
Abstract
N6-methyladenosine (m6A) is a prevalent internal RNA modification in higher eukaryotic cells. As the pivotal m6A regulator, RNA methyltransferase-like 3 (METTL3) is responsible for methyl group transfer in the progression of m6A modification. This epigenetic regulation contributes to the structure and functional regulation of RNA and further promotes tumorigenesis and tumor progression. Accumulating evidence has illustrated the pivotal roles of METTL3 in a variety of human cancers. Here, we systemically summarize the interaction between METTL3 and RNAs, and illustrate the multiple functions of METTL3 in human cancer. METLL3 is aberrantly expressed in a variety of tumors. Elevation of METTL3 is usually associated with rapid progression and poor prognosis of tumors. On the other hand, METTL3 may also function as a tumor suppressor in several cancers. Based on the tumor-promoting effect of METTL3, the possibility of applying METTL3 inhibitors is further discussed, which is expected to provide novel insights into antitumor therapy.
Similar content being viewed by others
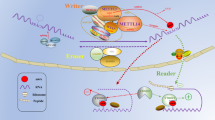


Introduction
Epigenetics promotes the functional plasticity of genome at multiple levels [1]. As the classical kinds of chemical modifications, 5-methylcytidine (m5C), 5-hydroxymethylcytidine (hm5C), N4-acetylcytidine (ac4C), and N6-methyladenosine (m6A) mainly participate in the epigenetic modification of RNAs [2]. Among the different kinds of modifications, m6A is the most common and effective modification in both coding and noncoding RNAs [3, 4]. The importance of m6A modification has been recognized in physiological and pathological processes [5,6,7]. Meanwhile, m6A modification also plays critical roles in yeast and plants [8, 9].
The dynamic progress of m6A modification is driven by the interactions between “writers”, “erasers”, and “readers” [10, 11]. The m6A deposition is primarily performed by “writers”, while the modification site is subsequently “read” by m6A recognition proteins or “erased” by m6A demethylases [12]. In particular, human N6-methyltransferase complex (MTC), which contains Methyltransferase-like 3 (METTL3) [13], METTL14 [14], Wilms tumor 1-associated protein (WTAP) [15], METTL16 [16], KIAA1429 [17], zinc finger CCCH-type containing 13 (ZC3H13) [18], RNA-binding motif protein 15 (RBM15) [19], and Cbl proto-oncogene like 1 (CBLL1) [20], is responsible for methyl group transfer. As the core component of MTC [21], METTL3 dominates the catalytic core and performs N6-methylase catalytic activity [22]. Dysregulation of METTL3 significantly affects the total m6A methylation level [23]. In addition, noncatalytic components of the complex also contribute to the RNA methylation progression. METTL14 assists to construct the RNA binding scaffold to promote the RNA binding ability of METTL3, thereby enhancing the catalytic effect of METTL3 [24]. In addition, WTAP can facilitate the nuclear speckle localization of METTL3 and METTL14 [24]. Apart from the essential components, recent studies have demonstrated that METTL16, KIAA1429, ZC3H13, RBM15 and HAKAI are involved in m6A modification in various ways [12].
It has been well recognized that RNA methylation influences the metabolic processes and functional regulation of RNA. As the critical component of MTC, METTL3 primarily affects post-transcriptional genetic modification (Fig. 1). Genetic modification leads to changes in biological processes, including cell growth, migration, differentiation and inflammatory response [25]. Recently, increasing studies have revealed the accumulation of m6A modification in human cancers, indicating the important role of METTL3 in tumorigenesis and tumor progression [26]. In this review, we systematically summarize the functions of METTL3 in different human malignancies and further discuss the potential of METTL3 inhibitors.

In the nucleus, METTL3 promotes mRNA splicing through recognizing the 3’UTR m6A sites on mRNA, thereby altering the mRNA structure. Moreover, METTL3 also transports from nucleus to the cytoplasm and further enhances the translation and degradation of mRNA
Reciprocal effects between METTL3 and RNAs in human cancers
Methyltransferase activity of METTL3 can be detected both in the nucleus and cytoplasm [27], suggesting that METTL3 could modulate the metabolism and function of RNAs in various ways. On the other hand, expression and functions of METTL3 can also be regulated by noncoding RNAs.
METTL3 regulates the maturation, transportation and translation of messenger RNA (mRNA)
METTL3 was involved in the regulation of mRNA, including the maturation, transportation and translation of mRNA [25]. Nucleus-localized METTL3 primarily promoted the maturation, splicing and transportation of mRNA. The abundant deposition of m6A in pre-mRNA was associated with the acceleration of pre-mRNA maturation [27]. After pre-mRNA production, methylation of spliced regions could affect the splicing of the pre-mRNA, thereby producing diverse sequences of mature mRNA [28]. In addition, increasing m6A on mature mRNA decreased the nuclear fraction of mRNA by promoting cytoplasmic transportation [17]. In the METTL3-enriched cytoplasm, METTL3 significantly enhanced mRNA translation by stabilizing mRNA [29]. Mechanistically, m6A modification frequently took place in the coding sequence (CDS), the 3′UTR and regions near the stop codons of mature mRNA [24]. Recognition proteins specifically recognized the m6A-abundant regions to stabilize mRNA and further enhance translation of mRNA in an m6A-dependent manner [24]. In addition, the interplay between specific transcription factor and METTL3 could also promote translation. For instance, functional interaction between METTL3 and eukaryotic translation initiation factor 3 subunit h (eIF3h) was required for enhanced translation and oncogenic transformation [30]. Apart from its promoting effect, METTL3 could also increase mRNA decay [31], revealing the dual effects of METTL3 on mature mRNA.
METTL3 promotes the maturation and activation of noncoding RNAs
m6A modification can regulate the maturation, transport, stability and degradation of noncoding RNAs, eventually affecting the biological function of tumor cells [32]. METTL3 was proved to promote the maturation and activation of microRNA (miRNA). m6A modification on the primary miRNA (pri-miRNA) directly facilitated the maturation of pri-miRNA, consequently promoting tumor progression [33]. For example, METTL3 modulated pri-miR221/222 maturation in an m6A-dependent manner in bladder cancer (BC). Mature miR221/222 then suppressed the expression of phosphate and tension homology deleted on chromosome 10 (PTEN) to accelerate tumor growth [34]. In addition, mature miR-1246 induced by METTL3 could activate the mitogen-activated protein kinase (MAPK) pathway by suppressing sprouty-related EVH1 domain protein 2 (SPRED2), which facilitated the invasion and distant metastasis of colorectal cancer (CRC) [35]. On the other hand, METTL3 could indirectly promoted miRNA activation by regulating long noncoding RNA (lncRNA). The activation of miR-1914-3p induced by hypermethylated lncMALAT1 distinctly enhanced the expression of YAP, leading to rapid progression and enhanced therapeutic resistance of non-small cell lung cancer (NSCLC) [36].
Noncoding RNAs regulate the expression and functions of METTL3
Noncoding RNAs, including miRNA and lncRNA, are involved in tumor progression by regulating METTL3. miRNA, which was regarded as specific transcription factors of METTL3, could decrease the expression and function of METTL3, thereby reversing the tumor-promoting effect of METTL3 [37,38,39]. lncRNA also participates in the regulation of METTL3. For example, the interaction between LINC00470 and METTL3 facilitated the degradation of PTEN mRNA and further contributed to the development and progression of gastric cancer (GC) [40]. In addition, lncRNA Rho GTPase activating protein 5 (ARHGAP5)-AS1 recruited METTL3 to enhance the stability of ARHGAP5 mRNA, eventually leading to the poor prognosis and chemoresistance of GC [41].
METTL3 mediates tumorigenesis via RNA methylation
An increasing number of studies have illustrated that METTL3 is involved in various aspects of tumor progression, including stemness maintenance, tumor growth, invasion, migration, and drug resistance.
