
Abstract
In severe trauma patients, coagulopathy is frequently observed in the acute phase of trauma. Trauma-induced coagulopathy is coagulopathy caused by the trauma itself. The pathophysiology of trauma-induced coagulopathy consists of coagulation activation, hyperfibrino(geno)lysis, and consumption coagulopathy. These pathophysiological mechanisms are the characteristics to DIC with the fibrinolytic phenotype.
Similar content being viewed by others

Background
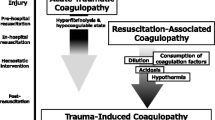
In severe trauma patients, coagulopathy is frequently observed in the acute phase of trauma, with profound effects on outcome [1–7]. This coagulopathy is caused by multiple factors associated with the trauma itself as well as certain interventions [8–12] and has been described with various terms. In this manuscript, we refer to the coagulopathy caused by diverse trauma-associated factors as “trauma-associated coagulopathy” and the coagulopathy caused by the trauma itself as “trauma-induced coagulopathy” (Fig. 1).

Trauma-associated coagulopathy and trauma-induced coagulopathy. Trauma-associated coagulopathy is caused by multiple factors and includes trauma-induced coagulopathy, which is caused by trauma itself.
Inconsistencies in the acute coagulopathy of trauma shock theory
Coagulation suppression by activated protein C?
In the acute coagulopathy of trauma shock (ACoTS) theory, trauma-shock stimulates release of soluble thrombomodulin (TM) from endothelial cells [13, 14]. Soluble TM binds to thrombin to form a thrombin-TM complex, which activates protein C [13, 14], which in turn suppresses prothrombinase complex (factor Va-factor Xa complex) activity and thrombin formation [13, 14].
However, TM is a receptor of thrombin and protein C on the endothelial cell surface and regulates the coagulation and complement system [15]. Soluble TM is formed via the limited proteolysis of TM by neutrophil elastase on the endothelial cell surface [16, 17], but it has not been confirmed that soluble TM is actively secreted by endothelial cells. However, the level of soluble TM correlates with the degree of endothelial injury [16, 17]. Furthermore, soluble TM has only 20% of activity of normal TM on the endothelial cell surface [18]. Under these circumstances, the anticoagulant property of the endothelium is impaired [16, 17]. Consequently, total anticoagulant activity of TM in vessels is impaired in the acute phase of trauma [17].
Hyperfibrinolysis by degradation of plasminogen activator inhibitor?
In the ACoTS theory, activated protein C decomposes plasminogen activator inhibitor (PAI) [13]. Because PAI suppresses fibrinolysis, PAI degradation accelerates fibrinolysis [13].
However, activated protein C level does not increase, because, as mentioned above, total TM activity in the vessel is impaired [17]. Furthermore, plasma PAI level does not increase immediately following trauma [19]. Chapman et al. [19] indicated that total plasma PAI in severe trauma patients with hyperfibrinolysis did not increase compared to that in healthy controls. Therefore, PAI degradation does not appear to play a significant role in the pathogenesis of hyperfibrinolysis in the acute phase of trauma [17, 19].
DIC phenotypes
We have repeatedly advocated that trauma-induced coagulopathy is a disseminated intravascular coagulation (DIC) with the fibrinolytic phenotype [12, 20–22]. However, it has been suggested that trauma-induced coagulopathy does not imply DIC [13]. We consider that this argument [13] might have resulted from a misunderstanding about DIC phenotypes.
DIC is divided into two phenotypes, the fibrinolytic and thrombotic phenotypes (Table 1) [20, 23, 24]. In critical care settings, sepsis-induced DIC is frequently observed, which is a representative of the thrombotic phenotype [24] and characterized by suppressed fibrinolysis with micro-vessel thrombosis and ischemic organ dysfunction [25]. However, trauma-induced coagulopathy, which is considered a type of DIC with the fibrinolytic phenotype, is markedly different from DIC with the thrombotic phenotype [12, 20, 21]. Coagulation activation is observed in both phenotypes of DIC. Plasma PAI suppresses fibrinolysis in DIC with the thrombotic phenotype, whereas fibrino(geno)lysis is activated by tissue-plasminogen activator (t-PA) in DIC with the fibrinolytic phenotype [24, 25]. Therefore, although sepsis-induced DIC does not lead to massive bleeding, trauma-induced DIC (fibrinolytic phenotype) in the acute phase of trauma contributes to massive bleeding and death [1–4].
