetate-rich (gluconeogenic) condition in E. coli and others.
In relation to the model development of the main metabolism, the accurate estimation of the cell growth rate is critical for the practical application point of view. In general, the cell growth rate may be expressed as a function of substrate such as Monod type model:
with:
where X and S are the cell and substrate concentrations, and μ is the specific cell growth rate. However, the saturation constant K s is small, and the dynamics depend on the maximum specific growth rate parameter μ m which is usually constant, resulting in the difficulty in estimating the reasonable cell growth rate by Monod type model and its modification. The importance of accurate estimation of the cell growth rate is more eminent for the dynamic simulation of the specific gene knockout mutants and for the effect of culture condition. Thus, it is desirable to be able to accurately predict the cell growth rate in general.
The cell growth rate is determined by the catabolic reactions such as ATP production as well as anabolic reactions under typical growth conditions. Once the main metabolic pathways could be appropriately modeled, the ATP production rate can be estimated. The model equations are established by the mass balance with kinetic equations for the main metabolic pathways (Figure 2). The solution to such ODEs can be used to compute the fluxes with respect to time. This enables us to compute the specific ATP production rate with respect to time such as:

Main metabolic pathways.
where v is the reaction rate of the pathway (Figure 2). OP NADH and \( O{P}_{{\mathrm{FADH}}_2} \) are the specific ATP production rate via oxidative phosphorylation by NADH and FADH2, respectively, and those may be expressed as:
where (P/O) and (P/O)' are the P/O ratios for NADH and FADH2, respectively, and those are most likely to be 2.5 and 1.5, respectively, under typical aerobic condition. Those may be considered as model parameters and can be adjusted by the experimental data [109].
Now, 13C-MFA shows the correlation between the specific ATP production rate and the specific cell growth rate [109,113-115]. This indicates that the above ν ATP can be used to estimate the specific growth rate, and in fact, it was shown that this approach allows us to estimate the cell growth rate and fluxes of the specific gene knockout mutant for E. coli to some extent [109,112].
In particular, in the case of anaerobic fermentation, NADH re-oxidation and substrate level phosphorylation for ATP generation are important, and ATP generation by acetate kinase (Ack) pathway is critical for survival in the case of using only xylose as a carbon source [116]. This may be simulated by the model with the cell growth rate taking into account the effect of ATP production rate as mentioned above.
Moreover, if the main metabolism was appropriately modeled, the specific CO2 production rate can be also estimated by:
where this can be used to estimate CO2 evolution rate (CER), and thus the cell yield may be estimated together with other metabolite production rates and the cell growth rate. Since CER can be measured in practice, this may be also validated by the experimental data.
In relation to NADH production as mentioned above, the specific NADPH production rate can be also estimated as:
where NADH is produced in many bacteria, while some bacteria such as E. coli produce NADPH at isocitrate dehydrogenase (ICDH). It has also been observed that the specific NADPH production rate is linearly correlated with the specific growth rate from the point of view of anabolism. This means that the flux from G6P to the oxidative PP pathway can be estimated as far as the oxidative PP pathway is dominant for NADPH production, once the specific growth rate was determined from the catabolic ATP production rate [109].
Metabolic regulation mechanisms to be incorporated in the kinetic model
As mentioned in the ‘Kinetic modeling and incorporation of metabolic regulation’ section, several efforts have been made for the appropriate kinetic models which can describe the metabolic regulation in response to genetic and/or environmental perturbations. Here, we consider the metabolic regulation mechanisms that have to be incorporated into the kinetic models, where the enzyme level regulation such as allosteric regulation may be incorporated into the kinetic rate expression, while the transcriptional regulation may be expressed as functions of transcription factors (TFs), where the activities of TFs may be considered to be functions of intracellular metabolites as will be mentioned next.
The cell system achieves the coupling between recognition and adjustment through TFs, whose activities respond to the culture environment, and regulate the expression of the associated genes. This combined recognition and adjustment forms the reaction networks that overarch the metabolic and genetic layers [5]. In general, fast action is made by the enzyme level regulation such as the feed-forward activation of pyruvate kinase (Pyk) by fructose 1,6-bisphosphate (FBP), and the feedback inhibition of phosphofruct kinase (Pfk) by phosphoenol pyruvate (PEP), a motif that enables a high level of the upstream metabolite to lower the level of the downstream metabolite [117] (Figure 3a). The slow action is made through the transcriptional regulation, where cAMP-Crp activates the expression of TCA cycle genes, while (FBP-inhibited) catabolite repressor/activator, Cra activates the expression of gluconeogenic pathway genes as well as some of the TCA cycle genes and the glyoxylate pathway genes, and represses the expression of the glycolysis genes in the case of E. coli (Figure 3b), which will be explained later.

Metabolic regulation of the main metabolism. (a) Enzyme level regulation of glycolysis. (b) Transcriptional and enzyme level regulations of the main metabolic pathways.
The levels of the flux-signaling metabolites become coupled, enabling a robust, coherent response of the TFs. The coherent behavior of the overall system is not established by a common transcriptional master regulator, but arises from the molecular interactions within the system itself [5]. It may be considered that the system of reactions of the lower glycolysis and the feed-forward activation of Pyk by FBP translate flux information into the concentration of FBP, and that this feed-forward activation affects the linearization of the glycolytic kinetics, where the glycolysis from FBP to PEP may be expressed as the reversible Michaelis-Menten (MM) equation, while Pyk may be expressed as the irreversible MM equation [118] such as:
where feed-forward activation of FBP on Pyk may be expressed by Monod-Wyman-Changeux (MWC) kinetics [118]. In fact, feed-forward regulation has been known to ensure the structural robustness against perturbations [117]. This mechanism may be conserved in many organisms. For example, in S. cerevisiae, sugar uptake rate is well correlated with the respiratory and fermentative pathways, or the specific ethanol production rate, and the similar relationship may be seen between the glycolytic flux and FBP [119,120], where Pyk is also feed-forward activation by FBP in S. cerevisiae [121].
In order to realize the above mechanism, the kinetic expression of Pyk (and also phosphoenolpyruvate carboxylase (Ppc)) must be a positive function of FBP such as:
where [・] denotes the concentration, and PEP and pyruvate (PYR) are the substrate and product of Pyk reaction, respectively. FBP is the allosteric activator, and p Pyk is the kinetic parameter vector for Pyk. The feed-forward activation of Pyk by FBP may be enhanced by the feedback inhibition of Pfk by PEP, where the kinetic expression for Pfk must be a negative function with respect to PEP:
where F6P and FBP are the substrate and product of Pfk reaction, respectively, while PEP is the allosteric inhibitor, and p Pfk is the parameter vector for Pfk reaction, namely, if the sugar uptake rate or the upper glycolytic flux were increased, FBP increases, and in turn allosterically activates Pyk, which decreases PEP concentration, and the feedback inhibition of Pfk by PEP is relaxed and causes further increase in the upper glycolytic flux (Figure 3a). In the nominal growth condition, the feedback inhibition of Pfk by PEP may not be important, but this may cause oscillatory behavior under certain condition [122].
Moreover, the increased pyruvate goes down through pyruvate dehydrogenase (PDH) to acetyl-coenzyme A (AcCoA), where AcCoA becomes homeostatic, namely, the increase in AcCoA activates Ppc reaction [123,124], thus reducing the upcoming Pyk-PDH fluxes, increases oxaloacetate (OAA), and activates citrate synthase (CS) reaction, and therefore activating the outgoing flux from AcCoA. In this way, AcCoA concentration decreases, forming the feed-back regulation against the initial increase in AcCoA (Figure 4). This mechanism can be realized by expressing the Ppc activity as a positive function of AcCoA as well as FBP such as:

Homeostasis of AcCoA by the activation of Ppc.
where PEP and OAA are the substrate and product of Ppc reaction, respectively, and FBP and AcCoA are the allosteric activators. In another view, this phenomenon may be considered as the feed-forward regulation in the sense that the repression of TCA cycle activity is detected by the increase in AcCoA, which causes the activation of the anaplerotic Ppc pathway, and backs up the precursor metabolite such as OAA since it is expected to be decreased due to deactivated TCA cycle.
