
Abstract
Traumatic injury is one of the leading causes of death, with uncontrolled hemorrhage from coagulation dysfunction as one of the main potentially preventable causes of the mortality. Hypothermia, acidosis, and resuscitative hemodilution have been considered as the significant contributors to coagulation manifestations following trauma, known as the lethal triad. Over the past decade, clinical observations showed that coagulopathy may be present as early as hospital admission in some severely injured trauma patients. The hemostatic dysfunction is associated with higher blood transfusion requirements, longer hospital stay, and higher mortality. The recognition of this early coagulopathy has initiated tremendous interest and effort in the trauma community to expand our understanding of the underlying pathophysiology and improve clinical treatments. This review discusses the current knowledge of coagulation complications following trauma.
Similar content being viewed by others
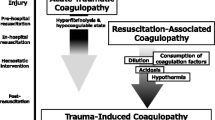


Background
Traumatic injury remains one of the leading causes of death, accounting for about 40% of prehospital death [1, 2]. Uncontrolled hemorrhage from coagulation dysfunction is one of the main potentially preventable causes of the mortality in both civilian and military settings [3–7]. Hypothermia, acidosis, and resuscitative hemodilution have been considered as the significant contributors to coagulation dysfunction after trauma. Over the past decade, clinical observations around the globe have independently shown that coagulopathy may be present as early as hospital admission in some trauma patients. The hemostatic manifestation is associated with increased blood transfusion requirements, longer hospital stay, and higher mortality [7–11]. The recognition of this early coagulopathy leads to the use of new terminology and proposed hypotheses [8, 12, 13]. However, to date, our understanding of the underlying mechanisms remains incomplete. This review summarizes the current knowledge of coagulation complications following trauma.
Coagulation process
Blood coagulation is an important physiological process, including a series of physical, biochemical and cellular responses after various stimuli. The essence of the process is the production of fibrin clots from fibrinogen (factor I), and thrombin plays a central role catalyzing the reaction [14]. Biochemically, blood clotting is initiated via the intrinsic and/or extrinsic pathways. The two pathways converge to form a common pathway to generate thrombin. The intrinsic pathway, or contact activation pathway, consists of the activations of factor VIII, IX, X, XI, XII and Xa complex, resulting in thrombin generation from precursor prothrombin (factor II). The extrinsic pathway is triggered by plasma factor VIIa binding with tissue factor (factor III) released from the injury sites. The factor VIIa/tissue factor complex, once produced, activates additional factor VII, initial thrombin, factor Xa complex, and platelets, resulting in the exponential thrombin burst for rapid clot formation [15]. This classic cascade model provides a biochemical description of the coagulation process and the basis for clinical assessments of coagulation; but it is now considered incomplete because it does not take into consideration the coagulation inhibition in plasma. Clinical standard plasma tests of prothrombin time (PT) and activated partial thromboplastin time (aPTT) reflect the overall enzyme activities involved in the extrinsic and intrinsic pathways, respectively.
The formation of fibrin clots is counterbalanced by its inhibitory and anti-coagulation processes. Circulating antithrombin III inhibits factor Xa and thrombin, with 2000-fold amplified effects by heparin [16]. Tissue factor pathway inhibitor inhibits factor Xa and eliminates the contribution of the extrinsic pathway to clot formation. Activated protein C, the product of the thrombomodulin-thrombin complex, inactivates prothrombinase and the intrinsic pathway [17]. In addition, fibrin clots, once formed, are subject to fibrinolysis by plasmin. Plasmin is generated from inactive protein plasminogen via tissue-type plasminogen activator (tPA) [18]. The activity of tPA can be inhibited by plasminogen activator inhibitors (PAI) [19, 20]. The fibrinolytic system is regulated through the generation of plasmin from the activities of tPA, PAI and an antiplasmin inhibitor. Under any normal physiological state, the blood coagulation status is a dynamic process and is the balance of clot formation, anti-coagulation and fibrinolysis.
Another description of the coagulation process is a cell-based model of hemostasis [21]. This model considers the process as three overlapping phases: initiation, amplification and propagation. All three phases are regulated by the properties of cell surfaces, receptors, and coagulation proteins. This model provides the basis of viscoelastic tests, such as thromboelastography (TEG) and rotational thromboelastometry (ROTEM), to profile the dynamic nature of the clotting process and guide resuscitation practice [22, 23].
Coagulation tests
Early traumatic coagulopathy has been defined by different measurements, including standard plasma tests of PT, aPTT, thrombin time, platelet counts, fibrinogen levels, and blood viscoelastic tests of clotting amplitudes and clot lysis [8, 22, 24–28]. At present,, there is no standard or globally accepted assay for diagnosing early traumatic coagulopathy, although prolonged PT has been used by many investigators to study trauma-induced coagulopathy.
