
Abstract
Fluxome analysis aims at the quantitative analysis of in vivo carbon fluxes in metabolic networks, i. e. intracellular activities of enzymes and pathways. It allows investigating the effects of genetic or environmental modifications and thus precisely provides a global perspective on the integrated genetic and metabolic regulation within the intact metabolic network. The experimental and computational approaches developed in this area have revealed fascinating insights into metabolic properties of various biological systems. Most of the comprehensive approaches for metabolic flux studies today involve isotopic tracer studies and GC-MS for measurement of the labeling pattern of metabolites. Initially developed and applied mainly in the field of biomedicine these GC-MS based metabolic flux approaches have been substantially extended and optimized during recent years and today display a key technology in metabolic physiology and biotechnology.
Similar content being viewed by others
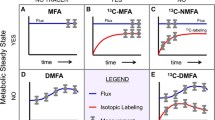


Background
The present review describes the use of stable isotopes, mainly 13C, for the analysis of metabolic fluxes. In all the approaches presented here, single or multiple labeled molecules, i.e. applied tracer substrates or analyzed metabolites, play an important role. Concerning the nomenclature of such molecules labeled with stable isotopes there are no strict IUPAC guidelines. Labeled molecules are identified by the number and position(s) of heavy isotopes in their constitutive atoms. Most commonly the following definitions and terms are used by researchers in the field of metabolic flux analysis. They are exemplified for the carbon isotopes 12C and 13C, but can be generally applied for other isotope labels as well. Positional isotopomers have an exactly determined labeling pattern with a specific number of 13C atoms in specific positions of the molecule. Different positional isotopomers thus exhibit identical global isotopic composition but differ by the position of the heavy atoms in the molecule. For example, [1-13C] pyruvate and [2-13C] pyruvate are positional isotopomers with the 13C label at carbon positions C1 and C2, respectively. Generally, 2n positional isotopomers are possible for a compound with n carbons. Mass isotopomers differ only by the number of heavy atoms in their molecules, resulting in different molecular weights. However, they do not differ by the position of the label. For example, [12C3] pyruvate, [13C-12C2] pyruvate, [13C2 12C] pyruvate and [13C3] pyruvate are different mass isotopomers. Usually, the lightest atoms are not specified and these mass isotopomers are identified as pyruvate, [13C] pyruvate, [13C2] pyruvate and [13C3] pyruvate, in agreement with the International Union of Chemistry. In mass spectrometric jargon, the corresponding different mass isotopomer fractions are referred to as M0, M1, M2 and M3 or as m, m+1, m+2 and m+3. When only one element of a compound with n such atoms of this element is labeled, the number of possible mass isotopomers is (n+1). Most mass isotopomers, except m and m+n, include multiple positional isotopomers. It should be noted that the number of positional isotopomers of symmetrical molecules such as succinate is less than 2n. However, chemically symmetrical molecules may be biologically asymmetrical because of the configuration of the active site of enzymes (e.g. the citrate synthase) or metabolic channeling [1]. The 13C labelling state of a molecule with n carbons can be also expressed as molar enrichment (ME) [2] or as summed fractional labelling (SFL) [3] describing the weighted sum of mass isotopomer fractions (Equation 1).
Introduction
Fluxome analysis aims at the quantitative analysis of in vivo carbon fluxes in metabolic networks. The experimental and computational approaches developed in this area have revealed fascinating insights into various biological systems. In addition they offered new possibilities as a rational basis for targeted strain improvement [4, 5]. Fluxome, i. e. metabolic flux analysis, therefore is in the core of metabolic engineering [6]. The most wide spread approaches for fluxome analysis are based on GC-MS measurement of labelling pattern of metabolites from the tracer studies performed. Initially developed and applied mainly in the field of biomedicine [7–10], these GC-MS based metabolic flux approaches have been substantially extended and optimized and emerged as a key technology in metabolic physiology and biotechnology [6, 11–15].
State-of art metabolic flux analysis is carried out by a comprehensive approach consisting of an experimental and a computational part (Figure 1). The organism of interest is hereby cultivated on a substrate, specifically labelled on certain positions with a stable isotope, mainly 13C. The labelling patterns of metabolites formed during cultivation are then measured, whereby, as stated above, GC-MS is the most used technique today. GC-MS can resolve single mass isotopomers of a compound differing by the number of labelled atoms and thus allows the measurement of mass isotopomer distributions. The mass isotopomer distribution of a compound can be obtained from the analysis of an ion cluster, which contains the intact carbon skeleton of the analyte. Similarly mass isotopomer distributions can be determined for certain parts of an analyte considering appropriate fragment ions containing only parts of its carbon skeleton. The mass isotopomer distribution can be used to calculate the average 13C enrichment of a molecule, called molar enrichment [2] or summed fractional labelling [16], via the weighted sum of mass isotopomer fractions. In selected cases, where a higher degree of labelling information is required GC-MS can be also applied to determine positional isotopomers [16, 17]. Like a fingerprint the GC-MS labelling data sensitively reflect the intracellular fluxes and thus can be used for their calculation. For flux calculation the different labelling data obtained are usually utilized to globally fit the unknown flux parameters by a computer flux model combining isotopomer and metabolite balancing [18–22]. For a given case the flux model contains all relevant reactions and pathways of the investigated metabolic network with full information on carbon transition in each of the reactions. Most suitable for the creation of such a flux model are modelling frameworks with a systematic and general approach for the quantitative description of the transfer of labelled 13C atoms in metabolic networks [14, 15]. Starting with a random initial guess for the free fluxes, the model computes the remaining dependent fluxes via stoichiometric mass balances and subsequently calculates the 13C labelling patterns of all compounds in the network for the given fluxes and compares them with the experimental labelling data. Applying an optimization algorithm the deviation of the labelling data between simulation and experiment is minimized by iterative variation of the free fluxes until the optimum fit is obtained. In combination with experimentally determined extracellular fluxes, absolute carbon fluxes throughout the network are obtained. With certain constraints GC-MS labelling data can also be used to calculate local flux ratios in the network [23, 24]. Combination of these flux ratios with a stoichiometric model and measured extracellular fluxes also leads to a distribution of absolute fluxes [25]. In recent years GC-MS has been successfully applied to study fluxes in bacteria [22, 23, 26], yeasts [18, 27, 28], fungi [29], mammalian cells [30, 31] or intact tissues [32, 33]. The wide spread use of GC-MS for metabolic flux analysis is due to different reasons. GC-MS allows very accurate quantification of labelling patterns resulting in high accuracy and small confidence intervals of the determined fluxes. Additionally, GC-MS usually provides rich information content of the labelling data so that important fine structures of the metabolic network, e. g. fluxes through parallel, bidirectional or cyclic fluxes, can be resolved. The high separation efficiency of GC can resolve the often complex biological mixtures. Moreover GC-MS systems are easy to operate and robust. The present review provides an actual insight into experimental and computational tools for GC-MS based metabolic flux analysis, thereby focussing on the currently most important approach which involves utilization of amino acid labelling patterns. Amino acids are often measured instead of the intermediary metabolites in the central metabolism since the amino acids in cell extracts and cell protein are much more abundant than their precursors [16] and provide extensive labelling information. Knowing the precursor-amino acid relationships it is easy to deduce the labelling patterns of the precursor metabolites from labelling patterns of the amino acids. For the different steps involved in metabolic flux analysis some practical notes and tips are addressed that seem crucial for proper flux measurements.

Strategy for 13C metabolic flux analysis including the experimental part with the tracer study and the GC-MS labelling analysis and the computational part with the simulation of the labelling data via an isotopomer model representing the investigated metabolic network. The flux estimation is based on minimizing the deviation (δ) between the measured and the simulated labelling data.
GC-MS instrumentation
GC-MS allows the measurement of various metabolites [34]. This comprises a number of volatiles such as ketones, aldehydes, alcohols, heterocyclic compounds, isocyanates, isothiocyanates, sulfides, lipids, and hydrocarbons up to 12 carbons, which all can be directly measured. Additionally, various non-volatile or semi-volatile metabolites including sugars, sugar-phosphates, sugar alcohols, organic acids, amino acids, lipids, peptides, long-chain alcohols, alkaloids, amines, amides, etc. are accessible after derivatization. For flux analysis amino acids, organic acids and sugar derivates are the most important compounds to be considered. Currently there is a wide range of instruments available varying in the type of ionization and the mass separation (Figure 2). Single-quadrupole mass spectrometers with electron impact (EI) ionization are the most often used type of instrument. Compared to other instruments they are relatively low cost and offer a range of advantages such as high robustness, high sensitivity and high accuracy of the measured labelling patterns.

Overview on different GC-MS instrumentation types. The most frequently used combination in the area of metabolic flux studies by isotope labelling is highlighted.