Gastrointestinal tumors
GC
Aggressive GC is generally accompanied with higher expression of METTL3, suggesting the oncogenic role of METTL3 in GC [42]. Elevation of METTL3 was proved to promote tumor growth, metastasis and therapeutic resistance in an m6A-dependent pattern [43]. METTL3 propelled tumor growth by inducing m6A deposition on the mRNA of oncogenes and rate-limiting enzymes of glucose metabolism. MYC and SEC62 were previously identified as oncogenes in GC. Abundant m6A deposition was not only detected on the component molecules of the MYC-targeted genes, but also contributed to the overexpression of MYC [44, 45]. Another study demonstrated that METTL3 promoted the stability of SEC62 mRNA in an m6A-mediated manner and further inhibited tumor cell apoptosis by suppressing the Bax-caspase3 pathway [46]. Apart from oncogenes, aerobic glycolysis was also activated in tumorigenesis [47]. Mechanistically, m6A modification enhanced the expression of hepatoma-derived growth factor (HDGF), which could activate solute carrier family 2 member 4 (GLUT4) and enolase 2 (ENO2) to potentiate aerobic glycolysis in tumor cells [48]. Moreover, the interaction between METTL3 and lncRNA LINC00470 promoted tumor growth by impairing the stability of PTEN mRNA [40].
Tumor metastasis and therapeutic resistance represent the characteristics of aggressive GC [43]. Epithelial mesenchymal transition (EMT) and angiogenesis provide proper conditions for cell mobility. Overexpression of zinc finger MYM-type containing 1 (ZMYM1) was induced by METTL3, thereby promoting the EMT process by suppressing the activation of E-cadherin [49]. Meanwhile, expression levels of EMT-related markers, especially growth factor independent 1 (GFI-1) and α-smooth muscle actin (α-SMA), were dramatically increased under the regulation of METTL3 [43]. The angiogenesis process, especially the proliferation of human umbilical vein endothelial cell (HUVEC) and tube formation, was correlated with overexpressed METTL3, subsequently promoting the invasion and distant migration of cancer cells [48]. The contribution of METTL3 to chemoresistance was verified as well. Mechanistically, METTL3 contributed to the stabilization of ARHGAP5 mRNA after being recruited by lncRNA ARHGAP5-AS1 and then induced chemoresistance [41].
On the other hand, METTL3 can suppress tumor progression under certain conditions. Xie et al. reported that METTL3 facilitated the m6A modification on basic leucine zipper ATF-like transcription factor 2 (BATF2) mRNA. Methylated BATF2 exerted tumor suppressive effects by stabilizing the p53 protein and inhibiting the phosphorylation of extracellular regulated kinase (ERK) [50]. In addition, METTL3-high GC cells preferred to respond to rapamycin (mTOR) inhibitors through m6A-DGCR8-dependent mechanism [51].
Hepatocellular cancer (HCC) and gallbladder cancer
Increased METTL3 is not only involved in tumorigenesis but is also related to rapid progression and poor prognosis of HCC [52, 53]. Mechanistically, METTL3 facilitated tumor progression by modulating suppressor of cytokine signaling 2 (SOCS2) [54], RAD52 motif 1 (RDM1) [55] and Snail [56]. Depending on the m6A modification, SOCS2 mRNA was functionally silenced, thereby promoting the proliferation, migration and stemness maintenance of HCC cells [54]. Hypermethylation of RDM1 induced by METTL3 suppressed the expression of RDM1, leading to the activation of the Ras/Raf/ERK pathway in tumor progression [55]. In addition, METTL3 accelerated the accumulation of Snail, which was essential for the persistence of oncogenic properties [56]. On the other hand, METTL3 also functioned on oncogenic noncoding RNAs [57]. Elevation of METTL3 contributed to enhance the expression of miR-6079 and lncRNA LINC00958, thereby potentiating aerobic glycolysis [57, 58]. Chemosensitivity of HCC cells modulated by METTL3 was also reported recently. Mechanically, METTL3 enhanced the stability of forkhead box O3 (FOXO3) mRNA in a METTL3-m6A-YTHDF1-dependent manner, subsequently promoting sorafenib sensitivity and inhibiting angiogenesis and autophagy-associated pathways [59].
Consistent with HCC, the tumor-promoting effect of METTL3 has been recently revealed in gallbladder cancer. METTL3 functionally reduced the protein level of PTEN via m6A-induced maturation of miR-92b-3p and subsequently activated the phosphatidylinositol 3′-kinase (PI3K)/protein kinase B (AKT) pathway [60].
CRC
Aberrant expression of METTL3 is considered to be a frequent event in the development of CRC, in which METTL3 mediates tumorigenesis by regulating target genes and pathways in an m6A-dependent manner. Li et al. reported that methylation of SRY-box 2 (SOX2) mRNA effectively prevented SOX2 mRNA from degradation, thereby provoking self-renewal, proliferation and migration of CRC cells [61]. In addition, upregulated cyclin E1 [62], activated miR-1246-SPRED2-MAPK axis [35] as well as inhibited SOCS2 [63] and yippee-like 5 (YPEL5) [31] were also induced by METTL3-catalyzed m6A modification. Rate-limiting enzymes of aerobic glycolysis were regulated by METTL3 as well. Overexpression of hexokinase 2 (HK2) and GLUT1 was attributed to the METTL3-m6A-IGF2BP2/3-dependent mechanism and further accelerated glycolysis to accelerate tumor growth [64]. Apart from the tumor-promoting effect, several studies had demonstrated the tumor suppressor role of METTL3 in cell migration, implying the role as a double-edged sword of METTL3 in CRC [65]. Moreover, METTL3 had dual effects on therapeutic resistance as well. Hypermethylation distinctly enhanced the general protein level of the p53 R273H mutant and leucine-rich repeat containing G protein-coupled receptor 5 (LGR5), thereby enhancing drug resistance [66, 67]. On the contrary, depletion of METTL3/14 strengthened the sensitivity to anti-PD-1 treatment through activating the interferon-γ (IFN-γ)/signal transducer and activator of transcription 1 (STAT1)/interferon regulatory factor 1 (IRF1) pathway [68].
Pancreatic cancer (PC)
Accumulated studies have identified METTL3 as an independent prognostic factor for PC [69]. Elevated expression of METTL3 enhanced tumor growth and metastasis by promoting maturation of miR-25-3 and activation of the PI3K/AKT pathway [70]. Meanwhile, hypermethylation contributed to chemo- and radio-resistance dependent on the dysregulation of MAPK cascades, ubiquitin modification and RNA process regulation [71]. Functional enrichment analysis further demonstrated that METTL3 could participate in the epinephrine stimulus response and neutrophil-mediated immune reaction, but the underlying mechanisms remain to be further studied [72].
Respiratory tumors
Nasopharyngeal cancer (NPC) and oral squamous cell carcinoma (OSCC)
It is well known that higher METTL3 is associated with advanced stage and distant metastasis, indicating the tumor-promoting role of METTL3 in NPC [73, 74]. METTL3 was reported to promote tumor growth and metastasis through functional regulation of NPC related genes. Zinc finger protein 750 (ZNF750) and enhancer of zeste homolog 2 (EZH2), which were identified as the tumor suppressor in NPC, could inhibit the growth and metastasis of NPC cells [75]. METTL3 contributed to the m6A modification of ZNF750 and consequently restrained cell apoptosis by inhibiting ZNF750/fibroblast growth factor 14 (FGF14) signaling [76]. METTL3 also inhibited the translation of EZH2, thereby increasing the expression of cyclin-dependent kinase inhibitor 1C (CDKN1C) to promote cell survival [74]. Moreover, Snail could promote tumor invasion and metastasis under the regulation of METTL3. Mechanistically, the mobility of NPC cells could be enhanced upregulated Snail through the METTL3-m6A-IGF2BP2-dependent mechanism [77].
Consistent with NPC, overexpression of METTL3 is associated with tumorigenesis of OSCC. Liu et al. revealed that METTL3 could promote OSCC growth and metastasis through the METTL3-m6A-IGF2BP1-BMI1 axis [78]. In addition, METTL3 strengthened the stability of MYC in a METTL3-m6A-YTHDF1-mediated manner, thereby stimulating tumor progression [79]. Taken together, METTL3 could play the pro-oncogenic role in OSCC.