Compared to plasma PT and aPTT, TEG and ROTEM provide more comprehensive description of coagulation status, including measurements of clotting formation time, clotting speed, clot strength, and fibrinolysis. This advantage has made its increased use in the diagnosis of trauma-induced coagulopathy, the prediction of massive transfusion and for guiding the transfusion of blood products [23, 29]. However, TEG and ROTEM have limited sensitivity in reflecting platelet dysfunction and moderate fibrinolysis [23, 30, 31]. A randomized controlled trial is warranted to validate TEG or ROTEM’s role in guiding massive transfusion protocols in trauma patients.
Coagulation complications after trauma
After traumatic injury, coagulation, anti-coagulation and fibrinolysis are disproportionally affected, leading to impaired hemostasis. The alterations have been found to be dynamic and multifactorial. For simplicity, it is helpful to describe the changes in three phases: 1) acute post-trauma phase, which occurs shortly, within hours, after trauma injury; 2) resuscitation phase, which occurs 24–48 h post trauma, when various resuscitation fluids may be used; and 3) later phase, which occurs days after trauma injury.
Acute post-trauma phase
Trauma-related coagulopathy has been considered to be primarily due to blood loss from injury, hemodilution from aggressive resuscitation, and the development of hypothermia and acidosis [32, 33]. During the last decade, clinical studies have shown that prolonged PT and aPTT prothrombin time were observed in some trauma patients at emergency room admission [8–10]. This hemostatic complication is independently associated with increased blood transfusion requirement and higher mortality than those with similar injury but without coagulopathy [8–10]. The recognition of this early coagulopathy prior to fluid resuscitation has initiated tremendous interest and effort to expand our understanding of trauma-related coagulopathy. As the results, new terminology has been created to describe the early developed coagulopathy, such as acute coagulopathy of trauma (ACT), acute traumatic coagulopathy (ATC), trauma induced coagulopathy (TIC), and early coagulopathy of trauma. Hypotheses have also been proposed to attempt to explain the underlying mechanisms.
One hypothesis is consumptive coagulopathy, a phenotypic variation of classic disseminated intravascular coagulation (DIC) [34]. Immediately after trauma, trauma injury exposes tissue factor, which is normally present inside tissues, to the circulation and initiates thrombin generation and clot formation. Platelets are activated through a network of regulated interconnecting cellular signals, including collagen in the sub-endothelial matrix binding to glycoprotein VI, von Willebrand Factor (vWF) and glycoprotein Ib [15]. The activation of platelets amplifies thrombin generation and the clotting process, causing the consumption of the coagulation factors. The most depleted factors are fibrinogen and factor V [35]. In addition, fibrinolysis is activated from the release of tissue plasminogen activator, which converts plasminogen to plasmin, into the circulation. Consequently, hypocoagulation and hyperfibrinolysis are developed in trauma patients.
Another hypothesis considers that activated protein C plays a central role in enhancing anti-coagulation [8, 24, 36, 37]. Based on this hypothesis, following severe trauma injury and hypoperfusion, thrombin is generated and binds thrombomodulin to form activated protein C. Activated protein C exerts its anticoagulant role by inhibiting factor Va and VIIIa and its hyperfibrinolytic role by inhibiting plasminogen activator inhibitor. Thus, activated protein C accounts for the hypocoagulation and hyperfibrinolysis characteristics observed in some trauma patients.
The third hypothesis focuses on trauma-induced neuro-hormonal and endothelial responses [38, 39]. Tissue injury from trauma induces sympathoadrenal responses and catecholamine release. Circulating catecholamine damages the endothelial glycocalyx and converts the endothelial function from antithrombotic to prothrombotic for local hemostasis. There is also a counterbalance mechanism of the anticoagulation and fibrinolytic responses in the blood to prevent this local response from extending beyond injury sites. However, this counterbalance mechanism is amplified after severe trauma injury, resulting in hypocoagulation and hyperfibrinolysis observed in some trauma patients.
To date, debates and controversies remain in these hypotheses [7, 13, 40]. Nevertheless, traumatic injury and shock related hypoperfusion have been widely accepted as the two important initiators of the early coagulopathy following trauma [24, 32, 41]. The severity of the trauma and the duration of shock appear to be positively related to the severity of coagulation dysfunction.
Resuscitation phase
The resuscitation phase covers the first few days (i.e., 24–48 h) after trauma injury. During this phase, metabolic acidosis and hypothermia may develop together with hemodilution from resuscitation fluids used to improve hemodynamics. These factors may further impair and amplify the already existing coagulopathy from the trauma injury [42–44].
Metabolic acidosis
Clinical acidosis is commonly observed in trauma patients due to hypo-perfusion from massive blood loss. Impaired clotting enzyme activities have demonstrated the effects of acidosis on coagulation. Acidotic trauma patients showed prolonged PT and aPTT and decreased coagulation factor levels. Quantitatively, when pH was reduced from 7.4 to 7.0 in vitro, the activities of factor VIIa and factor VIIa/TF on phospholipid vessels decreased by more than 90 and 60%, respectively [45]. When pH was reduced from 7.4 to 7.1 in pigs, thrombin generation decreased to 47% of the control values [46]. In thrombin generation kinetics, acidosis moderately inhibited the initiation phase of thrombin generation, but persistently and dramatically inhibited the propagation phase [46]. These data showed that acidosis more severely inhibited the activation of factor V, VIII, IX, X and the formation of factor Xase and the prothrombinase complex in the propagation phase, compared to the activation of factor VIIa/tissue factor complex in the initiation phase [46].
