
Abstract
Background
Buxus hyrcana (boxwood) is an endangered species in the Hyrcanian forests in the north of Iran. This tree is threatened by habitat loss but faces additional threats from the introduced disease the boxwood blight (caused by the fungus Calonectria pseudonaviculata syn. Cylindrocladium pseudonaviculatum, Cy. buxicola) and the potential effects of climate change. As wide range of genetic polymorphism is necessary to ensure successful adaptation to rapid climatic changes.
Methods
Genetic diversity and differentiation between 15 healthy and 15 infected trees of each of two populations were studied using RAPD and ISSR molecular markers.
Results
High-band polymorphism was found in pooled samples of B. hyrcana using both ISSR (58%) and RAPD (73%) markers. The ISSR data and the combined data set classified the trees into two groups. However, data from RAPD clustered the trees into three groups. These results indicate different degrees of genetic variation in the sequences of the tested B. hyrcana genomes targeted by the two marker types used. Genetic variation was found to be relatively high, with most of the diversity occurring within populations. Analyses of healthy versus infected pooled samples based on both marker types indicated that genetic diversity parameters were mostly higher in healthy trees.
Conclusions
Boxwood blight has had a major effect on B. hyrcana, killing individual stems quickly especially in dense populations and reducing population size (as observed in all populations). Considerable within-population diversity, and higher genetic variability in healthy trees than infected ones, suggested conservation efforts should focus on survivor trees in each population and consider the establishment of tree reservations. Propagation of plants from seeds is preferred, since it would include the widest range of genetic variation.
Similar content being viewed by others


Background
Buxus hyrcana is an evergreen shrub or small tree growing up to 1 to 12 m tall. It usually occurs as part of the understorey in the Hyrcanian forests of northern Iran. In the summer 2012, a boxwood blight disease was reported in the forests there (Mirabolfathy et al. 2013). To date, more than 70% of B. hyrcana trees in Hyrcanian forests have been infected by boxwood blight (Fig. 1). Boxwood blight, caused by Calonectria pseudonaviculata, was reported in England in the mid-1990s and then in New Zealand in 2002. Since the first reports, this disease has spread into European countries such as Austria, Belgium, Croatia, Czech Republic, Denmark, France, Georgia, Germany, Ireland, Italy, the Netherlands, Norway, Slovenia, Spain, Sweden, Switzerland and Turkey (Henricot et al. 2000; Brand 2005; Crepel and Inghelbrecht 2003; Saracchi et al. 2008; Pintos Varela et al. 2009; Cech et al. 2010; Gorgiladze et al. 2011; Akilli et al. 2012). Iran is the only Asian country that has reported this disease.

General view of B. hyrcana in Hyrcanian forests that have been infected by boxwood blight (caused by Calonectria pseudonaviculata, syn. Cylindrocladium pseudonaviculatum, Cy. Buxicola); Tuscatoc (right) and Escolac (left)
Devastating pest and disease epidemics have been reported in many parts of the world over the last 120 years, affecting trees of great economic and/or ecological importance (Boyd et al. 2013). American chestnut (Castanea dentata (Marshall) Borkh.) was decimated by a blight disease, caused by Cryphonectria parasitica Murr. Barr., and the species became effectively extinct from its native range in a period of 40 years (Choi and Nuss 1992). Dutch elm disease, caused by the fungus Ophiostoma ulmi, Buisman Nannt., and later by Ophiostoma novo-ulmi Brasier spread through native populations of elm (Ulmus spp.) trees in countries such as America, New Zealand and Europe causing widespread tree death (Comeau et al. 2015). Black sigatoka is a fungal (Mycosphaerella fijiensis) disease that affects the production and export of banana and plantain in various countries, with Grenada suffering complete loss of its plantations (Marin et al. 2003). Anthracnose fungi (usually Colletotrichum or Gloeosporium species) are a threat for many hardwood tree species (Berry 1985). Fusiform rust is a fungal (Cronartium quercuum f. sp. fusiforme) disease that affects the southern pines (Pinus spp.), leading to annual losses of millions of dollars for timber growers. Huge losses have occurred across Europe following ash (Fraxinus spp.) dieback caused by Hymenoscyphus fraxineus (Harper et al. 2016). These and many other fungal pathogens have serious effects on the future of tree species economically and ecologically.
Buxus hyrcana has been examined in a few biochemical (Ata et al. 2010) and ecological studies (Asadi et al. 2011; Asadi et al. 2012; Soleymanipoor and Esmailzadeh 2015; Kaviani and Negahdar 2016; Hosseinzadeh and Esmailzadeh 2017). Reports indicated an ecological range of box trees from sea level up to 1700-m elevation in mountain forests of north Iran (Soleymanipoor and Esmailzadeh 2015; Hosseinzadeh and Esmailzadeh 2017). However, until recently, the genetics of B. hyrcana had been little studied. Development of molecular methods has created new opportunities for Buxus research. Gandehari et al. (2013a, b) detected desirable genetic variation using random amplified polymorphic DNA (RAPD) and inter-simple sequence repeat (ISSR) markers in B. hyrcana populations. An amplified fragment length polymorphism (AFLP) marker was used to specify and distinguish the European and Asian Buxus taxa (Van Laere et al. 2011). Also, internal transcribed spacer regions and plastid ndhF sequences were used to describe the phylogenetic relationships (Von Balthazar et al. 2000). Thammina et al. (2014, 2016) studied the advantages of genic SSR markers and ploidy analysis for specifying the diversity (and, in some cases, for identifying accessions) of Buxus in the National Buxus Collection at the United State Department of Agriculture (USDA).
