
Abstract
Background
Philadelphia (Ph) chromosome results from the reciprocal translocation t(9;22)(q34.1;q11.2) and is diagnostic for chronic myeloid leukemia (CML). However, this translocation is also found in acute lymphoid leukemia (ALL), as well as in rare cases of acute myeloid leukemias (AML). Most patients with CML harbor either the e13a2 or the e14a2 BCR-ABL fusion product, while a small subset of the cases expresses e1a2 or e19a2 transcripts. Moreover, several atypical BCR-ABL1 transcripts, beside the most common e1a2, e13a2 and e14a2, have been described, mainly in patients with CML. However, ALL and de novo AML may also carry BCR-ABL1 atypical transcripts which will confer a poor prognosis.
Case presentation
A 78-years old male was admitted at our hospital with clinical and laboratory features allowing to make the diagnosis of AML. No evidence of a preceding CML (splenomegaly or basophilia) was found. The karyotype on G-banded metaphases was 46,XY, t(9;22)(q34;q11). While the molecular analysis was ongoing, the patient started treatment based on hydroxyurea followed by 5-aza-2′-deoxycytidine. The molecular biology analysis revealed the simultaneous presence of the common p190 e1a2 and the rare e6a2 isoforms. Because of persistent pancytopenia and presence of blasts, according to the molecular data, he was then switched to tyrosine kinase inhibitors (TKIs) treatment. Nevertheless, after 2 months, the patient was still refractory to second line treatment dying because of a pulmonary infection.
Conclusion
The atypical p190 e6a2 transcript seems to be associated in AML with aggressive disease. TKI therapy alone does not seem to control the disease. Prompt observations on these patients carrying rare BCR-ABL1 transcripts may help to establish optimal treatment approaches on these aggressive BCR-ABL1 phenotypes in different setting of patients.
Similar content being viewed by others


Background
The t(9;22) chromosome translocation originating the BCR-ABL1 fusion oncoprotein is detected in more than 95% of patients with CML representing the hallmark of the disease [1]. However, this translocation is also found in 10 to 20% of adults and in 2 to 5% of children with ALL, as well as in rare cases (1% approximately) of AML [2].
The breakpoint on chromosome 9 is localized between exons 1a, 1b and a2. In contrast, breakpoints on chromosome 22 occur within the BCR gene, consisting of 23 exons, thus resulting in several distinct fusion genes. Generally, the breakpoints location on BCR gene occur between b1-b5 exons, within a central region called major breakpoint cluster region (M-bcr), whereas rare cases occur within two other breakpoint cluster regions: minor (m-bcr), with the breakpoint between e1-e2 exons, and micro (μ-bcr) with rupture point between e19-e20 exons. Depending on the breakpoint of the ABL1 gene, the fusion protein has different molecular weight: p190, p210 and p230 proteins for m-bcr, M-bcr and μ-bcr, respectively [3]. Moreover, atypical BCR breakpoints outside the cluster regions have been described, involving splicing between whole exons, insertion of small sequences or genomic breakpoints within exons. For example, e8a2, e15a2 and e6a2 have been also described and their clinical significance is under investigation [2, 4, 5]. The atypical e6a2 BCR-ABL1 transcript produces a rare fusion protein of 185 kDa conferring a poor prognosis in CML due to its association with aggressive phenotype and early transformation, perhaps due to the lack of an important regulatory BCR sequence within the fusion proteins [6]. Here we report a rare case of de novo AML carrying the BCR-ABL1 transcript e6a2.
Case presentation
A 78-years old male was admitted to the Hematology of the University Hospital Sant’Andrea-Sapienza, because of worsening fatigue and abdominal pain. Written informed consent was obtained from the patient and the study was approved by our institutional review board.
The peripheral blood count showed hyperleucocytosis (WBC 118 × 109/L), anemia (hemoglobin 8.6 g/dl) and mild thrombocytopenia (98 × 109/L), with no associated splenomegaly. Peripheral blood smear showed hypercellularity with 90% blast cells.
The morphological examination of bone marrow (BM) aspirate showed 90% agranular blast cells of medium and large size (Fig. 1) and the immunophenotypic analysis performed on a FACScalibur flow cytometer using standard protocol revealed that blast cells were CD34+, CD117+, CD33+, CD13+, HLA-DR+, CD2+ MPO+/−, CD7+/− [7].