Pathophysiology of trauma-induced coagulopathy
Trauma-induced coagulopathy is generated by the following pathophysiological mechanisms:
-
1)
Coagulation activation
-
1.
Procoagulants in the systemic circulation
-
2.
Impairment of endogenous anticoagulant activity
-
3.
Thrombin generation in the systemic circulation
-
1.
-
2)
Hyper-fibrino(geno)lysis
-
1.
Acute release of t-PA-induced hyperfibrino(geno)lysis
-
2.
Coagulation activation-induced fibrino(geno)lysis
-
1.
-
3)
Consumption coagulopathy
Coagulation activation
Procoagulants in the systemic circulation
In severe trauma patients, particularly those with blunt trauma, massive tissue injury accelerates thrombin generation [3, 5–7]. Previous studies showed spontaneous thrombin generation in severe trauma by using non-stimulation thrombin generation assays (Fig. 2) [26, 27]. Shortly after trauma, various procoagulants are observed in the systemic circulation, which results in this spontaneous thrombin generation (Table 2).

Spontaneous thrombin generation in severe trauma. a Stimulated thrombin generation curve. Although the amount of thrombin generation was lower in the Trauma group than the control group, time to initiation of thrombin generation and time to peak thrombin generation were shorter in the trauma group than the control group, suggesting coagulation activation. b Non-stimulated thrombin generation curve. Spontaneous thrombin generation was observed in the trauma group but not in the control group, demonstrating the presence of circulating procoagulants in the systemic circulation of the trauma group. Blue line: control group; red line: trauma group. (Cited as Figure 5 in our previous manuscript [27] and adapted with permission from Wolters Kluwer Health, Inc.)
The platelet-derived microparticle is a well-known procoagulant in the acute phase of trauma [28–30], and several studies have indicated that various other cell-derived microparticles are subsequently released into the systemic circulation in the acute phase of trauma, such as the leukocyte-derived [30, 31], erythrocyte-derived [31], and endothelial-derived [30, 31] microparticles. Tissue factor is exposed on the membrane of certain microparticles [30, 32, 33]. Therefore, elevation of tissue factor antigen levels in the plasma reported in previous studies [34, 35] may reflect increase of tissue factor-exposing microparticles. Recently, brain-derived microparticles were detected in brain trauma animal models [32, 33]. These brain-derived microparticles expressed neuronal or glial cell markers, procoagulant phosphatidylserine, and tissue factor [32, 33]. In addition, other injured organs may possibly release microparticles in severe trauma.
Extracellular DNA and DNA-binding proteins, which are well known as damage-associated molecular patterns, are procoagulants observed in the systemic circulation shortly after trauma [36–44]. Histone and histone-complexed DNA fragments were detected in the systemic circulation shortly after trauma and induced inflammation, coagulation activation, and organ dysfunction [36, 37]. Furthermore, early release of high mobility group box nuclear protein 1 (HMGB-1) [41–44] and mitochondrial DNA [38–40] was observed in severe trauma and induced inflammation and coagulation activation.
In penetrating trauma, especially stab wounds, there is less tissue injury than that in blunt trauma. Therefore, procoagulant production and coagulation activation are far less than that immediately following blunt trauma.
Impairment of endogenous anticoagulant activity
In healthy subjects, coagulation activation is regulated by endogenous anticoagulants, such as antithrombin and the TM-protein C pathway. However, in severe trauma, the endogenous anticoagulant activities are immediately impaired and dysregulation of coagulation activation is observed [1–6, 45, 46].
Many studies reported an early decrease in antithrombin activity in severe trauma [1–4, 45], and thrombin generation assays showed a negative correlation between antithrombin activity and generated thrombin, regardless of a decrease in prothrombin concentration (Fig. 3) [26, 27]. This result indicates that decreased antithrombin activity causes dysregulation of thrombin generation [26, 27].