It is clear that species with limited genetic variation often cannot cope with the changing environments (Schaal et al. 1991) and that adaptive responses to stresses also depend on the level of remaining of genetic variation as well (Ayala and Kiger 1984). Therefore, knowledge of the distribution of genetic variation among and within host species populations is necessary for better understanding of ecosystem functioning. Such knowledge will also provide valuable insights into the direct and indirect effects of the pathogens introduced on native indigenous hosts, as well as into potential host maladaptation to climate change amplified epidemics of indigenous pathogens (Burdon & Thompson 1995). This knowledge is also of practical use for monitoring the diseases in tree hosts planted beyond their natural range.
This study aims to investigate the variation in genetic markers in relation to the disease status in B. hyrcana and compares the merits of two methods (RAPD and ISSR markers) for evaluating the genetic variation of the healthy and infected groups of B. hyrcana. The markers are two widely applicable techniques to identify relationships at the species and populations levels (Wali et al. 2007; Mehes-Smith et al. 2010; Phong et al. 2011; Zhiqiang et al. 2015; Elmeer et al. 2017), because they are rapid, simple to perform and inexpensive; they do not require prior knowledge of DNA sequences; and only a small amount of DNA is needed (Esselman et al. 1999). Selection of collection sites was difficult due to the limited numbers of healthy trees. Healthy and infected B. hyrcana trees belonging to two different local populations were evaluated for genetic variation using two types of marker based on DNA amplification (RAPD and ISSR). Furthermore, we investigated whether or not the methods are beneficial for exhibiting a link between the geographical origin of a given population and the evaluated genetic variation.
Materials and methods
Genomic DNA extraction from plant material
In total, 60 healthy (30 apparently free of boxwood blight) and infected (i.e. 30 with major branches dead) B. hyrcana were collected from Hyrcanian forests at two different localities (Escolac, 37° 01′ N and 49° 33′ E, and Toscatoc 36° 34′ N and 51° 44′ E). Choice of site was subject to finding at least 15 healthy trees per collection area. The sampling area in each site was approximately 10 km2 and distance between sampling trees was at least 20 m. The voucher numbers of deposited samples in TARI Herbarium are Iran36913 and Iran36914. Nuclear DNA was extracted from bud tissues of each individual and used for marker analysis (Dellaporta et al. 1983).
ISSR analysis
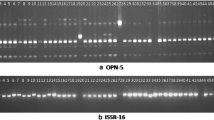
Polymerase chain reaction (PCR) was conducted according to Zietkiewicz et al. (1994). Six ISSR primers were initially screened but only four primers were used in the analysis (Table 1). The clear and reproducible banding patterns generated were used to evaluate genetic variation. The reaction conditions were optimised and mixtures were composed of 20 ng of DNA, 10× buffer (20 mM Tris-HCl pH 8.4; 50 mM KCl), 1 U TaqDNA polymerase, 2 mM MgCl2, 0.2 mM dNTP, and 0.8 μM primer. The PCR amplification protocol includes one cycle for 5 min at 94 °C and then is followed by 42 cycles for 30 s at 94 °C, 1 min at 51–53 °C (Table 1), 1 min at 72 °C and 5 min for final extension at 72 °C. Amplification productions (10 mL) are mixed with 5 mL bromophenol blue separated on 1.5% agarose gel and marked with 5 mL of SYBR Green and then photographed.
RAPD analysis
Polymerase chain reaction (PCR) was conducted according to Williams et al. (1990). Thirteen RAPD primers were initially screened, but only five primers were used in the analysis (Table 2). Only the clear and reproducible banding patterns were used to evaluate genetic variation. The reaction conditions were optimised, and mixtures were composed of 40 ng of DNA, 10× PCR reaction buffer (10 mM Tris-HCl, pH 9.0; 50 mM KCl; 1.5 mM MgCl2), 1 U TaqDNA polymerase, 0.1 mM dNTP, 0.4 μM primer and 1.2 mM MgCl2. The amplifications included 1 cycle of 5 min at 94 °C and following 45 cycles of 1 min at 94 °C, 2 min at 40 °C, 2 min at 72 °C and finally was extended for 7 min at 72 °C. Amplification products (10 mL) were mixed with 5 mL bromophenol blue separated on 1.5% agarose gel and were marked with 5 mL of SYBR Green and then photographed.
Data analysis
Amplification reactions from all individuals were scored; then, genetic variation statistics were computed using the GENAlEX 6.5 software for binary data (Peakall and Smouse 2012). The number of observed and private bands (number of bands unique to a single population), mean number of alleles (Na), effective number of alleles (Ne), Shannon’s information index (I), expected heterozygosity (He) and percentage of polymorphic loci (%P) were evaluated according to Nei (1978). According to Nei’s method (1978), the genetic distances were calculated and the similarity matrix was subjected to principal coordinate analysis (PCoA). Mantel (1967) was used for evaluating the relationship between the calculated distance matrices and the statistic tested for significance against 999 random permutations. The SIMJACARD code of the software package NTSYS-pc: 2.11 was used to estimate pairwise genetic similarity (Rohlf 2004). A similarity matrix based on the unweighted pair group method and arithmetic means (UPGMA) was generated using Jacard’s similarities and SHAN of NTSYS-pc to construct the dendrogram of all the 60 genotypes. Cophenetic matrices were calculated for characterising the correlations between the dendrograms and similarity matrices.