The bone marrow cell morphology of the patient at diagnosis. Agranular blast cells of medium and large size
A diagnosis of AML (M2) was established and the patient started cytoreduction with hydroxyurea obtaining after seven days of treatment a WBC count of 39 × 109/L.
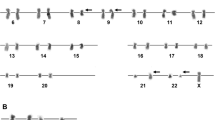
Conventional karyotyping was performed on the BM diagnostic aspirate after short-term culture and analyzed after G-banding. The description of the karyotype was made according to the International System for Human Cytogenetic Nomenclature. The cytogenetic analysis on G-banded metaphases disclosed a 46,XY,t(9;22)(q34;q11) karyotype. Then, interphase FISH experiments were carried out using BCR-ABL1 probes (Vysis) and demonstrated the presence of BCR-ABL1 fusion gene.
At the resolution of a pulmonary aspergillus infection treated with voriconazole, while the cytogenetic and molecular analyses were ongoing, the patient started treatment with 5-aza-2′-deoxycytidine (otherwise called decitabine, 20 mg/m2 for 5 days) for a total of two cycles. Subsequently, nested RT-PCR revealed the simultaneous presence of the common p190 e1a2 and the rare e6a2 isoforms (Fig. 2). PCR products, corresponding to BCR-ABL1 p190 amplification, were purified from agarose gel (QIAquick PCR Purification Kit - Qiagen) and directly sequenced on ABI/Prism 3130 Sequencer (Thermo Fisher Scientific) using BigDye® Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific). RQ-PCR for e6a2 BCR-ABL1 transcript analysis was performed using the forward primer Q-BCR_ex6_F (GATCCAACGACCAAGAACTCTCT), the reverse primer ENR561 and the ENP541 probe reported elsewhere [8]. The p190 e6a2 values were normalized on the number of ABL1 transcripts and expressed as the number of p190 e6a2 copies every 104 copies of ABL1. To assess molecular response after treatment, a real-time quantitative RT-PCR assay (RQ-PCR) for p190 e6a2 was designed by which we observed significant different kinetics through the e1a2 and e6a2 transcripts with consistent persistence of e6a2 [9].

Kinetics of BCR-ABL1 p190 e1a2 and e6a2 at diagnosis and during follow-up and treatment course of the AML patient
After the first, BM aspirated showed 70% blast cells and two transcripts e1a2 and e6a2 were respectively 3.09 and 5805.47/104 ABL1, while after second cycles blast cells were 20% and e1a2 and e6a2 5.71 and 5747.52.
Because of persistent pancytopenia and presence of blasts after two cycles of decitabine and in light of molecular data, the patient was then switched to TKI treatment.
The initial therapy consisted of imatinib 600 mg/day for two weeks that was subsequently reduced to 400 mg/day due to febrile neutropenia. After one month of imatinib, bone marrow showed 60% blast cells with small improvement of thrombocytopenia. Therefore, treatment was switched to dasatinib 100 mg/day, but it was discontinued five days later because of pulmonary embolism. At 10 days of TKI discontinuation, e1a2 and e6a2 were 0.17 and 9477.16/104 ABL1, respectively. After two months of continuous therapy with TKIs, bone marrow infiltration was present and the two transcripts were e1a2 1.6 and e6a2 23727.06/104 ABL1 (Fig. 2).
The patient, still refractory to second-line treatment, died of pulmonary infection.
Discussion
The AML with BCR-ABL1 fusion is a rare entity and has been included in the 2016 revised World Health Organization (WHO) as a provisional entity of myeloid neoplasm and acute leukemia [10]. The 2017 European LeukemiaNet risk stratification allocated AML with BCR-ABL1 to the adverse risk group [11]. In fact, the patients with BCR-ABL1 positive AML are often refractory to chemotherapy while the responses to imatinib are of limited duration [12]. A recent paper has reported that allogenic stem cell transplantation (allo-SCT) may improve outcome of younger patients [13]; however, given the small number of cases, the optimal therapy for AML with BCR-ABL1 has not been established yet. Beside common BCR-ABL1 transcripts, unusual transcripts have been also described in CML and acute leukemias. We report a rare case of AML characterized by the co-expression of the atypical e6a2 BCR-ABL1 transcript with the common e1a2 one. The e6a2 transcript is mainly observed in CML and sporadically in acute leukemia. In particular, so far about 24 e6a2 cases, in addition to our, have been described in the literature: 17 in CML (1 developed after chronic myelomonocytic leukemia -CMML-), 1 in a case of acute basophilic leukemia, 5 in AML (3 de novo and 2 cases developed after CMML and Myelofibrosis respectively) and 1 in an ALL patient [14]. Table 1 is a summary of the clinical features of e6a2 BCRABL-positive leukemia patients described so far. Notably, co-expression of e6a2 and e1a2 transcripts was previously described in only few cases of CML [15, 16]. Here we reported the first evidence of coexpression in AML.