Correlations between antithrombin activity and generated thrombin. Antithrombin was significantly negatively correlated with the ratio of the peak thrombin generation level to the factor II activity (ρ = −0.733, P < 0.001). Peak height/factor II ratio, peak thrombin generation level/factor II activity. a Antithrombin was significantly negatively correlated with the ratio of the generated thrombin amount to the factor II activity (ρ = −0.839, P < 0.001). ETP/factor II ratio, generated thrombin amount/factor II activity. (Cited as Figure 4 in our previous manuscript [27] and adapted with permission from Wolters Kluwer Health, Inc.)
Most of the TM in the body is found on vascular endothelial cells [47]. Therefore, evaluation of anticoagulation ability of TM is difficult. However, in severe trauma, endothelial damage leads to release of the TM on vascular endothelial cells as soluble TM in the systemic circulation [5, 6, 46]. Furthermore, plasma concentration of protein C decreases shortly after severe trauma [48–50]. Therefore, the anticoagulation ability of the TM-protein C pathway is impaired with resultant dysregulation of thrombin generation [2]. Although some investigators have advocated that activated protein C increases and suppresses coagulation [48–50], the increases in activated protein C (up to 10 ng/mL) did not reach sufficient concentrations to inhibit thrombin generation (70–80 ng/mL) [46, 50, 51].
Thrombin generation in the systemic circulation
The presence of procoagulants in the systemic circulation together with impairment of endogenous anticoagulant activities induces coagulation activation and thrombin generation [2, 27, 34, 35, 52, 53]. The half-life of thrombin is very short, which precludes measurement of plasma concentrations; therefore, other parameters have been used as evidence of thrombin generation in the systemic circulation. Soluble fibrin [2, 27] and fibrinopeptide A [34, 35, 52, 53] are considered to reflect active thrombin because these markers are formed as a result of the direct action of thrombin on fibrinogen, which is followed by fibrin formation. Early elevations of the plasma concentrations of these markers are evidence of thrombin generation in the systemic circulation and have been repeatedly reported [2, 27, 34, 35, 52, 53].
Hyperfibrino(geno)lysis
In severe trauma, hyperfibrino(geno)lysis, which is a combination of fibrinolysis and fibrinogenolysis, is frequently observed [1–7, 27, 34, 35, 45, 48–50, 52, 53]. This hyperfibrino(geno)lysis is caused by acute release of tissue-plasminogen activator (t-PA) and coagulation activation.
Shock-induced hyperfibrino(geno)lysis
One of the key enzymes in fibrino(geno)lysis is t-PA. t-PA catalyzes the cleavage of plasminogen to plasmin and thus initiates fibrin and fibrinogen degradation in plasma [54]. The main source of plasma t-PA is the Weibel-Palade body in the systemic vascular endothelial cells [54–56]. Severe shock (tissue hypoperfusion) stimulates the endothelial cells and induces release of t-PA from the Weibel-Palade bodies into the systemic circulation; this is called “acute release of t-PA” [55, 56]. Furthermore, the acute and massive t-PA release induces hyperfibrino(geno)lysis [3, 4, 12, 20, 57–62]. Thromboelastometry such as ROTEM® can detect acute release of t-PA as lysis of clots formed in test tubes [57–62].
Coagulation activation-induced fibrino(geno)lysis
In severe trauma, hyperfibrino(geno)lysis is frequently observed regardless of the presence of shock [3, 4, 63–69]. In particular, severe isolated head trauma, which is not usually complicated by hypotension, is a typical case in which hyperfibrino(geno)lysis may occur without shock [63, 66–69]. Hyperfibrino(geno)lysis without shock is induced by coagulation activation and is recognized by elevation of D-dimer and fibrin/fibrinogen degradation product (FDP) levels [3, 4, 63–69]. Kushimoto et al. reported [63] a correlative increase in fibrinogen degradation product and plasmin-α2 plasmin inhibitor complex levels. Furthermore, fibrinogen levels markedly decreased as a result of hyperfibrinogenolysis [63]. Many other studies reported that D-dimer and FDP levels increased not only in isolated head trauma [63, 66–69] but also in torso trauma regardless of the presence of shock [3, 4, 64].