The effects of acidosis on fibrinogen availability and metabolism were investigated in a swine model using stable isotope infusion and subsequent gas chromatograph mass spectrometry analysis [47]. Acidosis of pH 7.1 caused a 1.8-fold increase in fibrinogen breakdown rate compared to the control values but did not affect fibrinogen synthesis rate [47]. The accelerated consumption and unchanged production suggest a deficit of fibrinogen availability and support the supplementation of exogenous fibrinogen to improve hemostasis.
To restore coagulation function impaired by acidosis, bicarbonate solution was used to neutralize pH in a swine model after the induction of acidosis [48]. Acidosis of pH 7.1 depleted fibrinogen levels and platelets counts and impaired thrombin generation, clotting speed and clot strength [48]. The infusion of bicarbonate solution immediately corrected pH to 7.4. However, bicarbonate pH neutralization did not immediately recover the depleted substrate levels or coagulation dysfunction. Similar findings were observed when a different pH neutralizer, tris-hydroxymethyl-aminomethane, was used [49]. These findings demonstrated that acidosis-induced coagulopathy, once developed, cannot be corrected immediately by pH neutralization. Thus, the clinical focus of acidosis-induced coagulopathy should be on prevention instead of correction.
Hypothermia
The effects of hypothermia on the coagulation process have been estimated by cold-induced changes in standard clinical tests. Prolonged PT and aPTT have been shown in hypothermic patients and experimental animals, as well as plasma cooled in vitro [50–53]. The in vivo effects of temperature on thrombin generation kinetics were investigated in a swine model [46]. Hypothermia of 32 °C primarily inhibited the initiation phase of thrombin generation, involving the formation of factor VII/tissue factor complex [46]. The thrombin generation propagation phase, however, was not affected. Thus, compared to those observed in acidosis, hypothermia impairs the thrombin generation kinetics differently from acidosis.
Temperature effects on fibrinogen metabolism and availability were investigated in pigs with stable isotope infusion [54]. Hypothermia of 32 °C decreased fibrinogen synthesis rate by 50% of the control values, but fibrinogen breakdown rate remained unchanged [54]. Compared to the accelerated breakdown and unchanged synthesis by acidosis, hypothermia affects fibrinogen metabolism via different mechanisms. However, the decreased production and unchanged consumption by hypothermia indicate a similar outcome as acidosis: a potential deficit in fibrinogen availability.
Resuscitation
Following blood loss, fluid resuscitation is a routine clinical practice to restore tissue perfusion and hemodynamics. A variety of resuscitation fluids have been used around the world, with selections depending on availability, cost, and local clinical experience. Crystalloids, such as normal saline and lactated Ringer’s (LR) solution, are inexpensive and have been widely used for resuscitation [55–57]. Normal saline is a NaCl salt solution with an average pH of 5.0. LR has an average pH of 6.5 and has similar electrolytes to plasma, thus is considered as a more physiologically compatible fluid. In comparative trials of LR and normal saline in patients undergoing kidney transplant or aortic aneurysm repair, similar clinical outcomes of ICU stay, ventilation time, and complication incidence were observed in patients resuscitated with LR or normal saline, although patients with normal saline were more acidotic. In a rat model with moderate hemorrhage (36% of estimated total blood volume) and simultaneous resuscitation, normal saline and LR had equivalent survival rates [58]. However, LR resuscitation resulted in better survival after a massive hemorrhage (218% of estimated total blood volume) [58]. In a large animal model with femur fracture and 60% hemorrhage, normal saline and LR have similar effects on hemodynamics, oxygen metabolism and coagulation [59]. Normal saline required a larger resuscitation volume and was associated with poor acid base status and elevated serum potassium [59].
Colloids are highly effective at increasing the intravascular volume with a small volume increase in the interstitial space, compared with crystalloids. This volume-expanding advantage is logistically important in pre-hospital circumstances and in far forward battlefield conditions. Different colloids, such as albumin, gelatin, and hydrozyethyl starch, have been used clinically [60–63]. Although positive clinical outcomes have been reported in some clinical trials and animal studies, colloid resuscitation has been associated with a reduction in coagulation factors, platelet dysfunction and hemorrhagic complications [64–66]. In a swine model with traumatic hemorrhage, Hextend resuscitation caused severe reductions in coagulation factors, platelet counts and fibrinogen levels and impaired coagulation based on TEG. Those deteriorations persisted for the entire 6-h experimental duration,, whereas coagulation was restored 3 h after LR resuscitation [59].