Results
Of the six ISSR primers used in this study, four showed polymorphism. The number of polymorphic fragments ranged from 9 to 14, with an average of 9.5 per primer (Table 1). Thirteen RAPD primers were studied, and five of these showed worthwhile polymorphism. The number of polymorphic fragments ranged from 8 to 12, with a per-primer average of 9.8 (Table 1).
From across all analysed B. hyrcana trees (healthy and infected groups of two populations), genetic variation statistics are shown in Table 2. High band polymorphism was found in the pooled population samples of B. hyrcana using both ISSR (58%) and RAPD (73%) markers. Comparing the two marker systems, RAPDs (19.6%) showed higher genetic diversity statistics (He) than ISSRs (13.8%) (Table 2). Also, He was higher in the Toscatoc population than in the Escolac population using both ISSR and RAPD markers (Table 3). In contrast to ISSRs, the RAPD assays generated similar values of the genetic variation parameters for the studied populations. This indicated the suitability of ISSRs in population genetics research. Results of analyses of healthy versus infected trees in pooled across-populations samples are summarised in Table 4. Effective number of alleles, Shannon’s information index and expected heterozygosity were mostly higher in healthy trees using both marker types (Table 4).
UPGMA dendrograms were generated using ISSR and RAPD datasets to elucidate genetic diversity and population structure among genotypes (Additional files 1 and 2). In order to examine the relationships among the B. hyrcana genotypes, a combined UPGMA clustering was estimated based on the genetic similarity matrix by combining 87 polymorphic bands obtained from ISSR as well as RAPD data. The dendrogram delineated based on the combined data set (ISSR and RAPD) is shown in Fig. 2. Clusters of two groups of data points displaying a similar coefficient value (0.67) are apparent in this diagram. Thus, the combined data indicated similar results to the ISSR except for some minor changes in branch positions. The cophenetic correlation was r = 0.99, and this implies a very good fit. Group I included healthy and infected genotypes of the Toscatoc population with slight differences in branch positions as compared with the ISSR dendrogram (Fig. 2). Group II included genotypes similar to the ISSR clustering. Healthy and infected genotypes could not be differentiated from each other using either ISSR or RAPD dendrograms.

UPGMA dendrogram (based on combined ISSR and RAPD data) of the 60 healthy (h) and infected (i) B. hyrcana genotypes originating from two populations Tuscatoc (T) and Escolac (E) in the Hyrcanian forests
Results of principal component analyses (PCoA) using both the ISSR and RAPD data, in order to study further the genetic diversity among the B. hyrcana genotypes are shown in Fig. 3. A total of 72% variation was assigned to the first three components of PCoA using ISSRs, and 70% of the total variation was thus assigned using RAPDs. The first principal coordinate accounted for 42% of the total variation using ISSR data, and the Tuscatoc population was clearly separated from the Escolac population (Fig. 3a). The infected trees of Tuscatoc were partially separated from the healthy trees across the second principal coordinate, which explained 17% of the total variation. Results of the AMOVA using ISSR data implied that 34% of the genetic variation occurred between populations, 3% between healthy and infected trees, and most of the variation occurred within populations (Table 5).

Scatter diagram of the 60 healthy and infected B. hyrcana genotypes originating from two populations Tuscatoc and Escolac in Hyrcanian forests on the basis of data from ISSR (a), RAPD (b) and combined ISSR and RAPD (c). Healthy trees from Tuscatoc, black diamond; infected trees from Tuscatoc, white diamond; healthy trees from Escolac, black circle; infected trees from Escolac, white circle
According to the RAPD data only, the first principal coordinate (which explained 32% of the total variation) separated infected and healthy trees of the Tuscatoc population (Fig. 3b). The studied populations were partially separated, based on the second principal coordinate (which explained 24% of the total variation). Results of the AMOVA using RAPD data showed that 16% of the genetic variation exists between populations, 5% among healthy and infected trees and most of the marker variation occurred within populations (Table 5).
A combined PCoA using both the ISSR and RAPD datasets (Fig. 3c) was similar to the PCoA using only ISSRs data (Fig. 3a), i.e. genotypes were separated into two populations. The combined UPGMA clustering patterns and combined PCoA of genotypes were comparable (Figs. 2 and 3c respectively), and both partially separated the two populations, but the healthy and infected genotypes were not distinguished from each other. Mantel’s test showed no real correlation between two marker systems (r = 0.06). This result suggests that the two marker systems give somewhat different estimates of genetic relations among genotypes.
Discussion
This study aimed to compare the genetic variation of healthy and infected Buxus hyrcana trees originating from two populations in Hyrcanian forests using ISSR and RAPD markers. Both RAPD and ISSR markers have been widely applied in population genetic research of various species in not only wild (Dikshit et al. 2007; Yao et al. 2008; Chen et al. 2013) but also cultivated plants (Nagaoka and Ogihara 1997; Sikdar et al. 2010). Generally, all these studies have revealed the informativeness and efficiency of ISSR primers compared with RAPD primers.