Data reported so far indicate that the course of disease associated with e6a2a is particularly aggressive both in chronic and acute leukemias. In fact, within CML cases harboring this transcript, 6/17 developed an accelerated or blastic phase [6, 14, 17,18,19,20]. Furthermore, there was a prevalence of males on female (ratio 14:3) and the median age at diagnosis was 48 years old (range 18–76 years). Notably, the 3 de novo AML cases with e6a2 transcript were female and the age at diagnosis ranged from 53 to 55 years old [21,22,23]. These patients underwent allo-SCT in first CR after TKIs and chemotherapy. In these cases, dasatinib and nilotinib were effective treatments in inducing molecular remission after imatinib failure [22, 23] and against relapse after transplant maybe thanks to synergy between TKI and the graft versus-leukemia effect, reaching sustained response [21, 22]. They were alive at 24, 30 and 52 months from diagnosis [21,22,23]. On the contrary, the case here presented was older and not eligible to allo-SCT. The absence of prior clinical or laboratory evidences of CML (splenomegaly or basophilia) suggested a diagnosis of de novo AML with BCR-ABL1 [24]. He was resistant to decitabine and to TKIs and he died for disease shortly after the diagnosis.
Despite the clinical heterogeneity, all previously reported cases and the one here reported indicate that the atypical e6a2 transcript is associated with aggressive disease and underline the potential advantage of TKIs and allo-SCT in improving prognosis of these patients [6, 14, 15, 17,18,19,20, 25]. As suggested, the poor prognosis could be attributed to the partial loss of the Guanine Exchange Factor (GEF)/dbl-like domain [26]. This GEF/dbl-like domain is able to mediate the interaction with several Ras-like G proteins involved in proliferation, signalling and cell organization. Therefore, this truncation seems to determine an increased kinase activity with a greater transforming oncogenic potential. In our patient, the significant different kinetics of e1a2 and e6a2 transcripts quantified by RQ-PCR during the treatment further supports this hypothesis.
The case here described presents a diagnostic and therapeutic challenge since it shows the importance of combining conventional karyotype/FISH and appropriate genetic assays to detect rare BCR-ABL1 fusion transcripts. Furthermore, the different clearance of the e1a2 and e6a2 transcripts underlines the importance to design specific molecular assays for the evaluation of minimal residual disease and to choose the best therapeutic options.
Conclusions
In conclusion, hematological malignancies with the atypical e6a2 show an aggressive phenotype with an higher capacity of transformation to blast crisis in CML. Ph + AML is more rare, but with the same worse prognosis [15]. Therapeutic options include TKIs and allo-SCT. In fact, these patients can benefit from use of TKI, although this treatment seems to be more effective as salvage or maintenance therapy or even as bridging to transplant after a remission [15].
For patients not eligible to transplantation, the prognosis is even more unfavourable and a different approach should be considered. There is no standard of care and treatment of patients affected by Ph + AML, especially elderly, remains an area of unmet need. Notably, Shao et al. recently reported that dasatinib single agent may be an effective and tolerable induction therapy for AML patients with BCR-ABL1 and poor physical condition [27]. Hovewer, prognosis with low-intensity therapies is still poor and novel therapeutic strategies are needed. Future approaches may investigate role of Ras-Mek inhibitors, nowadays under investigation as targeted therapies in other diseases. Additional strategies can combine also novel agent: gentuzumab ozogamicin, tipifarnib, volasertib [28]. Moreover, interesting data arise from the use of ibrutinib [29] that inhibits AML blast proliferation, enhancing cytotoxic activities of cytarabine or daunorubicin.