With the emphasis on limiting crystalloids and increasing blood products, damage control resuscitation has been increasingly recognized and implemented in trauma care over the past decade [67–69]. Blood products, such as fresh frozen plasma (FFP), packed red blood cells (PRBC) and platelets, have been used for hemostatic resuscitation and hemodynamic resuscitation. As a proactive approach in damage control resuscitation, massive transfusion protocols quickly provide large amounts of blood products to critically injured and bleeding patients [70]. The selection and the order of the infusion of blood products in bleeding patients vary at different trauma centers [71, 72]. In both military and civilian trauma reports, higher ratios of plasma and platelets to PRBC appear to be more beneficial with improved survival [73, 74]. However, the use of blood components is also associated with increased risks in infection and organ failure [75–77]. The optimum ratios and doses of those blood products are still debatable.
Pharmaceutical hemostatic agents, such as fibrinogen concentrate, have been used as resuscitation to replenish fibrinogen levels. Among coagulation factors depleted after traumatic injury, fibrinogen is the first to drop to a critical level [47, 54, 78]. These findings support the notion of supplementing exogenous fibrinogen to restore coagulation function. The clinical use of fibrinogen concentrate has been shown in surgical patients to be efficacious, with improved clotting function and reduced transfusion requirements [79–83]. Large prospective clinical trials are on-going to investigate the efficacy of fibrinogen concentrate pre-hospital and in-hospital use in trauma patients.
Late post-trauma phase
During the late post-trauma phase, the systemic levels of cytokines and hormones increase, leading to endothelial cell activation. The activated endothelial cells, circulating cytokines and thrombin, lead to a slow transition of the endothelial cell phenotype from antithrombiotic to prothrombotic. The endothelial cell activation also down-regulates thrombomodulin and fibrinolysis. In addition, the fibrinogen levels increase several folds due to acute phase responses. Overall, the coagulation process at this phase becomes the prothrombotic state, predisposing patients to venous thromboembolism, which leads to patients requiring heparin or a newer anticoagulant drug.
Coagulation complications in sepsis
Coagulopathy in sepsis appears to be similar to the prothrombotic state observed in the late phase of trauma, although it is much less studied compared to trauma. During sepsis, the coagulation cascade is activated by inflammatory cytokine release and tissue factor [84–86]. Although the primary source of tissue factor remains unclear, it plays a key role in the activation of the coagulation cascade, via the binding of factor VII and the production of factor Xa for thrombin generation [86]. Inflammation also releases platelet activation factor to activate platelets, providing a surface for thrombin generation. In addition, pro-inflammatory cytokines are upregulated and play an important role in the suppression of anticoagulation. The enhanced prothrombic state and inhibited anticoagulation contribute to hypercoagulopathy and the development of DIC in sepsis [87]. As the severity of sepsis progresses, the dysfunctional coagulation leads to microvascular thrombosis and multiple organ dysfunction syndrome [85, 87].
Widespread intravascular activation of the coagulation system is the hallmark of DIC from various pathophysiological insults, such as sepsis. There are some similarities between DIC and early traumatic coagulopathy, including depleted coagulation factors and increased fibrinolysis [27, 34]. However, histological examination did not show disseminated clot formation in trauma patients [88]. The underlying mechanisms contributing to the development of sepsis DIC and trauma-induced coagulopathy remain unclear.
Conclusion
Coagulation complications after trauma have been considered to be attributed to hypothermia, acidosis and hemodilution from blood loss and resuscitation. Clinical findings over the last decade have expanded our knowledge of this topic to shortly after trauma injury. Hemostatic manifestations may be present at hospital admission in some severely injured trauma patients, with mortality 3 to 4 times higher than those without coagulation complications. This recognition has led to the use of new terminology and the generation of some hypotheses in the trauma community. However, the underlying mechanisms related to the development of coagulation complications after trauma remain unclear. Continuing research effort and large clinical trials are warranted to improve our understanding and to facilitate the search of effective treatments for coagulation complications after trauma.
Abbreviations
- ACT:
-
Acute coagulopathy of trauma
- aPTT:
-
Activated partial thromboplastin time
- ATC:
-
Acute traumatic coagulopathy
- DIC:
-
Disseminated intravascular coagulation
- FFP:
-
Fresh frozen plasma
- LR:
-
Lactated Ringer’s
- PAI:
-
Plasminogen activator inhibitors
- PRBC:
-
Packed red blood cells
- PT:
-
Prothrombin time
- ROTEM:
-
Rotational thromboelastometry
- TEG:
-
Thromboelastography
- TIC:
-
Trauma induced coagulopathy
- tPA:
-
Tissue-type plasminogen activator
- vWF:
-
von Willebrand Factor.
References
Krug EG, Sharma GK, Lozano R. The global burden of injuries. Am J Public Health. 2000;90(4):523–6.
Kauvar DS, Lefering R, Wade CE. Impact of hemorrhage on trauma outcome: an overview of epidemiology, clinical presentations, and therapeutic considerations. J Trauma. 2006;60(6 Suppl):S3–S11.
