
Abstract
Background
A feature of myeloproliferative neoplasia is transforming to more aggressive and malignant myeloid neoplasia, including acute myeloid leukemia. Different pathogenesis mechanisms participate in transformation, including transformation of existing potential preleukemic clones, since JAK2-mutant myeloproliferative neoplasms often transform to JAK2 wild-type acute myeloid leukemia.
Case presentation
Here, we present an 80 year old man with a JAK2-V617F mutant primary myelofibrosis. After 10 months the disease transform into a Philadelphia chromosome positive acute myeloid leukemia, detecting the cytogenetic aberration; t(9;22)(q34;q22) encoding the rare BCR-ABL1 fusion gene; e6a2. The patient had treatment response to tyrosine kinases, illustrating the potential benefits of such approach in treating these patients subset.
Conclusion
The case illustrates the potential of leukemic transformation to Philadelphia chromosome positive myeloid malignancies from potential existing preleukemic clones, and the awareness of such an evolution among patients with myeloproliferative neoplasms. Tyrosine kinases have potential effect also in patients presenting without chronic myeloid leukemia and with rare BCR-ABL1 fusion transcripts, and should probably be a part of the treatment approach.
Similar content being viewed by others


Introduction
The Philadelphia chromosome (Ph) is a diagnostic feature for chronic myeloid leukemia (CML); Ph+ CML is identified by the genetic translocation t(9;22)(q34;q11.2) [1], that involve the fusion of the Abelson oncogene (ABL1) with the breakpoint cluster region (BCR) gene. The malignant transformation is hence caused by the acquisition of the fusion tyrosine kinase BCR-ABL1 in a hematopoietic stem cell, pivotal in transforming of the stem cell into a leukemic stem cell (LSC) that self-renews, proliferates, and differentiates to give rise to Ph+ acute myeloid leukemia (AML), acute lymphoblastic leukemia (ALL), or more frequently CML [2, 3]. The complete carcinogenesis of BCR-ABL1 is incompletely understood, since as many as 10% of healthy individuals may have this fusion transcript [4], presence of low level of BCR-ABL1 transcripts seems to increase with increasing age [5], and in those individuals that develop BCR-ABL1 positive CML nearly half of the patients have additional mutations frequently found in myeloid disorders [6]. However, Ph+ AML developing after a previous Ph− myeloproliferative neoplasia (MPN) or myelodysplastic syndrome (MDS) is rarely described. In this report we describe a patient with JAK2V617F positive primary myelofibrosis (PMF) that progressed to secondary Ph+ AML with gain of the rare BCR-ABL1 fusion transcript e6a2.
Case report
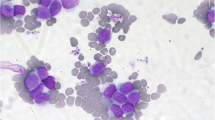
The patient was an 80 years old man whose previous medical record included diabetes mellitus type 2, atrial fibrillation, cerebrovascular disease, polymyalgia rheumatica and osteoporosis. His regular prescriptions included metformin, warfarin and prednisolone. He was admitted to hospital with a hematoma at his right thigh after a minor trauma. At clinical examination palpable splenomegaly at inspiration was detected. Standard peripheral blood tests revealed hemoglobin (Hgb) 10.3 g/dL (normal range 13.4–17.0), platelets > 2000 × 109/L (150–450), white blood cell count (WBC) 23× 109/L [4,5,6,7,8,9,10,11] and lactate dehydrogenase (LDH) 366 U/L (115–255). Microscopy of the peripheral blood smear revealed a leukoerythroblastic picture including nucleated erythrocytes and promyelocytes as well as myelocytes but no blasts. The bone marrow (BM) smear demonstrated increased cellularity with increased megakaryocytes and 4% myeloblasts; the BM biopsy confirmed this and showed in addition focal bundles of reticulin fibers and in addition proliferation of megakaryocytes with classic atypia, including small size and hypolobulation. There was reduced myelopoiesis, although without evidence of proliferation of immature cells (Fig. 1). Mutational analysis for JAK2V617F derived from peripheral blood mononuclear cell (PBMC) was positive with an allele burden of 0.6%, and a real time polymerase chain reaction (RT-qPCR) detecting the most common BCR-ABL1 fusions; e13a2/e14a2/e1a2/e19a2, was negative. Hence, our patient fulfilled all WHO major criteria for a Ph− myeloproliferative neoplasia; namely primary myelofibrosis (PMF); with (i) megakaryocytic proliferation and reticulin fibrosis, (ii) the presence of JAK2 mutation and (iii) not fulfilling the criteria for other myeloid malignancies. In addition to all the five minor criteria were also fulfilled with (i) anemia, (ii) leukocytosis, (iii) palpable splenomegaly, (iv) increased LDH and (v) leukoerythroblastosis [7].