GC separation
The sample solution (usually between 0.1 to 1 μL) is injected into the GC inlet where it is vaporized and directed onto the chromatographic column by the carrier gas (usually helium). The sample molecules are separated through different interactions between the carrier gas phase and the stationary phase. To obtain the highest resolution in GC-MS analysis, the capillary column should be carefully chosen. The best general purpose phases are dimethylsiloxane (DB-1 or equivalent) or 5 % phenyl/95%dimethlysiloxane (DB-5 or equivalent). These non-polar phases exhibit an excellent separation capability for many compounds and a generally low column bleeding. Polar columns (DB-Wax or equivalent) are useful for selected derivates such as more polar alkylated compounds. The high separation capacity of GC is illustrated in Figure 3 for a sample containing various amino acids and related compounds.

Total ion current (TIC) spectrum of a sample with TBDMS-derivatized metabolites. The separation of the totally 28 compounds is performed on a HP5-MS column (60 m, 250 μm inner diameter, Hewlett-Packard, Avondale, PA).
Ionization
The outlet of the GC column is connected via a heated transfer line with the ion source of the MS where the compounds eluting from the column are ionized. The most frequently used ionization method is electron impact (EI) ionization (Figure 4). Hereby the interference of the sample stream with a crossing electron beam leads to the loss of an electron from the sample molecules. The resulting ions with one electron missing, i.e. the molecular ions, give the total mass of each analyte. Due to the large amount of energy imparted to the molecular ion it usually fragments, producing further smaller ions with characteristic relative abundances that provide a 'fingerprint' for that molecular structure (Figure 5A). Often the molecular ion itself is not observed. The different fragment ions may contain different parts of the carbon skeleton of the analyte. In the given example for TBDMS-derivatized alanine the ion cluster [M-57]+at m/z 260 results from loss of a C4H9 group from the derivatization residue and contains all three alanine carbon atoms, whereas the ion cluster [M-85]+ at m/z 232 only contains the carbon atoms C2 and C3 of alanine due to additional loss of a CO-group including the C1 atom of alanine. Chemical ionization (CI) as alternative ionization method plays a minor role for flux analysis, but has proven valuable in selected cases such as labelling analysis of glucose aldonitrile pentaacetate in different medical flux studies [35–38].

Schematic view of the ion source based on electron impact ionization and the quadrupole mass filter typically found in a GC-MS instrument.

Mass spectrum of TBDMS3-alanine derived by electron impact ionization in GC-MS analysis: Naturally labeled alanine with a mass isotopomer distribution resulting from the nautrally occurring isotopes (A), alanine from the cell protein of S. cerevisiae cultivated on [1-13C] glucose (B), alanine from the cell protein of lysine producing C. glutamium cultivated on [1-13C] glucose (C). The monoisotopic mass of the molecular ion, which itself is not detected, is 317. The structures of valuable ion clusters for labeling analysis in metabolic flux studies (m/z 260, m/z 232) are additionally.
Mass separation
Through a slit the positive ions from the ion source enter into the mass separation part. Figure 3 shows the most wide spread type of mass separator in GC-MS analysis, a quadrupole, composed of four parallel cylindrical rods (about 25 cm length each), whereby opposite rods are electrically connected. The mass separation is based on the motion of ions in an oscillating electric field created through voltage variation between the rods. At a certain voltage, only analyte molecules of a distinct mass to charge (m/z) ratio (resonance ions) can pass and enter the detector, whereas ions exhibiting a different m/z ratio (non-resonance ions) are subjected to oscillations causing their collision with the rods. Within a few seconds the whole mass range available (usually 50 – 850 m/z) can be fully scanned. This short time interval required for a full scan is important, due to the typically narrow peaks eluting from the GC column that have to be scanned several times each (Figure 3). The sensitivity of a quadrupole detector can be significantly enhanced by selected ion monitoring (SIM), whereby only selected masses are sequentially measured with frequencies of 0.1 to 2 seconds each. The number of collected ions for the selected masses is increased up to several thousand folds in comparison to the scan mode. Quadrupole MS spectra can be easily compared with commercial or laboratory databases facilitating the identification of novel compounds and the validation that a signal is not interfered through isobaric overlay. In most cases the nominal-mass information provided by such instruments is sufficient, since the compounds analyzed are a priori known.
Other mass separators available are time-of-flight (TOF) mass separators or ion traps. In TOF instruments, ions formed in the ion source are accelerated in a short electric field to a high kinetic energy, whereby ions with lower masses reach higher velocities and need a shorter time to reach the detector. The mass to charge ratio of each ion is determined from the time elapsed from ion formation to ion arrival at the detector. This type of mass separation in GC-TOF-MS systems offers a greater mass accuracy as compared to a conventional quadrupole. This, however, is normally not required for the labelling measurement and does therefore not compensate for the relatively high price of such instruments. Only the high scan speed possible allowing multiple scanning of single peaks during their elution and shortening of analysis time [39] could be beneficial for high-throughput applications or the analysis of rather complex samples. Alternatively, ion traps can be applied for mass separation. A drawback of ion traps to be used for flux analysis is the comparatively low accuracy for the labelling measurement resulting in a higher uncertainty of the determined fluxes. Relative errors of around 1 % for the most abundant mass peaks [40] are about two to tenfold higher as compared to that of quadrupole instruments [15]. An advantage, however, is the possibility to use the ion trap as a multi stage MSn mass spectrometer which can provide more detailed information on the labelling pattern. This might be useful in cases, where a high degree of labelling information is needed to determine the fluxes of interest [41]. Similarly triple-quadrupole instruments can be run as multistage mass spectrometers. A further variant, useful e.g. for isotope labelling studies in humans or animals, is isotope ratio (IR) GC-MS [42, 43]. It is based on electron impact ionization with maximized ionization probability. IR GC-MS exhibits an extremely high precision of ± 0.00001 % for the isotope ratio measurement and is optimal to quantify low label enrichment [44]. It is, however, limited to the analysis of gases of high volatility and low reactivity such as CO2, N2 or SO2. The analytes of interest are transformed into one of these gases before introduction into the instrument. Usually this provides information only about the specific labelling enrichment, i.e. the relative abundance of 13C atoms in the entire molecule. More detailed information on number and position of 13C can be obtained, if all carbon atoms are isolated position specific prior to measurement as recently shown for position specific 13C analysis of methyl palmitate through pyrolytic fragmentation [45].
Labelling analysis with GC-MS
The analysis of the labelling pattern is a central step of metabolic flux analysis. It has to be ensured that the labelling patterns are not affected by the sampling, sample pre-treatment or the GC-MS analysis itself. The major compounds analyzed by GC-MS for flux analysis are amino acids [21, 22, 35, 36], organic acids [35, 36, 46], sugars [2, 37, 47], lipids and fatty acids [48]. Moreover, mass distributions of polymers and their building blocks were assessed. Examples are glycogen [37, 49], cell protein [16, 18, 24, 50], or DNA[51].
Sampling and sample pre-treatment
Depending on the experiment, the analytes of interest are cellular polymers (analysis of balanced growth in chemostat or batch processes), free intracellular metabolites (analysis of process dynamics) or extracellular metabolites in the medium (analysis of production phases with reduced growth or without growth). Exemplified for amino acids, the different experimental steps of sampling and sample pre-treatment for subsequent labelling analysis by GC-MS are given in Figure 6. Free intracellular amino acids are extracted from the cells, whereby immediate quenching is important to prevent changes in the labelling pattern during sampling. Due to the high sensitivity of GC-MS only 1 mg cell dry mass are required to obtain labelling patterns from free intracellular amino acids [52]. Various quenching and fast sampling methods described in the literature for metabolome analysis can be applied here. For bacteria fast filtration with cellulose nitrate or polyamide filters (0.2 μm pore size) is suitable [53], whereas yeast cells can be efficiently quenched with cold buffered methanol [54]. The quenched cells should be washed to prevent interference with compounds contained in the medium. Metabolite extraction can be performed by incubation in boiling ethanol or in boiling water. After separation from cell debris the extract is lyophilized to concentrate the metabolites and remove the water. For the analysis of amino acids from the cell protein cells are harvested by centrifugation or filtration, washed and subsequently hydrolyzed by 24 h incubation at 110°C in 6 M HCl. During hydrolysis asparagine and glutamine are deaminated to aspartate and glutamate. Cysteine, methionine and tryptophan are destroyed through oxidation. If the hydrolysis time is shortened to about 8 h, signals can be also obtained for e.g. cysteine or methionine, but at the cost of a generally low yield. The hydrolysate is neutralized by addition of 6 M NaOH, whereby often the required volume of NaOH is less than the volume of HCl due to HCl evaporation during the hydrolysis. The pH should be checked, because the subsequent derivatization is significantly disturbed by an alkaline pH. The neutralized hydrolysate is clarified by filtration, whereby a stable glycerol-free filter material has to be chosen. Glycerol would be leached out of the filter and interfere in the later derivatization and GC-MS analysis. Finally the clarified hydrolysate is lyophilized. Extracellular metabolites are harvested by centrifugation followed directly by lyophilization.

Experimental protocols for sampling and processing of amino acids in yeast and bacterial cell extracts, culture supernatant and biomass (protein) hydrolysate for GC-MS analysis.