Prompt observations of patients carrying rare BCR-ABL1 transcripts associated with unfavourable prognosis remain mandatory to include these patients in more aggressive treatment strategies. In vitro studies are necessary to clarify the transforming property and the mechanisms of resistance of e6a2 transcript and to design novel target therapeutic agents.
Abbreviations
- ALL:
-
Acute lymphoid leukemia
- AML:
-
Acute myeloid leukemia
- BM:
-
Bone marrow
- BMT:
-
Bone marrow transplantation
- CM:
-
Chronic myeloid leukemia
- DAC:
-
Dacogen
- DAS:
-
Dasatinib
- FUO:
-
Fever of unknown origin
- GEF:
-
Guanine exchange factor
- IFI:
-
Invasive fungal disease
- M-bcr:
-
Major breakpoint cluster region
- m-bcr:
-
Micro breakpoint cluster region
- m-bcr:
-
minor breakpoint cluster region
- MRD:
-
Minimal residual disease
- Ph:
-
Philadelphia
- RQ-PCR:
-
Real-time quantitative RT-PCR
- TKIs:
-
Tyrosine kinase inhibitors
- WBC:
-
White blood cells
References
Carter BZ, Mak DH, Cortes J, Andreeff M. The elusive chronic myeloid leukemia stem cell: does it matter and how do we eliminate it? SeminHematol. 2010;47(4):362–70.
Hochhaus A, Reiter A, Skladny H, Melo JV, Sick C, Berger U, Guo JQ, Arlinghaus RB, Hehlmann R, Goldman JM, Cross NC. A novel BCR-ABL fusion gene (e6a2) in a patient with Philadelphia chromosome-negative chronic myelogenous leukemia. Blood. 1996;88(6):2236–40.
Melo JV. The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype. Blood. 1996;88(7):2375–84.
Branford S, Rudzki Z, Hughes TP. A novel BCR±ABL transcript (e8a2) with the insertion of an inverted sequence of ABL intron 1b in a patient with Philadelphia-positive chronic myeloid leukaemia. Br J Haematol. 2000;109(3):635–7.
Moreno Mdel P, Cortinas MN, Bonomi R, Cardeza A. UriarteMdelR. A novel BCR-ABL fusion transcript (e15a2) in 2 patients with atypicalchronicmyeloproliferativesyndrome. Blood. 2001;97(11):3668–9.
Schultheis B, Wang L, Clark RE, Melo JV. BCR-ABL with an e6a2 fusion in aCML patient diagnosed in blast crisis. Leukemia. 2003;17:2054–5.
Montillo M, Tedeschi A, Pagano L, Venditti A, Ferrara F, Fabris P, Martino B, Musso M, De Rosa G, Specchia G, Monaco M, Sparaventi G, Spadea A, Palmas A, Deplano W, Manna A, Melillo L, Miraglia E, Mirto S, Mandelli F. Feasibility of peripheral blood stem cell rescue as intensification in elderly patients with acute myelocytic leukaemia: a pilot study from the Gimema group. Gruppo Italiano Malattie Ematologiche Maligne Dell'Adulto. Br J Haematol 2000;111(1):334–337.
Gabert J, Beillard E, van der Velden VH, Bi W, Grimwade D, Pallisgaard N, Barbany G, Cazzaniga G, Cayuela JM, Cavé H, Pane F, Aerts JL, De Micheli D, Thirion X, Pradel V, González M, Viehmann S, Malec M, Saglio G, van Dongen JJ. Standardization and quality control studies of 'real-time' quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia - a Europe against Cancer program. Leukemia. 2003;17(12):2318–57.
Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, Wermke M, Bornhäuser M, Ritter M, Neubauer A, Ehninger G, Illmer T. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002;99(12):4326–35.
Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, Bloomfield D, Cazzola M, Vardiman JW. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–405.
Döhner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA, Levine RL, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz M, Sierra J, Tallman MS, Tien HF, Wei AH, Löwenberg B, Bloomfield CD. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–47.
Soupir CP, Vergilio JA, Dal Cin P, Muzikansky A, Kantarjian H, Jones D, Hasserjian RP. Philadelphia chromosome-positive acute myeloid leukemia: a rare aggressive leukemia with clinicopathologic features distinct from chronic myeloid leukemia in myeloid blast crisis. Am J ClinPathol. 2007;127(4):642–50.