Eastridge BJ, Mabry RL, Seguin P, Cantrell J, Tops T, Uribe P, et al. Death on the battlefield (2001–2011): implications for the future of combat casualty care. J Trauma Acute Care Surg. 2012;73(6 Suppl 5):S431–7.
Sauaia A, Moore FA, Moore EE, Moser KS, Brennan R, Read RA, et al. Epidemiology of trauma deaths: a reassessment. J Trauma. 1995;38(2):185–93.
Engels PT, Rezende-Neto JB, Al Mahroos M, Scarpelini S, Rizoli SB, Tien HC. The natural history of trauma-related coagulopathy: implications for treatment. J Trauma. 2011;71(5 Suppl 1):S448–55.
Heckbert SR, Vedder NB, Hoffman W, Winn RK, Hudson LD, Jurkovich GJ, et al. Outcome after hemorrhagic shock in trauma patients. J Trauma. 1998;45(3):545–9.
Gonzalez E, Moore EE, Moore HB, Chapman MP, Silliman CC, Banerjee A. Trauma-induced coagulopathy: an institution’s 35 year perspective on practice and research. Scand J Surg. 2014;103(2):89–103.
Brohi K, Singh J, Heron M, Coats T. Acute traumatic coagulopathy. J Trauma. 2003;54(6):1127–30.
MacLeod JB, Lynn M, McKenney MG, Cohn SM, Murtha M. Early coagulopathy predicts mortality in trauma. J Trauma. 2003;55(1):39–44.
Maegele M, Lefering R, Yucel N, Tjardes T, Rixen D, Paffrath T, et al. Early coagulopathy in multiple injury: an analysis from the German Trauma Registry on 8724 patients. Injury. 2007;38(3):298–304.
Park MS, Martini WZ, Dubick MA, Salinas J, Butenas S, Kheirabadi BS, et al. Thromboelastography as a better indicator of hypercoagulable state after injury than prothrombin time or activated partial thromboplastin time. J Trauma. 2009;67(2):266–75.
Armand R, Hess JR. Treating coagulopathy in trauma patients. Transfus Med Rev. 2003;17(3):223–31.
Palmer L, Martin L. Traumatic coagulopathy--part 1: Pathophysiology and diagnosis. J Vet Emerg Cri care. 2014;24(1):63–74.
Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost. 2003;1(7):1504–14.
Loscalzo J, Schafer AI. Thrombosis and Hemorrhage. Philadelphia: Lippincott Williams & Wilkins; 2003.
Lindahl U, Backstrom G, Thunberg L, Leder IG. Evidence for a 3-O-sulfated D-glucosamine residue in the antithrombin-binding sequence of heparin. Proc Natl Acad Sci U S A. 1980;77(11):6551–5.
Lu D, Kalafatis M, Mann KG, Long GL. Comparison of activated protein C/protein S-mediated inactivation of human factor VIII and factor V. Blood. 1996;87(11):4708–17.
Robbins KC, Summaria L, Hsieh B, Shah RJ. The peptide chains of human plasmin. Mechanism of activation of human plasminogen to plasmin. J Biol Chem. 1967;242(10):2333–42.
Sprengers ED, Kluft C. Plasminogen activator inhibitors. Blood. 1987;69(2):381–7.
Nicoloso G, Hauert J, Kruithof EK, Van Melle G, Bachmann F. Fibrinolysis in normal subjects--comparison between plasminogen activator inhibitor and other components of the fibrinolytic system. Thromb Haemost. 1988;59(2):299–303.
Hoffman M, Monroe 3rd DM. A cell-based model of hemostasis. Thromb Haemost. 2001;85(6):958–65.
Schochl H, Nienaber U, Hofer G, Voelckel W, Jambor C, Scharbert G, et al. Goal-directed coagulation management of major trauma patients using thromboelastometry (ROTEM)-guided administration of fibrinogen concentrate and prothrombin complex concentrate. Crit Care (London, England). 2010;14(2):R55.
Rugeri L, Levrat A, David JS, Delecroix E, Floccard B, Gros A, et al. Diagnosis of early coagulation abnormalities in trauma patients by rotation thrombelastography. J Thromb Haemost. 2007;5(2):289–95.
Davenport R, Manson J, De’Ath H, Platton S, Coates A, Allard S, et al. Functional definition and characterization of acute traumatic coagulopathy. Crit Care Med. 2011;39(12):2652–8.
Yuan S, Ferrell C, Chandler WL. Comparing the prothrombin time INR versus the APTT to evaluate the coagulopathy of acute trauma. Thromb Res. 2007;120(1):29–37.
Schochl H, Voelckel W, Maegele M, Solomon C. Trauma-associated hyperfibrinolysis. Hamostaseologie. 2012;32(1):22–7.
Sawamura A, Hayakawa M, Gando S, Kubota N, Sugano M, Wada T, et al. Disseminated intravascular coagulation with a fibrinolytic phenotype at an early phase of trauma predicts mortality. Thromb Res. 2009;124(5):608–13.