Derivatization
In order for a compound to be analyzed by GC-MS it must be volatile and thermally stable. Most of the metabolites utilized for flux analysis are polar or even charged and thus not sufficiently volatile to be directly analyzed, so that derivatization is typically required. This holds for amino acids [16, 21, 24, 50], organic acids [32, 35] or sugars [19, 31, 37, 55, 56]. For this purpose a number of straightforward derivatization reactions involving silylation, alkylation or acetylation are available (Table 1). The reactions are mainly simple, one-pot conversions with high yield. More details on derivatization protocols are given in recent review articles [57–62]. For the analysis of amino acids, silylation with N-methyl-N-t-butyldimethylsilyl-trifluoro-acetamide (MBDSTFA) is especially useful, since it leads to derivates which exhibit high signal intensity for the [M-57] fragment ions. These originate from loss of a t-butyl group from the derivatization residue, contain the entire carbon skeleton of the analyte and are thus valuable for flux calculation [63]. It should be noted that many reagents used are unstable in the presence of water, so that the aqueous samples, as described above, have to be dried and re-dissolved in organic solvent prior to derivatization. It is also possible to directly add the reagent to the lyophilized sample, using an additional solvent such as dimethylformamide, however, improves the conversion.
Labelling information
The mass isotopomer distribution of a compound can be directly taken from an ion cluster containing the entire carbon backbone of the analyte. In many cases these mass isotopomer distributions sensitively reflect the flux parameters of interest. This can be illustrated for the problem of quantifying the flux partitioning at the glucose 6-phosphate node between the pentose phosphate pathway (PPP) and glycolysis, important pathways in many microorganisms. With [1-13C] glucose as tracer substrate the labelled carbon atom is completely released as CO2 in the PPP, whereas the label is completely conserved through the different glycolytic reactions (Figure 7). The mass isotopomer distribution of pyruvate, receiving carbon from both pathways, thus significantly changes with a variation of the flux partitioning. As revealed by metabolic simulations an increase of the relative flux into the PPP is reflected by a decrease of the fraction of single labelled pyruvate and an increase of the fraction of non-labelled pyruvate. GC-MS measurement of the labelling of pyruvate or pyruvate derived metabolites such as alanine allows precise estimation of PPP and glycolytic flux. This becomes obvious from mass spectra of TBDMS-derivatized alanine from tracer experiments of a lysine producing Corynebacterium glutamicum mutant (Figure 5B) and Saccharomyces cerevisiae (Figure 4C) cultivated on 99 % [1-13C] glucose in batch culture. In both cases the heavier mass isotopomer fractions, e.g. at the ion cluster m/z 260, are increased due to the enrichment with 13C. In comparison with S. cerevisiae the fraction of single labelled alanine is markedly reduced in C. glutamicum revealing a significantly higher flux through the PPP in this organism, which is related to the increased NADPH demand for lysine production. Similarly mass isotopomer distributions of other metabolites give access to various other flux parameters, such as dual pathways in amino acid biosynthesis [64], bidirectional fluxes around the pyruvate node [64] or parallel pathways in different cellular compartments [18] and different flux ratios [24]. In other studies, summed fractional labelling data have been utilized for the analysis of fluxes [65, 66]. Hereby, the lower degree of information for each single compound was compensated by the consideration of different ion fragments containing different parts of the carbon skeleton for the analytes [67]. More detailed information on the labelling of a compound can be obtained with GC-MS via additional analysis of fragment ions, which contain only specific parts of the carbon skeleton of the analyte. As example, the entire positional isotopomer distribution of pyruvate can be determined by GC-MS via analysis of four ion clusters of its methyl-ester derivate [17] or via GC-MS analysis of different ions of pyruvate derived amino acids valine and alanine [16]. More complex protocols may be required for analytes containing more carbon atoms, thus exhibiting a substantially increased number of possible positional isotopomers. The determination of 24 out of 32 positional isotopomers of glutamate involves chemical and enzymatic synthesis of five different derivates of glutamate prior to GC-MS analysis [36]. Also other protocols for resolution of positional isotopomer pools are rather laborious and include various steps for purification and chemical or enzymatic conversion of the analytes [35, 49, 68, 69] which impedes the broad and routine use of such techniques. It should be noticed that the resolution of single positional isotopomer pools not necessarily leads to an increase in information for flux quantification [67, 70], but in selected cases single isotopomer pools might be required to calculate all fluxes of interest. A major tool to obtain additional fragments towards positional isotopomer analysis is targeted fragmentation using MS/MS, also called tandem MS, where selected ions can be isolated, subsequently fragmented and the fragments can be analyzed for their mass distribution. The potential of such approaches for flux analysis is underlined by recent studies [71, 72].

Quantification of the flux partitioning between pentose phosphate pathway (PPP) and glycolysis: Carbon transfer from [1-13C] glucose in the underlying metabolic reactions (A), Influence of a variation of the relative flux into the PPP on the relative abundance of non labelled (M0) and single labelled (M1) pyruvate as determined by simulation with an isotopomer model (B).
Correction for natural isotopes
To calculate the 13C labelling of a molecule from a GC-MS measurement, the data have to be corrected for natural isotopes. Hereby, atoms of the analyte and of added derivatization residues have to be considered. Natural isotopes typically occurring in GC-MS are carbon, hydrogen, nitrogen, oxygen and silicium (Table 2). Mathematically, the presence of natural isotopes can be considered by correction matrices [17, 73–75]. Straightforward is the 'addition' of the natural isotopes to the simulated data during flux calculation and the direct comparison with the measurement data [17]. Alternatively the natural isotopes can be 'subtracted' from the measurement signals to obtain the labelling of the carbon skeleton of the analyte and compare this value with the corresponding simulated data during flux calculation [76].
Guidelines for optimal labelling measurement
Quantitative accuracy of the measurement is crucial for mass isotopomer analysis. Several sources of error may potentially affect the measurement result and should therefore be taken into account. First the signals have to be checked for purity, i.e. that isobaric interference of the analytes with co-eluting compounds does not occur. For the consistency check of GC-MS mass isotopomer distributions several efficient software tools have been developed [25, 76, 77]. A check can also be done experimentally by an additional cultivation on naturally labelled substrate in parallel to the tracer experiment and comparison of the mass isotopomer distributions from this study with theoretically expected values, calculated from the natural abundance of isotopes or experimental values from pure standard compounds. Another potential cause of inaccuracy is incomplete resolution of adjacent ions due to ion scattering and peak tailing. Therefore the mass spectrometer (lens system, quadrupole pre-filter, quadrupole rods, detector) has to be tuned to enable optimum mass-resolving capacity [50]. To obtain optimal signal intensity and avoid interference the signal for each mass isotopomer should be collected in SIM mode from the maximum of each mass peak. The m/z value at the peak maximum typically deviates from an integer value due to underlying atom masses contained in the ion. Nonlinearity of the detector response at different mass isotopomer ratios or different analyte amounts may further lead to false results [78]. Whereas such effects are observed for fatty acid methyl esters, mass isotopomer ratios of naturally labelled TBDMS-amino acids were not affected over a concentration range of 3 orders of magnitude [50]. Interference with background noise leads to false results for low abundance signals which therefore should not be considered for flux calculation. The same holds for signals above the saturation of the detector which underestimate the relative abundance of the most prominent mass isotopomer fractions. Such effects can be visualized by an inhomogeneous peak composition [50]. Reduction of the electron multiplier voltage (EMV) at the detector or sample dilution may compensate for such effects. One should further consider that the efficiency of electron multipliers can significantly decrease with life time. This can lead to a drift of the measured labelling pattern, i.e. mass isotopomer ratio, with increasing EMV or sample amount (Figure 8A). In such cases the multiplier should be changed. The high separation capacity of GC leads to isotope discrimination, as shown by the elution behaviour of the different mass isotopomers of TBDMS3-glutamate (Figure 8B). To adequately consider all mass isotopomers of a compound and obtain meaningful results, the average mass spectrum of the whole peak range has to be considered. This leads to a significantly reduced signal-to-noise ratio that excludes the use of labelling data from low abundance fragments [50].

Influence of the life-time of the electron multiplier (A) and of isotope discrimination effects during the GC separation (B) on GC-MS labelling analysis. The effect of the electron multiplier is exemplified for the ratio between the single and the non labelled mass isotopomer fraction of naturally labelled TBDMS2-alanine whereby the dashed line represents the theoretical value and the experimental values result from measurement using an electron multiplier with extended life time and a new electron multiplier, respectively. The isotope discrimination effects are given for the different mass isotopomers of TBDMS3-glutamate.
Experimental design, modelling and flux calculation
Metabolic flux analysis with GC-MS is usually performed as stationary flux analysis, whereby the metabolism is in metabolic and isotopic steady-state. Application examples are chemostat cultures [41] or batch cultures with balanced growth [4, 23, 66]. Additionally novel approaches have been developed to study fluxes also under non-steady-state conditions [79].