As the glucose uptake rate increases, the TCA cycle flux tends to increase by the increased OAA and AcCoA, and then NADH is overproduced. The accumulated NADH inhibits CS and ICDH allosterically [125], forming feedback regulation, and thus results in AcCoA accumulation, which in turn causes acetate overflow metabolism. This enzyme level regulation by NADH in the TCA cycle can be verified by incorporating NADH oxidase (NOX) [126] or nicotinic acid [125], whereby activating TCA cycle. This effect is more enhanced under arcA mutant [127]. In the long run, the expression of TCA cycle genes is eventually repressed by the transcriptional regulation by cAMP-Crp toward steady state as will be explained later. The inhibitory effect of NADH on CS and ICDH may be expressed explicitly in the rate equation, but the problem is that the estimation of NADH/NAD+ pool is difficult without detailed proper modeling of the respiratory chain, which is not easy at this stage.
The typical growth condition changes from glucose-rich to acetate-rich in the batch culture. This requires a significant reorganization of the central metabolism from glycolysis to gluconeogenesis. Although the molecular mechanism underlying the metabolic transition from glucose to acetate has been extensively investigated in E. coli [128], its dynamics have been poorly understood. Since it is critical for the cell to efficiently and quickly reprogram the metabolism under the changing environmental condition, the cell must have the elaborate managing system.
The expression of the reaction rate for Ppc is the function of both FBP and AcCoA as mentioned above, which then enables us to simulate the ultrasensitive regulation of anapleurosis [129], namely, after glucose depletion, FBP concentration decreases accordingly, where Ppc and Pyk activities decrease in turn by the allosteric regulation, and PEP consumption is almost completely turned off. These make PEP concentration to be increased, and this buildup of PEP is kept during certain period [114], and this may serve to quickly uptake the glucose by PTS if it becomes available again [129]. This mechanism is important for the fed-batch culture compensated by DO-stat or pH-stat, where carbon limitation often occurs periodically, and the uptake of carbon source can be made quickly and efficiently without delay. Such phenomenon can be simulated by the model as mentioned above as compared to the case without feed-forward regulation mechanism. This feed-forward regulation mechanism is also important for the modeling and simulation of lactic acid bacteria, where lactate dehydrogenase (LDH) as well as Pyk is also activated by FBP, thus producing lactate quickly and lowering the pH around the cell as soon as the glucose is available [130]. Although the kinetic model for lactic acid bacteria has been developed previously [131], the above mechanism is not incorporated, and thus the simulation result does not properly reflect the real characteristics.
Moreover, after glucose depletion, FBP level drops, and thus Ppc activity decreases, while PEP carboxykinase (Pck) activity is activated by the activated Cra caused by the decreased FBP. This reveals the mechanism of avoiding the futile cycling caused by Ppc and Pck during gluconeogenic phase [129], where ATP generation becomes important. During the active glycolysis with enough sugars available, this futile cycling occurs, and loses ATP without efficient use for the compensation of the flexible metabolic fluxes and the metabolic regulation [123]. This may be simulated by the appropriate models taking into account both enzymatic and transcriptional regulations.
Now, the enzyme level regulation in the glycolysis made by Pyk and Pfk as well as FBP and PEP as mentioned above keeps increasing the substrate uptake rate, where this makes the system unstable. This is counterbalanced by the transcriptional regulation by cAMP-Crp, where cAMP level decreases due to the lower level of phosphorylated EIIA (EIIA-P), and lower activity of adenylate cyclase (Cya) at higher glucose consumption rate. Since ptsG which encodes EIIBC of PTS is under control of cAMP-Crp, the glucose uptake rate is repressed by the lower level of cAMP-Crp (Figure 3b). Thus, the transcriptional repression of PTS by cAMP-Crp must be incorporated into the model to realize such feedback regulation for the glucose uptake rate. The molecular mechanism for catabolite regulation has been illustrated by several researchers [132-136], and the PTS and catabolic regulation have been modeled by several researchers [5,109,111,137].
Moreover, the TCA cycle is transcriptionally repressed by the lower level of cAMP-Crp as the glucose consumption rate was increased. In the continuous culture (Chemostat), the effect of the cell growth rate or the dilution rate on the metabolism can be appropriately simulated for E. coli [112]. Namely, as the specific growth rate was increased, FBP increases due to the increase in the fluxes of the upper glycolysis and Cra decreases. Moreover, PEP concentration decreases as the PTS flux increases, since PEP is the co-substrate of PTS, and in turn EIIA-P decreases, resulting in the decrease in Cya activity and cAMP-Crp level. The decrease in cAMP-Crp causes the repression of the TCA cycle, and acetate overflow metabolism occurs as the specific growth rate increases. The trend of the simulation result by reflecting the above mechanism [112] is the similar as the experimental data [105] (Figure 5).

In E. coli, acetate is formed from AcCoA by Pta-Ack and from pyruvate by pyruvate oxidase, Pox [128]. Acetate can be metabolized to AcCoA either by the reversed reactions of Pta-Ack or by acetyl-coenzyme A synthetase (Acs). Acetate formation has been known to be due to metabolic imbalance, where the rate of AcCoA formation via glycolysis surpasses the capacity of the TCA cycle in E. coli [138]. Pox and Acs may be expressed as functions of the sigma factor (σ38) RpoS, but it may be difficult to predict the behavior of RpoS, while Acs may be expressed as a function of cAMP-Crp, where Acs is activated by cAMP-Crp during gluconeogenic phase [112].
Among intracellular metabolites, α-keto acid such as αKG turns to be a master regulator for catabolite regulation and co-ordination of different regulations [139]. Namely, when favored carbon source such as glucose was depleted, αKG level fall, and cAMP increases to stimulate other carbon catabolic machinery. Namely, when preferred carbon source such as glucose is abundant, the cell growth rate becomes higher with lower cAMP level, while if it is scarce, the cell growth rate declines with higher cAMP level. In particular, under nitrogen (N)-limitation, αKG accumulates due to decreased activity of glutamate dehydrogenase (GDH) and inhibits carbon assimilation, where there is less need for carbon-catabolic enzymes, and more demand for those involved in such nutrient assimilation. On the other hand, when anabolic nutrient such as ammonia is in excess, αKG concentration decreases due to activated GDH, producing glutamate (Glu) from αKG, cAMP level increases, and carbon catabolic enzymes increases to accelerate carbon assimilation. Namely, αKG coordinates the catabolic (C)-regulation and N-regulation by inhibiting EI of PTS [140] or cAMP via Cya [58,141]. Moreover, the physiological function of cAMP signaling goes beyond simply enabling hierarchical utilization of carbon sources as will be mentioned later but also controls the function of the proteome [139,142]. In order to model such phenomenon, EI of PTS has to be expressed as the inhibition by αKG, or Cya has to be expressed as the inhibition by keto acids such as OAA and PYR as well as αKG, where the modeling for nitrogen regulation will be mentioned later.
In the case of biofuels production from cellulosic biomass, the hydrolyzed biomass contains multiple sugars, and those sugars are selectively assimilated with catabolite repression depending on the type of microorganism used [143,144]. The metabolic regulation differs depending on the carbon sources used.
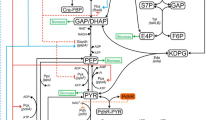
Glycerol has been paid recent attention for the production of biofuels and biochemicals, since it is a by-product of the biodiesel production [145-149]. In E. coli, glycerol is transported and phosphorylated to produce dihydroxy acetone phosphate (DHAP) of the central metabolism via the pathway encoded by glpF, glpK, and glpD, where ATP (or in certain cases PEP) is used for the phosphorylation at glycerol kinase (GlpK) reaction, while NADH is produced at glycerol 3-phosphate dehydrogenase (G3PDH) reaction under aerobic condition (Figure 6). These genes are under catabolic regulation by cAMP-Crp, so that glycerol is assimilated after glucose was depleted if glucose co-exists. NADH production at G3PDH becomes important for the biofuels production under anaerobic condition affecting NADH/NAD+ balance for dehydrogenase reactions. In the case of using glycerol as a single carbon source, cAMP-Crp increases due to the increase in the phosphorylated EIIAGlc, where cAMP-Crp induces glpFKD genes. Since FBP concentration decreases in the case of using glycerol as a carbon source, Cra is activated, and this together with upregulation of cAMP-Crp causes pckA gene as well as TCA cycle genes to be upregulated [150,151]. The kinetic expressions for the glycerol uptake pathways have been proposed [152]. This together with the inclusion of PTS and the transcriptional regulation as mentioned above enables the simulation for the case of using multiple carbon sources such as glucose and glycerol. Moreover, the enhancement of the TCA cycle caused by the increase in cAMP-Crp can be also simulated in the case of using glycerol as a single carbon source.