Lazarevic VL, Labopin M, Depei W, Yakoub-Agha I, Huynh A, Ljungman P, Schaap N, Cornelissen JJ, Maillard N, Pioltelli P, Gedde-Dahl T, Lenhoff S, Houhou M, Esteve J, Mohty M, Nagler A. Relatively favorable outcome after allogeneic stem cell transplantation for BCR-ABL1-positive AML: a survey from the acute leukemia working party of the European society for blood and marrow transplantation (EBMT). Am J Hematol. 2018;93(1):31–9.
Zagaria A, Anelli L, Coccaro N, Tota G, Casieri P, Cellamare A, Impera L, Brunetti C, Minervini A, Minervini CF, Delia M, Cumbo C, Orsini P, Specchia G, Albano F. BCR-ABL1 e6a2 transcript in chronic myeloid leukemia: biological features and molecular monitoring by droplet digital PCR. Virchows Arch. 2015;467(3):357–63.
Langabeer SE, Crampe M, Kelly J, Fadalla K, Connaghan G, Conneally E. Nilotinib and allogeneic stem cell transplantation in a chronic myeloid leukemia patient with e6a2 and e1a2 BCR-ABL transcripts. Leuk Res. 2010;34(8):e204–5.
Roti G, La Starza R, Gorello P, Gottardi E, Crescenzi B, Martelli MF. Mecucci C. e6a2 BCR/ABL1 fusion with cryptic der(9)t(9;22) deletions in a patient with chronic myeloid leukemia. Haematologica. 2005;90:1139–41.
Vefring HK, Gruber FX, Wee L, Hovland R, Hjorth-Hansen H, Gedde Dahl T, Meyer P. Chronic myelogenous leukemia with the e6a2 BCR-ABL and lacking imatinib response: presentation of two cases. ActaHaematol. 2009;122:11–6.
Beel KA, Lemmens J, Vranckx H, Maertens J, Vandenberghe P. CML with e6a2 BCR-ABL1 transcript: an aggressive entity? Ann Hematol. 2011;90:1241–3.
RohonP DM, Calabkova L, Mojzikova R, Katrincsakova B, Rusinakova Z, Lapcikova A, Raida L, Faber E, Jarosova M, Divoky V, Indrak K. Identification of e6a2 BCR-ABL fusion in a Philadelphia positive CML with marked basophilia: implications for treatment strategy. Biomed Pap Med FacUnivPalacky Olomouc Czech Repub. 2011;155:187–90.
Crampe M, Haslam K, Groarke E, Kelleher E, O'Shea D, Conneally E, Langabeer SE. Chronic myeloid leukemia with an e6a2 BCR-ABL1 fusion transcript: cooperating mutations at blast crisis and molecular monitoring. Case Rep Hematol. 2017;2017:9071702.
Harada Y, Nishiwaki S, Sugimoto T, Onodera K, Goto T, Sato T, Kamoshita S, Kawashima N, Seto A, Okuno S, Yamamoto S, Iwasaki T, Ozawa Y, Miyamura K, Akatsuka Y, Sugiura I. Successful treatment with allogeneic stem cell transplantation followed by DLI and TKIs for e6a2 BCR-ABL-positive acute myeloid leukaemia: a case report and literature review. Medicine (Baltimore). 2017;96(50):e9160.
Ritchie DS, McBean M, Westerman DA, Kovalenko S, Seymour JF, Dobrovic A. Complete molecular response of e6a2 BCRABL-positive acute myeloid leukemia toimatinib then dasatinib. Blood. 2008;111:2896–8.
Corm S, Renneville A, Rad-Quesnel E, Grardel N, Preudhomme C, Quesnel B. Successful treatment of imatinib-resistant acute megakaryoblastic leukemia with e6a2 BCR/ABL: use of dasatinib and reduced-conditioning stem-cell transplantation. Leukemia. 2007;21:2376–7.
Konoplev S, Yin CC, Kornblau SM, Kantarjian HM, Konopleva M, Andreeff M, Lu G, Zuo Z, Luthra R, Medeiros LJ, Bueso-Ramos CE. Molecular characterization of de novo Philadelphia chromosome-positive acute myeloid leukemia. Leuk Lymphoma. 2013;54(1):138–44.