Considerable genetic marker variation for B. hyrcana was observed based on the mean values of the He for each marker type (ISSR: 0.138 and RAPD: 0.196). The reason for the variation detected within populations may be related to genetic structure, which is probably due to heterozygosity resulting from cross-pollination of B. hyrcana (Lazaro and Traveset 2006). However, average levels of genetic variation within either healthy or infected B. hyrcana populations included in this study are lower than those reported by Ghandehari et al. (2013a, b) in populations not infected by boxwood blight in Hyrcanian forests using ISSR (0.34) and RAPD (0.25). A possibility is that the differences were due to genetic drift caused by the high degree of tree mortality and corresponding reduction in population size. The loss of genetic variation within natural populations may occur through bottlenecks, namely severe reductions of population size over a relatively short period. Bottlenecks may determine reductions of within-population genetic diversity owing to the loss of alleles through genetic drift or random fluctuations in allele frequencies (Spielman et al. 2004). McDonald et al. (1998) studied the impact of an oak wilt epidemic on the genetic structure of a live oak (Quercus fusiformis Small) population in Texas, USA, by allozyme comparison of pre-epidemic and post-epidemic (survivors of a wilt epidemic) trees. They demonstrated that disease affects the genetic structure of a natural host population.
Results from the current study revealed that more genetic variation occurred within populations of B. hyrcana than between populations using both ISSR (66% and 34%, respectively) and RAPD markers (84% and 16%, respectively). These observations are consistent with earlier studies on B. hyrcana in Hyrcanian forests (Ghandehari et al. 2013a, b) and on other tree and shrub species that are characterised by high genetic variation within populations (Hamrick et al. 1992). As quoted by Hamrick and Godt (1996), reproductive biology is one of the most important factors for specifying the genetic structure of plant populations. They showed that 10–20% of the genetic variation occurring among populations is typical of outcrossing plant species, while 50% of the variation occurring among populations is typical of self-pollinating species. Therefore, partial inbreeding can explain the observed genetic variation among studied populations. Buxus hyrcana is an ambophilous (i.e. pollinated by wind and insects) species and is an outcrosser that shows partial self-compatibility based on studies of flowering and pollination biology (Von Balthazar and Endress 2002). Lazaro Traveset 2006).
The genetic correlation among the genotypes, specified by ISSR and RAPD markers, is partially related to the geographical origin of B. hyrcana genotypes, especially with ISSR data. Previous results for various Picea species (Nkongolo et al. 2005; Tayefeh Aliakbarkhani et al. 2015), Angelica sinensis (Oliv.) Diels (Mei et al. 2007), Dalbergia oliveri Prain (Phong et al. 2011), Ginkgo biloba L. (Mei et al. 2014), Tectona grandis L.f. (Narayanan et al. 2015), Salix spp. (Trybush et al. 2008; Zhai et al. 2016; Sulima et al. 2018), and Morus alba L. (Saha et al. 2016) have consistently indicated that genotypes with different geographic origins are quite different genetically as well. The geographical separation of the two boxwood populations through ISSRs can be explained by mesoclimatic differences, restricted pollination and seed dispersal (by gravity) in an understorey species or small tree species (Shah et al. 2008; Jenkins et al. 2010; Zeng et al. 2012). More importantly, the current study aimed to find whether or not different marker systems reflect different aspects of genetic relationships. Due to higher number of alleles per locus and a moderate value of He, a higher level of polymorphism (ISSR 58% and RAPD 73%) was observed using RAPDs. Differences in genetic relationships (based on the two marker systems studied) may simply reflect differences in the levels of polymorphism detected by each marker system. Discordance between different marker systems may or may not be very informative for understanding genetic relationships within a study group.
The mean values of genetic diversity parameters (Ne and He) of healthy trees (pooled from the two populations) were higher than in the trees infected by boxwood blight based on both ISSR and RAPD data even though AMOVA showed a low degree of genetic differentiation between healthy and infected B. hyrcana groups (ISSR: 5%, RAPD: 13%). Houston and Houston (2000) indicated that genetic parameters of resistant and susceptible trees to beech bark disease using isozyme data were similar, while the heterozygosity observed was actually higher in the susceptible population. They postulated that the larger within-stand heterozygote deficits in these resistant groups of trees led to underlying genetic differences between resistant and susceptible populations which are related to adaptation, tree vigour and stand history. Studies have also shown that infection by fungal pathogens may change significantly the frequencies at which particular resistance genes occur (e.g. Burdon and Thompson 1995; Webster et al. 1986). According to the study by Burdon and Thompson (1995), a rust epidemic (caused by Melampsora lini Ehrenb. Lev.) changed significantly the profile of resistant genes in a natural population of wild flax (Linum marginale A. Cum. ex Planch.). Although the resistance structure of the flax population changed, its resistance to the predominant pathogen genotypes did not improve. These results indicated that the effects of selection may be unforeseeable. Also, the effects of fungal disease including chestnut blight, Dutch elm disease and Gilbertson root rot (caused by Phellinus weirii, Murrill) on the composition of plant communities have been reported in the western mountain forests of North America (Harper 1990; Holah et al. 1997). In addition, a study of isozyme variation among nine populations of Ozark chinkapin (Castanea pumila (L.) Mill.) threatened by their susceptibility to chestnut blight (Cryphonectria parasitica) revealed higher genetic variation (He = 0.227) compared to other Castanea species (He = 0.115) on the North American continent that were not susceptible to infection by this disease (Dane and Hawkins 1999; Huang et al. 1994a, 1994b; Huang et al. 1998). According to these studies, populations with high levels of genetic variation and unusual alleles should be subject to further study by conservation biologists in order to capture as much of the genetic variation of the species as possible.