Glycerol-, fructose-, and xylose-assimilating pathways (a,b).
In the case of using fructose, it is transported by fructose-PTS, which has its own HPr-like protein domain called FPr. Namely, the phosphate of PEP is first transferred to EI (as EI-P), but then this phosphate is transferred to FPr instead of HPr, and in turn the phosphate is transferred via fructose specific EIIAFru and EIIBCFru, and phosphorylates fructose, where phosphorylated fructose becomes fructose 1-phosphate (F1P) [153]. The fruBKA operon is under control of cAMP-Crp, and thus glucose is preferentially consumed by glucose PTS when glucose co-exists. On the other hand, this operon is repressed by Cra [154]. Because of this, cra gene knockout enables co-consumption of glucose and fructose with fructose to be consumed faster as compared to glucose [155], where activated FruB in cra mutant competes with HPr (for glucose phosphorylation) for the phosphate of EI-P. Since phosphorylation of EIIAGlc via HPr becomes lower [156], the glucose uptake rate decreases as compared to the wild-type strain [155]. This phenomenon may be also simulated by the similar expression as glucose-PTS but compete the phosphate of EI with glucose-PTS (Figure 6).
In the case of using xylose as a carbon source, it is transported either by an ATP-dependent high affinity ABC transporter encoded by xylFGH or ATP-independent low affinity proton symporter encoded by xylE [157,158]. In the case of xylose utilization, the transcription factor XylR regulates xylAB/xylFGH [159], where xylR is under control of cAMP-Crp, and then catabolite repression occurs when glucose co-exists, where glucose is preferentially consumed first. The kinetic model for xylose uptake pathways as well as Entner-Doudoroff (ED) pathways has been proposed for Zymomonas mobilis [160], and thus it is necessary to incorporate the activation of the transporter by cAMP-Crp, and this can be made for the catabolite repression when co-exist with glucose [112] (Figure 6).
Modeling for the peripheral metabolism
Although it is critical to consider the main metabolism for the metabolic regulation as well as for the cell growth rate, the peripheral metabolic pathways become important for the practical applications such as amino acids fermentation.
The kinetic model for lysine synthetic pathways from OAA in the TCA cycle has been proposed [161], and this can be used to apply sensitivity analysis such as metabolic control analysis (MCA) to identify the limiting pathways in Corynebacterium glutamicum [162]. This investigation revealed that lysine production is primarily controlled by aspartokinase (Ask) and lysine permease. This was verified by the experiment using the recombinant strain overexpressing Ask, resulting in the significant increase in lysine production, although that flux did not increase as much as would be expected by MCA [162].
Shikimic acid production and aromatic amino acids production may be also simulated based on the formation of the precursor metabolites such as erythrose 4-phosphate (E4P) and PEP in the central metabolism [111]. Other amino acid fermentation may be also simulated using dynamic metabolic models [111,137,163].
Modeling for the metabolism under oxygen limitation
Most of the biofuels and biochemical productions by microbes is made by the fermentation under anaerobic condition, and thus it is important to properly model such fermentation as well as under aerobic condition, where the latter is often employed for the enhancement of the cell growth rate before anaerobic condition to improve the productivity of the target metabolites.
Although the modeling and computer simulation of a microbial cell cultivated under anaerobic condition such as lactate fermentation [131], and acetone-butanol-ethanol fermentation [164-166] has been proposed by several researchers, the regulatory mechanisms are rarely incorporated. Moreover, cofactor balance such as NADH balance becomes important under anaerobic condition, and thus it may be better to appropriately incorporate in the model equations. However, this is not so easy without proper modeling of the respiratory pathways under different oxygen concentration.
In order to properly model the metabolic transition from aerobic to anaerobic conditions, the roles of global regulators such as ArcA/B and Fnr must be properly incorporated, where the effect of dissolved oxygen concentration on the activation of such TFs has been reported [167,168], and this may be taken into account for the simulation under microaerobic conditions. In particular, it is important to properly simulate the behavior at the branch point of PYR, where the reaction rate through PDH, ν PDH must be the negative function of ArcA (or phosphorylated ArcA, ArcA-P), while the reaction rate through pyruvate formate lyase (Pfl), ν Pfl is the positive function with respect to ArcA and Fnr, where PYR is converted to formate (FOR) and AcCoA (Figure 7). Moreover, ethanol-forming pathway from AcCoA, alcohol dehydrogenase (ADH) must be activated where NADH is required for this reaction. As for TCA cycle, α-ketoglutarate dehydrogenase (KGDH) may be repressed by ArcA, while formate reductase (Frd) is activated by Fnr, thus the TCA cycle will be branched under anaerobic conditions. Some attempt has been made to estimate the fluxes in relation to such global regulators [169].

Anoxic regulation of the metabolic pathways by ArcA and Fnr.
Modeling for the nitrogen regulation
Next to carbon (C) catabolite regulation, the nitrogen (N) regulation is also important [170], and the silicon-cell models have been developed based on experimental kinetic data for the enzymes, involved that predict the flux of assimilation of extracellular ammonia into glutamate in E. coli.
Glutamate (Glu) and glutamine play key roles in cellular metabolism and serve as precursors of protein synthesis. Glutamate can be synthesized from two different pathways such as by one simple step reaction catalyzed by glutamate dehydrogenase (GDH) from αKG, and by glutamine synthetase (GS) and glutamate synthase (GOGAT) [171]. GS is active during low ammonium concentration while GDH is active at higher ammonium concentrations, where GS has a higher affinity than GDH for ammonia (K m = 0.1 and 1.1 mM, respectively) [172,173]. The activity of GS was controlled by PII which acts in response to the concentration of glutamine and αKG [174,175]. AmtB is used for NH3 for transport when the ammonia concentration is lower than 0.05 μM [176], while the AmtB will be blocked when it was higher than 50 μM.
The activation (adenylylation) and inactivation (deadenylylation) of GS depends on C/N ratio such as αKG (C)-to-glutamine (N) ratio. In the case where C/N ratio was higher, GS will be adenylylated, otherwise, GS is deadenylylated. Sensing of the C/N ratio for GS adenylylation involves the protein PII, which is in two forms such as urydylylated PI-UMP and deurydylylated PII. Urydylylation/deurydylylation of PII catalyzed by UT/UR enzymes are promoted by glutamine (Figure 8).

Ammonia assimilation pathways.
The model developed by Bruggeman et al. [173] combined metabolic regulation with signal transduction through the covalent modification of PII and GS by urydylyl transferase (UTase) and adenylyl transferase (ATase). It shows that the regulation is distributed between the two modes of regulation. However, the model may be incomplete in the sense that αKG pool size was assumed to be constant, while it changes significantly during nitrogen perturbations, where it is not only the substrate for ammonia assimilation but also a regulator of the GS covalent modification cascade [177]. Moreover, it is important to capture the interdependence of metabolite pools and growth, where metabolite pool size of αKG affects the glucose uptake by inhibiting EI of PTS [140].
In order to see the effect of ammonium assimilation, the main metabolic pathway must be considered. This model involves GDH, GS, and GOGAT pathways together with nitrogen regulation mechanism. At present, several kinetic models have been proposed for ammonium assimilation [173,176,178], but little has been analyzed for the relationship between cell growth rate and NADPH production rate in relation to ammonium assimilation. Moreover, it is strongly desirable to combine the models for catabolite regulation and nitrogen regulation in order to simulate the coordinated regulation between C- and N-regulations via the dynamic behavior of intracellular metabolite αKG.