In the acute phase of trauma, plasma PAI activity has not yet increased enough [19]. Therefore, although trauma-induced coagulation activation reactively causes fibrino(geno)lysis, the fibrino(geno)lysis is not suppressed by PAI [19, 65]. Furthermore, non-suppressed fibrino(geno)lysis consumes α2-plasmin inhibitor and the consumption of α2-plasmin inhibitor accelerates the dysregulation of fibrino(geno)lysis [35, 52, 63, 65, 70].
Consumption coagulopathy
As mentioned above, in severe trauma, coagulation activation and hyperfibrino(geno)lysis are simultaneously observed. Therefore, various coagulation factors and platelets are consumed in the acute phase of trauma [1, 64, 71–75]. Consumption of coagulation factors has been repeatedly reported because this phenomenon is easy to evaluate by measurement of the coagulation factors [1, 64, 71–75]. The plasma fibrinogen level decreases more frequently and earlier than the levels of other routinely measured coagulation parameters (prothrombin time, activated partial thromboplastin time, and platelet count) [1]. Furthermore, infusion or transfusion leads more readily to dilution of fibrinogen than the other coagulation factors [10, 76]. The other coagulation factors cannot compensate for the role of fibrinogen as a unique precursor of fibrin [77, 78]; therefore, decreased fibrinogen may lead to massive bleeding and poor outcome [1, 64, 71, 72]. Other coagulation factor activities also decrease correlatively with the severity of trauma [73–75]. Of these, factor V activity decreases more than the other factor activities [73–75]. Together with decreased fibrinogen levels, decreased factor V levels were detected in patients at accident sites [74]. Platelet counts are seldom reduced to a critical level (<100 × 109/L) in patients on arrival at emergency departments and decrease slower than do fibrinogen levels [1].
Conclusions
The pathophysiology of trauma-induced coagulopathy consists of coagulation activation, hyperfibrino(geno)lysis, and consumption coagulopathy. These pathophysiological mechanisms are characteristic to DIC with the fibrinolytic phenotype.
Abbreviations
- DIC:
-
Disseminated intravascular coagulation
- PAI:
-
Plasminogen activator inhibitor
- t-PA:
-
Tissue-plasminogen activator
- HMGB-1:
-
High mobility group box nuclear protein 1, TM, thrombomodulin
- FDP:
-
Fibrin/fibrinogen degradation products.
References
Hayakawa M, Gando S, Ono Y, Wada T, Yanagida Y, Sawamura A. Fibrinogen level deteriorates before other routine coagulation parameters and massive transfusion in the early phase of severe trauma: a retrospective observational study. Semin Thromb Hemost. 2015;41:35–42.
Yanagida Y, Gando S, Sawamura A, Hayakawa M, Uegaki S, Kubota N, et al. Normal prothrombinase activity, increased systemic thrombin activity, and lower antithrombin levels in patients with disseminated intravascular coagulation at an early phase of trauma: comparison with acute coagulopathy of trauma-shock. Surgery. 2013;154:48–57.
Hayakawa M, Sawamura A, Gando S, Kubota N, Uegaki S, Shimojima H, et al. Disseminated intravascular coagulation at an early phase of trauma is associated with consumption coagulopathy and excessive fibrinolysis both by plasmin and neutrophil elastase. Surgery. 2011;149:221–30.
Sawamura A, Hayakawa M, Gando S, Kubota N, Sugano M, Wada T, et al. Disseminated intravascular coagulation with a fibrinolytic phenotype at an early phase of trauma predicts mortality. Thromb Res. 2009;124:608–13.
Gando S, Nakanishi Y, Kameue T, Nanzaki S. Soluble thrombomodulin increases in patients with disseminated intravascular coagulation and in those with multiple organ dysfunction syndrome after trauma: role of neutrophil elastase. J Trauma. 1995;39:660–4.