Conclusion
Boxwood blight has had a major effect on B. hyrcana, killing individual stems quickly especially in dense populations and reducing population size in all populations. Considerable within-population genetic diversity, and generally higher genetic diversity in healthy genotypes compared with infected ones, suggested conservation efforts should focus on survivor trees in every population and consider the establishment of tree reserves. Propagation of plants from seeds is preferred, since it would include the widest range of genetic variation.
The present study compares the markers analysis among the B. hyrcana genotypes. Low correlation between the marker types cautions against reliance on a single marker technique in B. hyrcana. This result suggests that the two marker systems give different estimates of genetic relations among genotypes. The genetic correlation among the genotypes, specified by ISSR and RAPD markers, is partially related to the geographical origin of B. hyrcana genotypes, especially with ISSR data.
In order to understand mechanisms for maintaining genetic variation in B. hyrcana and protect this endangered species, it is necessary to perform more studies in the field of pollination biology, parentage analysis using co-dominant markers and levels of polymorphism among populations with different level of hazards. Moreover, investigation on influence of blight disease on physiological and genetic features of susceptibility of B. hyrcana can lead to the identification of several novel mechanisms that may be useful for developing resistance to biotic stresses.
Abbreviations
- %P:
-
Percentage of polymorphic bands
- He :
-
Expected heterozygosity
- I:
-
Shannon’s information Index
- Na :
-
Average number of observed bands
- Nb :
-
Number of bands
- Ne :
-
Effective number of bands
- Nf :
-
Number of frequent bands freq. ≥ 5%
- Np :
-
Number of private bands
References
Akilli, S., Katircioglu, Y. Z., Zor, K., & Maden, S. (2012). First report of box blight caused by Cylindrocladium pseudonaviculatum in the Eastern Black Sea region of Turkey. New Disease Reports, 25, 23.
Asadi, H., Hosseini, S. M., Esmailzadeh, O., & Ahmadi, A. (2011). Flora, life form and chorological study of box tree (Buxus hyrcanus Pojark.) sites in Khybus protected forest, Mazandaran. Journal of Plant Biology, 8, 27–40.
Asadi, H., Hosseini, S. M., Esmailzadeh, O., & Baskin, C. C. (2012). Persistent soil seed banks in old-growth Hyrcanian box tree (Buxus hyrcana) stands in northern Iran. Ecological Research, 27, 23–33.
Ata, A., Iverson, C. D., Kalhari, K. S., Akhter, S., Betteridge, J., Meshkatalsadat, M. H., Orhan, I., & Sener, B. (2010). Triterpenoidal alkaloids from Buxus hyrcana and their enzyme inhibitory, anti-fungal and anti-leishmanial activities. Phytochemistry, 71, 1780–1786. https://doi.org/10.1016/j.phytochem.
Ayala, F., & Kiger, J. (1984). Modern genetics. V. 3. California: The Benjamin/Cummings Publishing Company, Inc.
Berry, F. H. (1985). Anthracnose diseases of eastern hardwoods. Forest Insect & Disease Leaflet 133. Washington: USDA Forest Service.
Boyd, I. L., Freer-Smith, P. H., Gilligan, C. A., & Godfray, H. C. (2013). The consequence of tree pests and diseases for ecosystem services. Science, 342, 1235773. https://doi.org/10.1126/science.1235773.
Brand, T. (2005). Auftreten von Cylindrocladium buxicola B. Henricot an Buchsbaum in Nordwest-Deutschland [Outbreak of Cylindrocladium buxicola B. Henricot on boxwood in Northwest-Germany]. Nachrichtenblatt des Deutschen Pflanzenschutzdienstes, 57, 237–240.
Burdon, J. J., & Thompson, J. N. (1995). Changed patterns of resistance in a population of Linum marginale attacked by the rust pathogen Melampsora lini. Journal of Ecology, 83, 199–206.
Cech, T., Diminic, D., & Heungens, K. (2010). Cylindrocladium buxicola causes common box blight in Croatia. New Disease Reports, 22, 9. https://doi.org/10.1111/j.1365-3059.2010.02361.x.
Chen, S. Y., Dai, T. X., Chang, Y. T., Wang, S. S., Ou, S. L., Chuang, W. L., Cheng, C. Y., Lin, Y. H., Lin, L. Y., & Ku, H. M. (2013). Genetic diversity among Ocimum species based on ISSR, RAPD and SRAP markers. Australian Journal of Crop Science, 7, 1463–1471.
Choi, G. H., & Nuss, D. L. (1992). Hypovirulence of chestnut blight fungus conferred by an infectious viral cDNA. Science, 257, 800–803.
Crepel, C., & Inghelbrecht, S. (2003). First report of blight on Buxus spp. caused by Cylindrocladium buxicola in Belgium. Plant Disease, 87, 1539. https://doi.org/10.1094/PDIS.2003.87.12.1539A.
Comeau, A. M., Dufour, J., Bouvet, G. F., Jacobi, V., Nigg, M., Henrissat, B., Laroche, J., Levesque, R. C., & Bernier, L. (2015). Functional annotation of the Ophiostoma novo-ulmi genome: insights into the phytopathogenicity of the fungal agent of Dutch elm disease. Genome Biology and Evolution, 7(2), 410–30.
Dane, F., & Hawkins, L. K. (1999). Genetic variation and population structure of Castanea pumila var. ozarkensis. Journal of the American Society for Horticultural Science, 124, 666–670.
Dellaporta, S. L., Wood, J., & Hicks, J. B. (1983). A plant DNA minipreparation: version II. Plant Molecular Biology Reporter, 1, 19–21.