Network topology
Generally, metabolic flux analysis by isotope labelling experiments requires a known topology of the biochemical network. Topological phenomena, such as metabolite channelling, compartmentation or zonation, mainly observed in eukaryotic systems, may influence the fate and the labelling pattern of metabolites in the network and must be considered, when interpreting obtained labelling data [80]. Metabolite channelling can prevent label scrambling of symmetric intermediates through orientation-conserved biocatalytic reactions as found for mitochondrial TCA cycle enzymes in mammalian cells [81] and yeast [82] and for pentose phosphate pathway (PPP) enzymes in yeast [83]. The compartments in eukaryotic cells lead to spatially separated metabolite pools and reactions, which has significant impact on labelling patterns. As example amino acid metabolism in yeast may occur in the cytosol, the mitochondrium or in both compartments, whereby the pathway location may vary between different species [84, 85] or with cultivation conditions [18]. In tissue cultures zonation of metabolic activities may lead to heterogeneous labelling patterns as described for triose phosphate pools in rat liver [86]. The significant impact of metabolite channelling, compartmentation or zonation on labelling patterns, requesting their consideration in flux analysis, can be exploited by using isotope labelling studies to unravel these important features in biological systems [87]. Evidence for isotope effects on rate and equilibrium of metabolic reactions has not been gained, so that it appears justified to neglect them [16].
The majority of GC-MS based flux approaches utilizes amino acid labelling patterns [16, 50, 85, 88–90]. Knowing the precursor-amino acid relationships it is easy to deduce the labelling patterns of the precursor metabolites from labelling patterns of the amino acids. The relationship between precursor compounds from glycolysis, pentose phosphate pathway and TCA cycle and the amino acids for E. coli, C. glutamicum, B. subtilis, P. putida and S. cerevisiae is given in Figure 9. Except for lysine the synthesis of all amino acids is identical in all these microorganisms [91]. In E. coli, B. subtilis and P. putida one lysine biosynthetic route is available each from oxaloacetate and pyruvate, respectively, which includes a symmetric intermediate that results in label scrambling. Bakers's yeast utilizes a-ketoglutarate and acetyl-CoA as lysine precursors. C. glutamicum possesses two alternative pathways for lysine biosynthesis, whereby the in vivo activity of the two branches can vary with cultivation conditions or genetic modifications [92]. One should note that serine and glycine may not exclusively originate from their well known precursor 3-phosphoglycerate, but can also be formed from oxaloacetate via the threonine pathway and threonine aldolase [4, 66].

Relationship between the carbon skeleton of amino acids and the carbon skeleton of their metabolic precursors for the anabolic pathways in E. coli, S. cerevisiae, C. glutamicum and B. subtilis. The data are partly taken from [130].
Choice of tracer substrates
The resolution of intracellular fluxes is strongly dependent on the 13C-labeling strategy. In GC-MS based approaches the use of [1-13C] glucose as well as the use of a mixture of [13C6] glucose and unlabeled glucose has proven useful to resolve important fluxes throughout the central metabolism in different organisms. Concerning the central metabolic pathways, a mixture of [13C6] glucose and unlabeled glucose is particularly useful to resolve fluxes downstream of PEP and some exchange fluxes that result in C-C bond cleavages, whereas the use of [1-13C] glucose is valuable for resolving the upper part of metabolism, in particular the oxidative PP pathway, glycolysis and the Entner-Doudoroff pathway [24]. Combining [U-13C] glucose and [1-13C] glucose, either in two separate experiments [19, 21, 24] or as a substrate mixture [20] may lead to an even better resolution of fluxes in the network. The resolution of a particular flux, however, depends on the network topology itself so general guidelines cannot be given. Questions concerning the determinability and the predicted accuracy of a certain flux together with an optimal experimental approach can be effectively answered by computer based experimental design [64, 70, 93]. One should note that the flux calculation is rather sensitive to the mixing ratio of two differently labelled compounds applied at the same time, e.g. [13C6] glucose and naturally labelled glucose. This mixing ratio should be exactly known, considering the facts that during storage some labelled compounds are hygroscopic. For glucose this can be taken into account by 8 h drying at 80°C prior to weighting.
Flux calculation, statistics and goodness of fit
The flux parameters are usually estimated from the 13C tracer studies data by minimization of the deviation between experimental and simulated labelling data using numerical routines [15] Hereby, the free flux parameters of interest, such as flux partitioning ratios or reversibilities are varied by the optimization function starting from initial values until an acceptable agreement between experimental and simulated labelling patterns is achieved. Different optimization functions such as gradient or adaptive random search functions are available. To increase the probability of the identification of a global optimum for the solution, the convergence is usually tested for different initial guesses. As error criterion a weighted sum of least squares (SLS) is typically used (Equation 2).
By this, the differences between experimental (rexp) and calculated (rcalc) labelling data, e. g. molar fractions of mass isotopomers ratios, are normalized and the resulting relative experimental errors of the corresponding MS measurements (sr,i) are used for weighting. As a result, data with relatively small error contribute to a higher extent, whereas the influence of data with a relatively high error and uncertainty has only a minor influence on the overall optimization result. Statistical analysis is of great importance in order to identify whether differences observed for intracellular flux distributions between different experiments can be really attributed to strain or condition specific differences. Such analysis can be performed by a Monte-Carlo approach including multiple parameter estimation runs with statistical variation of the experimental data [94, 95]. The statistical variation is done such that random errors are added to the data sets, assuming a normal distribution of measurement errors around previously obtained mean values. Subsequently, multiple parameter estimations are carried out for each scenario, yielding multiple flux distributions with a corresponding mean value and a standard deviation for each intracellular flux parameter, from which confidence limits for the single parameters can be calculated. The high accuracy of intracellular fluxes determined by GC-MS based flux analysis is illustrated by a phase plane plot for different mutant strains of C. glutamicum (Figure 10). Based on previous experimental data [21] 250 independent estimates for the PPP and the TCA cycle flux with statistically varied experimental data for each strain show that, despite the strains differ only gradually in the corresponding fluxes, clear differentiation is possible. This is a great advantage as compared to NMR based flux approaches resulting in much higher uncertainty of intracellular fluxes [21]. Flux analysis using GC-MS allows a high goodness of fit, if all critical steps of the comprehensive approach are carefully addressed [19, 21, 22]. High deviation in selected mass isotopomer distributions leaves the estimated fluxes questionable. Such deviations may point at underlying errors in the performed study. Potential sources of errors include the labelling measurement itself, assumptions in the network topology, e.g. on available pathways, or carbon transfer in the underlying reactions, or simply errors in the complex metabolic model. Simulated and experimental labelling data as well as statistics on the calculated fluxes display important information that should always be included in the presentation of flux studies.

Statistical analysis of metabolic fluxes using a Monte-Carlo approach exemplified for flux through major NADPH generating pathways, the pentose phosphate pathway (PPP) and the TCA cycle. The calculation is based on a previous flux study of different lysine producing strains of C. glutamicum [21] and represents 250 independent flux estimations with statistically varied experimental data for each of the five strains shown.
Metabolic flux studies using GC-MS
The following chapter shows the application of GC-MS to metabolic flux analysis by different examples and should illustrate the broad application potential of such approaches in different fields of research. Whereas some of the examples focus on flux quantification of only a single or a few selected reactions, others aim at the analysis of larger parts of the metabolism.
Bacteria, yeasts and fungi
GC-MS based metabolic flux analysis has been applied to various microorganisms. This includes several flux studies of E. coli, one of the most prominent model organisms. As example, different mutants were studied at miniaturized scale providing information on gene function [23]. Metabolic flux analysis utilizing GC-MS was used also to investigate the adaptation of E. coli to the loss of key metabolic enzymes [96], the function of soluble and membrane bound transhydrogenase [97] or growth on different substrates [26]. Additionally, the effect of single gene deletion on metabolic fluxes has been studied applying a combination of GC-MS and NMR for labelling analysis [26, 98–102]. A number of studies have been carried out for the industrial amino acid producer C. glutamicum, providing important knowledge for rational strain improvement. This microorganism has been investigated with respect to the influence of different substrates in batch culture [19, 22], fluxes in chemostat culture under substrate limitation [41], the comparison of different lysine producing mutants [4, 21, 103] and different phases of a production process [20]. Further prokaryotes studied on the level of intracellular carbon flux using GC-MS based approaches are Bacillus subtilis [79, 104], Bacillus clausii [105]or Streptomyces noursei [106]. In another study flux ratio analysis was applied to compare fluxes in Pseudomonas putida, Zymononas mobilis, Sinorhizobium meliloti, Rhodobacter sphaeroides and Lactococcus versutus [107]. Among yeasts studied on the level of intracellular fluxes by GC-MS are Saccharomyces cerevisiae with studies on the influence of growth rate [18, 27], or the comparison of different mutants [66], several members of the genus Pichia [84, 88, 108], Phaffia rhodozyma [28] or different other species [109]. Flux studies with fungi comprise e. g. biotechnologically relevant species such as Penicillium chrysogenum [29], Aspergillus nidulans [65] or Aspergillus niger [95].