Conclusions
Completeness of the model may not be necessary for it to improve predictions or rationalizations. The uncertainty of the model or the simulation result comes either from uncertainty of the model parameters, uncertainty of the model structure due to ambiguity in the selection of rate laws, or uncertainty caused by neglecting the regulation mechanism or neglecting cofactor balances etc. The importance of the above factors depends on the strains and culture conditions. It is useful in understanding cellular mechanism, and the process towards the development of the whole cell metabolism could well pay off. The appropriate model will be of immense value to the following:
-
Biotechnologists aiming to improve fermentation performances such as the yield and productivity of the target metabolite,
-
Microbial engineers aiming to design novel microbes able to capture available carbon and produce bio-fuels and biochemicals,
-
Basic scientists aiming to understand the metabolic regulation system in the living organisms, which can be used for the synthetic biology, and
-
Systems biologists aiming to advance the science of modeling.
The modeling approach will greatly exceed the importance of the microbial genome sequencing projects, as it will be much closer to understanding biological function and will have widespread practical application.
In the present article, it is stressed the importance of incorporating the enzyme level and transcriptional regulations appropriately in the kinetic model to predict the cell's growth characteristics under environmental and/or genetic perturbations. The drawback of the kinetic modeling is the increase in the kinetic model parameters as the system becomes large, and thus it may be difficult to expand to genome-scale. The reasonable idea may be to consider the kinetic modeling only for the main metabolism, and the simplified model may be considered for the peripheral metabolisms.
Moreover, it is quite important to combine the catabolic regulation model with nitrogen regulation model for the coordination between C- and N-regulations, where the intracellular pool sizes of α-keto acids play important roles affecting PTS and cAMP level.
The simulation result based on the model developed must be verified by experiments, or the simulation result may give hint for additional experimental design. In this way, modeling approach together with experimental works contributes to the innovation for the efficient design of the cell factories for biofuels and biochemical production.
Abbreviations
- 6PG:
-
6-phosphogluconate
- FBA:
-
Flux balance analysis
- MFA:
-
Metabolic flux analysis
- ED pathway:
-
Entner-Doudoroff pathway
- TCA cycle:
-
Tricarboxylic acid cycle
- PTS:
-
Phosphotransferase system
- Crp:
-
cAMP receptor protein
- Cya:
-
Adenylate cyclase
- EI:
-
Enzyme I
- EII:
-
Enzyme II
- EIIAGlc-P:
-
Phosphorylated EIIAGlc
- HPr:
-
Histidine-phosphorylatable protein
- Cra:
-
Catabolite repressor/activator
- Arc system:
-
Anoxic respiration control system
- Fnr:
-
Fumarate and nitrate reduction
- Ack:
-
Acetate kinase
- Acs:
-
Acetyl coenzyme A synthetase
- ADH:
-
Alcohol dehydrogenase
- Ask:
-
Aspartokinase
- CS:
-
Citrate synthase
- Eno:
-
Enolase
- Fbp:
-
Fructose bisphosphatase
- Frd:
-
Fumarate reductase
- Fum:
-
Fumarase
- GAPDH:
-
Glyceraldehyde-3-phosphate dehydrogenase
- GDH:
-
Glutamate dehydrogenase
- Glk:
-
Glucokinase
- GlpK:
-
Glycerol kinase
- GOGAT:
-
Glutamate synthase
- G3PDH:
-
Glycerol 3-phophate dehydrogenase
- G6PDH:
-
Glucose-6-phosphate dehydrogenase
- GS:
-
Glutamine synthetase
- ICDH:
-
Isocitrate dehydrogenase
- Icl:
-
Isocitrate lyase
- KGDH:
-
Ketoglutarate dehydrogenase
- LDH:
-
Lactate dehydrogenase
- MDH:
-
Malate dehydrogenase
- Mez:
-
Malic enzyme
- MS:
-
Malate synthase
- Pck:
-
Phosphoenolpyruvate carboxykinase
- PDHc:
-
Pyruvate dehydrogenase complex
- Pfk:
-
Phosphofructokinase
- Pfl:
-
Pyruvate formate-lyase
- 6PGDH:
-
6-phosphogluconate dehydrogenase
- Pgi:
-
Phosphoglucose isomerase
- Pgk:
-
phosphoglycerate kinase
- Pox:
-
Pyruvate oxidase
- Ppc:
-
Phosphoenolpyruvate carboxylase
- Pps:
-
Phosphoenolpyruvate synthase
- Pta:
-
Phosphotransacetylase
- Pyk:
-
Pyruvate kinase
- Rpe:
-
Ribulose phosphate epimerase
- Rpi:
-
Ribose phosphate isomerase
- SCS:
-
Succinyl-CoA synthetase
- SDH:
-
Succinate dehydrogenase
- Tal:
-
Transaldolase
- TktA:
-
Transketolase I
- TktB:
-
Transketolase II
- AcCoA:
-
Acetyl-coenzyme A
- ACE:
-
Acetate
- AcP:
-
Acetyl phosphate
- CIT:
-
Citrate
- DHAP:
-
Dihydroxy acetone phosphate
- E4P:
-
Erythrose-4-phosphate
- ETH:
-
Ethanol
- F1P:
-
Fructose-1-phosphate
- F6P:
-
Fructose-6-phosphate
- FBP:
-
Fructose-1,6-bisphosphate
- FOR:
-
Formate
- G6P:
-
Glucose-6-phosphate
- GAP:
-
Glycelaldehyde-3-phosphate
- GLC:
-
Glucose
- GLU:
-
Glutamate
- GOX:
-
Glyoxylate
- ICT:
-
Isocitrate
- LAC:
-
Lactate
- MAL:
-
Malate
- OAA:
-
Oxaloacetate
- PEP:
-
Phosphoenol pyruvate
- PYR:
-
Pyruvate
- R5P:
-
Ribose-5-phosphate
- RU5P:
-
Ribulose-5-phosphate
- S7P:
-
Sedoheptulose-7-phosphate
- SUC:
-
Succinate
- X5P:
-
Xylulose-5-phosphate
- αKG:
-
α-ketoglutarate
References
Shimizu K (2014) Microbial production of biofuels and biochemicals from biomass. NOVA publ, Co, New York
Kitano H (2002) Systems biology: a brief overview. Science 295:1662–1664
Kitano H (2002) Computational systems biology. Nature 420:206–210
Stelling J (2004) Mathematical models in microbial systems biology. Curr Opin Microbiol 7:513–518
Kotte O, Zaugg JB, Heinemann M (2010) Bacterial adaptation through distributed sensing of metabolic fluxes. Mol Sys Biol 6:355
Vemuri GN, Aristidou A (2005) Metabolic engineering in the -omics era: elucidating and modulating regulatory networks. Microbiol Mol Biol Rev 69:197–216
Shimizu K (2014) Regulation systems of bacteria such as Escherichia coli in response to nutrient limitation and environmental stresses. Metabolites 4:1–35
Matsuoka Y, Shimizu K (2011) Metabolic regulation in Escherichia coli in response to culture environments via global regulators. Biotechnol J 6:1330–1341
Chuvukov V, Gerosa L, Kochanowski K, Sauer U (2014) Coordination of microbial metabolism. Nat Rev 12:327–340
Selinger DW, Wright MA, Church GM (2003) On the complete determination of biological systems. Trends Biotechnol 21:251–254
Machado D, Costa R, Rocha M, Ferreira E, Tidor B, Rocha I (2011) Modeling formalisms in systems biology. AMP Expre 1:1–34
Almquist J, Cvijovic M, Hatzimanikatis V, Nielsen J, Jirstrand M (2014) Kinetic models in industrial biotechnology-improving cell factory performance. Metabolic Eng 24:38–60
Costa RS, Machado D, Rocha I, Pereira EC (2011) Critical perspective on the consequences of the limited availability of kinetic data in metabolic dynamic modeling. IET Syst Biol 5:157–163
Ashyraliyev M, Fomekong-Nanfack Y, Kaandorp JA, Blom JG (2009) Systems biology: parameter estimation for biochemical models. FEBS J 276:886–902
Cvijovic M, Bordel S, Nielsen J (2011) Mathematical models of cell factories: moving towards the core of industrial biotechnology. Microb Biotechnol 4:572–584