Histopathological examination in hematoxylin-eosin. a: BM at the initial diagnosis of PMF, showing hypercellularity with some fat cells left (circle), clusters of mature granulocytes (closed arrow), singular and groups of atypical megakaryocytes (open arrow). b: BM at follow up, showing maximal cellularity without any fat cells left, no organized hematopoiesis with only singular mature granulocytes, a few atypical megakaryocytes, and clusters of immature cells and blast cells (asterisk). (Pictures: Friedemann Leh, Department of Pathology, Haukeland University Hospital)
Cytostatic treatment with hydroxyurea was initiated at a dose of 2500 mg daily. The dose was reduced after some weeks due to severe headache. During the next 6 months the platelet count was reduced by hydroxyurea, although with difficulties in achieving satisfactory platelet counts without imposing neutropenia as a side effect. A shift of treatment to anagrelide (1 mg/day) was attempted, however had to be disrupted due to unacceptable side effects with headache, heart palpitations and back pain.
Seven months after the diagnosis of PMF the routine peripheral blood smear showed an increasing blast percentage and flow-cytometric analysis verified 22% immature cells. However, a BM biopsy demonstrated only 7% blasts. The diagnosis of PMF was therefore maintained and hydroxyurea continued.
Six weeks later the patient was admitted to hospital because of increasing anemia (Hgb 6.7 g/dL), leukocytosis (25.2 × 109/L) and CRP 35 mg/L. Peripheral blood smear showed 43% myeloblasts, confirmed by flow cytometric analysis. BM biopsy demonstrated a hypercellular BM without organized hematopoiesis, absence of erythropoiesis and increased myelopoiesis with relatively few mature granulocytes, focal nodes of immature cells and blast cells and significantly increased amount of reticulin fibers (Fig. 1). The findings were consistent with transformation from PMF to AML.
Surprisingly the cytogenetic analysis by conventional G-banding detected the Philadelphia chromosome with the translocation t(9;22)(q34;q22) in all ten metaphases analyzed. This was confirmed by BCR-ABL1 fusion in 57% of the cells by fluorescent in situ hybridization (FISH) analysis, (Fig. 2) and to be the BCR-ABL1 e6a2 transcript variant by sequencing of positive product from reverse transcriptase PCR. RT-qPCR confirmed the existence of an e6a2 BCR-ABL1 transcript, with a BCR-ABL1/GUSB ratio of 69%. Retrospectively, the e6a2 transcript was also detected at the initial diagnosis of PMF, although only with a BCR-ABL1/GUSB ratio of 14% (Fig. 3). The JAK2V617F mutation could not be detected at the point of AML diagnosis.