Dikshit, H. K., Jhang, T., Singh, N. K., Koundal, K. R., Bansal, K. C., Chandra, N., Tickoo, J. L., & Sharma, T. R. (2007). Genetic differentiation of Vigna species by RAPD, URP and SSR markers. Biologia Plantarum, 51, 451–457. https://doi.org/10.1007/s10535-007-0095-8.
Elmeer, K., Alghanem, M., Al-Latifi, L., & Alhemairi, H. (2017). Efficiency of RAPD and ISSR markers for the detection of polymorphisms and genetic relationships in date palm. Biotechnology, 16, 19–26. https://doi.org/10.3923/biotech.2017.19.26.
Esselman, E. J., Jianqiang, L., Crawford, D. J., Winduss, J. L., & Wolfe, A. D. (1999). Clonal diversity in the rare Calamagrostis porter ssp. Insperata (Poaceae): comparative results for allozymes and random amplified polymorphic DNA (RAPD) and inter simple sequence repeat (ISSR) markers. Molecular Ecology, 8, 443–451.
Ghandehari, V., Ahmadikhah, A., & Payamnoor, V. (2013a). Genetic diversity of Buxus hyrcana populations in north of Iran using ISSR markers. Iranian Journal of Rangelands and Forests Plant Breeding and Genetic Research, 21, 11–19.
Ghandehari, V., Ahmadikhah, A., & Payamnoor, V. (2013b). Investigation of inter- and intra-population genetic diversity of Buxus hyrcana in forests of north of Iran using RAPD molecular markers. Iranian Journal of Forest, 5, 207–218.
Gorgiladze, L., Meparishvili, G., Sikharulidze, Z., Natsarishvili, K., & Davitadze, R. (2011). First report of box blight caused by Cylindrocladium buxicola in Georgia. New Disease Reports, 23, 24.
Hamrick, J. L., & Godt, M. J. W. (1996). Conservation genetics of endemic plant species. In J. C. Avise & J. L. Hamrick (Eds.), Conservation genetics: Case histories from nature (pp. 281–304). New York: Chapman and Hall.
Hamrick, J. L., Godt, M. J. W., & Sherman-Broyles, S. L. (1992). Factors influencing levels of genetic diversity in woody plant species. New Forests, 6, 95–124.
Harper, J. L. (1990). Pest, pathogens and plant communities: an introduction. In J. J. Burdon & S. R. Leather (Eds.), Pests, pathogens and plant communities (pp. 3–14). Oxford: Blackwell Scientific Publications.
Harper, A. L., McKinney, L. V., Nielsen, L. R., Havlickova, L., Li, Y., Trick, M., Fraser, F., Wang, L., Fellgett, A., Sollars, E. S., Janacek, S. H., Downie, J. A., Buggs, R. J., Kjær, E. D., & Bancroft, I. (2016). Molecular markers for tolerance of European ash (Fraxinus excelsior) to dieback disease identified using associative transcriptomics. Scientific Reports, 6, 19335. https://doi.org/10.1038/srep19335.
Henricot, B., Pérez Sierra, A., & Prior, C. (2000). A new blight disease on Buxus in the UK caused by the fungus Cylindrocladium. Plant Pathology, 49, 805. https://doi.org/10.1046/j.1365-3059.2000.00508.x.
Holah, J. C., Wilson, M. V., & Hansen, E. M. (1997). Impacts of a native root-rotting pathogen on successional development of old-growth Douglas-fir forests. Oecologia, 111, 429–433.
Hosseinzadeh, S., & Esmailzadeh, O. (2017). Floristic study of Buxus hyrcana stands in the western forests of Haraz district, Amol. Iranian Journal of Applied Ecology, 6, 1–13.
Houston, D. B., & Houston, D. R. (2000). Allozyme genetic diversity among Fagus grandifolia trees resistant or susceptible to beech bark disease in natural populations. Canadian Journal of Forest Research, 30, 778–789. https://doi.org/10.1139/x99-252.
Huang, H., Dane, F., & Kubisiak, T. L. (1998). Allozyme and RAPD analysis of the genetic diversity and geographic variation in wild populations of the American chestnut. American Journal of Botany, 85, 1013–1021.
Huang, H., Dane, F., & Norton, J. D. (1994a). Genetic analysis of 11 polymorphic isozyme loci in chestnut species and characterization of chestnut cultivars by multi-locus allozyme genotypes. Journal of American Society for Horticultural Science, 119, 840–849.
Huang, H., Dane, F., & Norton, J. D. (1994b). Allozyme diversity in Chinese, Seguin and American chestnut (Castanea spp.). Theoretical Applied Genetics, 88, 981–985.
Jenkins, D. G., Carey, M., Czerniewska, J., Fletcher, J., Hether, T., & Jones, A. (2010). A meta-analysis of isolation by distance: relic or reference standard for landscape genetics? Ecography, 33, 315–320.
Kaviani, B., & Negahdar, N. (2016). Effects of biological nano-fertilizer on the morphological, physiological and proliferation traits and quality of Buxus hyrcana Pojark. Bangladesh Journal of Botany, 45, 1135–1142.
Lazaro, A., & Traveset, A. (2006). Reproductive success of the endangered shrub Buxus balearica Lam. (Buxaceae): pollen limitation, and inbreeding and outbreeding depression. Plant Systematics and Evolution, 261, 117–128.
Mantel, N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Research, 27, 209–220.