Lung cancer
METTL3 has been previously identified as a potential target for the treatment of NSCLC. The aberrant expression of METTL3 contributes to the tumorigenesis of NSCLC in multiple ways. METTL3-mediated m6A modification potentiated translation of YAP mRNA in a METTL3-m6A-YTHDF3-dependent manner, subsequently promoting the generation of cancer stem cells [36]. METTL3 installed m6A deposition on lncRNA ABHD11-AS1 and then enhanced aerobic glycolysis to propel tumor progression [80]. Additionally, the functional activation of METTL3 could promote rapid tumor growth and EMT, which dependent of the activation of PI3K/AKT pathways and the overexpression of EZH2 [38, 80]. In particular, elimination of miR-143-3p and vasohibin-1 (VASH1) induced by METTL3 resulted in enhanced brain metastasis [81]. Moreover, METTL3 could positively mediate the autophagy related pathway and further induce gefitinib resistance, indicating the potential role of METTL3 in the treatment of NSCLC [82].
In addition to NSCLC, dataset analysis revealed an upregulated METTL3 in lung adenocarcinoma (LUAD), which was correlated with poor prognosis of patients [83]. Although METTL3 is regarded as a potential biomarker of LUAD, the specific mechanisms remain to be further explored.
Urological tumors
Renal cell carcinoma
The frequent alteration of METTL3 was reported in clear cell renal cell carcinoma (ccRCC), implying that METTL3 had potential predictive values in ccRCC [84]. Moreover, the close connection between METTL3 and critical biological processes was newly identified, including EMT, oxygen homeostasis, leukocyte migration, and so on [85, 86]. Since the underlying mechanisms are insufficient so far, it is necessary to conduct functional studies to explore the underlying mechanisms of METTL3 in ccRCC.
BC
Higher METTL3 is parallel with poor prognosis of patients with BC, suggesting the prognostic value and tumor promoting effect of METTL3 in BC [87]. It was demonstrated that METTL3 promoted rapid tumor growth, aggressive invasion, and self-renewal maintenance through different mechanisms. Abundant m6A deposition on the 3’UTR of CUB domain containing protein 1 (CDCP1) mRNA stimulated cell proliferation and transformation through the METTL3-m6A-YTHDF1 axis both in vitro and in vivo [88]. Similarly, the m6A modification within the 3’UTR of adhesive molecule integrin alpha-6 (ITGA6) mRNA permitted ITGA6 expression in an YTHDF1/3-dependent manner, thereby modulating the aggressive phenotype of BC [89]. In addition, METTL3 positively regulated the expression of endogenous AF4/FMR2 family member 4 (AFF4). The rapid tumor growth and aggressive invasion were ascribed to the AFF4/NF-κB/MYC pathway induced by METTL3, while self-renewal maintenance of BC stem cells was performed by the METTL3-AFF4-SOX2 axis [90, 91]. Tumor suppressor genes, such as SET domain containing 7 (SETD7) and Kruppel-like factor 4 (KLF4), are rapidly degraded under the regulation of METTL3 [92]. Furthermore, maturation of pri-miR221/222 associated with PTEN inhibition was conducted by METTL3, leading to poor prognosis in BC patients [34].
Prostate cancer (PCa)
METTL3 acts as an oncogene in PCa by promoting the pathogenesis and metastasis of tumor [93]. Mechanistically, METTL3 distinctly enhanced the expression of MYC, leading to the development and progression of PCa [94]. Besides, the METTL3-lymphoid enhancer-binding factor 1 (LEF1) axis activated the Wnt pathway in a METTL3-m6A-IGF2BP2-dependent manner, thereby promoting cell proliferation and migratory ability [95]. In addition, depletion of METTL3 disrupted the proliferation and immortality of tumor cells by inhibiting GLI1 in the sonic hedgehog (SHH) pathway [96]. Apart from tumor growth, bone metastasis was positively correlated with higher level of METTL3. Activation of human antigen R (HuR) induced by METTL3 resulted in the stability of integrin β1 (ITGB1) mRNA, thereby potentiating bone metastasis of PCa [97].
Neurological tumors
Glioblastoma (GBM)
m6A modification is definitively involved in the tumorigenesis of GBM, but the roles of METTL3 are controversial. Methylation enrichment analysis revealed that m6A deposition was usually concentrated in the transcripts mediating cell growth, self-renewal and metabolic regulation pathways [98]. Mechanistically, upregulated METTL3 maintained the activation of glioblastoma stem cell (GSC) through regulating the RNA editing enzyme and the YTHDF2-mediated RNA decay [99]. In addition, METTL3 also mediated tumorigenesis of GBM independent of the methylase catalysis. Direct interaction between METTL3 and histone-mediated modification elevated the translation of oncogenes, including SOX2, spalt-like transcription factor 2 (SALL2), oligodendrocyte lineage transcription factor 2 (OLIG2) and POU class 3 homeobox 2 (POU3F2) [99]. On the other hand, METTL3 enhanced resistance to γ-irradiation by regulating m6A modification of SOX2, indicating the important role of METTL3 in therapeutic resistance [100].
A negative correlation between GSC and m6A modification was recently illustrated. Depletion of METTL3/14 in turn enhanced the cell proliferation and self-renewal ability, thereby strengthening the tumorigenic properties of GSC [101]. METTL3 could also impair the proliferation and mobility of glioma cells, indicating the dual role of METTL3 in GBM [102].
Gynecologic tumors
Breast cancer
Previous studies reported that METTL3 was able to promote breast cancer cell proliferation by regulating the expression of BCL-2, hepatitis B X-interacting protein (HBXIP) and p21 through m6A [37, 103, 104]. Therapeutic resistance of breast cancer was also dependent on the m6A-based epitranscriptomic mechanism. Adriamycin resistance derived from METTL3 induced the maturation of pri-miRNA-221-3p [105], while tamoxifen resistance arose from METTL3-mediated overexpression of adenylate kinase 4 (AK4) [106]. Conversely, poor prognosis of the triple-negative breast cancer (TNBC) was associated with lower expression of METTL3, suggesting the tumor suppressing role of METTL3 [107]. Mechanistically, METTL3 inhibited the mobility of TNBC cells and adhesion to the cell extracellular matrix (ECM) by increasing m6A modification of collagen type III alpha Mechanistically, METTL3 inhibited the mobility of TNBC cells and adhesion to the cell extracellular matrix (ECM) by increasing m6A modification of collagen type III alpha 1 chain (COL3A1) [107]. Taken together, the differential functions of METTL3 were assessed in breast cancer, and various functions of METTL3 warrant further verification.
Ovarian cancer and endometrial cancer
METTL3 has been reported to promote tumor progression of ovarian cancer. METTL3 induced m6A modification in the transcripts of target genes in endometrioid epithelial ovarian cancer, including eIF3c, AXL, colony stimulating factor 1 (CSF-1), frizzled class receptor 10 (FZD10) and so on [108]. Upregulated AXL induced by methylation specifically promoted EMT to accelerate tumor progression [109]. METTL3 also participated in the regulation of oncogenic pathways in ovarian cancer. METTL3 downregulated the BCL-2-related apoptotic pathway to resist the apoptosis of ovarian cancer cells [110]. In addition, METTL3 mediated the activation of the AKT pathway via facilitating the maturation of miR-126-5p and further enhanced inhibition of PTEN, which was targeted by miR-126-5p [111].
Compared with ovarian cancer, METTL3 plays as a tumor suppressor in the pathogenesis of endometrial cancer. Mechanistically, reduced METTL3 could lead to activation of the AKT pathway, which promoted rapid proliferation of endometrial cancer cells [112].
Cervical cancer (CC)
METTL3 is identified as an independent prognostic factor in CC due to its distinct correlation with tumor progression and poor survival of patients [113, 114]. Increased METTL3 could bring to the rapid growth of CC through different mechanisms. Overexpression of METTL3 stabilized RAB2B mRNA to enhance cell proliferation in an IGF2BP3-dependent manner [115]. In addition, METTL3 was involved in m6A-regulated glycolysis, which was one of the critical hallmarks of tumor growth. METTL3 enhanced the stability of pyruvate dehydrogenase kinase 4 (PDK4) and HK2 mRNAs in a METTL3-m6A-YTHDF1-dependent manner, ultimately promoting tumor growth and chemoresistance [116].