Sauer U (2006) Metabolic networks in motion: 13C-based flux analysis. Mol Syst Anal 2:62
Long CP, Antoniewicz MR (2014) Metabolic flux analysis of Escherichia coli knockouts: lessons from the Keio collection and future outlook. Curr Opin Biotechnol 28:127–133
Quek LE, Nielsen LK (2014) Steady-State 13C Fluxomics Using OpenFLUX. In: Krömer JO, Nielsen LK, Blank LM (eds) Metabolic flux analysis: methods and protocols, vol. 1191, Springer, New York, 209-224
Shimizu K (2004) Metabolic flux analysis based on 13C-labeling experiments and integration of the information with gene and protein expression patterns. Adv Biochem Eng Biotechnol 91:1–49
Shimizu K (2013) Bacterial cellular metabolic systems. Woodhead Publ Ltd., Oxford
Matsuoka Y, Shimizu K (2014) 13C-Metabolic flux analysis for Escherichia coli. In: Krömer JO, Nielsen LK, Blank LM (eds) Metabolic flux analysis: methods and protocols, vol. 1191, Springer, New York, 261-289
Shimizu K (2009) Toward systematic metabolic engineering based on the analysis of metabolic regulation by the integration of different levels of information. Biochem Eng J 46:235–251
Wittman C (2007) Fluxome analysis using GC-MS. Microb Cell Fact 6:6
Herrgard MJ, Lee B-S, Portnoy V, Palsson BO (2006) Integrated analysis of regulatory and metabolic networks reveals novel regulatory mechanisms in Saccharomyces cerevisiae. Genome Res 16:627–635
O'Brien EJ, Lerman JA, Chang RL, Hyduke DR, Palsson BO (2013) Genome-scale models of metabolism and gene expression extend and refine growth phenotype prediction. Mol Sys Biol 9:693
Schuetz R, Kuepfer SU (2007) Systematic evaluation of objective functions forpredicting intracellular fluxes in Escherichia coli. Mol Syst Biol 3:119
Schuetz R, Zamboni N, Zampieri M, Heinemann M, Sauer U (2012) Multidimensional optimality of microbial metabolism. Science 336:601–604
Burgard AP, Pharkya P, Maranas CD (2003) Optknock: a bilevel programming framework for identifying gene knockout strategies for microbial strain optimization. Biotechnol Bioeng 84:647–657
Pharkya P, Maranas CD (2006) An optimization framework for identifying reaction activation/inhibition or elimination candidates for overproduction in microbial systems. Metab Eng 8:1–13
Ranganathan S, Suthers PF, Maranas CD (2010) OptForce: an optimization procedure for identifying all genetic manipulations leading to targeted overproductions. Plos Comput Biol 6:e1000744
Rocha I, Maia P, Evangelista P, Vilaca P, Soares S, Pinto JP, Nielsen J, Patil KR, Ferreira EC, Rocha M (2010) OptFlux: an open-source software platform for in silico metabolic engineering. BMC Syst Biol 4:45
Choon YW, Mohamad MS, Deris S, Illias RM, Chong CK, Chai LE, Omatu S, Corchado JM (2014) Differential bees flux balance analysis with OptKnock for in silico microbial strains optimization. PLoS One 9:e102744
Pharkya P, Burgard AP, Maranas CD (2014) OptStrain: a computational framework for redesign of microbial production systems. Genom Res 14:2367–2376
Yang L, Cluett WR, Mahadevan R (2011) EMILiO: a fast algorithm for genome-scale strain design. Metab Eng 13:272–281
Cotten C, Reed JL (2013) Constraint-based strain design using continuous modifications (CosMos) of flux bounds finds new strategies for metabolic engineering. Biotechnol J 8:595–604
Ibarra RU, Edwards JS, Palsson BO (2002) Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420:186–189
Segrè D, Vitkup D, Church GM (2002) Analysis of optimality in natural and perturbed metabolic networks. Proc Natl Acad Sci U S A 99:15112–15117
Rark JM, Kim TY, Lee SY (2009) Constraints-based genome-scale metabolic simulation for systems metabolic engineering. Biotechnol Adv 27:979–988
Zomorrodi AR, Suthers PF, Ranganathan S, Maranas CD (2012) Mathematical optimization applications in metabolic networks. Metab Eng 14:672–686
Covert MW, Xiao N, Chen TJ, Karr JR (2008) Integrating metabolic, transcriptional regulatory and signal transduction models in Escherichia coli. Bioinformatics 24:2044–2050
Meadows AL, Karnik R, Lam H, Forestell S, Snedecor B (2010) Application of dynamic flux balance analysis to an industrial Escherichia coli fermentation. Metab Eng 12:150–160
Feng X, Xu Y, Chen Y, Tang YJ (2012) MicrobesFlux: a web platform for drafting metabolic models from the KEGG database. BMC Syst Biol 6:94
Zhuang K, Izallalen M, Mouser P, Richter H, Risso C, Mahadevan R, Lovley DR (2011) Genome-scale dynamic modeling of the competition between Rhodoferax and Geobacter in anoxic subsurface environments. Isme J 5:305–316
Salimi F, Zhuang K, Mahadevan R (2010) Genome-scale metabolic modeling of a clostridial co-culture for consolidated bioprocessing. Biotechnol J 5:726–738
Jamshidi N, Palsson BØ (2008) Formulating genome-scale kinetic models in the post-genome era. Mol Syst Biol 4:171
Jamshidi N, Palsson BØ (2010) Mass action stoichiometric simulation models: incorporating kinetics and regulation into stoichiometric models. Biophys J 98:175–185
Smallbone K, Simeonidis E, Broomhead DS, Kell DB (2007) Something from nothing - bridging the gap between constraint-based and kinetic modelling. FEBS J 274:5576–5585
Smallbone K, Simeonidis E, Swainston N, Mendes P (2010) Towards a genome-scale kinetic model of cellular metabolism. BMC Syst Biol 4:6
Fleming RM, Thiele I, Provan G, Nasheuer HP (2010) Integrated stoichiometric, thermodynamic and kinetic modelling of steady state metabolism. J Theor Biol 264:683–692
Antoniewicz MR (2013) Dynamic metabolic flux analysis-tools for probing transient states of metabolic networks. Curr Opin Biotechnol 24:973–978
Hoffner K, Harwood SM, Barton PI (2013) A reliable simulator for dynamic flux balance analysis. Biotechnol Bioeng 110:792–802
Mahadevan R, Edwards JS, Doyle FJ (2002) Dynamic flux balance analysis of diauxic growth in Escherichia coli. Biophys J 83:1331–1340
Hanly TJ, Henson MA (2011) Dynamic flux balance modeling of microbial co-cultures for efficient batch fermentation of glucose and xylose mixtures. Biotechnol Bioeng 108:376–385
Hanly TJ, Urello M, Henson MA (2012) Dynamic flux balance modeling of S. cerevisiae and E. coli co-cultures for efficient consumption of glucose/xylose mixtures. Appl Microbiol Biotechnol 93:2529–2541
Hanly TJ, Henson MA (2013) Dynamic metabolic modeling of a microaerobic yeast co-culture: predicting and optimizing ethanol production from glucose/xylose mixtures. Biotechnol Biofuels 6:44
Chowdhury A, Zomorrodi AR, Maranas CD (2014) k-OptForce: integrating kinetics with flux balance analysis for strain design. PLoS Comput Biol 10:e1003487
Klumpp S, Hwa T (2008) Growth-rate dependent partitioning of RNA polymerases in bacteria. PNAS USA 105:20245–20250
Klumpp S, Zhang Z, Hwa T (2009) Growth-rate dependent global effects on gene expression in bacteria. Cell 139:1366–1375
Valgepea K, Adamberg K, Seiman A, Vilu R (2013) Escherichia coli achieves faster growth by increasing catalytic and translational rates of proteins. Mol Biosyst 9:2344–2358