Gando S, Kameue T, Matsuda N, Hayakawa M, Ishitani T, Morimoto Y, et al. Combined activation of coagulation and inflammation has an important role in multiple organ dysfunction and poor outcome after severe trauma. Thromb Haemost. 2002;88:943–9.
Engelman DT, Gabram SG, Allen L, Ens GE, Jacobs LM. Hypercoagulability following multiple trauma. World J Surg. 1996;20:5–10.
Martini WZ, Pusateri AE, Uscilowicz JM, Delgado AV, Holcomb JB. Independent contributions of hypothermia and acidosis to coagulopathy in swine. J Trauma. 2005;58:1002–9. discussion 9-10.
Martini WZ. Coagulopathy by hypothermia and acidosis: mechanisms of thrombin generation and fibrinogen availability. J Trauma. 2009;67:202–8. discussion 8-9.
Hiippala S. Replacement of massive blood loss. Vox Sang. 1998;74 Suppl 2:399–407.
Ho AM, Karmakar MK, Dion PW. Are we giving enough coagulation factors during major trauma resuscitation? Am J Surg. 2005;190:479–84.
Gando S, Hayakawa M. Pathophysiology of trauma-induced coagulopathy and management of critical bleeding requiring massive transfusion. Semin Thromb Hemost. 2016;42:155–65.
Hess JR, Brohi K, Dutton RP, Hauser CJ, Holcomb JB, Kluger Y, et al. The coagulopathy of trauma: a review of mechanisms. J Trauma. 2008;65:748–54.
Frith D, Davenport R, Brohi K. Acute traumatic coagulopathy. Curr Opin Anaesthesiol. 2012;25:229–34.
Weiler H. Regulation of inflammation by the protein C system. Crit Care Med. 2010;38:S18–25.
Ishii H, Uchiyama H, Kazama M. Soluble thrombomodulin antigen in conditioned medium is increased by damage of endothelial cells. Thromb Haemost. 1991;65:618–23.
Gando S, Wada H, Thachil J, Scientific Standardization Committee on DIC of the International Society on Thrombosis and Haemostasis. Differentiating disseminated intravascular coagulation (DIC) with the fibrinolytic phenotype from coagulopathy of trauma and acute coagulopathy of trauma-shock (COT/ACOTS). J Thromb Haemost. 2013;11:826–35.
Ohlin AK, Larsson K, Hansson M. Soluble thrombomodulin activity and soluble thrombomodulin antigen in plasma. J Thromb Haemost. 2005;3:976–82.
Chapman MP, Moore EE, Moore HB, Gonzalez E, Gamboni F, Chandler JG, et al. Overwhelming tPA release, not PAI-1 degradation, is responsible for hyperfibrinolysis in severely injured trauma patients. J Trauma Acute Care Surg. 2016;80:16–23. discussion -5.
Gando S, Otomo Y. Local hemostasis, immunothrombosis, and systemic disseminated intravascular coagulation in trauma and traumatic shock. Crit Care. 2015;19:72.
Gando S. Hemostasis and thrombosis in trauma patients. Semin Thromb Hemost. 2015;41:26–34.
Gando S, Sawamura A, Hayakawa M. Trauma, shock, and disseminated intravascular coagulation: lessons from the classical literature. Ann Surg. 2011;254:10–9.
Gando S, Wada H, Kim HK, Kurosawa S, Nielsen JD, Thachil J, et al. Comparison of disseminated intravascular coagulation in trauma with coagulopathy of trauma/acute coagulopathy of trauma-shock. J Thromb Haemost. 2012;10:2593–5.
Asakura H. Classifying types of disseminated intravascular coagulation: clinical and animal models. J Intensive Care. 2014;2:20.
Taylor Jr FB, Toh CH, Hoots WK, Wada H, Levi M, Scientific Subcommittee on Disseminated Intravascular Coagulation of the International Society on T, et al. Towards definition, clinical and laboratory criteria, and a scoring system for disseminated intravascular coagulation. Thromb Haemost. 2001;86:1327–30.