Mammalian cell cultures, tissues, animals and humans
Isotopic tracer experiments with GC-MS labelling analysis are utilized in the biomedical area field since many years [12, 13]. Flux studies in this field significantly contributed to our current understanding on metabolic function in mammalian cell lines or organs or on the underlying metabolism related to diseases [110]. The inherent complexity of the networks with different compartments or tissue zones, metabolite channelling and complex dilution effects of the labelling often restricts the determination by a tracer experiment to selected flux parameters [15]. Flux studies of mammalian cell cultures comprise e.g. the comparison of different cell lines [30], the influence of butyrate on differentiation of adenocarcinoma cells [55], or the response of fasted rat hepatocytes to different substrates [31]. Extensive work has been carried out on quantifying fluxes in the central metabolism of tissue cultures. This involves studies on the pentose phosphate pathway in human hepatoma cells [111], or on anaplerosis, cataplerosis and the TCA cycle in perfused rat liver [35, 36, 112], and rat heart [32, 113, 114]. Another important application of stable isotopes and GC-MS is the in vivo quantification of polymer biosynthesis as exemplified by studies on DNA synthesis in rats [51, 115, 116], protein turn-over in humans [117] or lipidogenesis in humans [118]. Hereby, a mathematical approach is applied that allows the calculation of the labelling of the actual precursor molecules of a particular polymer [119–121].
Outlook
Due to high robustness, sensitivity and versatility, GC-MS based approaches will play a central role as routine technology for future analysis of metabolic fluxes in various biological systems. The use of stable isotopes in combination with GC-MS for metabolic flux analysis will establish as important method in actual fields such as functional genomics, systems biology, and pharmaceutical research for drug development [122, 123]. The compatibility with miniaturized cultivation tools will hereby allow the application to broad sets of mutants or cultivation conditions [24, 103, 124]. In addition to GC-MS also other mass spectrometry techniques, recently coming into focus, will be important tools for flux analysis. This involves LC-MS with excellent characteristics for dynamic flux measurements via labelling patterns of intracellular metabolites [71, 125–127] or MALDI-TOF MS with ultra-fast measurement at low experimental effort [128] providing an outstanding innovation for high-throughput flux analysis e.g. of mutant libraries or culture conditions [124].
References
Brunengraber H, Kelleher JK, Des Rosiers C: Applications of mass isotopomer analysis to nutrition research. Annu Rev Nutr. 1997, 17: 559-596. 10.1146/annurev.nutr.17.1.559.
Kelleher JK: Estimating gluconeogenesis with [U-13C]glucose: molecular condensation requires a molecular approach. Am J Physiol. 1999, 277 (3 Pt 1): E395-400.
Christensen B, Thykaer J, Nielsen J: Metabolic characterization of high- and low-yielding strains of Penicillium chrysogenum. Appl Microbiol Biotechnol. 2000, 54 (2): 212-217. 10.1007/s002530000371.
Becker J, Klopprogge C, Zelder O, Heinzle E, Wittmann C: Amplified expression of fructose 1,6-bisphosphatase in Corynebacterium glutamicum increases in vivo flux through the pentose phosphate pathway and lysine production on different carbon sources. Appl Environ Microbiol. 2005, 71 (12): 8587-8596. 10.1128/AEM.71.12.8587-8596.2005.
Becker J, Klopprogge C, Zelder O, Wittmann C: Metabolic flux engineering of L-lysine production in Corynebacterium glutamicum - overexpression and modification of glucose 6-phosphate dehydrogenase. J Biotechnol. 2006, submitted:
Christensen B, Nielsen J: Metabolic network analysis. A powerful tool in metabolic engineering. Adv Biochem Eng Biotechnol. 2000, 66: 209-231.
Lapidot A, Nissim I: Regulation of pool sizes and turnover rates of amino acids in humans: 15N-glycine and 15N-alanine single-dose experiments using gas chromatography-mass spectrometry analysis. Metabolism. 1980, 29 (3): 230-239. 10.1016/0026-0495(80)90064-5.
Nissim I, Yudkoff M, Yang W, Terwilliger T, Segal S: Rapid gas chromatographic-mass spectrometric analysis of [15N]urea: application to human metabolic studies. Clin Chim Acta. 1981, 109 (3): 295-304. 10.1016/0009-8981(81)90315-6.
Tsalikian E, Howard C, Gerich JE, Haymond MW: Increased leucine flux in short-term fasted human subjects: evidence for increased proteolysis. Am J Physiol. 1984, 247 (3 Pt 1): E323-7.
Nissim I, Lapidot A: Dynamic aspects of amino acid metabolism in alloxan-induced diabetes and insulin-treated rabbits: in vivo studies with 15N and gas chromatography-mass spectrometry. Biochem Med Metab Biol. 1986, 35 (1): 88-100. 10.1016/0885-4505(86)90063-0.
Des Rosiers C, Chatham JC: Myocardial phenotyping using isotopomer analysis of metabolic fluxes. Biochem Soc Trans. 2005, 33 (Pt 6): 1413-1417.
Des Rosiers C, Lloyd S, Comte B, Chatham JC: A critical perspective of the use of 13C-isotopomer analysis by GCMS and NMR as applied to cardiac metabolism. Metab Eng. 2004, 6 (1): 44-58. 10.1016/j.ymben.2003.10.004.
Kelleher JK: Flux estimation using isotopic tracers: common ground for metabolic physiology and metabolic engineering. Metab Eng. 2001, 3 (2): 100-110. 10.1006/mben.2001.0185.
Wiechert W: 13C metabolic flux analysis. Metab Eng. 2001, 3 (3): 195-206. 10.1006/mben.2001.0187.
Wittmann C: Metabolic flux analysis using mass spectrometry. Adv Biochem Eng Biotechnol. 2002, 74: 39-64.
Christensen B, Nielsen J: Isotopomer analysis using GC-MS. Metab Eng. 1999, 1 (4): 282-290. 10.1006/mben.1999.0117.
Wittmann C, Heinzle E: Mass spectrometry for metabolic flux analysis. Biotechnol Bioeng. 1999, 62 (6): 739-750. 10.1002/(SICI)1097-0290(19990320)62:6<739::AID-BIT13>3.0.CO;2-E.
Frick O, Wittmann C: Characterization of the metabolic shift between oxidative and fermentative growth in Saccharomyces cerevisiae by comparative 13C flux analysis. Microb Cell Fact. 2005, 4: 30-10.1186/1475-2859-4-30.
Kiefer P, Heinzle E, Zelder O, Wittmann C: Comparative metabolic flux analysis of lysine-producing Corynebacterium glutamicum cultured on glucose or fructose. Appl Environ Microbiol. 2004, 70 (1): 229-239. 10.1128/AEM.70.1.229-239.2004.
Kromer JO, Sorgenfrei O, Klopprogge K, Heinzle E, Wittmann C: In-depth profiling of lysine-producing Corynebacterium glutamicum by combined analysis of the transcriptome, metabolome, and fluxome. J Bacteriol. 2004, 186 (6): 1769-1784. 10.1128/JB.186.6.1769-1784.2004.
Wittmann C, Heinzle E: Genealogy profiling through strain improvement by using metabolic network analysis: metabolic flux genealogy of several generations of lysine-producing corynebacteria. Appl Environ Microbiol. 2002, 68 (12): 5843-5859. 10.1128/AEM.68.12.5843-5859.2002.
Wittmann C, Kiefer P, Zelder O: Metabolic fluxes in Corynebacterium glutamicum during lysine production with sucrose as carbon source. Appl Environ Microbiol. 2004, 70 (12): 7277-7287. 10.1128/AEM.70.12.7277-7287.2004.
Fischer E, Sauer U: Metabolic flux profiling of Escherichia coli mutants in central carbon metabolism using GC-MS. Eur J Biochem. 2003, 270 (5): 880-891. 10.1046/j.1432-1033.2003.03448.x.
Fischer E, Zamboni N, Sauer U: High-throughput metabolic flux analysis based on gas chromatography-mass spectrometry derived 13C constraints. Anal Biochem. 2004, 325 (2): 308-316. 10.1016/j.ab.2003.10.036.
Zamboni N, Fischer E, Sauer U: FiatFlux--a software for metabolic flux analysis from 13C-glucose experiments. BMC Bioinformatics. 2005, 6: 209-10.1186/1471-2105-6-209.
Zhao J, Shimizu K: Metabolic flux analysis of Escherichia coli K12 grown on 13C-labeled acetate and glucose using GC-MS and powerful flux calculation method. J Biotechnol. 2003, 101 (2): 101-117. 10.1016/S0168-1656(02)00316-4.
Blank LM, Sauer U: TCA cycle activity in Saccharomyces cerevisiae is a function of the environmentally determined specific growth and glucose uptake rates. Microbiology. 2004, 150 (Pt 4): 1085-1093. 10.1099/mic.0.26845-0.
Cannizzaro C, Christensen B, Nielsen J, von Stockar U: Metabolic network analysis on Phaffia rhodozyma yeast using 13C-labeled glucose and gas chromatography-mass spectrometry. Metab Eng. 2004, 6 (4): 340-351. 10.1016/j.ymben.2004.06.001.
Thykaer J, Christensen B, Nielsen J: Metabolic network analysis of an adipoyl-7-ADCA-producing strain of Penicillium chrysogenum: elucidation of adipate degradation. Metab Eng. 2002, 4 (2): 151-158. 10.1006/mben.2001.0218.
Lin YY, Cheng WB, Wright CE: Glucose metabolism in mammalian cells as determined by mass isotopomer analysis. Anal Biochem. 1993, 209 (2): 267-273. 10.1006/abio.1993.1118.