Harcomb WR, Delaney NF, Leiby N, Klitgord N, Marx CJ (2013) The ability of flux balance analysis to predict evolution of central metabolism scales with the initial distance to the optimum. PLoS Comput Biol 9:e1003091
Edwards JS, Covert MW, Palsson BØ (2002) Metabolic modelling of microbes: the flux-balance approach. Environ Microbiol 4:133–140
Karr JR, Sanghvi JC, Macklin DN, Gutschow MW, Jacobs JM, Bolival B Jr, Assad-Garcia N, Glass JI, Covert MW (2012) A whole-cell computational model predicts phenotype from genotype. Cell 150:389–401
Gunawardera J (2012) Silicon dreams of cells into symbols. Nature 30:838–840
Freddolino PL, Tavazoie S (2012) The dawn of virtual cell biology. Cell 150:248–250
Tomita M (2001) Whole-cell simulation: a grand challenge of the 21st century. Trends in Biotech 19:205–210
Heinrich R, Rapoport TA (1974) A linear steady-state treatment of enzymatic chains. General properties, control and effector strength. Eur J Biochem 42:89–95
van Riel NA (2006) Dynamic modelling and analysis of biochemical networks: mechanism-based models and model-based experiments. Brief Bioinform 7:364–374
Heijnen JJ (2005) Approximative kinetic formats used in metabolic network modeling. Biotechnol Bioeng 91:534–545
Wu L, Wang WM, van Winden WA, van Gulik WM, Heijnen JJ (2004) A new framework for the estimation of control parameters in metabolic pathways using lin-log kinetics. Eur J Biochem 271:3348–3359
del Rosario RCH, Mendoza E, Voit EO (2008) Challenges in lin-log modelling of glycolysis in Lactococcus lactis. Iet Syst Biol 2:136–149
Hatzimanikatis V, Emmerling M, Sauer U, Bailey JE (1998) Application of mathematical tools for metabolic design of microbial ethanol production. Biotechnol Bioeng 58:154–161
Wang L, Hatzimanikatis V (2006) Metabolic engineering under uncertainty-II: analysis of yeast metabolism. Metab Eng 8:142–159
Pozo C, Marín-Sanguino A, Alves R, Guillén-Gosálbez G, Jiménez L, Sorribas A (2011) Steady-state global optimization of metabolic non-linear dynamic models through recasting into power-law canonical models. BMC Syst Biol 5:137
Sorribas A, Hernandez-Bermejo B, Vilaprinyo E, Alves R (2007) Cooperativity and saturation in biochemical networks: a saturable formalism using Taylor series approximations. Biotechnol Bioeng 97:1259–1277
Liebermeister W, Klipp E (2006) Bringing metabolic networks to life: convenience rate law and thermodynamic constraints. Theor Biol Med Model 3:41
Kim JI, Song HS, Sunkara SR, Lali A, Ramkrishna D (2012) Exacting predictions by cybernetic model confirmed experimentally: steady state multiplicity in the chemostat. Biotechnol Prog 28:1160–1166
Covert MW, Palsson BØ (2002) Transcriptional regulation in constraints-based metabolic models of Escherichia coli. J Biol Chem 277:28058–28064
Covert MW, Palsson BØ (2003) Constraints-based models: regulation of gene expression reduces the steady-state solution space. J Theor Biol 221:309–325
Covert MW, Schilling CH, Famili I, Edwards JS, Goryanin II, Selkov E, Palsson BØ (2001) Metabolic modeling of microbial strains in silico. Trends Biochem Sci 26:179–186
Herrgård MJ, Fong SS, Palsson BØ (2006) Identification of genome-scale metabolic network models using experimentally measured flux profiles. Plos Comput Biol 2:676–686
Song HS, Morgan JA, Ramkrishna D (2009) Systematic development of hybrid cybernetic models: application to recombinant yeast co-consuming glucose and xylose. Biotechnol Bioeng 103:984–1002
Ramkrishna D, Kompala DS, Tsao GT (1987) Are microbes optimal strategists. Biotechnol Progr 3:121–126
Varner J, Ramkrishna D (1999) Metabolic engineering from a cybernetic perspective. 1. Theoretical preliminaries. Biotechnol Prog 15:407–425
Young JD (2005) A system-level mathematical description of metabolic regulation combining aspects of elementary mode analysis with cybernetic control laws. PhD thesis, Purdue University
Young JD, Henne KL, Morgan JA, Konopka AE, Ramkrishna D (2008) Integrating cybernetic modeling with pathway analysis provides a dynamic, systems-level description of metabolic control. Biotechnol Bioeng 100:542–559
Schuster S, Fell DA, Dandekar T (2000) A general definition of metabolic pathways useful for systematic organization and analysis of complex metabolic networks. Nat Biotechnol 18:326–332
Kim JW, Dang CV (2005) Multifaceted roles of glycolytic enzymes. Trends Biocem Sci 30:142–150
Kim JI, Varner JD, Ramkrishna D (2008) A hybrid model of anaerobic E. coli GJT001: combination of elementary flux modes and cybernetic variables. Biotechnol Prog 24:993–1006
Teusink B, Passarge J, Reijenga CA, Esgalhado E, van der Weijden CC, Schepper M, Walsh MC, Bakker BM, van Dam K, Westerhoff HV, Snoep JL (2000) Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. Eur J Biochem 267:5313–5329
Chakrabarti A, Miskovic L, Soh KC, Hatzimanikatis V (2013) Towards kinetic modeling of genome-scale metabolic networks without sacrificing stoichiometric, thermodynamic and physiological constraints. Biotechnol J 8:1043–1057
Hatzimanikatis V, Bailey JE (1996) MCA has more to say. J Theor Biol 182:233–242
Smallbone K, Messiha HL, Carroll KM, Winder CL, Malys N, Dunn WB, Murabito E, Swainston N, Dada JO, Khan F, Pir P, Simeonidis E, Spasić I, Wishart J, Weichart D, Hayes NW, Jameson D, Broomhead DS, Oliver SG, Gaskell SJ, McCarthy JE, Paton NW, Westerhoff HV, Kell DB, Mendes P (2013) A model of yeast glycolysis based on a consistent kinetic characterisation of all its enzymes. FEBS Lett 587:2832–2841
Stanford NJ, Lubitz T, Smallbone K, Klipp E, Mendes P, Liebermeister W (2013) Systematic construction of kinetic models from genome-scale metabolic networks. PLoS One 8:e79195
Savageau MA (1970) Biochemical systems analysis. 3. Dynamic solutions using a power-law approximation. J Theor Biol 26:215–226
Voit Eberhard O (2013) Biochemical systems theory: a review. ISRN Biomathematics 2013:897658
Dräger A, Kronfeld M, Ziller MJ, Supper J, Planatscher H, Magnus JB, Oldiges M, Kohlbacher O, Zell A (2009) Modeling metabolic networks in C. glutamicum: a comparison of rate laws in combination with various parameter optimization strategies. BMC Syst Biol 3:5
Costa RS, Machado D, Rocha I, Ferreira EC (2010) Hybrid dynamic modeling of Escherichia coli central metabolic network combining Michaelis-Menten and approximate kinetic equations. Biosystems 100:150–157
Rizk ML, Liao JC (2009) Ensemble modeling for aromatic production in Escherichia coli. PLoS One 4:e6903
Tan YK, Liao JC (2012) Metabolic ensemble modeling for strain engineers. Biotechnol J 7:343–353
Contador CA, Rizk ML, Asenjo JA, Liao JC (2009) Ensemble modeling for strain development of L-lysine-producing Escherichia coli. Metab Eng 11(4–5):221–233
Dean JT, Rizk ML, TanY DKM, Liao JC (2010) Ensemble modeling of hepatic fatty acid metabolism with a synthetic glyoxylate shunt. Biophys J 98:1385–1395
Lee Y, Lafontaine Rivera JG, Liao JC (2014) Ensemble modeling for robustness analysis in engineering non-native metabolic pathways. Metab Eng 25:63–71
Khazaei T, McGuigan A, Mahadevan R (2012) Ensemble modeling of cancer metabolism. Front Physiol 3:135