Hematological malignancies
Acute myeloid leukemia (AML)
AML is an aggressive hematological malignancy (HM) characterized by various genetic abnormalities and epigenetic dysregulation [117, 118]. Compared with normal progenitor cells, METTL3 was more abundant in AML cells, coupled with declined cell differentiation and apoptosis both in vitro and in vivo [119, 120]. Mechanistically, increased expression of METTL3 promoted the translation of MYC, BCL-2 and PTEN mRNAs, while depressing the differentiation-promoting effect of AKT [119].
Other types of hematological malignancies
Recent studies demonstrate that METTL3 participates in the development and progression of B-cell-derived hematological malignancies. Aberrant expression of METTL3 in acute B lymphoblastic leukemia (B-ALL) was profiled recently. Low expression of METTL3 was found in the ETV6/RUNX1 (E/R)-positive cohort and associated with high recurrence rate [121]. Compared with B-ALL, upregulated METTL3 was identified in B cell lymphoma. In contrast to ALL, m6A modification was enriched in the regulation pathways of cell division and RNA metabolism but also referred to favorable survival of mantle cell lymphoma (MCL) [122]. In addition, elevation of METTL3 in diffuse large B-cell lymphoma (DLBCL) promoted the expression of pigment epithelium-derived factor (PEDF) transcripts, thereby activating the Wnt pathway to accelerate cell proliferation [123]. Since particular mechanism of METTL3 is not yet sufficient at present, further studies on METTL3 in HM still needed.
Head and neck squamous cell carcinoma and thyroid carcinoma
Emerging studies have demonstrated the pivotal role of METTL3 in head and neck squamous cell carcinoma (HNSCC) and thyroid carcinoma (TC). Dataset analysis revealed higher expression level of METTL3 in HNSCC, which was associated with poor OS and advanced tumor grade [124]. Similarly, METTL3 was highly expressed and closely associated with poor prognosis of TC [125]. Mechanistically, METTL3 regulated the expression of the HNF1 homeobox A (HNF1A) in a METTL3-m6A-IGF2BP2-dependent manner, eventually enhancing the migratory ability of tumor cells and activating the Wnt pathway [125].
Targeting METTL3 in antitumor therapy
Based on the diverse functions of METTL3, targeting METTL3 may bring a novel perspective for individualized therapy of cancer. Meanwhile, the development of METTL3 inhibitors is feasible depending on structural and functional features. The functional domain of METTL3 can be considered as the target of inhibitors [126]. In particular, the cofactor S-adenosyl-L-methionine (SAM) in METTL3 was responsible for methyl group transfer, in which the competitive binding of small molecular complexes could effectively reduce the activity of methyltransferase [127]. From another perspective, METTL3 was commonly related to drug resistance in tumors. Chemoresistance induced by METTL3 were detected in several kinds of cancers [41, 66, 82, 105], indicating that functional inhibition of METTL3 might restore the chemosensitivity of tumor cells [127]. In addition, depletion of METTL3/14 could strengthen the therapeutic effect of anti-PD-1 therapy through activating the IFN pathway [68]. Therefore, targeting METTL3 may be regarded as a promising approach of tumor targeted therapy.
Discussion
Multiple functions of METTL3-mediated m6A modification have been determined in the pathogenesis and progression of tumors (Table 1), which is realized through regulating the expression and function of target genes (Fig. 2). Given the tumor-promoting effects of METTL3, targeting METTL3 brings a bright future for tumor targeted therapy. Nevertheless, more investigations are still required to explore the novel functions of METTL3.

METTL3 regulates tumorigenesis and tumor progression by targeting downstream substrates. METTL3 is involved in many cellular processes, including tumor growth (a), therapeutic response (b), and metastasis (c)
Previous studies suggest that METTL3 usually regulates target genes in an m6A-dependent manner. In addition, biological functions of METTL3 can be independent of the methylase catalytic activity. For instance, the direct combination of METTL3 and ribosomes promoted the interaction between METTL3 and translation initiation, thereby enhancing the translation of mRNA and promoting tumor progression of lung cancer [128]. In addition, METTL3 also exhibited co-transcriptional interactions via regulating histone modification [129], indicating that METTL3 could interact with other types of epigenetic modification. On the other hand, mechanistic studies on METTL3 remain insufficient. For example, enrichment analysis uncovered the importance of METTL3 in glucose and lipid metabolism [130], but the specific mechanisms of METTL3 in tumor lipid metabolism were in infancy. Furthermore, mechanistic studies of METTL3 in some types of cancer, such as ccRCC, HNSCC and TC, were incomplete. Taken together, underlying mechanisms of METTL3 warrant further investigation.
In addition to exploring the novel functions of METTL3, fundamental researches also aim to achieve clinical transformation. Based on the tumor-promoting effect of METTL3, targeting METTL3 is expected to be an effective strategy for anti-tumor therapy. Functional inhibition of METTL3 was found to restore chemosensitivity of tumor cells in vitro, implying that inhibition of METTL3 might have potential value in vivo. In other words, application of METTL3 inhibitor is possible to produce anti-tumor effect and provide a solution for patients with refractory features. Investigations of METTL3-targeted therapies are an irresistible trend.
Conclusions
The importance of METTL3 in tumor progression has been broadly identified in human cancer. METTL3 mainly promotes cell proliferation, invasion, migration, metabolic reprogramming, and drug resistance in cancer. Further investigations on the underling mechanisms and targeted inhibitors of METTL3 are of great significance for deeper understanding of the relationship between m6A modification and human cancer.
Availability of data and materials
Not applicable.
Abbreviations
- ac4C:
-
N4-acetylcytidine
- AFF4:
-
AF4/FMR2 family member 4
- AKT:
-
Protein kinase B
- AK4:
-
Adenylate kinase 4
- AML:
-
Acute myeloid leukemia
- ARHGAP5:
-
Rho GTPase activating protein 5
- B-ALL:
-
Acute B lymphoblastic leukemia
- BATF2:
-
Basic leucine zipper ATF-like transcription factor 2
- BC:
-
Bladder cancer
- CBLL1:
-
Cbl proto-oncogene like 1
- CC:
-
Cervical cancer
- ccRCC:
-
Clear cell renal cell carcinoma
- CDCP1:
-
CUB domain containing protein 1
- CDKN1C:
-
Cyclin-dependent kinase inhibitor 1C
- CDS:
-
Coding sequence
- COL3A1:
-
Collagen type III alpha 1 chain
- CRC:
-
Colorectal cancer
- CSF-1:
-
Colony stimulating factor 1
- DLBCL:
-
Diffuse large B-cell lymphoma
- ECM:
-
Cell-extracellular matrix
- eIF3h:
-
Eukaryotic translation initiation factor 3 subunit h
- EMT:
-
Epithelial mesenchymal transition
- ENO2:
-
Enolase 2
- E/R:
-
ETV6/RUNX1
- ERK:
-
Extracellular-regulated kinase
- EZH2:
-
Enhancer of zeste homolog 2
- FGF14:
-
Fibroblast growth factor 14
- FOXO3:
-
Forkhead box O3
- FZD10:
-
Frizzled class receptor 10
- GBM:
-
Glioblastoma
- GC:
-
Gastric cancer
- GFI-1:
-
Growth factor independent 1
- GLUT4:
-