Chromosome analysis for detection of Philadelphia chromosome t(9; 22) in the patient. G-banding of diagnostic sample revealed a t(9;22)(q34;q11) (a) resulting in fusion of BCR and ABL verified by double fusion FISH from Vysis (b) and multiples RT-PCR (c). The multiplex (1 + 2) showed a 796 bp internal control (BCR) and 1051 bp BCR-ABL1 product generated by primer-combination BCR exon 1 and ABL1 exon 3 (2). The size gave suspicion of an e6a2 fusion verified by sanger sequencing using primer combination 2 (reverse sequence). (Picture: Randi Hovland, Haukeland University Hospital)

Development of genetically, hematological and biochemical values during the follow up of the patient. The diagnosis of AML is set to time point 0, and the negative values indicate days before the AML diagnosis, and the positive value days after the AML diagnosis
We started treatment with dasatinib 100 mg once daily combined with hydroxyurea (500 mg/day) for the first 22 days and valproic acid (300 mg + 600 mg/day) for the first 20 days [8]. Peripheral blood smears after 11 and 19 days showed no myeloblasts. The BCR-ABL1/GUSB ratio fell from 52 to 7.6% after 3 months (Fig. 3). Because of increasing fatigability, the patient was referred to an echocardiography that showed a pericardial effusion of 1.8 cm at the level of the right atrium. This was regarded as an adverse effect of dasatinib [9], and serous effusions trigged by dasatinib is suggested to be predictive for therapy efficiency in CML [10, 11]. Due to risk of recurrent pericardial effusion combined with the general condition of the patient dasatinib therapy was discontinued after a treatment period of approximately 4 months.
Five days after dasatinib discontinuation, treatment with imatinib 400 mg daily was started, and a repeated echocardiography 2 weeks later demonstrated reduction in the pericardial effusion to 1.0 cm. At his last visit 2 weeks after initiating imatinib the patient reported that he had gradually improved. He experienced two episodes of diarrhea, but no other side effects of imatinib. Peripheral blood test showed Hgb 11.5 g/dL, WBC 5.3 × 109/L, neutrophils 2.8 × 109/L, and platelets 161 × 109/L. He continued imatinib therapy for 6 months and had detectable although stable levels of BCR-ABL1/GUSB ratio measured by qPCR during this period (Fig. 3). Thereafter the patient developed increasing abdominal pain and diarrhea. A CT scan demonstrated a tumor in the pancreatic head, radiological consistent with adenocarcinoma. The patient was considered inoperable and unable to tolerate chemotherapy, and the tumor was not biopsied. He continued the imatininb treatment for an additional period of few weeks and died shortly thereafter.
Discussion
Myeloproliferative neoplasms (MPNs) comprise a group of clonal stem cell disorders characterized by a high prevalence of mutations in one of the three genes JAK2, CALR or MPL [12], overproduction of mature blood cells, and variable rates of transformation to AML [13]. MPN comprise a wider spectrum of mutations, and the combination of mutations seems to predict progression rate to more aggressive myeloid neoplasms like AML [14]. Our patient was initially diagnosed with PMF, as he fulfilled all the major and minor diagnostic criteria as stated by the WHO guidelines [7]. He received cytoreductive treatment for lowering of the platelets count [15]. Accordingly, his platelet counts fell, and he had no sign of thrombotic disease.
The risk of transformation to more aggressive myeloid malignancies should always be kept in mind during follow up of for MPN patients. Older age by itself seems to be the main independent risk factor for transformation [16], although genotoxic therapy also seems to increase the risk [16]. The risk is higher for patients with PMF than for patients diagnosed with polycythemia vera (PV) and essential thrombocytosis (ET). Our patient developed increasing thrombocytopenia and raising WBC counts during follow up (Fig. 3), and a blood smear demonstrated an increasing blasts count. Development of AML was therefore suspected and verified by bone marrow examination showing a blast count > 20% [7] (Fig. 1). Hence, the patient fulfilled the criteria for secondary AML (s-AML). Surprisingly, the karyotyping demonstrated the presence of a Ph chromosome, confirmed by FISH analysis. Hence the myeloid malignancy was classified as Ph+. The Ph chromosome is a hallmark for CML, where > 97% of the patients present this translocation. However, it is believed that a low fraction (~ 1%) of AML patients also has this features [17]. Consequently, this is defined as an own entity with prognostic impact in the new recommendations from European Leukemia NET [18]. Although, the distinction between CML in blast phase and AML or bi-lineage acute leukemia (BI-AL) is not straightforward. Patients with Ph+ AML seem to have distinct morphological, clinical and genetical features distinguishing them from CML in blast phase [19, 20], while immunophenotypic features distinguish Ph+ AML from Ph + ALL and Ph + BI-AL.