Khodayari A, Zomorrodi AR, Liao JC, Maranas CD (2014) A kinetic model of Escherichia coli core metabolism satisfying multiple sets of mutant flux data. Metab Eng 25:50–62
Ishii N, Nakahigashi K, Baba T, Robert M, Soga T, Kanai A, Hirasawa T, Naba M, Hirai K, Hoque A, Ho PY, Kakazu Y, Sugawara K, Igarashi S, Harada S, Masuda T, Sugiyama N, Togashi T, Hasegawa M, Takai Y, Yugi K, Arakawa K, Iwata N, Toya Y, Nakayama Y, Nishioka T, Shimizu K, Mori H, Tomita M (2007) Multiple high-throughput analyses monitor the response of E. coli to perturbations. Science 316:593–597
Rizzi M, Baltes M, Theobald U, Reuss M (1997) In vivo analysis of metabolic dynamic in Saccharomyces cerevisiae: II. Mathematical model. Biotechnol Bioeng 55:592–608
Theobald U, Mailinger W, Baltes M, Rizzi M, Reuss M (1997) In vivo analysis of metabolic dynamic in Saccharomyces cerevisiae: I. Experimental observations. Biotechnol Bioeng 55:305–316
Chassagnole C, Noisommit-Rizzi N, Schmid JW, Mauch K, Reuss M (2002) Dynamic modeling of the central carbon metabolism of Escherichia coli. Biotechnol Bioeng 79:53–73
Kadir TA, Mannan AA, Kierzek AM, McFadden J, Shimizu K (2010) Modeling and simulation of the main metabolism in Escherichia coli and its several single-gene knockout mutants with experimental verification. Microb Cell Fact 9:88
Peskov K, Mogilevskaya E, Demin O (2012) Kinetic modelling of central carbon metabolism in Escherichia coli. FEBS J 279:3374–3385
Usuda Y, Nishio Y, Iwatani S, Van Dien SJ, Imaizumi A, Shimbo K, Kageyama N, Iwahata D, Miyano H, Matsui K (2010) Dynamic modeling of Escherichia coli metabolic and regulatory systems for amino-acid production. J Biotechnol 147:17–30
Matsuoka Y, Shimizu K (2013) Catabolite regulation analysis of Escherichia coli for acetate overflow mechanism and co-consumption of multiple sugars based on systems biology approach using computer simulation. J Biotechnol 168:155–173
Yao R, Hirose Y, Sarkar D, Nakahigashi K, Ye Q, Shimizu K (2011) Catabolic regulation analysis of Escherichia coli and its crp, mlc, mgsA, pgi and ptsG mutants. Microb Cell Fact 10:67
Toya Y, Ishii N, Nakahigashi K, Hirasawa T, Soga T, Tomita T, Shimizu K (2010) 13C-metabolic flux analysis for batch culture of Escherichia coli and its Pyk and Pgi gene knockout mutants based on mass isotopomer distribution of intracellular metabolites. Biotechnol Prog 26:975–992
Toya Y, Nakahigashi K, Tomita M, Shimizu K (2012) Metabolic regulation analysis of wild-type and arcA mutant Escherichia coli under nitrate conditions using different levels of omics data. Mol Biosyst 8:2593–2604
Hasona A, Kim Y, Healy FG, Ingram LO, Shanmugam KT (2004) Pyruvate formate lyase and acetate kinase are essential for anaerobic growth of Escherichia coli on xylose. J Bacteriol 186:7593–7600
Kremling A, Bettenbrock K, Gilles ED (2008) A feed-forward loop guarantees robust behavior in Escherichia coli carbohydrate uptake. Bioinformatics 24:704–710
Kochanowski K, Volkmer B, Gerosa L, Haverkorn van Rijsewijk BR, Schmidt A, Heinemann M (2013) Functioning of a metabolic flux sensor in Escherichia coli. Proc Natl Acad Sci U S A 110:1130–1135
Huberts DH, Niebel B, Heinemann M (2012) A flux-sensing mechanism could regulate the switch between respiration and fermentation. FEMS Yeast Res 12:118–128
Christen S, Sauer U (2011) Intracellular characterization of aerobic glucose metabolism in seven yeast species by 13C flux analysis and metabolomics. FEMS Yeast Res 11:263–272
Boels E, Hollenberg CP (1997) The molecular genetics of hexose transport in yeasts. FEMS Microbiol Rev 21:85–111
Ricci JCD (1996) Influence of phosphenolpyruvate on the dynamic behavior of phosphofructokinase of Escherichia coli. J Theor Biol 178:145–150
Yang C, Hua Q, Baba T, Mori H, Shimizu K (2003) Analysis of Escherichia coli anaprelotic metabolism and its regulation mechanisms from the metabolic responses to altered dilution rates and phosphoenolpyruvate carboxykinase knockout. Biotechnol Bioeng 84:129–144
Lee B, Yen J, Yang L, Liao JC (1999) Incorporating qualitative knowledge in enzyme kinetic models using fuzzy logic. Biotechnol Bioeng 63:722–729
Nizam SA, Zhu JF, Ho PY, Shimizu K (2009) Effects of arcA and arcB genes knockout on the metabolism in Escherichia coli under aerobic condition. Biochem Eng J 44:240–250
Vemuri GN, Eiteman MA, Altman E (2006) Increased recombinant protein production in Escherichia coli strains with overexpressed water-forming NADH oxidase and a deleted ArcA regulatory protein. Biotechnol Bioeng 94:538–542
Vemuri GN, Altman E, Sangurdekar DP, Khodursky AB, Eiteman MA (2006) Overflow metabolism in Escherichia coli during steady-state growth: transcriptional regulation and effect of the redox ratio. Appl Environ Microbiol 72:3653–3661
Wolfe AJ (2005) The acetate switch. Microbiol Mol Biol Rev 69:12–50
Xu YF, Amador-Noguez D, Reaves ML, Feng XJ, Rabinowitz JD (2012) Ultrasensitive regulation of anapleurosis via allosteric activation of PEP carboxylase. Nat Chem Biol 8:562–568
Voit E, Neves AR, Santos H (2006) The intricate side of systems biology. Proc Natl Acad Sci U S A 103:9452–9457
Hoefnagel MHN, Starrenburg MJC, Martens DE, Hugenholtz J, Kleerebezem M, Van Swam II, Bongers R, Westerhoff HV, Snoep JL (2002) Metabolic engineering of lactic acid bacteria, the combined approach: kinetic modeling, metabolic control and experimental analysis. Microbiol 148:1003–1013
Kremling A, Jahreis K, Lengeler JW, Gilles ED (2000) The organization of metabolic reaction networks: a signal-oriented approach to cellular models. Metab Eng 2:190–200
Kremling A, Gilles ED (2001) The organization of metabolic reaction networks. II. Signal processing in hierarchical structured functional units. Metab Eng 3:138–150
Kremlng A, Fischer S, Sauter T, Bettenbrock K, Gilles ED (2004) Time hierarchies in the Escherichia coli carbohydrate uptake and metabolism. BioSystems 73:57–71
Sauter T, Gilles ED (2004) Modeling and experimental validation of the signal transduction via the Escherichia coli sucrose phospho transferase system. J Biotech 110:181–199
Bettenbrock K, Fischer S, Kremling A, Jahreis K, Sauter T, Gilles ED (2006) A quantitative approach to catabolite repression in Escherichia coli. J Biol Chem 281:2578–2584
Nishio Y, Usuda Y, Matsui K, Kurata H (2008) Computer-aided rational design of the phosphotransferase system for enhanced glucose uptake in Escherichia coli. Mol Syst Biol 4:160
Majewski RA, Domach MM (1990) Simple constrained-optimization view of acetate overflow in Escherichia coli. Biotech Bioeng 35:732–738
Rabinowitz J, Silhavy TJ (2012) Metabolite turns master regulator. Nature 500:283–284
Doucette CD, Schwab DJ, Wingreen NS, Rabinowitz JD (2011) Alpha-ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nat Chem Biol 7:894–901
Scott M, Gunderson CW, Mateescu EM, Zhang Z, Hwa T (2010) Interdependence of cell growth and gene expression: origins and consequences. Science 330:1099–1102
You C, Okano H, Hui S, Zhang Z, Kim M, Gunderson CW, Wang Y-P, Lenz P, Yan D, Hwa T (2013) Coordination of bacterial proteome with metabolism by cyclic AMP signaling. Nature 500:301–306