Marin, D. H., Romero, R. A., Guzman, M., & Sutton, T. B. (2003). Black sigatoka: An increasing threat to banana cultivation. Plant Disease, 87, 208–222.
McDonald, B. A., Bellamy, B. K., Zhan, J., & Appel, D. N. (1998). The effect of an oak wilt epidemic on the genetic structure of a Texas live oak population. Canadian Journal of Botany, 76, 1900–1907.
Mehes-Smith, M., Michael, P., & Nkongolo, K. (2010). Species-diagnostic and species-specific DNA sequences evenly distributed throughout pine and spruce chromosomes. Genome, 53, 769–777. https://doi.org/10.1139/G10-065.
Mei, Z., Khan, A., Zeng, W., & Fu, J. (2014). DNA fingerprints of living fossil Ginkgo biloba by using ISSR and improved RAPD analysis. Biochemical Systematics and Ecology, 57, 332–337.
Mei, Z., Zhang, C., Khan, A., Zhu, Y., Tania, M., & Fu, J. (2007). Efficiency of improved RAPD and ISSR markers in assessing genetic diversity and relationships in Angelica sinensis (Oliv.) Diels varieties of China. Journal of Tropical Forest Science, 19, 218–225.
Mirabolfathy, M., Ahangaran, Y., Lombard, L., & Crous, P. W. (2013). Leaf blight of Buxus sempervirens in northern forests of Iran caused by Calonectria pseudonaviculata. Plant Disease, 97, 1121.
Nagaoka, T., & Ogihara, Y. (1997). Applicability of inter-simple sequence repeat polymorphisms in wheat for use as DNA markers in comparison to RFLP and RAPD markers. Theoretical Applied Genetics., 94, 597–602. https://doi.org/10.1007/s001220050456.
Narayanan, C., Wali, S. A., Shukla, N., Kumar, R., Mandal, A. K., & Ansari, S. A. (2015). RAPD and ISSR markers for molecular characterization of Teak (Tectona grandis) plus trees. Electronic Journal of Biotechnology, 18, 96–102.
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89, 583–590.
Nkongolo, K. K., Michael, P., & Demers, T. (2005). Application of ISSR, RAPD, and cytological markers to the certification of Picea mariana, P. glauca, and P. engelmannii trees, and their putative hybrids. Genome, 48, 302–311.
Peakall, R., & Smouse, P. E. (2012). GenAlEx 6.5: Genetic analysis in Excel. Population genetic software for teaching and research: an update. Bioinformatics, 28, 2537–2539.
Phong, D. T., Hien, V. T., Thanh, T. T., & Tang, D. V. (2011). Comparison of RAPD and ISSR markers for assessment of genetic diversity among endangered rare Dalbergia oliveri (Fabaceae) genotypes in Vietnam. Genetics and Molecular Research, 6, 2382–2393. https://doi.org/10.4238/2011.October.6.3.
Pintos Varela, C., Gonzáles Penalta, B., Mansilla Vázquez, J. P., & Aguin Casal, O. (2009). First report of Cylindrocladium buxicola on Buxus sempervirens in Spain. Plant Disease, 93, 670. https://doi.org/10.1094/PDIS-93-6-0670B.
Rohlf, J. (2004). NTSYS-PC Version 2.11T. Numerical Taxonomy and Multivariate Analysis System. Applied Bioestastistics, Inc.
Saha, S., Adhikari, S., Dey, T., & Ghosh, P. (2016). RAPD and ISSR based evaluation of genetic stability of micropropagated plantlets of Morus alba L. variety S-1. Meta Gene, 7, 7–15.
Saracchi, M., Rocchi, F., Pizzatti, C., & Cortesi, P. (2008). Box blight, a new disease of Buxus in Italy caused by Cylindrocladium buxicola. Journal of Plant Pathology, 90, 581–583.
Schaal, B. A., Leverich, W. J., & Rogstad, S. H. (1991). A comparison of methods for assessing genetic variation in plant conservation biology. In D. A. Falk & K. E. Holsinger (Eds.), Genetics and conservation of rare plants (pp. 123–134). New York: Oxford University Press.
Shah, A., Li, D. Z., Gao, L. M., Li, H. T., & Möller, M. (2008). Genetic diversity within and among populations of the endangered species Taxus fuana (Taxaceae) from Pakistan and implications for its conservation. Biochemical Systematics and Ecology, 36, 183–193. https://doi.org/10.1016/j.bse.2007.09.012.
Sikdar, B., Bhattacharya, M., Mukherjee, A., Banerjee, A., Ghosh, E., Ghosh, B., & Roy, S. C. (2010). Genetic diversity in important members of Cucurbitaceae using isozyme, RAPD and ISSR markers. Biologia Plantarum, 54, 135–140. https://doi.org/10.1007/s10535-010-0021-3.
Soleymanipoor, S., & Esmailzadeh, O. (2015). Flora, life form and chorology of box trees (Buxus hyrcana Pojark.) habitats in forests of Farim area of Sari. Taxonomic and Biosystematics Journal, 23, 39–56.
Spielman, D., Brook, B. W., & Frankham, R. (2004). Most species are not driven to extinction before genetic factors impact them. Proceedings of the National Academy of Sciences of USA, 101, 15261–15264.
Sulima, P., Prinz, K., & Przyborowski, J. A. (2018). Genetic diversity and genetic relationships of purple willow (Salix purpurea L.) from natural locations. International Journal of Molecular Sciences, 19, 105. https://doi.org/10.3390/ijms19010105.