In contrast to CML in blast phase, in which the disease mandatory harbors a BCR-ABL1 fusion, AML transformation following a JAK2V617F-positive MPN commonly lacks the JAK2V617F mutation and thus presents with a JAK2 wild-type leukemia [21, 22]. This was also the case in our patient, as the JAK2V617F mutation detected at diagnosis of PMF was no longer detected at the diagnosis of AML. Contrariwise, the BCR-ABL1 e6a2 fusion gene detected at the diagnosis of AML was only detected retrospectively, and thus represents a minor cell subset at the time of PMF diagnosis. Taken together, these results indicate that the BCR-ABL1 positive AML clone arose from a JAK2 wild type cells and not from the clone giving rice to the initial PMF.
The JAK2V617F mutation allelic burden was low at the time of diagnosis, however low allelic mutation burden are not uncommon in PMF [23,24,25], in fact low JAK2V617F allele burden in PMF is associated with a more aggressive disease and poor overall survival [23, 24]. It has been postulated that an overriding JAK2V617F-negative clone conferring a more aggressive disease phenotype could be present [23,24,25,26], and for our patient we retrospectively detected this clone as being Ph+.
Our patient presented with the very rare BCR-ABL1 transcript e6a2 that has been reported only for a few patients, and we have summarized these findings in Table 1. To the best of our knowledge this is the first report of this rare transcript arising in a previously JAK2V617F mutated patients. Clinically, CML cells with this variant e6a2 BCR-ABL1 fusion transcript, often present in advanced stage with an aggressive disease, including presentation in blast phase and with AML. However, occasionally good responses to TKI have also been documented, a rare phenomenon reported in the case of Ph+ AML. For our patient, first dasatinib and later imatinib, resulted in considerable reduction of the BCR-ABL1 transcript (Fig. 3). There is no evidence to support the preference for one TKI in Ph+ AML [18], although given the broader spectrum of kinase inhibition with dasatinib compared to other TKIs, some authors suggest dasatinib to be the TKI of choice [27].
Our patient developed a tumor in the pancreatic head. The tumor was not biopsied as the patient was considered unable to tolerate chemotherapy or surgery, and the diagnosis of pancreatic tumor was based on findings by CT scan, and considered radiological consistent with adenocarcinoma. Autopsy was not performed, so we can for sure not rule out a myeloid sarcoma as an uncommon manifestation of AML, although this seem unlikely. Interestingly, patient with MPNs probably have a modest, although significant, increased risk of secondary malignancies [28], as also detected in the present patient.
To the best of our knowledge this is the first report of a secondary Ph+ AML arising from previously JAK2 mutated MPN. The very rare BCR-ABL1 transcript e6a2 was detected in the transformed AML cells, and this transcript is associated with an aggressive phenotype. The AML arouse possibly from a more resistant but less robust primitive ancestral clone. The case illustrates the importance of new genetic evaluation also in s-AML since TKIs can be a treatment approach for such patients.
Abbreviations
- ABL1:
-
Abelson
- ALL:
-
Acute lymphoblastic leukemia
- AML:
-
Acute myeloid leukemia
- ASCT:
-
Allogenic stem cell transplantation
- BCR:
-
Breakpoint cluster region
- BI-ALL:
-
Bi-lineage acute leukemia (BI-AL)
- BM:
-
Bone marrow
- CRP:
-
C-reactive protein
- CML:
-
Chronic myeloid leukemia
- CMML:
-
Chronic myelomonocytic leukemia
- CMR:
-
Complete molecular response
- CT:
-
Computed tomography
- ET:
-
Essential thrombocytosis
- F:
-
Female
- FISH:
-
Fluorescent in situ hybridization
- Hgb:
-
Hemoglobin
- LDH:
-
Lactate dehydrogenase
- LSC:
-
Leukemic stem cell
- M:
-
Male
- MPN:
-
Myeloproliferative neoplasia
- PBMC:
-
Peripheral blood mononuclear cell
- PCR:
-
Polymerase chain reaction
- Ph:
-
Philadelphia chromosome
- PMF:
-
Primary myelofibrosis
- PV:
-
Polycythemia vera
- RT-qPCR:
-
Quantitative real time polymerase chain reaction
- s-AML:
-
Secondary acute myeloid leukemia
- TKI:
-
Tyrosine kinase inhibitor
- WBC:
-
White blood cells
- WHO:
-
World Health Organization
References
Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243(5405):290–3.