Dunbar NM, Chandler WL. Thrombin generation in trauma patients. Transfusion. 2009;49:2652–60.
Hayakawa M, Gando S, Ono Y, Wada T, Yanagida Y, Sawamura A, et al. Noble-Collip Drum trauma induces disseminated intravascular coagulation but not acute coagulopathy of trauma-shock. Shock. 2015;43:261–7.
Park MS, Xue A, Spears GM, Halling TM, Ferrara MJ, Kuntz MM, et al. Thrombin generation and procoagulant microparticle profiles after acute trauma: a prospective cohort study. J Trauma Acute Care Surg. 2015;79:726–31.
Park MS, Owen BA, Ballinger BA, Sarr MG, Schiller HJ, Zietlow SP, et al. Quantification of hypercoagulable state after blunt trauma: microparticle and thrombin generation are increased relative to injury severity, while standard markers are not. Surgery. 2012;151:831–6.
Nekludov M, Mobarrez F, Gryth D, Bellander BM, Wallen H. Formation of microparticles in the injured brain of patients with severe isolated traumatic brain injury. J Neurotrauma. 2014;31:1927–33.
Matijevic N, Wang YW, Wade CE, Holcomb JB, Cotton BA, Schreiber MA, et al. Cellular microparticle and thrombogram phenotypes in the Prospective Observational Multicenter Major Trauma Transfusion (PROMMTT) study: correlation with coagulopathy. Thromb Res. 2014;134:652–8.
Yasui H, Donahue DL, Walsh M, Castellino FJ, Ploplis VA. Early coagulation events induce acute lung injury in a rat model of blunt traumatic brain injury. Am J Physiol Lung Cell Mol Physiol. 2016;311:L74–86.
Tian Y, Salsbery B, Wang M, Yuan H, Yang J, Zhao Z, et al. Brain-derived microparticles induce systemic coagulation in a murine model of traumatic brain injury. Blood. 2015;125:2151–9.
Gando S, Kameue T, Nanzaki S, Hayakawa T, Nakanishi Y. Participation of tissue factor and thrombin in posttraumatic systemic inflammatory syndrome. Crit Care Med. 1997;25:1820–6.
Gando S, Nanzaki S, Kemmotsu O. Coagulofibrinolytic changes after isolated head injury are not different from those in trauma patients without head injury. J Trauma. 1999;46:1070–6. discussion 6-7.
Abrams ST, Zhang N, Manson J, Liu T, Dart C, Baluwa F, et al. Circulating histones are mediators of trauma-associated lung injury. Am J Respir Crit Care Med. 2013;187:160–9.
Johansson PI, Windelov NA, Rasmussen LS, Sorensen AM, Ostrowski SR. Blood levels of histone-complexed DNA fragments are associated with coagulopathy, inflammation and endothelial damage early after trauma. J Emerg Trauma Shock. 2013;6:171–5.
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, et al. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7.
Yamanouchi S, Kudo D, Yamada M, Miyagawa N, Furukawa H, Kushimoto S. Plasma mitochondrial DNA levels in patients with trauma and severe sepsis: time course and the association with clinical status. J Crit Care. 2013;28:1027–31.
Simmons JD, Lee YL, Mulekar S, Kuck JL, Brevard SB, Gonzalez RP, et al. Elevated levels of plasma mitochondrial DNA DAMPs are linked to clinical outcome in severely injured human subjects. Ann Surg. 2013;258:591–6. discussion 6-8.
Giannoudis PV, Mallina R, Harwood P, Perry S, Sante ED, Pape HC. Pattern of release and relationship between HMGB-1 and IL-6 following blunt trauma. Injury. 2010;41:1323–7.
Wang XW, Karki A, Du DY, Zhao XJ, Xiang XY, Lu ZQ. Plasma levels of high mobility group box 1 increase in patients with posttraumatic stress disorder after severe blunt chest trauma: a prospective cohort study. J Surg Res. 2015;193:308–15.