Marin S, Lee WN, Bassilian S, Lim S, Boros LG, Centelles JJ, FernAndez-Novell JM, Guinovart JJ, Cascante M: Dynamic profiling of the glucose metabolic network in fasted rat hepatocytes using [1,2-13C2]glucose. Biochem J. 2004, 381 (Pt 1): 287-294.
Comte B, Vincent G, Bouchard B, Jette M, Cordeau S, Rosiers CD: A 13C mass isotopomer study of anaplerotic pyruvate carboxylation in perfused rat hearts. J Biol Chem. 1997, 272 (42): 26125-26131. 10.1074/jbc.272.42.26125.
Khairallah M, Labarthe F, Bouchard B, Danialou G, Petrof BJ, Des Rosiers C: Profiling substrate fluxes in the isolated working mouse heart using 13C-labeled substrates: focusing on the origin and fate of pyruvate and citrate carbons. Am J Physiol Heart Circ Physiol. 2004, 286 (4): H1461-70. 10.1152/ajpheart.00942.2003.
Villas-Boas SG, Mas S, Akesson M, Smedsgaard J, Nielsen J: Mass spectrometry in metabolome analysis. Mass Spectrom Rev. 2005, 24 (5): 613-646. 10.1002/mas.20032.
Des Rosiers C, Di Donato L, Comte B, Laplante A, Marcoux C, David F, Fernandez CA, Brunengraber H: Isotopomer analysis of citric acid cycle and gluconeogenesis in rat liver. Reversibility of isocitrate dehydrogenase and involvement of ATP-citrate lyase in gluconeogenesis. J Biol Chem. 1995, 270 (17): 10027-10036. 10.1074/jbc.270.17.10037.
Di Donato L, Des Rosiers C, Montgomery JA, David F, Garneau M, Brunengraber H: Rates of gluconeogenesis and citric acid cycle in perfused livers, assessed from the mass spectrometric assay of the 13C labeling pattern of glutamate. J Biol Chem. 1993, 268 (6): 4170-4180.
Hellerstein MK, Neese RA, Linfoot P, Christiansen M, Turner S, Letscher A: Hepatic gluconeogenic fluxes and glycogen turnover during fasting in humans. A stable isotope study. J Clin Invest. 1997, 100 (5): 1305-1319.
Hellerstein MK, Neese RA, Schwarz JM, Turner S, Faix D, Wu K: Altered fluxes responsible for reduced hepatic glucose production and gluconeogenesis by exogenous glucose in rats. Am J Physiol. 1997, 272 (1 Pt 1): E163-72.
Davis SC, Makarov AA, Hughes JD: Supersonic molecular beam-hyperthermal surface ionisation coupled with time-of-flight mass spectrometry applied to trace level detection of polynuclear aromatic hydrocarbons in drinking water for reduced sample preparation and analysis time. Rapid Commun Mass Spectrom. 1999, 13 (4): 247-250. 10.1002/(SICI)1097-0231(19990228)13:4<247::AID-RCM448>3.0.CO;2-U.
Klapa MI, Aon JC, Stephanopoulos G: Ion-trap mass spectrometry used in combination with gas chromatography for high-resolution metabolic flux determination. Biotechniques. 2003, 34 (4): 832-6, 838, 840 passim.
Klapa MI, Aon JC, Stephanopoulos G: Systematic quantification of complex metabolic flux networks using stable isotopes and mass spectrometry. Eur J Biochem. 2003, 270 (17): 3525-3542. 10.1046/j.1432-1033.2003.03732.x.
Meier-Augenstein W: Use of gas chromatography-combustion-isotope ratio mass spectrometry in nutrition and metabolic research. Curr Opin Clin Nutr Metab Care. 1999, 2 (6): 465-470. 10.1097/00075197-199911000-00005.
Meier-Augenstein W: Applied gas chromatography coupled to isotope ratio mass spectrometry. J Chromatogr A. 1999, 842 (1-2): 351-371. 10.1016/S0021-9673(98)01057-7.
Thompson GN, Pacy PJ, Ford GC, Halliday D: Practical considerations in the use of stable isotope labelled compounds as tracers in clinical studies. Biomed Environ Mass Spectrom. 1989, 18 (5): 321-327. 10.1002/bms.1200180507.
Corso TN, Brenna JT: High-precision position-specific isotope analysis. Proc Natl Acad Sci U S A. 1997, 94 (4): 1049-1053. 10.1073/pnas.94.4.1049.
Nissim I, Nissim I, Yudkoff M: Carbon flux through tricarboxylic acid cycle in rat renal tubules. Biochim Biophys Acta. 1990, 1033 (2): 194-200.
Katz J, Wals PA, Lee WN: Determination of pathways of glycogen synthesis and the dilution of the three-carbon pool with [U-13C]glucose. Proc Natl Acad Sci U S A. 1991, 88 (6): 2103-2107. 10.1073/pnas.88.6.2103.
Hellerstein MK, Schwarz JM, Neese RA: Regulation of hepatic de novo lipogenesis in humans. Annu Rev Nutr. 1996, 16: 523-557.
Katz J, Wals P, Lee WN: Isotopomer studies of gluconeogenesis and the Krebs cycle with 13C-labeled lactate. J Biol Chem. 1993, 268 (34): 25509-25521.
Dauner M, Sauer U: GC-MS analysis of amino acids rapidly provides rich information for isotopomer balancing. Biotechnol Prog. 2000, 16 (4): 642-649. 10.1021/bp000058h.
Macallan DC, Fullerton CA, Neese RA, Haddock K, Park SS, Hellerstein MK: Measurement of cell proliferation by labeling of DNA with stable isotope-labeled glucose: studies in vitro, in animals, and in humans. Proc Natl Acad Sci U S A. 1998, 95 (2): 708-713. 10.1073/pnas.95.2.708.
Wittmann C, Hans M, Heinzle E: In vivo analysis of intracellular amino acid labelings by GC/MS. Anal Biochem. 2002, 307 (2): 379-382. 10.1016/S0003-2697(02)00030-1.
Wittmann C, Krömer JO, Kiefer P, Binz T, Heinzle E: Impact of the cold shock phenomenon on quantification of intracellular metabolites in bacteria. Anal Biochem. 2004, 327 (1): 135-139. 10.1016/j.ab.2004.01.002.
Hans MA, Heinzle E, Wittmann C: Quantification of intracellular amino acids in batch cultures of Saccharomyces cerevisiae. Appl Microbiol Biotechnol. 2001, 56 (5-6): 776-779. 10.1007/s002530100708.
Boren J, Lee WN, Bassilian S, Centelles JJ, Lim S, Ahmed S, Boros LG, Cascante M: The stable isotope-based dynamic metabolic profile of butyrate-induced HT29 cell differentiation. J Biol Chem. 2003, 278 (31): 28395-28402. 10.1074/jbc.M302932200.
Arnoldi L, Valsecchi G, Magni F, Monti LD, Piatti PM, Costa S, Kienle MG: Gluconeogenesis in isolated rat hepatocytes evaluated by gas chromatography/mass spectrometry using deuterated water. J Mass Spectrom. 1998, 33 (5): 444-452. 10.1002/(SICI)1096-9888(199805)33:5<444::AID-JMS651>3.0.CO;2-B.
Fox A: Carbohydrate profiling of bacteria by gas chromatography-mass spectrometry and their trace detection in complex matrices by gas chromatography-tandem mass spectrometry. J Chromatogr A. 1999, 843 (1-2): 287-300. 10.1016/S0021-9673(98)00884-X.
Halket JM, Waterman D, Przyborowska AM, Patel RK, Fraser PD, Bramley PM: Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J Exp Bot. 2005, 56 (410): 219-243. 10.1093/jxb/eri069.
Halket JM, Zaikin VG: Derivatization in mass spectrometry--1. Silylation. Eur J Mass Spectrom (Chichester, Eng). 2003, 9 (1): 1-21. 10.1255/ejms.527.
Segura J, Ventura R, Jurado C: Derivatization procedures for gas chromatographic-mass spectrometric determination of xenobiotics in biological samples, with special attention to drugs of abuse and doping agents. J Chromatogr B Biomed Sci Appl. 1998, 713 (1): 61-90. 10.1016/S0378-4347(98)00089-9.
Toyo'oka T: Use of derivatization to improve the chromatographic properties and detection selectivity of physiologically important carboxylic acids. J Chromatogr B Biomed Appl. 1995, 671 (1-2): 91-112. 10.1016/0378-4347(94)00559-N.
Wasels R, Belleville F: Gas chromatographic-mass spectrometric procedures used for the identification and determination of morphine, codeine and 6-monoacetylmorphine. J Chromatogr A. 1994, 674 (1-2): 225-234. 10.1016/0021-9673(94)85227-8.
Kitson FG, Larsen B, Mc Even CN: Gas Chromatography and Mass Spectrometry: A Practical Guide. 1996, San Diego , Academic Press
Wittmann C, Heinzle E: Modeling and experimental design for metabolic flux analysis of lysine-producing Corynebacteria by mass spectrometry. Metab Eng. 2001, 3 (2): 173-191. 10.1006/mben.2000.0178.