Holyoake TL, Vetrie D. The chronic myeloid leukemia stem cell: stemming the tide of persistence. Blood. 2017;129(12):1595–606.
Tarafdar A, Hopcroft LE, Gallipoli P, Pellicano F, Cassels J, Hair A, et al. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood. 2017;129(2):199–208.
Ismail SINR, Yousef AM, Ghanim MT. Incidence of bcr-abl fusion transcripts in healthy individuals. Mol Med Rep. 2014;4:1271–6.
Biernaux C, Sels A, Huez G, Stryckmans P. Very low level of major BCR-ABL expression in blood of some healthy individuals. Bone Marrow Transplant. 1996;17(Suppl 3):S45–7.
Schmidt M, Rinke J, Schafer V, Schnittger S, Kohlmann A, Obstfelder E, et al. Molecular-defined clonal evolution in patients with chronic myeloid leukemia independent of the BCR-ABL status. Leukemia. 2014;28(12):2292–9.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Heo SKNE, Yoon DJ, Jo JC, Park JH, Kim H. Dasatinib accelerates valproic acid-induced acute myeloid leukemia cell death by regulation of differentiation capacity. PLoS One. 2014;9(2):e98859.
Hjorth-Hansen H, Stenke L, Soderlund S, Dreimane A, Ehrencrona H, Gedde-Dahl T, et al. Dasatinib induces fast and deep responses in newly diagnosed chronic myeloid leukaemia patients in chronic phase: clinical results from a randomised phase-2 study (NordCML006). Eur J Haematol. 2015;94(3):243–50.
Dasatinib SP. Large granular lymphocytosis, and pleural effusion: useful or adverse effect? Crit Rev Oncol Hamatol. 2014;89(2):242–7.
Breccia MAG. Pleural/pericardic effusions during dasatinib treatment: incidence, management and risk factors associated to their development. Expert Opin Drug Saf. 2010;9(5):713–21.
Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med. 2013;369(25):2379–90.
Spivak JL. Myeloproliferative neoplasms. N Engl J Med. 2017;377(9):895–6.
Patel U LR, Medeiros LJ, Patel KP. Diagnostic, Prognostic, and Predictive Utility of Recurrent Somatic Mutations in Myeloid Neoplasms. Clin Lymphoma Myeloma Leuk. 2017;Jul:62–74.
Reikvam H, Tiu RV. Venous thromboembolism in patients with essential thrombocythemia and polycythemia vera. Leukemia. 2012;26(4):563–71.
Finazzi G, Caruso V, Marchioli R, Capnist G, Chisesi T, Finelli C, et al. Acute leukemia in polycythemia vera: an analysis of 1638 patients enrolled in a prospective observational study. Blood. 2005;105(7):2664–70.
Grimwade D, Ivey A, Huntly BJ. Molecular landscape of acute myeloid leukemia in younger adults and its clinical relevance. Blood. 2016;127(1):29–41.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47.
Soupir CP, Vergilio JA, Dal Cin P, Muzikansky A, Kantarjian H, Jones D, et al. Philadelphia chromosome-positive acute myeloid leukemia: a rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am J Clin Pathol. 2007;127(4):642–50.
Nacheva EP, Grace CD, Brazma D, Gancheva K, Howard-Reeves J, Rai L, et al. Does BCR/ABL1 positive acute myeloid leukaemia exist? Br J Haematol. 2013;161(4):541–50.
Theocharides A, Boissinot M, Girodon F, Garand R, Teo SS, Lippert E, et al. Leukemic blasts in transformed JAK2-V617F-positive myeloproliferative disorders are frequently negative for the JAK2-V617F mutation. Blood. 2007;110(1):375–9.
Campbell PJ, Baxter EJ, Beer PA, Scott LM, Bench AJ, Huntly BJ, et al. Mutation of JAK2 in the myeloproliferative disorders: timing, clonality studies, cytogenetic associations, and role in leukemic transformation. Blood. 2006;108(10):3548–55.