Wang XW, Karki A, Zhao XJ, Xiang XY, Lu ZQ. High plasma levels of high mobility group box 1 is associated with the risk of sepsis in severe blunt chest trauma patients: a prospective cohort study. J Cardiothorac Surg. 2014;9:133.
Cohen MJ, Brohi K, Calfee CS, Rahn P, Chesebro BB, Christiaans SC, et al. Early release of high mobility group box nuclear protein 1 after severe trauma in humans: role of injury severity and tissue hypoperfusion. Crit Care. 2009;13:R174.
Oshiro A, Yanagida Y, Gando S, Henzan N, Takahashi I, Makise H. Hemostasis during the early stages of trauma: comparison with disseminated intravascular coagulation. Crit Care. 2014;18:R61.
Johansson PI, Sorensen AM, Perner A, Welling KL, Wanscher M, Larsen CF, et al. High sCD40L levels early after trauma are associated with enhanced shock, sympathoadrenal activation, tissue and endothelial damage, coagulopathy and mortality. J Thromb Haemost. 2012;10:207–16.
Reinhart K, Bayer O, Brunkhorst F, Meisner M. Markers of endothelial damage in organ dysfunction and sepsis. Crit Care Med. 2002;30:S302–12.
Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma. 2008;64:1211–7. discussion 7.
Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg. 2007;245:812–8.
Cohen MJ, Call M, Nelson M, Calfee CS, Esmon CT, Brohi K, et al. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann Surg. 2012;255:379–85.
Liaw PC, Ferrell G, Esmon CT. A monoclonal antibody against activated protein C allows rapid detection of activated protein C in plasma and reveals a calcium ion dependent epitope involved in factor Va inactivation. J Thromb Haemost. 2003;1:662–70.
Gando S, Tedo I, Kubota M. Posttrauma coagulation and fibrinolysis. Crit Care Med. 1992;20:594–600.
Gando S, Nanzaki S, Sasaki S, Kemmotsu O. Significant correlations between tissue factor and thrombin markers in trauma and septic patients with disseminated intravascular coagulation. Thromb Haemost. 1998;79:1111–5.
Huber D, Cramer EM, Kaufmann JE, Meda P, Masse JM, Kruithof EK, et al. Tissue-type plasminogen activator (t-PA) is stored in Weibel-Palade bodies in human endothelial cells both in vitro and in vivo. Blood. 2002;99:3637–45.
Lowenstein CJ, Morrell CN, Yamakuchi M. Regulation of Weibel-Palade body exocytosis. Trends Cardiovasc Med. 2005;15:302–8.
Kooistra T, Schrauwen Y, Arts J, Emeis JJ. Regulation of endothelial cell t-PA synthesis and release. Int J Hematol. 1994;59:233–55.
Levrat A, Gros A, Rugeri L, Inaba K, Floccard B, Negrier C, et al. Evaluation of rotation thrombelastography for the diagnosis of hyperfibrinolysis in trauma patients. Br J Anaesth. 2008;100:792–7.
Kutcher ME, Cripps MW, McCreery RC, Crane IM, Greenberg MD, Cachola LM, et al. Criteria for empiric treatment of hyperfibrinolysis after trauma. J Trauma Acute Care Surg. 2012;73:87–93.
Theusinger OM, Baulig W, Seifert B, Muller SM, Mariotti S, Spahn DR. Changes in coagulation in standard laboratory tests and ROTEM in trauma patients between on-scene and arrival in the emergency department. Anesth Analg. 2015;120:627–35.
Theusinger OM, Wanner GA, Emmert MY, Billeter A, Eismon J, Seifert B, et al. Hyperfibrinolysis diagnosed by rotational thromboelastometry (ROTEM) is associated with higher mortality in patients with severe trauma. Anesth Analg. 2011;113:1003–12.
Kashuk JL, Moore EE, Sawyer M, Wohlauer M, Pezold M, Barnett C, et al. Primary fibrinolysis is integral in the pathogenesis of the acute coagulopathy of trauma. Ann Surg. 2010;252:434–42. discussion 43-4.
Schochl H, Frietsch T, Pavelka M, Jambor C. Hyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J Trauma. 2009;67:125–31.