David H, Krogh AM, Roca C, Akesson M, Nielsen J: CreA influences the metabolic fluxes of Aspergillus nidulans during growth on glucose and xylose. Microbiology. 2005, 151 (Pt 7): 2209-2221. 10.1099/mic.0.27787-0.
Gombert AK, Moreira dos Santos M, Christensen B, Nielsen J: Network identification and flux quantification in the central metabolism of Saccharomyces cerevisiae under different conditions of glucose repression. J Bacteriol. 2001, 183 (4): 1441-1451. 10.1128/JB.183.4.1441-1451.2001.
Christensen B, Gombert AK, Nielsen J: Analysis of flux estimates based on 13C-labelling experiments. Eur J Biochem. 2002, 269 (11): 2795-2800. 10.1046/j.1432-1033.2002.02959.x.
Beylot M, David F, Brunengraber H: Determination of the 13C-labeling pattern of glutamate by gas chromatography-mass spectrometry. Anal Biochem. 1993, 212 (2): 532-536. 10.1006/abio.1993.1364.
Beylot M, Previs SF, David F, Brunengraber H: Determination of the 13C-labeling pattern of glucose by gas chromatography-mass spectrometry. Anal Biochem. 1993, 212 (2): 526-531. 10.1006/abio.1993.1363.
Wiechert W, Mollney M, Isermann N, Wurzel M, de Graaf AA: Bidirectional reaction steps in metabolic networks: III. Explicit solution and analysis of isotopomer labeling systems. Biotechnol Bioeng. 1999, 66 (2): 69-85. 10.1002/(SICI)1097-0290(1999)66:2<69::AID-BIT1>3.0.CO;2-6.
Kiefer P, Nicolas C, Letisse F, Portais JC: Determination of carbon labeling distribution of intracellular metabolites from single fragment ions by ion chromatography tandem mass spectrometry. Anal Biochem. 2007, 360 (2): 182-188. 10.1016/j.ab.2006.06.032.
van Dam JC, Eman MR, Frank J, Lange HC, van Dedem GWK, Heijnen JJ: Analysis of glycolytic intermediates in Saccharomyces cerevisiae using anion exchange chromatography and electrospray ionization with tandem mass spectrometric detection. Anal Chim Acta. 2002, 460: 209–218-10.1016/S0003-2670(02)00240-4.
Fernandez CA, Des Rosiers C, Previs SF, David F, Brunengraber H: Correction of 13C mass isotopomer distributions for natural stable isotope abundance. J Mass Spectrom. 1996, 31 (3): 255-262. 10.1002/(SICI)1096-9888(199603)31:3<255::AID-JMS290>3.0.CO;2-3.
Lee WN, Byerley LO, Bergner EA, Edmond J: Mass isotopomer analysis: theoretical and practical considerations. Biol Mass Spectrom. 1991, 20 (8): 451-458. 10.1002/bms.1200200804.
van Winden WA, Wittmann C, Heinzle E, Heijnen JJ: Correcting mass isotopomer distributions for naturally occurring isotopes. Biotechnol Bioeng. 2002, 80 (4): 477-479. 10.1002/bit.10393.
Wahl SA, Dauner M, Wiechert W: New tools for mass isotopomer data evaluation in 13C flux analysis: mass isotope correction, data consistency checking, and precursor relationships. Biotechnol Bioeng. 2004, 85 (3): 259-268. 10.1002/bit.10909.
Talwar P, Wittmann C, Lengauer T, Heinzle E: Software tool for automated processing of 13C labeling data from mass spectrometric spectra. Biotechniques. 2003, 35 (6): 1214-1215.
Fagerquist CK, Neese RA, Hellerstein MK: Molecular ion fragmentation and its effects on mass isotopomer abundances of fatty acid methyl esters ionized by electron impact. J Am Soc Mass Spectrom. 1999, 10 (5): 430-439. 10.1016/S1044-0305(99)00003-3.
Zamboni N, Fischer E, Muffler A, Wyss M, Hohmann HP, Sauer U: Transient expression and flux changes during a shift from high to low riboflavin production in continuous cultures of Bacillus subtilis. Biotechnol Bioeng. 2005, 89 (2): 219-232. 10.1002/bit.20338.
van Winden W, Verheijen P, Heijnen S: Possible pitfalls of flux calculations based on 13C-labeling. Metab Eng. 2001, 3 (2): 151-162. 10.1006/mben.2000.0174.
Sherry AD, Sumegi B, Miller B, Cottam GL, Gavva S, Jones JG, Malloy CR: Orientation-conserved transfer of symmetric Krebs cycle intermediates in mammalian tissue. Biochemistry. 1994, 33 (20): 6268-6275. 10.1021/bi00186a029.
Sumegi B, Sherry AD, Malloy CR, Srere PA: Evidence for orientation-conserved transfer in the TCA cycle in Saccharomyces cerevisiae: 13C NMR studies. Biochemistry. 1993, 32 (47): 12725-12729. 10.1021/bi00210a022.
Debnam PM, Shearer G, Blackwood L, Kohl DH: Evidence for channeling of intermediates in the oxidative pentose phosphate pathway by soybean and pea nodule extracts, yeast extracts, and purified yeast enzymes. Eur J Biochem. 1997, 246 (2): 283-290. 10.1111/j.1432-1033.1997.00283.x.
Fiaux J, Cakar ZP, Sonderegger M, Wuthrich K, Szyperski T, Sauer U: Metabolic-flux profiling of the yeasts Saccharomyces cerevisiae and Pichia stipitis. Eukaryot Cell. 2003, 2 (1): 170-180. 10.1128/EC.2.1.170-180.2003.
Maaheimo H, Fiaux J, Cakar ZP, Bailey JE, Sauer U, Szyperski T: Central carbon metabolism of Saccharomyces cerevisiae explored by biosynthetic fractional 13C labeling of common amino acids. Eur J Biochem. 2001, 268 (8): 2464-2479. 10.1046/j.1432-1327.2001.02126.x.
Previs SF, Hallowell PT, Neimanis KD, David F, Brunengraber H: Limitations of the mass isotopomer distribution analysis of glucose to study gluconeogenesis. Heterogeneity of glucose labeling in incubated hepatocytes. J Biol Chem. 1998, 273 (27): 16853-16859. 10.1074/jbc.273.27.16853.
Lapidot A, Gopher A: Quantitation of metabolic compartmentation in hyperammonemic brain by natural abundance 13C-NMR detection of 13C-15N coupling patterns and isotopic shifts. Eur J Biochem. 1997, 243 (3): 597-604. 10.1111/j.1432-1033.1997.00597.x.
Sola A, Maaheimo H, Ylonen K, Ferrer P, Szyperski T: Amino acid biosynthesis and metabolic flux profiling of Pichia pastoris. Eur J Biochem. 2004, 271 (12): 2462-2470. 10.1111/j.1432-1033.2004.04176.x.
Szyperski T: Biosynthetically directed fractional 13C-labeling of proteinogenic amino acids. An efficient analytical tool to investigate intermediary metabolism. Eur J Biochem. 1995, 232 (2): 433-448. 10.1111/j.1432-1033.1995.tb20829.x.
Szyperski T, Glaser RW, Hochuli M, Fiaux J, Sauer U, Bailey JE, Wuthrich K: Bioreaction network topology and metabolic flux ratio analysis by biosynthetic fractional 13C labeling and two-dimensional NMR spectroscopy. Metab Eng. 1999, 1 (3): 189-197. 10.1006/mben.1999.0116.
KEGG: Kyoto Encyclopedia of Genes and Genomes. 2007
Sonntag K, Eggeling L, De Graaf AA, Sahm H: Flux partitioning in the split pathway of lysine synthesis in Corynebacterium glutamicum. Quantification by 13C- and 1H-NMR spectroscopy. Eur J Biochem. 1993, 213 (3): 1325-1331. 10.1111/j.1432-1033.1993.tb17884.x.
Wiechert W, Mollney M, Petersen S, de Graaf AA: A universal framework for 13C metabolic flux analysis. Metab Eng. 2001, 3 (3): 265-283. 10.1006/mben.2001.0188.
Mollney M, Wiechert W, Kownatzki D, de Graaf AA: Bidirectional reaction steps in metabolic networks: IV. Optimal design of isotopomer labeling experiments. Biotechnol Bioeng. 1999, 66 (2): 86-103. 10.1002/(SICI)1097-0290(1999)66:2<86::AID-BIT2>3.0.CO;2-A.
Schmidt K, Norregaard LC, Pedersen B, Meissner A, Duus JO, Nielsen JO, Villadsen J: Quantification of intracellular metabolic fluxes from fractional enrichment and 13C-13C coupling constraints on the isotopomer distribution in labeled biomass components. Metab Eng. 1999, 1 (2): 166-179. 10.1006/mben.1999.0114.
Fong SS, Nanchen A, Palsson BO, Sauer U: Latent pathway activation and increased pathway capacity enable Escherichia coli adaptation to loss of key metabolic enzymes. J Biol Chem. 2006, 281 (12): 8024-8033. 10.1074/jbc.M510016200.
Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E: The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J Biol Chem. 2004, 279 (8): 6613-6619. 10.1074/jbc.M311657200.
Li M, Ho PY, Yao S, Shimizu K: Effect of lpdA gene knockout on the metabolism in Escherichia coli based on enzyme activities, intracellular metabolite concentrations and metabolic flux analysis by 13C-labeling experiments. J Biotechnol. 2006, 122 (2): 254-266. 10.1016/j.jbiotec.2005.09.016.
Zhu T, Phalakornkule C, Ghosh S, Grossmann IE, Koepsel RR, Ataai MM, Domach MM: A metabolic network analysis & NMR experiment design tool with user interface-driven model construction for depth-first search analysis. Metab Eng. 2003, 5 (2): 74-85. 10.1016/S1096-7176(03)00023-5.
Shimizu K: Metabolic flux analysis based on 13C-labeling experiments and integration of the information with gene and protein expression patterns. Adv Biochem Eng Biotechnol. 2004, 91: 1-49.
Peng L, Arauzo-Bravo MJ, Shimizu K: Metabolic flux analysis for a ppc mutant Escherichia coli based on 13C-labelling experiments together with enzyme activity assays and intracellular metabolite measurements. FEMS Microbiol Lett. 2004, 235 (1): 17-23. 10.1111/j.1574-6968.2004.tb09562.x.
Al Zaid Siddiquee K, Arauzo-Bravo MJ, Shimizu K: Metabolic flux analysis of pykF gene knockout Escherichia coli based on 13C-labeling experiments together with measurements of enzyme activities and intracellular metabolite concentrations. Appl Microbiol Biotechnol. 2004, 63 (4): 407-417. 10.1007/s00253-003-1357-9.
Wittmann C, Kim HM, Heinzle E: Metabolic network analysis of lysine producing Corynebacterium glutamicum at a miniaturized scale. Biotechnol Bioeng. 2004, 87 (1): 1-6. 10.1002/bit.20103.
Fischer E, Sauer U: Large-scale in vivo flux analysis shows rigidity and suboptimal performance of Bacillus subtilis metabolism. Nat Genet. 2005, 37 (6): 636-640. 10.1038/ng1555.
Neese RA, Misell LM, Turner S, Chu A, Kim J, Cesar D, Hoh R, Antelo F, Strawford A, McCune JM, Christiansen M, Hellerstein MK: Measurement in vivo of proliferation rates of slow turnover cells by 2H2O labeling of the deoxyribose moiety of DNA. Proc Natl Acad Sci U S A. 2002, 99 (24): 15345-15350. 10.1073/pnas.232551499.
Jonsbu E, Christensen B, Nielsen J: Changes of in vivo fluxes through central metabolic pathways during the production of nystatin by Streptomyces noursei in batch culture. Appl Microbiol Biotechnol. 2001, 56 (1-2): 93-100. 10.1007/s002530100613.
Fuhrer T, Fischer E, Sauer U: Experimental identification and quantification of glucose metabolism in seven bacterial species. J Bacteriol. 2005, 187 (5): 1581-1590. 10.1128/JB.187.5.1581-1590.2005.
Fredlund E, Blank LM, Schnurer J, Sauer U, Passoth V: Oxygen- and glucose-dependent regulation of central carbon metabolism in Pichia anomala. Appl Environ Microbiol. 2004, 70 (10): 5905-5911. 10.1128/AEM.70.10.5905-5911.2004.
Blank LM, Lehmbeck F, Sauer U: Metabolic-flux and network analysis in fourteen hemiascomycetous yeasts. FEMS Yeast Res. 2005, 5 (6-7): 545-558. 10.1016/j.femsyr.2004.09.008.
Lee K, Berthiaume F, Stephanopoulos GN, Yarmush ML: Metabolic flux analysis: a powerful tool for monitoring tissue function. Tissue Eng. 1999, 5 (4): 347-368. 10.1089/ten.1999.5.347.
Lee WN, Boros LG, Puigjaner J, Bassilian S, Lim S, Cascante M: Mass isotopomer study of the nonoxidative pathways of the pentose cycle with [1,2-13C2]glucose. Am J Physiol. 1998, 274 (5 Pt 1): E843-51.
Des Rosiers C, Fernandez CA, David F, Brunengraber H: Reversibility of the mitochondrial isocitrate dehydrogenase reaction in the perfused rat liver. Evidence from isotopomer analysis of citric acid cycle intermediates. J Biol Chem. 1994, 269 (44): 27179-27182.
Poirier M, Vincent G, Reszko AE, Bouchard B, Kelleher JK, Brunengraber H, Des Rosiers C: Probing the link between citrate and malonyl-CoA in perfused rat hearts. Am J Physiol Heart Circ Physiol. 2002, 283 (4): H1379-86.
Vincent G, Comte B, Poirier M, Rosiers CD: Citrate release by perfused rat hearts: a window on mitochondrial cataplerosis. Am J Physiol Endocrinol Metab. 2000, 278 (5): E846-56.
Kim SJ, Turner S, Killion S, Hellerstein MK: In vivo measurement of DNA synthesis rates of colon epithelial cells in carcinogenesis. Biochem Biophys Res Commun. 2005, 331 (1): 203-209. 10.1016/j.bbrc.2005.02.195.
Collins ML, Eng S, Hoh R, Hellerstein MK: Measurement of mitochondrial DNA synthesis in vivo using a stable isotope-mass spectrometric technique. J Appl Physiol. 2003, 94 (6): 2203-2211.
Busch R, Kim YK, Neese RA, Schade-Serin V, Collins M, Awada M, Gardner JL, Beysen C, Marino ME, Misell LM, Hellerstein MK: Measurement of protein turnover rates by heavy water labeling of nonessential amino acids. Biochim Biophys Acta. 2006, 1760 (5): 730-744.
Siler SQ, Neese RA, Hellerstein MK: De novo lipogenesis, lipid kinetics, and whole-body lipid balances in humans after acute alcohol consumption. Am J Clin Nutr. 1999, 70 (5): 928-936.
Hellerstein M: Methods for measuring polymerisation biosynthesis: three general solutions to the problem of the "true precursor". Diabetes Nutr Metab. 2000, 13 (1): 46-60.
Hellerstein MK, Neese RA: Mass isotopomer distribution analysis at eight years: theoretical, analytic, and experimental considerations. Am J Physiol. 1999, 276 (6 Pt 1): E1146-70.
Papageorgopoulos C, Caldwell K, Shackleton C, Schweingrubber H, Hellerstein MK: Measuring protein synthesis by mass isotopomer distribution analysis (MIDA). Anal Biochem. 1999, 267 (1): 1-16. 10.1006/abio.1998.2958.
Hellerstein MK, Murphy E: Stable isotope-mass spectrometric measurements of molecular fluxes in vivo: emerging applications in drug development. Curr Opin Mol Ther. 2004, 6 (3): 249-264.
Turner SM, Murphy EJ, Neese RA, Antelo F, Thomas T, Agarwal A, Go C, Hellerstein MK: Measurement of TG synthesis and turnover in vivo by 2H2O incorporation into the glycerol moiety and application of MIDA. Am J Physiol Endocrinol Metab. 2003, 285 (4): E790-803.
Sauer U: High-throughput phenomics: experimental methods for mapping fluxomes. Curr Opin Biotechnol. 2004, 15 (1): 58-63. 10.1016/j.copbio.2003.11.001.
Matsuda F, Morino K, Ano R, Kuzawa M, Wakasa K, Miyagawa H: Metabolic flux analysis of the phenylpropanoid pathway in elicitor-treated potato tuber tissue. Plant Cell Physiol. 2005, 46 (3): 454-466. 10.1093/pcp/pci042.
Oldiges M, Kunze M, Degenring D, Sprenger GA, Takors R: Stimulation, monitoring, and analysis of pathway dynamics by metabolic profiling in the aromatic amino acid pathway. Biotechnol Prog. 2004, 20 (6): 1623-1633. 10.1021/bp0498746.
van Winden WA, van Dam JC, Ras C, Kleijn RJ, Vinke JL, van Gulik WM, Heijnen JJ: Metabolic-flux analysis of Saccharomyces cerevisiae CEN.PK113-7D based on mass isotopomer measurements of 13C-labeled primary metabolites. FEMS Yeast Res. 2005, 5 (6-7): 559-568. 10.1016/j.femsyr.2004.10.007.
Wittmann C, Heinzle E: Application of MALDI-TOF MS to lysine-producing Corynebacterium glutamicum: a novel approach for metabolic flux analysis. Eur J Biochem. 2001, 268 (8): 2441-2455. 10.1046/j.1432-1327.2001.02129.x.
Rosman KJR, Taylor PDP: Isotopic composition of the elements. Pure Appl Chern. 1998, 70: 217-231.
Szyperski T: 13C-NMR, MS and metabolic flux balancing in biotechnology research. Q Rev Biophys. 1998, 31 (1): 41-106. 10.1017/S0033583598003412.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wittmann, C. Fluxome analysis using GC-MS. Microb Cell Fact 6, 6 (2007). https://doi.org/10.1186/1475-2859-6-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2859-6-6