Solute carrier family 2 member 4
- GSC:
-
Glioblastoma stem cell
- HBXIP:
-
Hepatitis B X-interacting protein
- HCC:
-
Hepatocellular carcinoma
- HDGF:
-
Hepatoma-derived growth factor
- HK2:
-
Hexokinase 2
- HM:
-
Hematological malignancy
- hm5C:
-
5-hydroxymethylcytidine
- HNF1A:
-
HNF1 homeobox A
- HNSCC:
-
Head and neck squamous cell carcinoma
- HuR:
-
Human antigen R
- HUVECs:
-
Human umbilical vein endothelial cells
- IFN-γ:
-
Interferon-γ
- IRF1:
-
Interferon regulatory factor 1
- ITGA6:
-
Integrin alpha-6
- ITGB1:
-
Integrin β1
- KLF4:
-
Kruppel-like factor 4
- LEF1:
-
Lymphoid enhancer-binding factor 1
- LGR5:
-
Leucine-rich repeat-containing G protein-coupled receptor 5
- lncRNA:
-
Long non-coding RNA
- LUAD:
-
Lung adenocarcinoma
- m6A:
-
N6-adenosine methylation
- MAPK:
-
Mitogen-activated protein kinase
- m5C:
-
Cytosine hydroxylation
- MCL:
-
Mantle cell lymphoma
- METTL3:
-
Methyltransferase-like 3
- miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
- MTC:
-
N6-methyltransferase complex
- mTOR:
-
Mammalian target of the rapamycin
- NPC:
-
Nasopharyngeal carcinoma
- NSCLC:
-
Non-small cell lung cancer
- OLIG2:
-
Oligodendrocyte lineage transcription factor 2
- OSCC:
-
Oral squamous cell carcinoma
- PC:
-
Pancreatic cancer
- PCa:
-
Prostate cancer
- PDK4:
-
Pyruvate dehydrogenase kinase 4
- PEDF:
-
Pigment epithelium-derived factor
- PI3K:
-
Phosphatidylinositol 3'-kinase
- Pri-miRNA:
-
Primary miRNA
- POU3F2:
-
POU class 3 homeobox 2
- PTEN:
-
Phosphate and tension homology deleted on chromsome ten
- RBM15:
-
RNA-binding motif protein 15
- RDM1:
-
RAD52 motif 1
- SALL2:
-
Spalt-like transcription factor 2
- SAM:
-
S-adenosyl-L-methionine
- SETD7:
-
SET domain containing 7
- SHH:
-
Sonic hedgehog
- SOCS2:
-
Suppressor of cytokine signaling 2
- SOX2:
-
SRY-box 2
- SPRED2:
-
Sprouty-related EVH1 domain protein 2
- STAT1:
-
Signal transducer and activator of transcription 1
- TC:
-
Thyroid carcinoma
- TNBC:
-
Triple-negative breast cancer
- VASH1:
-
Vasohibin-1
- WTAP:
-
Wilms tumor 1-associated protein
- YPEL5:
-
Yippee-like 5
- ZC3H13:
-
Zinc finger CCCH-type containing 13
- ZMYM1:
-
Zinc finger MYM-type containing 1
- ZNF750:
-
Zinc finger protein 750
- α-SMA:
-
α-smooth muscle actin
References
Zheng HX, Zhang XS, Sui N. Advances in the profiling of N (6)-methyladenosine (m (6) a) modifications. Biotechnol Adv. 2020;45:107656.
Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet. 2013;29(2):108–15.
Sun WJ, Li JH, Liu S, Wu J, Zhou H, Qu LH, et al. RMBase: a resource for decoding the landscape of RNA modifications from high-throughput sequencing data. Nucleic Acids Res. 2016;44(D1):D259–65.
Yang X, Hu X, Liu J, Wang R, Zhang C, Han F, et al. N6-methyladenine modification in noncoding RNAs and its function in cancer. Biomark Res. 2020;8(1):61. https://doi.org/10.1186/s40364-020-00244-x.
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6(1):74.
Weng H, Huang H, Chen J. RNA N (6)-Methyladenosine modification in Normal and malignant hematopoiesis. Adv Exp Med Biol. 2019;1143:75–93.
Chen YT, Shen JY, Chen DP, Wu CF, Guo R, Zhang PP, et al. Identification of cross-talk between m (6) a and 5mC regulators associated with onco-immunogenic features and prognosis across 33 cancer types. J Hematol Oncol. 2020;13(1):22. https://doi.org/10.1186/s13045-020-00854-w.
Liang Z, Riaz A, Chachar S, Ding Y, Du H, Gu X. Epigenetic modifications of mRNA and DNA in plants. Mol Plant. 2020;13(1):14–30.
Jenjaroenpun P, Wongsurawat T, Wadley TD, Wassenaar TM, Liu J, Dai Q, et al. Decoding the epitranscriptional landscape from native RNA sequences. Nucleic Acids Res. 2021;49(2):e7.
Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–7.
Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, et al. ALKBH5-dependent m6A demethylation controls splicing and stability of long 3′-UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A. 2018;115(2):E325–33.
Zhao Y, Shi Y, Shen H, Xie W. m (6) A-binding proteins: the emerging crucial performers in epigenetics. J Hematol Oncol. 2020;13(1):35.
Schumann U, Shafik A, Preiss T. METTL3 gains R/W access to the Epitranscriptome. Mol Cell. 2016;62(3):323–4. https://doi.org/10.1016/j.molcel.2016.04.024.
Chen H, Gu L, Orellana EA, Wang Y, Guo J, Liu Q, et al. METTL4 is an snRNA m (6) am methyltransferase that regulates RNA splicing. Cell Res. 2020;30(6):544–7.
Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, et al. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–89.
Koh CWQ, Goh YT, Goh WSS. Atlas of quantitative single-base-resolution N (6)-methyl-adenine methylomes. Nat Commun. 2019;10(1):5636.
Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, et al. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8(1):284–96.
Knuckles P, Lence T, Haussmann IU, Jacob D, Kreim N, Carl SH, et al. Zc3h13/Flacc is required for adenosine methylation by bridging the mRNA-binding factor Rbm15/Spenito to the m (6) a machinery component Wtap/Fl (2) d. Genes Dev. 2018;32(5–6):415–29.
Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, et al. m (6) a RNA methylation promotes XIST-mediated transcriptional repression. Nature. 2016;537(7620):369–73. https://doi.org/10.1038/nature19342.
Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, et al. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002;4(3):222–31.
Zhang C, Chen Y, Sun B, Wang L, Yang Y, Ma D, et al. m (6) a modulates haematopoietic stem and progenitor cell specification. Nature. 2017;549(7671):273–6.
Scholler E, Weichmann F, Treiber T, Ringle S, Treiber N, Flatley A, et al. Interactions, localization, and phosphorylation of the m (6) a generating METTL3-METTL14-WTAP complex. RNA. 2018;24(4):499–512.
Lan Q, Liu PY, Haase J, Bell JL, Huttelmaier S, Liu T. The critical role of RNA m (6) a methylation in Cancer. Cancer Res. 2019;79(7):1285–92.
Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell. 2017;169(7):1187–200.
Liu S, Zhuo L, Wang J, Zhang Q, Li Q, Li G, et al. METTL3 plays multiple functions in biological processes. Am J Cancer Res. 2020;10(6):1631–46.
Shen H, Lan Y, Zhao Y, Shi Y, Jin J, Xie W. The emerging roles of N6-methyladenosine RNA methylation in human cancers. Biomark Res. 2020;8:24.
Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol. 2017;18(1):31–42.
Geula S, Moshitch-Moshkovitz S, Dominissini D, Mansour AA, Kol N, Salmon-Divon M, et al. Stem cells. m6A mRNA methylation facilitates resolution of naive pluripotency toward differentiation. Science. 2015;347(6225):1002–6.
Wang S, Chai P, Jia R, Jia R. Novel insights on m (6) a RNA methylation in tumorigenesis: a double-edged sword. Mol Cancer. 2018;17(1):101.
Choe J, Lin S, Zhang W, Liu Q, Wang L, Ramirez-Moya J, et al. mRNA circularization by METTL3-eIF3h enhances translation and promotes oncogenesis. Nature. 2018;561(7724):556–60.
Zhou D, Tang W, Xu Y, Xu Y, Xu B, Fu S, et al. METTL3/YTHDF2 m6A axis accelerates colorectal carcinogenesis through epigenetically suppressing YPEL5. Mol Oncol. 2021. https://doi.org/10.1002/1878-0261.12898.
Ma S, Chen C, Ji X, Liu J, Zhou Q, Wang G, et al. The interplay between m6A RNA methylation and noncoding RNA in cancer. J Hematol Oncol. 2019;12(1):121.
Alarcon CR, Lee H, Goodarzi H, Halberg N, Tavazoie SF. N6-methyladenosine marks primary microRNAs for processing. Nature. 2015;519(7544):482–5.