Kushimoto S, Shibata Y, Yamamoto Y. Implications of fibrinogenolysis in patients with closed head injury. J Neurotrauma. 2003;20:357–63.
Hayakawa M, Maekawa K, Kushimoto S, Kato H, Sasaki J, Ogura H, et al. High D-dimer levels predict a poor outcome in patients with severe trauma, even with high fibrinogen levels on arrival: a multicenter retrospective study. Shock. 2016;45:308–14.
Hayakawa M, Gando S, Ieko M, Honma Y, Homma T, Yanagida Y, et al. Massive amounts of tissue factor induce fibrinogenolysis without tissue hypoperfusion in rats. Shock. 2013;39:514–9.
Goodnight SH, Kenoyer G, Rapaport SI, Patch MJ, Lee JA, Kurze T. Defibrination after brain-tissue destruction: a serious complication of head injury. N Engl J Med. 1974;290:1043–7.
Saggar V, Mittal RS, Vyas MC. Hemostatic abnormalities in patients with closed head injuries and their role in predicting early mortality. J Neurotrauma. 2009;26:1665–8.
Tian HL, Chen H, Wu BS, Cao HL, Xu T, Hu J, et al. D-dimer as a predictor of progressive hemorrhagic injury in patients with traumatic brain injury: analysis of 194 cases. Neurosurg Rev. 2010;33:359–65. discussion 65-6.
Hijazi N, Abu Fanne R, Abramovitch R, Yarovoi S, Higazi M, Abdeen S, et al. Endogenous plasminogen activators mediate progressive intracerebral hemorrhage after traumatic brain injury in mice. Blood. 2015;125:2558–67.
Risberg B, Medegard A, Heideman M, Gyzander E, Bundsen P, Oden M, et al. Early activation of humoral proteolytic systems in patients with multiple trauma. Crit Care Med. 1986;14:917–25.
Schochl H, Cotton B, Inaba K, Nienaber U, Fischer H, Voelckel W, et al. FIBTEM provides early prediction of massive transfusion in trauma. Crit Care. 2011;15:R265.
Inaba K, Karamanos E, Lustenberger T, Schochl H, Shulman I, Nelson J, et al. Impact of fibrinogen levels on outcomes after acute injury in patients requiring a massive transfusion. J Am Coll Surg. 2013;216:290–7.
Rizoli SB, Scarpelini S, Callum J, Nascimento B, Mann KG, Pinto R, et al. Clotting factor deficiency in early trauma-associated coagulopathy. J Trauma. 2011;71:S427–34.
Floccard B, Rugeri L, Faure A, Saint Denis M, Boyle EM, Peguet O, et al. Early coagulopathy in trauma patients: an on-scene and hospital admission study. Injury. 2012;43:26–32.
Jansen JO, Scarpelini S, Pinto R, Tien HC, Callum J, Rizoli SB. Hypoperfusion in severely injured trauma patients is associated with reduced coagulation factor activity. J Trauma. 2011;71:S435–40.
McLoughlin TM, Fontana JL, Alving B, Mongan PD, Bunger R. Profound normovolemic hemodilution: hemostatic effects in patients and in a porcine model. Anesth Analg. 1996;83:459–65.
Sorensen B, Larsen OH, Rea CJ, Tang M, Foley JH, Fenger-Eriksen C. Fibrinogen as a hemostatic agent. Semin Thromb Hemost. 2012;38:268–73.
Lowe GD, Rumley A, Mackie IJ. Plasma fibrinogen. Ann Clin Biochem. 2004;41:430–40.
Acknowledgements
Not applicable
Funding
No funding to declare.
Availability of data and materials
Not applicable.
Authors’ contributions
MH wrote the manuscript and approved the final manuscript.
Competing interests
The author declares that he/she has no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hayakawa, M. Pathophysiology of trauma-induced coagulopathy: disseminated intravascular coagulation with the fibrinolytic phenotype. j intensive care 5, 14 (2017). https://doi.org/10.1186/s40560-016-0200-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40560-016-0200-1