Vinuselvi P, Kim MK, Lee SK, Ghim C-M (2012) Rewiring carbon catabolite repression for microbial cell factory. BMB Rep 45(2):59–70
Gorke B, Stulke J (2008) Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nature Rev Microbiol 6:613–24
Vasudevan P, Briggs M (2008) Biodiesel production-current state of the art and challenges. J Ind Microbiol Biotechnol 35:421–430
Dharmadi Y, Murarka A, Gonzalez R (2006) Anaerobic fermentation of glycerol by Escherichia coli: a new platform for metabolic engineering. Biotechnol Bioeng 94:821–829
Clomburg JM, Gonzalez R (2013) Anaerobic fermentation of glycerol: a platform for renewable fuels and chemicals. Trends Biotechnol 31:20–28
Almeida JRM, Fávaro LCL, Betania F, Quirino BF (2012) Biodiesel biorefinery: opportunities and challenges for microbial production of fuels and chemicals from glycerol waste. Biotechnol for Biofuels 5:48
Martínez-Gómez K, Flores N, Castañeda HM, Martínez-Batallar G, Hernández-Chávez G, Ramírez OT, Gosset G, Encarnación S, Bolivar F (2012) New insights into Escherichia coli metabolism: carbon scavenging, acetate metabolism and carbon recycling responses during growth on glycerol. Micob Cell Fact 11:46
Oh MK, Liao JC (2000) Gene expression profiling by DNA microarrays and metabolic fluxes in Escherichia coli. Biotechnol Prog 16:278–286
Peng L, Shimizu K (2003) Global metabolic regulation analysis for Escherichia coli K12 based on protein expression by 2-dimensional electrophoresis and enzyme activity measurement. Appl Microbiol Biotechnol 61:163–178
Cintolesi A, Clomburg JM, Rigou V, Zygourakis K, Gonzalez R (2012) Quantitative analysis of the fermentative metabolism of glycerol in Escherichia coli. Biotechnol Bioeng 109:187–198
Saier MH, Ramseier TM (1996) The catabolite repressor/activator (Cra) protein of enteric bacteria. Journal of Bacteriology 178:3411–3417
Kornberg HL (2001) Routes for fructose utilization by Escherichia coli. J Mol Microbiol Biotechnol 3:355–359
Yao R, Shimizu K (2013) Recent progress in metabolic engineering for the production of biofuels and biochemicals from renewable sources with particular emphasis on catabolite regulation and its modulation. Process Biochem 48:1409–1417
Crasnier-Mednansky M, Park MC, Studley WK, Saier MH Jr (1997) Cra-mediated regulations of Escherichia coli adenylate cyclase. Microbiology 143:785–792
Griffith JK, Baker ME, Rouch DA, Page MG, Skurray RA, Paulsen IT, Chater KF, Baldwin SA, Henderson PJ (1992) Membrane transport proteins: implications of sequence comparisons. Curr Opin Cell Biol 4:684–695
Sumiya M, Davis EO, Packman LC, McDonald TP, Henderson PJ (1995) Molecular genetics of a receptor protein for d-xylose, encoded by the gene xylF, in Escherichia coli. Receptors Channels 3:117–128
Song S, Park C (1997) Organization and regulation of the d-xylose operons in Escherichia coli K-12: XylR acts as a transcriptional activator. J Bacteriol 179:7025–7032
Altintas MM, Eddy CK, Zhang M, McMillan JD, Kompala DS (2006) Kinetic modeling to optimize pentose fermentation in Zymomonas mobilis. Biotechnol Bioeng 94:273–295
Yang C, Hua Q, Shimizu K (1999) Development of a kinetic model for L-lysine biosynthesis in Corynebacterium glutamicum and its application to metabolic control analysis. J Biosci Bioeng 88:393–403
Hua Q, Yang C, Shimizu K (2000) Metabolic control analysis for lysine synthesis using Corynebacterium glutamicum and experimental verification. J Biosci Bioeng 90:184–192
Nishio Y, Ogishima S, Ichikawa M, Yamada Y, Usuda Y, Masuda T, Tanaka H (2013) Analysis of L-glutamic acid fermentation by using a dynamic metabolic simulation model of Escherichia coli. BMC Sys Biol 7:92
Li R-D, Li Y-Y, Lu L-Y, Ren C, Li Y-X, Liu L (2011) An improved kinetic model for the acetone-butanol-etahnol pathway of Clostridium acetobutyricum and model-based perturbation analysis. BMC Sys Biol 5:S12
Shinto H, Tashiro Y, Kobayashi G, Sekiguchi T, Hanai T, Kuriya Y, Okamoto M, Sonomoto K (2007) Kinetic modeling and sensitivity analysis of acetone-butanol-ethanol production. J Biotechnol 131:45–56
Shinto H, Tashiro Y, Kobayashi G, Sekiguchi T, Hanai T, Kuriya Y, Okamoto M, Sonomoto K (2008) Kinetic study of substrate dependency for higher butanol production in acetone-butanol-ethanol fermentation. Proc Biochem 43:1452–1461
Alexeeva S, Hellingwerf KJ, de Mattos JT (2003) Requirement of ArcA for redox regulation in Escherichia coli under microaerobic but not anaerobic or aerobic conditions. J Bacteriol 185:204–209
Shalel-Levanon S, San K-Y, Bennett GN (2005) Effect of oxygen, and ArcA and FNR regulators on the expression of genes related to the electron transfer chain and the TCA cycle in Escherichia coli. Metab Eng 7:364–374
Cox SJ, Levanon SS, Bennett GN, San K-Y (2005) Genetically constrained metabolic flux analysis. Metab Eng 7:445–456
van Heeswijk WC, Westerhoff HV, Boogerd FC (2013) Nitrogen assimilation in Escherichia coli: putting molecular data into a systems perspective. Microbiol Mol Biol Rev 77:628–695
Rhee SG, Chock PB, Stadtman ER (1985) Glutamine synthetase from Escherichia coli. Methods Enzymol 113:213–241
Sakamoto N, Kotre AM, Savageau MA (1975) Glutamate dehydrogenase from Escherichia coli: purification and properties. J Bacteriol 124:775–783
Bruggeman FJ, Boogerd FC, Westerhoff HV (2005) The multifarious short-term regulation of ammonium assimilation of Ecsherichia coli: dissection using an in silico replica. FEBS J 272:1965–1985
Atkinson MR, Blauwkamp TA, Bondarenko V, Studitsky V, Ninfa AJ (2002) Activation of the glnA, glnK, and nac promoters as Escherichia coli undergoes the transition from nitrogen excess growth to nitrogen starvation. J Bacteriol 184:5358–5363
Reitzer L (2003) Nitrogen assimilation and global regulation in Escherichia coli. Annu Rev Microbiol 57:155–176
Ma H, Boogerd FC, Goryanin I (2009) Modelling nitrogen assimilation of Escherichia coli at low ammonium concentration. J Biotechnol 144:175–183
Yuan J, Doucette CD, Fowler WU, Feng XJ, Piazza M, Rabitz HA, Wingreen NS, Rabinowitz JD (2009) Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol Syst Biol 5:302
Lodeiro A, Melgarejo A (2008) Robustness in Escherichia coli glutamate and glutamine synthesis studied by a kinetic mode. J Biol Phys 34:91–106
Acknowledgements
The discussion on the virtual microbe is also made with Prof. H. Westerhoff of the University of Manchester, Prof. J. McFadden of the Surrey University in UK, and the UK-Japan systems biology project funded by JST (Japan) and BBSRC (UK).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YM and KS both contributed for the preparation of the manuscript. Both authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Matsuoka, Y., Shimizu, K. Current status and future perspectives of kinetic modeling for the cell metabolism with incorporation of the metabolic regulation mechanism. Bioresour. Bioprocess. 2, 4 (2015). https://doi.org/10.1186/s40643-014-0031-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40643-014-0031-7