Han J, Wang JZ, Yang X, Yu H, Zhou R, Lu HC, et al. METTL3 promote tumor proliferation of bladder cancer by accelerating pri-miR221/222 maturation in m6A-dependent manner. Mol Cancer. 2019;18(1):110.
Peng W, Li J, Chen R, Gu Q, Yang P, Qian W, et al. Upregulated METTL3 promotes metastasis of colorectal Cancer via miR-1246/SPRED2/MAPK signaling pathway. J Exp Clin Cancer Res. 2019;38(1):393.
Jin D, Guo J, Wu Y, Du J, Yang L, Wang X, et al. m (6) a mRNA methylation initiated by METTL3 directly promotes YAP translation and increases YAP activity by regulating the MALAT1-miR-1914-3p-YAP axis to induce NSCLC drug resistance and metastasis. J Hematol Oncol. 2021;14(1):32.
Cai X, Wang X, Cao C, Gao Y, Zhang S, Yang Z, et al. HBXIP-elevated methyltransferase METTL3 promotes the progression of breast cancer via inhibiting tumor suppressor let-7g. Cancer Lett. 2018;415:11–9.
Wei W, Huo B, Shi X. miR-600 inhibits lung cancer via downregulating the expression of METTL3. Cancer Manag Res. 2019;11:1177–87.
Chen T, Hao YJ, Zhang Y, Li MM, Wang M, Han W, et al. Zhou Q: m (6) a RNA methylation is regulated by microRNAs and promotes reprogramming to pluripotency. Cell Stem Cell. 2015;16(3):289–301.
Yan J, Huang X, Zhang X, Chen Z, Ye C, Xiang W, et al. LncRNA LINC00470 promotes the degradation of PTEN mRNA to facilitate malignant behavior in gastric cancer cells. Biochem Biophys Res Commun. 2020;521(4):887–93.
Zhu L, Zhu Y, Han S, Chen M, Song P, Dai D, et al. Impaired autophagic degradation of lncRNA ARHGAP5-AS1 promotes chemoresistance in gastric cancer. Cell Death Dis. 2019;10(6):383.
Wang Q, Geng W, Guo H, Wang Z, Xu K, Chen C, et al. Emerging role of RNA methyltransferase METTL3 in gastrointestinal cancer. J Hematol Oncol. 2020;13(1):57.
Liu T, Yang S, Sui J, Xu SY, Cheng YP, Shen B, et al. Dysregulated N6-methyladenosine methylation writer METTL3 contributes to the proliferation and migration of gastric cancer. J Cell Physiol. 2020;235(1):548–62.
Yang DD, Chen ZH, Yu K, Lu JH, Wu QN, Wang Y, et al. METTL3 promotes the progression of gastric Cancer via targeting the MYC pathway. Front Oncol. 2020;10:115.
Yang Z, Jiang X, Li D, Jiang X. HBXIP promotes gastric cancer via METTL3-mediated MYC mRNA m6A modification. Aging (Albany NY). 2020;12(24):24967–82.
He H, Wu W, Sun Z, Chai L. MiR-4429 prevented gastric cancer progression through targeting METTL3 to inhibit m (6) A-caused stabilization of SEC62. Biochem Biophys Res Commun. 2019;517(4):581–7.
Wang XH, Jiang ZH, Yang HM, Zhang Y, Xu LH. Hypoxia-induced FOXO4/LDHA axis modulates gastric cancer cell glycolysis and progression. Clin Transl Med. 2021;11(1):e279.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m (6) a modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69(7):1193–205.
Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18(1):142.
Xie JW, Huang XB, Chen QY, Ma YB, Zhao YJ, Liu LC, et al. Li P: m (6) a modification-mediated BATF2 acts as a tumor suppressor in gastric cancer through inhibition of ERK signaling. Mol Cancer. 2020;19(1):114.
Sun Y, Li S, Yu W, Zhao Z, Gao J, Chen C, et al. N (6)-methyladenosine-dependent pri-miR-17-92 maturation suppresses PTEN/TMEM127 and promotes sensitivity to everolimus in gastric cancer. Cell Death Dis. 2020;11(10):836.
Liu GM, Zeng HD, Zhang CY, Xu JW. Identification of METTL3 as an adverse prognostic biomarker in hepatocellular carcinoma. Dig Dis Sci. 2020.
Wu X, Zhang X, Tao L, Dai X, Chen P. Prognostic value of an m6A RNA methylation regulator-based signature in patients with hepatocellular carcinoma. Biomed Res Int. 2020;2020:2053902.
Chen M, Wei L, Law CT, Tsang FH, Shen J, Cheng CL, et al. RNA N6-methyladenosine methyltransferase-like 3 promotes liver cancer progression through YTHDF2-dependent posttranscriptional silencing of SOCS2. Hepatology. 2018;67(6):2254–70.
Chen SL, Liu LL, Wang CH, Lu SX, Yang X, He YF, et al. Loss of RDM1 enhances hepatocellular carcinoma progression via p53 and Ras/Raf/ERK pathways. Mol Oncol. 2020;14(2):373–86.
Xu H, Wang H, Zhao W, Fu S, Li Y, Ni W, et al. SUMO1 modification of methyltransferase-like 3 promotes tumor progression via regulating snail mRNA homeostasis in hepatocellular carcinoma. Theranostics. 2020;10(13):5671–86. https://doi.org/10.7150/thno.42539.
Zuo X, Chen Z, Gao W, Zhang Y, Wang J, Wang J, et al. M6A-mediated upregulation of LINC00958 increases lipogenesis and acts as a nanotherapeutic target in hepatocellular carcinoma. J Hematol Oncol. 2020;13(1):5. https://doi.org/10.1186/s13045-019-0839-x.
Lin Y, Wei X, Jian Z, Zhang X. METTL3 expression is associated with glycolysis metabolism and sensitivity to glycolytic stress in hepatocellular carcinoma. Cancer Med. 2020;9(8):2859–67.
Lin Z, Niu Y, Wan A, Chen D, Liang H, Chen X, et al. RNA m (6) a methylation regulates sorafenib resistance in liver cancer through FOXO3-mediated autophagy. EMBO J. 2020;39(12):e103181.
Lin R, Zhan M, Yang L, Wang H, Shen H, Huang S, et al. Deoxycholic acid modulates the progression of gallbladder cancer through N (6)-methyladenosine-dependent microRNA maturation. Oncogene. 2020;39(26):4983–5000.
Li T, Hu PS, Zuo Z, Lin JF, Li X, Wu QN, et al. METTL3 facilitates tumor progression via an m (6) A-IGF2BP2-dependent mechanism in colorectal carcinoma. Mol Cancer. 2019;18(1):112.
Zhu W, Si Y, Xu J, Lin Y, Wang JZ, Cao M, et al. Methyltransferase like 3 promotes colorectal cancer proliferation by stabilizing CCNE1 mRNA in an m6A-dependent manner. J Cell Mol Med. 2020;24(6):3521–33.
Xu J, Chen Q, Tian K, Liang R, Chen T, Gong A, et al. m6A methyltransferase METTL3 maintains colon cancer tumorigenicity by suppressing SOCS2 to promote cell proliferation. Oncol Rep. 2020;44(3):973–86.
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X, et al. Hong J: m (6) A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer. 2020;19(1):72.
Deng R, Cheng Y, Ye S, Zhang J, Huang R, Li P, et al. Deng Y: m (6) a methyltransferase METTL3 suppresses colorectal cancer proliferation and migration through p38/ERK pathways. Onco Targets Ther. 2019;12:4391–402.
Zhang Y, Kang M, Zhang B, Meng F, Song J, Kaneko H, et al. Tang B: m (6) a modification-mediated CBX8 induction regulates stemness and chemosensitivity of colon cancer via upregulation of LGR5. Mol Cancer. 2019;18(1):185.
Uddin MB, Roy KR, Hosain SB, Khiste SK, Hill RA, Jois SD, et al. An N (6)-methyladenosine at the transited codon 273 of p53 pre-mRNA promotes the expression of R273H mutant protein and drug resistance of cancer cells. Biochem Pharmacol. 2019;160:134–45.