Tefferi A, Lasho TL, Huang J, Finke C, Mesa RA, Li CY, et al. Low JAK2V617F allele burden in primary myelofibrosis, compared to either a higher allele burden or unmutated status, is associated with inferior overall and leukemia-free survival. Leukemia. 2008;22(4):756–61.
Vannucchi AM, Pieri L, Guglielmelli P. JAK2 allele burden in the myeloproliferative neoplasms: effects on phenotype, prognosis and change with treatment. Ther Adv Hematol. 2011;2(1):21–32.
Lippert E, Mansier O, Migeon M, Denys B, Nilsson A, Rosmond C, et al. Clinical and biological characterization of patients with low (0.1-2%) JAK2V617F allele burden at diagnosis. Haematologica. 2014;99(7):e98–101.
Guglielmelli P, Lasho TL, Rotunno G, Score J, Mannarelli C, Pancrazzi A, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. 2014;28(9):1804–10.
Klaeger S, Heinzlmeir S, Wilhelm M, Polzer H, Vick B, Koenig PA, et al. The target landscape of clinical kinase drugs. Science. 2017;358(6367).
Brunner AM, Hobbs G, Jalbut MM, Neuberg DS, Fathi AT. A population-based analysis of second malignancies among patients with myeloproliferative neoplasms in the SEER database. Leuk Lymphoma. 2016;57(5):1197–200.
Dupont M, Jourdan E, Chiesa J. Identification of E6A2 BCR-ABL fusion in a Philadelphia-positive CML. Leukemia. 2000;14(11):2011–2.
Beel KA, Lemmens J, Vranckx H, Maertens J, Vandenberghe P. CML with e6a2 BCR-ABL1 transcript: an aggressive entity? Ann Hematol. 2011;90(10):1241–3.
Ritchie DS, McBean M, Westerman DA, Kovalenko S, Seymour JF, Dobrovic A. Complete molecular response of e6a2 BCR-ABL-positive acute myeloid leukemia to imatinib then dasatinib. Blood. 2008;111(5):2896–8.
Colla S, Sammarelli G, Voltolini S, Crugnola M, Sebastio P, Giuliani N. e6a2 BCR-ABL transcript in chronic myeloid leukemia: is it associated with aggressive disease? Haematologica. 2004;89(5):611–3.
Schultheis B, Wang L, Clark RE, Melo JV. BCR-ABL with an e6a2 fusion in a CML patient diagnosed in blast crisis. Leukemia. 2003;17(10):2054–5.
Hochhaus A, Reiter A, Skladny H, Melo JV, Sick C, Berger U, et al. A novel BCR-ABL fusion gene (e6a2) in a patient with Philadelphia chromosome-negative chronic myelogenous leukemia. Blood. 1996;88(6):2236–40.
Harada Y, Nishiwaki S, Sugimoto T, Onodera K, Goto T, Sato T, et al. Successful treatment with allogeneic stem cell transplantation followed by DLI and TKIs for e6a2 BCR-ABL-positive acute myeloid leukaemia: a case report and literature review. Medicine (Baltimore). 2017;96(50):e9160.
Crampe M, Haslam K, Groarke E, Kelleher E, O'Shea D, Conneally E, et al. Chronic myeloid leukemia with an e6a2 BCR-ABL1 fusion transcript: cooperating mutations at blast crisis and molecular monitoring. Case Rep Hematol. 2017;2017:9071702.
Popovici C, Cailleres S, David M, Lafage-Pochitaloff M, Sainty D, Mozziconacci MJ. E6a2 BCR-ABL fusion with BCR exon 5-deleted transcript in a Philadelphia positive CML responsive to Imatinib. Leuk Lymphoma. 2005;46(9):1375–7.
Roti G, La Starza R, Gorello P, Gottardi E, Crescenzi B, Martelli MF, et al. e6a2 BCR/ABL1 fusion with cryptic der(9)t(9;22) deletions in a patient with chronic myeloid leukemia. Haematologica. 2005;90(8):1139–41.