Tayefeh Aliakbarkhani, S., Akbari, M., Hassan khah, A., Talaie, A., & Fattahi Moghadam, M. R. (2015). Phenotypic and genotypic variation in Iranian pistachios. Journal of Genetic Engineering and Biotechnology, 13, 235–241.
Thammina, C., Olsen, R. T., Malapi-Wight, M., Crouch, J. A., & Pooler, M. R. (2014). Development of polymorphic genic-SSR markers by cDNA library sequencing in boxwood, Buxus spp. (Buxaceae). Applications in Plant Sciences, 2. https://doi.org/10.3732/apps.1400095.
Thammina, C. S., Olsen, R. T., Kramer, M., & Pooler, M. R. (2016). Genetic relationships of boxwood (Buxus L.) accessions based on genic simple sequence repeat markers. Genetic Resources and Crop Evolusion, 64, 1281–1293. https://doi.org/10.1007/s10722-016-0436-6.
Trybush, S. O., Jahodov, Š., Macalpine, W., & Karp, A. (2008). A genetic study of a Salix germplasm resource reveals new insights into relationships among sub-genera, sections and species. Bioenergy Research, 1, 67–79.
Van Laere, K., Hermans, D., Leus, L., & Van Huylenbroeck, J. (2011). Genetic relationships in European and Asiatic Buxus species based on AFLP markers, genome sizes and chromosome numbers. Plant Systematics and Evolution, 293, 1–11.
Von Balthazar, M., & Endress, P. K. (2002). Reproductive structures and systematics of Buxaceae. Botanical Journal of the Linnean Society, 140, 193–228.
Von Balthazar, M., Endress, P. K., & Qiu, Y. L. (2000). Phylogenetic relationships in Buxaceae based on nuclear internal transcribed spacers and plastid ndhF sequences. International Journal of Plant Sciences, 161, 785–792.
Wali, S. A., Shukla, N., Kumar, R., Mandai, A. K., & Ansari, S. (2007). RAPD and ISSR markers for molecular characterization of teak (Tectona grandis) plus trees. Journal of Tropical Forest Science, 19, 218–225.
Webster, R. K., Saghai-Maroof, M. A., & Allard, R. W. (1986). Evolutionary response of barley composite cross II to Rhynchosporium secalis analyzed by pathogenic complexity and by gene by race relationships. Phytopathology, 76, 661–668.
Williams, J. G. K., Kubelik, A. R., Livak, K. J., Rafalski, J. A., & Tingey, S. V. (1990). DNA polymorphisms amplified by arbitrary primers are useful as genetic markers. Nucleic Acids Research, 18, 6531–6535.
Yao, H., Zhao, Y., Chen, D. F., Chen, J. K., & Zhao, T. S. (2008). ISSR primer screening and preliminary evaluation of genetic diversity in wild populations of Glycyrrhiza uralensis. Biologia Plantarum, 52, 117–120. https://doi.org/10.1007/s10535-008-0022-7.
Zeng, X., Michalski, S. G., Fischer, M., & Durka, W. (2012). Species diversity and population density affect genetic structure and gene dispersal in a subtropical understory shrub. Journal of Plant Ecology, 5, 270–278. https://doi.org/10.1093/jpe/rtr029.
Zhai, F., Mao, J., Liu, J., Peng, X., Han, L., & Sun, Z. (2016). Male and female subpopulations of Salix viminalis present high genetic diversity and high long-term migration rates between them. Frontiers in Plant Science, 7, 330.
Zhiqiang, M., Chun, Z., Asaduzzaman, K. M., Ye, Z., Mousumi, T., Peiyi, L., & Junjiang, F. (2015). Efficiency of improved RAPD and ISSR markers in assessing genetic diversity and relationships in Angelica sinensis (Oliv.) Diels varieties of China. Electronic Journal of Biotechnology, 18, 96–102. https://doi.org/10.1016/j.ejbt.2014.12.006.
Zietkiewicz, E., Rafalski, A., & La buda, D. (1994). Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics, 20, 176–183.
Acknowledgements
This work was supported by the Agricultural Research, Education and Extension Organization, and Research Institute of Forests and Rangelands (RIFR), Iran; Project no. 14-09-09-9354-93007.
Funding
None.
Availability of data and materials
Please contact author for data requests.
Author information
Authors and Affiliations
Contributions
PSS planned and directed the study, analysed the data and interpretation of results and drafted the manuscript. HJ was responsible for the correction of manuscript. LR was responsible for the production of data. MA was responsible for the collection of field specimens. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional File 1:
UPGMA dendrogram (based on ISSR marker) of the 60 healthy (h) and infected (i) B. hyrcana genotypes originating from two populations Tuscatoc (T) and Escolac (E) in the Hyrcanian forests based on ISSR marker. (JPG 1188 kb)
Additional File 2:
UPGMA dendrogram of the 60 healthy (h) and infected (i) B. hyrcana genotypes originating from two populations Tuscatoc (T) and Escolac (E) in Hyrcanian forests based on RAPD marker. (JPG 1184 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

{kind=link}
{kind=link}
Cite this article
Salehi Shanjani, P., Javadi, H., Rasoulzadeh, L. et al. Evaluation of genetic differentiation among healthy and infected Buxus hyrcana with boxwood blight using RAPD and ISSR markers. N.Z. j. of For. Sci. 48, 15 (2018). https://doi.org/10.1186/s40490-018-0120-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40490-018-0120-z