Carroll RC, Craft RM, Langdon RJ, Clanton CR, Snider CC, Wellons DD, et al. Early evaluation of acute traumatic coagulopathy by thrombelastography. Transl Res. 2009;154(1):34–9.
Schochl H, Frietsch T, Pavelka M, Jambor C. Hyperfibrinolysis after major trauma: differential diagnosis of lysis patterns and prognostic value of thrombelastometry. J Trauma. 2009;67(1):125–31.
Raza I, Davenport R, Rourke C, Platton S, Manson J, Spoors C, et al. The incidence and magnitude of fibrinolytic activation in trauma patients. J Thromb Haemost. 2013;11(2):307–14.
Veigas PV, Callum J, Rizoli S, Nascimento B, da Luz LT. A systematic review on the rotational thrombelastometry (ROTEM(R)) values for the diagnosis of coagulopathy, prediction and guidance of blood transfusion and prediction of mortality in trauma patients. Scand J Trauma Resusc Emerg Med. 2016;24(1):114.
Frith D, Goslings JC, Gaarder C, Maegele M, Cohen MJ, Allard S, et al. Definition and drivers of acute traumatic coagulopathy: clinical and experimental investigations. J Thromb Haemost. 2010;8(9):1919–25.
Moore EE, Thomas G. Orr Memorial Lecture. Staged laparotomy for the hypothermia, acidosis, and coagulopathy syndrome. Am J Surg. 1996;172(5):405–10.
Gando S, Sawamura A, Hayakawa M. Trauma, shock, and disseminated intravascular coagulation: lessons from the classical literature. Ann Surg. 2011;254(1):10–9.
Shaz BH, Winkler AM, James AB, Hillyer CD, MacLeod JB. Pathophysiology of early trauma-induced coagulopathy: emerging evidence for hemodilution and coagulation factor depletion. J Trauma. 2011;70(6):1401–7.
Brohi K, Cohen MJ, Ganter MT, Matthay MA, Mackersie RC, Pittet JF. Acute traumatic coagulopathy: initiated by hypoperfusion: modulated through the protein C pathway? Ann Surg. 2007;245(5):812–8.
Bouillon B, Brohi K, Hess JR, Holcomb JB, Parr MJ, Hoyt DB. Educational initiative on critical bleeding in trauma: Chicago, July 11–13, 2008. J Trauma. 2010;68(1):225–30.
Johansson PI, Ostrowski SR. Acute coagulopathy of trauma: balancing progressive catecholamine induced endothelial activation and damage by fluid phase anticoagulation. Med Hypotheses. 2010;75(6):564–7.
Johansson PI, Sorensen AM, Perner A, Welling KL, Wanscher M, Larsen CF, et al. Disseminated intravascular coagulation or acute coagulopathy of trauma shock early after trauma? An observational study. Crit Care (London, England). 2011;15(6):R272.
Cap A, Hunt BJ. The pathogenesis of traumatic coagulopathy. Anaesthesia. 2015;70 Suppl 1:96–101. e132–104.
Brohi K, Cohen MJ, Ganter MT, Schultz MJ, Levi M, Mackersie RC, et al. Acute coagulopathy of trauma: hypoperfusion induces systemic anticoagulation and hyperfibrinolysis. J Trauma. 2008;64(5):1211–7. discussion 1217.
Martini WZ. Coagulopathy by hypothermia and acidosis: mechanisms of thrombin generation and fibrinogen availability. J Trauma. 2009;67(1):202–8. discussion 208–209.
Martini WZ. Fibrinogen metabolic responses to trauma. Scand J Trauma Resusc Emerg Med. 2009;17(1):2.
Ganter MT, Pittet JF. New insights into acute coagulopathy in trauma patients. Best Pract Res Clin Anaesthesiol. 2010;24(1):15–25.
Meng ZH, Wolberg AS, Monroe 3rd DM, Hoffman M. The effect of temperature and pH on the activity of factor VIIa: implications for the efficacy of high-dose factor VIIa in hypothermic and acidotic patients. J Trauma. 2003;55(5):886–91.
Martini WZ, Pusateri AE, Uscilowicz JM, Delgado AV, Holcomb JB. Independent contributions of hypothermia and acidosis to coagulopathy in swine. J Trauma. 2005;58(5):1002–9. discussion 1009–1010.
Martini WZ, Holcomb JB. Acidosis and coagulopathy: the differential effects on fibrinogen synthesis and breakdown in pigs. Ann Surg. 2007;246(5):831–5.
Martini WZ, Dubick MA, Pusateri AE, Park MS, Ryan KL, Holcomb JB. Does bicarbonate correct coagulation function impaired by acidosis in swine? J Trauma. 2006;61(1):99–106.
Martini WZ, Dubick MA, Wade CE, Holcomb JB. Evaluation of tris-hydroxymethylaminomethane on reversing coagulation abnormalities caused by acidosis in pigs. Crit Care Med. 2007;35(6):1568–74.