Schnittger S, Bacher U, Kern W, Haferlach T, Hertenstein B, Haferlach C. A new case with rare e6a2 BCR-ABL fusion transcript developing two new resistance mutations during imatinib mesylate, which were replaced by T315I after subsequent dasatinib treatment. Leukemia. 2008;22(4):856–8.
Vefring HK, Gruber FX, Wee L, Hovland R, Hjorth-Hansen H, Gedde Dahl T, et al. Chronic myelogenous leukemia with the e6a2 BCR-ABL and lacking imatinib response: presentation of two cases. Acta Haematol. 2009;122(1):11–6.
Langabeer SE, Crampe M, Kelly J, Fadalla K, Connaghan G, Conneally E. Nilotinib and allogeneic stem cell transplantation in a chronic myeloid leukemia patient with e6a2 and e1a2 BCR-ABL transcripts. Leuk Res. 2010;34(8):e204–5.
Hayette S, Tigaud I, Thomas X, French M, Perrin MC, Nicolini F, et al. Identification of a rare e6a2 BCR-ABL fusion gene during the disease progression of chronic myelomonocytic leukemia: a case report. Leukemia. 2004;18(10):1735–6.
Yao J, Douer D, Wang L, Arcila ME, Nafa K, Chiu A. A case of acute myeloid leukemia with e6a2 BCR-ABL fusion transcript acquired after progressing from chronic myelomonocytic leukemia. Leuk Res Rep. 2017;7:17–9.
Zagaria A, Anelli L, Coccaro N, Tota G, Casieri P, Cellamare A, et al. BCR-ABL1 e6a2 transcript in chronic myeloid leukemia: biological features and molecular monitoring by droplet digital PCR. Virchows Arch. 2015;467(3):357–63.
Tanimoto K, Yamamoto A, Yoshinobu A. Successful treatment of CML with e6a2 BCR/ABL for a long time: Only use of tyrosine kinase inhibitor. Annual Meeting of the Japnese Society of Hematology. 2013;Abstract 61.
Corm S, Renneville A, Rad-Quesnel E, Grardel N, Preudhomme C, Quesnel B. Successful treatment of imatinib-resistant acute megakaryoblastic leukemia with e6a2 BCR/ABL: use of dasatinib and reduced-conditioning stem-cell transplantation. Leukemia. 2007;21(11):2376–7.
Rohon P, Divoka M, Calabkova L, Mojzikova R, Katrincsakova B, Rusinakova Z, et al. Identification of e6a2 BCR-ABL fusion in a Philadelphia-positive CML with marked basophilia: implications for treatment strategy. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2011;155(2):187–90.
Breccia M, Cannella L, Diverio D, Streponi P, Nanni M, Stefanizzi C, et al. Isolated thrombocytosis as first sign of chronic myeloid leukemia with e6a2 BCR/ABL fusion transcript, JAK2 negativity and complete response to imatinib. Leuk Res. 2008;32(1):177–80.
Acknowledgements
We appreciate the collaboration and technical support from staff from the University of Bergen, Haukealnd University Hospital and the University Hospital of Northern Norway.
Funding
The study received support from The University of Bergen and Helse Vest and The Norwegian Cancer Society.
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
MKB, KL and HR were the primary contributors in writing the manuscript. MKB, RH, FL and HR created the illustrations. KL, ØB, BTG and HR contributed to patient care. RH, IJL and MV interpreted and analyzed the patient data and genetic tests. FL analyzed and interpreted the histological examination. RH, FL, ØB and BTG critically revised the first version. HR initiated and coordinated the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Sampling for bio banking was approved by the local Ethics Committee (Region III, University of Bergen, Norway) and sample collected after written informed consent.
Consent for publication
Written informed consent for research and publication from the patients was obtained.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Brattås, M.K., Lilleeng, K., Hovland, R. et al. Philadelphia chromosome positive AML arising from JAK2-positive myelofibrosis. Biomark Res 6, 33 (2018). https://doi.org/10.1186/s40364-018-0147-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40364-018-0147-6