Wang L, Hui H, Agrawal K, Kang Y, Li N, Tang R, et al. m (6) a RNA methyltransferases METTL3/14 regulate immune responses to anti-PD-1 therapy. EMBO J. 2020;39(20):e104514.
Hou J, Wang Z, Li H, Zhang H, Luo L. Gene signature and identification of clinical trait-related m (6) a regulators in pancreatic Cancer. Front Genet. 2020;11:522.
Zhang J, Bai R, Li M, Ye H, Wu C, Wang C, et al. Excessive miR-25-3p maturation via N (6)-methyladenosine stimulated by cigarette smoke promotes pancreatic cancer progression. Nat Commun. 2019;10(1):1858.
Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52(2):621–9.
Xia T, Wu X, Cao M, Zhang P, Shi G, Zhang J, et al. The RNA m6A methyltransferase METTL3 promotes pancreatic cancer cell proliferation and invasion. Pathol Res Pract. 2019;215(11):152666.
Liu ZF, Yang J, Wei SP, Luo XG, Jiang QS, Chen T, et al. Upregulated METTL3 in nasopharyngeal carcinoma enhances the motility of cancer cells. Kaohsiung J Med Sci. 2020;36(11):895–903.
Meng QZ, Cong CH, Li XJ, Zhu F, Zhao X, Chen FW. METTL3 promotes the progression of nasopharyngeal carcinoma through mediating M6A modification of EZH2. Eur Rev Med Pharmacol Sci. 2020;24(8):4328–36.
Yang H, Pan L, Xu C, Zhang Y, Li K, Chen S, et al. Overexpression of tumor suppressor gene ZNF750 inhibits oral squamous cell carcinoma metastasis. Oncol Lett. 2017;14(5):5591–6.
Zhang P, He Q, Lei Y, Li Y, Wen X, Hong M, et al. m (6) A-mediated ZNF750 repression facilitates nasopharyngeal carcinoma progression. Cell Death Dis. 2018;9(12):1169.
Yu X, Zhao H, Cao Z. The m6A methyltransferase METTL3 aggravates the progression of nasopharyngeal carcinoma through inducing EMT by m6A-modified snail mRNA. Minerva Med. 2020. https://doi.org/10.23736/S0026-4806.20.06653-7.
Liu L, Wu Y, Li Q, Liang J, He Q, Zhao L, et al. METTL3 promotes tumorigenesis and metastasis through BMI1 m (6) a methylation in Oral squamous cell carcinoma. Mol Ther. 2020;28(10):2177–90. https://doi.org/10.1016/j.ymthe.2020.06.024.
Zhao W, Cui Y, Liu L, Ma X, Qi X, Wang Y, et al. METTL3 facilitates Oral squamous cell carcinoma tumorigenesis by enhancing c-Myc stability via YTHDF1-mediated m (6) a modification. Mol Ther Nucleic Acids. 2020;20:1–12.
Chen WW, Qi JW, Hang Y, Wu JX, Zhou XX, Chen JZ, et al. Simvastatin is beneficial to lung cancer progression by inducing METTL3-induced m6A modification on EZH2 mRNA. Eur Rev Med Pharmacol Sci. 2020;24(8):4263–70.
Wang H, Deng Q, Lv Z, Ling Y, Hou X, Chen Z, et al. N6-methyladenosine induced miR-143-3p promotes the brain metastasis of lung cancer via regulation of VASH1. Mol Cancer. 2019;18(1):181.
Liu S, Li Q, Li G, Zhang Q, Zhuo L, Han X, et al. The mechanism of m (6) a methyltransferase METTL3-mediated autophagy in reversing gefitinib resistance in NSCLC cells by beta-elemene. Cell Death Dis. 2020;11(11):969.
Li F, Wang H, Huang H, Zhang L, Wang D, Wan Y. m6A RNA methylation regulators participate in the malignant progression and have clinical prognostic value in lung adenocarcinoma. Front Genet. 2020;11:994.
Chen J, Yu K, Zhong G, Shen W. Identification of a m (6) a RNA methylation regulators-based signature for predicting the prognosis of clear cell renal carcinoma. Cancer Cell Int. 2020;20:157.
Zhou J, Wang J, Hong B, Ma K, Xie H, Li L, et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma - a retrospective study using TCGA database. Aging (Albany NY). 2019;11(6):1633–47.
Wang J, Zhang C, He W, Gou X. Effect of m (6) a RNA methylation regulators on malignant progression and prognosis in renal clear cell carcinoma. Front Oncol. 2020;10:3.
Han Y, Zheng Q, Tian Y, Ji Z, Ye H. Identification of a nine-gene panel as a prognostic indicator for recurrence with muscle-invasive bladder cancer. J Surg Oncol. 2019;119(8):1145–54.
Yang F, Jin H, Que B, Chao Y, Zhang H, Ying X, et al. Dynamic m (6) a mRNA methylation reveals the role of METTL3-m (6) A-CDCP1 signaling axis in chemical carcinogenesis. Oncogene. 2019;38(24):4755–72.
Jin H, Ying X, Que B, Wang X, Chao Y, Zhang H, et al. N (6)-methyladenosine modification of ITGA6 mRNA promotes the development and progression of bladder cancer. EBioMedicine. 2019;47:195–207.
Cheng M, Sheng L, Gao Q, Xiong Q, Zhang H, Wu M, et al. The m (6) a methyltransferase METTL3 promotes bladder cancer progression via AFF4/NF-kappaB/MYC signaling network. Oncogene. 2019;38(19):3667–80.
Gao Q, Zheng J, Ni Z, Sun P, Yang C, Cheng M, et al. The m (6) a methylation-regulated AFF4 promotes self-renewal of bladder Cancer stem cells. Stem Cells Int. 2020;2020:8849218.
Xie H, Li J, Ying Y, Yan H, Jin K, Ma X, et al. METTL3/YTHDF2 m (6) a axis promotes tumorigenesis by degrading SETD7 and KLF4 mRNAs in bladder cancer. J Cell Mol Med. 2020;24(7):4092–104.
Tao Z, Zhao Y, Chen X. Role of methyltransferase-like enzyme 3 and methyltransferase-like enzyme 14 in urological cancers. Peer J. 2020;8:e9589.
Yuan Y, Du Y, Wang L, Liu X. The M6A methyltransferase METTL3 promotes the development and progression of prostate carcinoma via mediating MYC methylation. J Cancer. 2020;11(12):3588–95. https://doi.org/10.7150/jca.42338.
Ma XX, Cao ZG, Zhao SL. m6A methyltransferase METTL3 promotes the progression of prostate cancer via m6A-modified LEF1. Eur Rev Med Pharmacol Sci. 2020;24(7):3565–71.
Cai J, Yang F, Zhan H, Situ J, Li W, Mao Y, et al. RNA m (6) a methyltransferase METTL3 promotes the growth of prostate Cancer by regulating hedgehog pathway. Onco Targets Ther. 2019;12:9143–52.
Li E, Wei B, Wang X, Kang R. METTL3 enhances cell adhesion through stabilizing integrin beta1 mRNA via an m6A-HuR-dependent mechanism in prostatic carcinoma. Am J Cancer Res. 2020;10(3):1012–25.
Li F, Yi Y, Miao Y, Long W, Long T, Chen S, et al. N (6)-Methyladenosine modulates nonsense-mediated mRNA decay in human Glioblastoma. Cancer Res. 2019;79(22):5785–98.
Visvanathan A, Patil V, Abdulla S, Hoheisel JD, Somasundaram K. N (6)-Methyladenosine Landscape of Glioma Stem-Like Cells: METTL3 Is Essential for the Expression of Actively Transcribed Genes and Sustenance of the Oncogenic Signaling. Genes. 2019;10(2):141.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m (6) a modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37(4):522–33.
Cui Q, Shi H, Ye P, Li L, Qu Q, Sun G, et al. Shi Y: m (6) a RNA methylation regulates the self-renewal and tumorigenesis of Glioblastoma stem cells. Cell Rep. 2017;18(11):2622–34.