Steinemann S, Shackford SR, Davis JW. Implications of admission hypothermia in trauma patients. J Trauma. 1990;30(2):200–2.
Staab DB, Sorensen VJ, Fath JJ, Raman SB, Horst HM, Obeid FN. Coagulation defects resulting from ambient temperature-induced hypothermia. J Trauma. 1994;36(5):634–8.
Watts DD, Trask A, Soeken K, Perdue P, Dols S, Kaufmann C. Hypothermic coagulopathy in trauma: effect of varying levels of hypothermia on enzyme speed, platelet function, and fibrinolytic activity. J Trauma. 1998;44(5):846–54.
Rohrer MJ, Natale AM. Effect of hypothermia on the coagulation cascade. Crit Care Med. 1992;20(10):1402–5.
Martini WZ. The effects of hypothermia on fibrinogen metabolism and coagulation function in swine. Metabolism. 2007;56(2):214–21.
Mullins RJ, editor. Management of Shock. Philadelphia: Appleton Lange; 1996.
Maier RV. Shock. In: Greenfield LJ, Mulholland MW, Oldham KT, Zelenock GB, Lillemoe KD, editors. Surgery: Scientific Principles and Practice. 2nd ed. Philadelphia: Lippincott-Raven; 1997. p. 182–215.
Spoerke N, Michalek J, Schreiber M, Brasel KJ, Vercruysse G, MacLeod J, et al. Crystalloid resuscitation improves survival in trauma patients receiving low ratios of fresh frozen plasma to packed red blood cells. J Trauma. 2011;71(2 Suppl 3):S380–3.
Healey MA, Davis RE, Liu FC, Loomis WH, Hoyt DB. Lactated ringer’s is superior to normal saline in a model of massive hemorrhage and resuscitation. J Trauma. 1998;45(5):894–9.
Martini WZ, Dubick MA, Blackbourne LH. Comparisons of lactated Ringer’s and Hextend resuscitation on hemodynamics and coagulation following femur injury and severe hemorrhage in pigs. J Trauma Acute Care Surg. 2013;74(3):732–40.
Munsch CM, MacIntyre E, Machin SJ, Mackie IJ, Treasure T. Hydroxyethyl starch: an alternative to plasma for postoperative volume expansion after cardiac surgery. Br J Surg. 1988;75(7):675–8.
Moggio RA, Rha CC, Somberg ED, Praeger PI, Pooley RW, Reed GE. Hemodynamic comparison of albumin and hydroxyethyl starch in postoperative cardiac surgery patients. Crit Care Med. 1983;11(12):943–5.
Choi SJ, Ahn HJ, Chung SS, Kim MH, Choi DH, Lee SM, et al. Hemostatic and electrolyte effects of hydroxyethyl starches in patients undergoing posterior lumbar interbody fusion using pedicle screws and cages. Spine (Phila Pa 1976). 2010;35(7):829–34.
Gandhi SD, Weiskopf RB, Jungheinrich C, Koorn R, Miller D, Shangraw RE, et al. Volume replacement therapy during major orthopedic surgery using Voluven (hydroxyethyl starch 130/0.4) or hetastarch. Anesthesiology. 2007;106(6):1120–7.
Stump DC, Strauss RG, Henriksen RA, Petersen RE, Saunders R. Effects of hydroxyethyl starch on blood coagulation, particularly factor VIII. Transfusion. 1985;25(4):349–54.
Franz A, Braunlich P, Gamsjager T, Felfernig M, Gustorff B, Kozek-Langenecker SA. The effects of hydroxyethyl starches of varying molecular weights on platelet function. Anesth Analg. 2001;92(6):1402–7.
Moskowitz DM, Shander A, Javidroozi M, Klein JJ, Perelman SI, Nemeth J, et al. Postoperative blood loss and transfusion associated with use of Hextend in cardiac surgery patients at a blood conservation center. Transfusion. 2008;48(4):768–75.
Duchesne JC, Kimonis K, Marr AB, Rennie KV, Wahl G, Wells JE, et al. Damage control resuscitation in combination with damage control laparotomy: a survival advantage. J Trauma. 2010;69(1):46–52.
Beekley AC. Damage control resuscitation: a sensible approach to the exsanguinating surgical patient. Crit Care Med. 2008;36(7 Suppl):S267–74.
Nunez TC, Cotton BA. Transfusion therapy in hemorrhagic shock. Curr Opin Crit Care. 2009;15(6):536–41.
Malone DL, Hess JR, Fingerhut A. Massive transfusion practices around the globe and a suggestion for a common massive transfusion protocol. J Trauma. 2006;60(6 Suppl):S91–6.
Cotton BA, Gunter OL, Isbell J, Au BK, Robertson AM, Morris JA, et al. Damage control hematology: the impact of a trauma exsanguination protocol on survival and blood product utilization. J Trauma. 2008;64(5):1177–82.