Li F, Zhang C, Zhang G. m6A RNA methylation controls proliferation of human Glioma cells by influencing cell apoptosis. Cytogenet Genome Res. 2019;159(3):119–25.
Wang H, Xu B, Shi J. N6-methyladenosine METTL3 promotes the breast cancer progression via targeting Bcl-2. Gene. 2020;722:144076.
Cheng L, Zhang X, Huang YZ, Zhu YL, Xu LY, Li Z, et al. Metformin exhibits antiproliferation activity in breast cancer via miR-483-3p/METTL3/m (6) a/p21 pathway. Oncogenesis. 2021;10(1):7.
Pan X, Hong X, Li S, Meng P, Xiao F. METTL3 promotes adriamycin resistance in MCF-7 breast cancer cells by accelerating pri-microRNA-221-3p maturation in a m6A-dependent manner. Exp Mol Med. 2021;53(1):91–102.
Liu X, Gonzalez G, Dai X, Miao W, Yuan J, Huang M, et al. Adenylate kinase 4 modulates the resistance of breast Cancer cells to Tamoxifen through an m (6) A-based Epitranscriptomic mechanism. Mol Ther. 2020;28(12):2593–604.
Shi Y, Zheng C, Jin Y, Bao B, Wang D, Hou K, et al. Reduced expression of METTL3 promotes metastasis of triple-negative breast Cancer by m6A methylation-mediated COL3A1 up-regulation. Front Oncol. 2020;10:1126.
Ma Z, Li Q, Liu P, Dong W, Zuo Y. METTL3 regulates m6A in endometrioid epithelial ovarian cancer independently of METTl14 and WTAP. Cell Biol Int. 2020;44(12):2524–31.
Hua W, Zhao Y, Jin X, Yu D, He J, Xie D, et al. METTL3 promotes ovarian carcinoma growth and invasion through the regulation of AXL translation and epithelial to mesenchymal transition. Gynecol Oncol. 2018;151(2):356–65.
Liang S, Guan H, Lin X, Li N, Geng F, Li J. METTL3 serves an oncogenic role in human ovarian cancer cells partially via the AKT signaling pathway. Oncol Lett. 2020;19(4):3197–204.
Bi X, Lv X, Liu D, Guo H, Yao G, Wang L, et al. METTL3-mediated maturation of miR-126-5p promotes ovarian cancer progression via PTEN-mediated PI3K/Akt/mTOR pathway. Cancer Gene Ther. 2020. https://doi.org/10.1038/s41417-020-00222-3.
Liu J, Eckert MA, Harada BT, Liu SM, Lu Z, Yu K, et al. m (6) a mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol. 2018;20(9):1074–83.
Wu F, Zhang Y, Fang Y, Ma S, Zheng H, Liu K, et al. Elevated expression of inhibitor of apoptosis-stimulating protein of p53 (iASPP) and methyltransferase-like 3 (METTL3) correlate with poor prognosis in FIGO Ib1-IIa squamous cell cervical Cancer. J Cancer. 2020;11(9):2382–9.
Wang Q, Guo X, Li L, Gao Z, Su X, Ji M, et al. N (6)-methyladenosine METTL3 promotes cervical cancer tumorigenesis and Warburg effect through YTHDF1/HK2 modification. Cell Death Dis. 2020;11(10):911.
Hu Y, Li Y, Huang Y, Jin Z, Wang C, Wang H, et al. METTL3 regulates the malignancy of cervical cancer via post-transcriptional regulation of RAB2B. Eur J Pharmacol. 2020;879:173134.
Li Z, Peng Y, Li J, Chen Z, Chen F, Tu J, et al. N (6)-methyladenosine regulates glycolysis of cancer cells through PDK4. Nat Commun. 2020;11(1):2578.
Zhou X, Fang X, Jiang Y, Geng L, Li X, Li Y, et al. Klotho, an anti-aging gene, acts as a tumor suppressor and inhibitor of IGF-1R signaling in diffuse large B cell lymphoma. J Hematol Oncol. 2017;10(1):37.
Zhou X, Zhan L, Huang K, Wang X. The functions and clinical significance of circRNAs in hematological malignancies. J Hematol Oncol. 2020;13(1):138.
Vu LP, Pickering BF, Cheng Y, Zaccara S, Nguyen D, Minuesa G, et al. The N (6)-methyladenosine (m (6) a)-forming enzyme METTL3 controls myeloid differentiation of normal hematopoietic and leukemia cells. Nat Med. 2017;23(11):1369–76.
Guirguis AA, Liddicoat BJ, Dawson MA. The old and the new: DNA and RNA methylation in normal and malignant hematopoiesis. Exp Hematol. 2020;90:1–11.
Sun C, Chang L, Liu C, Chen X, Zhu X. The study of METTL3 and METTL14 expressions in childhood ETV6/RUNX1-positive acute lymphoblastic leukemia. Mol Genet Genomic Med. 2019;7(10):e00933.
Zhang W, He X, Hu J, Yang P, Liu C, Wang J, et al. Dysregulation of N (6)-methyladenosine regulators predicts poor patient survival in mantle cell lymphoma. Oncol Lett. 2019;18(4):3682–90.
Cheng Y, Fu Y, Wang Y, Wang J. The m6A methyltransferase METTL3 is functionally implicated in DLBCL development by regulating m6A modification in PEDF. Front Genet. 2020;11:955.
Zhao X, Cui L. Development and validation of a m (6) a RNA methylation regulators-based signature for predicting the prognosis of head and neck squamous cell carcinoma. Am J Cancer Res. 2019;9(10):2156–69.
Wang K, Jiang L, Zhang Y, Chen C. Progression of thyroid carcinoma is promoted by the m6A methyltransferase METTL3 through regulating m (6) a methylation on TCF1. Onco Targets Ther. 2020;13:1605–12.
Zeng C, Huang W, Li Y, Weng H. Roles of METTL3 in cancer: mechanisms and therapeutic targeting. J Hematol Oncol. 2020;13(1):117.
Bedi RK, Huang D, Eberle SA, Wiedmer L, Sledz P, Caflisch A. Small-molecule inhibitors of METTL3, the major human Epitranscriptomic writer. Med Chem. 2020;15(9):744–8.
Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m (6) a methyltransferase METTL3 promotes translation in human Cancer cells. Mol Cell. 2016;62(3):335–45.
Li Y, Xia L, Tan K, Ye X, Zuo Z, Li M, et al. N (6)-Methyladenosine co-transcriptionally directs the demethylation of histone H3K9me2. Nat Genet. 2020;52(9):870–7.
Zhao Z, Meng J, Su R, Zhang J, Chen J, Ma X, et al. Epitranscriptomics in liver disease: basic concepts and therapeutic potential. J Hepatol. 2020;73(3):664–79.
Acknowledgments
Not applicable.
Funding
This study was supported by National Natural Science Foundation (No.81800194, No.82070203, No.81770210, No.81473486 and No.81270598); Key Research and Development Program of Shandong Province (No.2018CXGC1213); Development Project of Youth Innovation Teams in Colleges and Universities of Shandong Province (No.2020KJL006); China Postdoctoral Science Foundation (No.2020 M672103); Technology Development Projects of Shandong Province (No.2017GSF18189); Translational Research Grant of NCRCH (No.2021WWB02, No.2020ZKMB01); Shandong Provincial Natural Science Foundation (No.ZR2018BH011); Technology Development Project of Jinan City (No.201805065); Taishan Scholars Program of Shandong Province; Shandong Provincial Engineering Research Center of Lymphoma; Academic Promotion Programme of Shandong First Medical University (No. 2019QL018, No.2020RC006).
Author information
Authors and Affiliations
Contributions
XW and XZ provided direction and guidance throughout the preparation of this manuscript. YC, RF, TL and XC wrote and edited the manuscript. XW and XZ reviewed and revised the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors consent to publication.
Competing interests
The authors declare that they have no potential conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Cai, Y., Feng, R., Lu, T. et al. Novel insights into the m6A-RNA methyltransferase METTL3 in cancer. Biomark Res 9, 27 (2021). https://doi.org/10.1186/s40364-021-00278-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-021-00278-9