Kashuk JL, Moore EE, Johnson JL, Haenel J, Wilson M, Moore JB, et al. Postinjury life threatening coagulopathy: is 1:1 fresh frozen plasma:packed red blood cells the answer? J Trauma. 2008;65(2):261–70.
Holcomb JB, Fox EE, Wade CE. The PRospective Observational Multicenter Major Trauma Transfusion (PROMMTT) study. J Trauma Acute Care Surg. 2013;75(1 Suppl 1):S1–2.
Holcomb JB, Tilley BC, Baraniuk S, Fox EE, Wade CE, Podbielski JM, et al. Transfusion of plasma, platelets, and red blood cells in a 1:1:1 vs a 1:1:2 ratio and mortality in patients with severe trauma: the PROPPR randomized clinical trial. JAMA. 2015;313(5):471–82.
Sarani B, Dunkman WJ, Dean L, Sonnad S, Rohrbach JI, Gracias VH. Transfusion of fresh frozen plasma in critically ill surgical patients is associated with an increased risk of infection. Crit Care Med. 2008;36(4):1114–8.
Dunne JR, Riddle MS, Danko J, Hayden R, Petersen K. Blood transfusion is associated with infection and increased resource utilization in combat casualties. Am Surg. 2006;72(7):619–25.
Vamvakas EC. Platelet transfusion and postoperative infection in cardiac surgery. Transfusion. 2007;47(2):352–4. author reply 354–6.
Hiippala ST, Myllyla GJ, Vahtera EM. Hemostatic factors and replacement of major blood loss with plasma-poor red cell concentrates. Anesth Analg. 1995;81(2):360–5.
Fenger-Eriksen C, Jensen TM, Kristensen BS, Jensen KM, Tonnesen E, Ingerslev J, et al. Fibrinogen substitution improves whole blood clot firmness after dilution with hydroxyethyl starch in bleeding patients undergoing radical cystectomy: a randomized, placebo-controlled clinical trial. J Thromb Haemost. 2009;7(5):795–802.
Yamamoto K, Usui A, Takamatsu J. Fibrinogen concentrate administration attributes to significant reductions of blood loss and transfusion requirements in thoracic aneurysm repair. J Cardiothorac Surg. 2014;9(1):90.
Karlsson M, Ternstrom L, Hyllner M, Baghaei F, Flinck A, Skrtic S, et al. Prophylactic fibrinogen infusion reduces bleeding after coronary artery bypass surgery. A prospective randomised pilot study. Thromb Haemost. 2009;102(1):137–44.
Sadeghi M, Atefyekta R, Azimaraghi O, Marashi SM, Aghajani Y, Ghadimi F, et al. A randomized, double blind trial of prophylactic fibrinogen to reduce bleeding in cardiac surgery. Brazil J Anesth. 2014;64(4):253–7.
Innerhofer P, Westermann I, Tauber H, Breitkopf R, Fries D, Kastenberger T, et al. The exclusive use of coagulation factor concentrates enables reversal of coagulopathy and decreases transfusion rates in patients with major blunt trauma. Injury. 2013;44(2):209–16.
Cavaillon JM, Adib-Conquy M, Fitting C, Adrie C, Payen D. Cytokine cascade in sepsis. Scand J Iinfect Dis. 2003;35(9):535–44.
O’Brien M. The reciprocal relationship between inflammation and coagulation. Top Companion Anim Med. 2012;27(2):46–52.
Tsao CM, Ho ST, Wu CC. Coagulation abnormalities in sepsis. Acta Anaesthesiol Taiwan. 2015;53(1):16–22.
Semeraro N, Ammollo CT, Semeraro F, Colucci M. Sepsis-associated disseminated intravascular coagulation and thromboembolic disease. Mediterr J Hematol Infect Dis. 2010;2(3):e2010024.
Rizoli S, Nascimento Jr B, Key N, Tien HC, Muraca S, Pinto R, et al. Disseminated intravascular coagulopathy in the first 24 h after trauma: the association between ISTH score and anatomopathologic evidence. J Trauma. 2011;71(5 Suppl 1):S441–7.
Acknowledgements
The opinions or assertions contained herein are the private views of the author and are not to be construed as official or as reflecting the views of the U.S. Department of the Army or the U.S. Department of Defense
Funding
There was no foundation or external funding for this article.
Availability of data and material
Not applicable.
Authors’ contributions
The author drafted the manuscript.
Competing interests
The author declares no conflict of interest according to the guidelines of the International Committee of Medical Journal Editors.
Consent for publication
All of our studies mentioned in this review were approved by the Institutional Animal Care and Use Committee of the US Army Institute of Surgical Research and were conducted in compliance with the Animal Welfare Act and the Animal Welfare Regulations in accordance with the principles of the Guide for the Care and Use of Laboratory Animals.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Martini, W.Z. Coagulation complications following trauma. Military Med Res 3, 35 (2016). https://doi.org/10.1186/s40779-016-0105-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40779-016-0105-2