Yao J, Douer D, Wang L, Arcila ME, Nafa K, Chiu A. A case of acute myeloid leukemia with e6a2 BCR-ABL fusion transcript acquired after progressing from chronic myelomonocytic leukemia. Leuk Res Rep. 2017;7:17–9.
Popovici C, Cailleres S, David M, Lafage-Pochitaloff M, Sainty D, Mozziconacci MJ. E6a2 BCR-ABL fusion with BCR exon 5-deleted transcript in a Philadelphia positive CML responsive to imatinib. Leuk Lymphoma. 2005;46:1375–7.
Shao X, Chen D, Xu P, Peng M, Guan C, Xie P, Yuan C, Chen B. Primary Philadelphia chromosome positive acute myeloid leukemia: A case report. Medicine (Baltimore). 2018 Nov;97(44):e12949.
Erba HP. Finding the optimal combination therapy for the treatment of newly diagnosed AML in older patients unfit for intensive therapy. Leuk Res. 2015;Feb;39(2):183–91.
Rushworth SA, Murray MY, Zaitseva L, Bowles KM, MacEwan DJ. Identification of Bruton's tyrosine kinase as a therapeutic target in acute myeloid leukemia. Blood. 2014;123(8):1229–38.
Dupont M, Jourdan E, Chiesa J. Identification of E6A2 BCR-ABL fusion in a Philadelphia-positive CML. Leukemia. 2000;14:2011–2.
Colla S, Sammarelli G, Voltolini S, Crugnola M, Sebastio P. Giuliani N. e6a2 BCR-ABL transcript in chronic myeloid leukemia: is it associated with aggressive disease? Haematologica. 2004;89:611–3.
Hayette S, Tigaud I, Thomas X, French M, Perrin MC, Nicolini F, Michallet M, Magaud JP. Identification of a rare e6a2 BCR-ABL fusion gene during the disease progression of chronic myelomonocytic leukemia: a case report. Leukemia. 2004;18:1735–6.
Grégoire MJ, Latger-Cannard V, Staal A, Bologna S, Leotard B, Rault JP, Béry-Dexheimer M, Jonveaux P. Identification of an acute basophilic leukaemia carrying a rare e6a2 BCR-ABL transcript. ActaHaematol. 2006;116:216–8.
Burmeister T, Schwartz S, Taubald A, Jost E, Lipp T, Schneller F, Diedrich H, Thomssen H, Mey UJ, Eucker J, Rieder H, Gökbuget N, Hoelzer D, Thiel E. Atypical BCR-ABL mRNA transcripts in adult acute lymphoblastic leukemia. Haematologica. 2007;92:1699–702.
Breccia M, Cannella L, Diverio D, Streponi P, Nanni M, Stefanizzi C, Natalino F, Mecarocci S, Alimena G. Isolated thrombocytosis as first sign of chronic myeloid leukemia with e6a2 BCR/ABL fusion transcript, JAK2 negativity and complete response to imatinib. Leuk Res. 2008;32:177–80.
Schnittger S, Bacher U, Kern W, Haferlach T, Hertenstein B, Haferlach C. A new case with rare e6a2 BCR-ABL fusion transcript developing two new resistance mutations during imatinibmesylate, which were replaced by T315I after subsequent dasatinib treatment. Leukemia. 2008;22:856–8.
Torres F, Ivanova-Dragoeva A, Pereira M, Veiga J, Rodrigues AS, Sousa AB, Tavares P, Fernandes AR. An e6a2 BCR-ABL fusion transcript in a CML patient having an iliac chloroma at initial presentation. Leuk Lymphoma. 2007;48:1034–7.
Brattås MK, Lilleeng K, Hovland R, Lægreid IJ, Vorland M, Leh F, Bruserud Ø, Gjertsen BT, Reikvam H. Philadelphia chromosome positive AML arising from JAK2- positive myelofibrosis. Biomark Res. 2018;6:33.
Acknowledgements
The authors thank the team of the laboratory of the immunophenotype, molecular biology and cytogenetics of the Sant’Andrea Hospital for the diagnostic contribution and D. Calef and Dr. V. Gianfelici for editing the manuscript.
Funding
This research was funded by Sapienza University (C26A14CPRB, C26A158NJR, RM116154EC670667 RP11715C7D08BDC2, and RM11715C7D01147A) grants, by Fondazione Internazionale D’AMATO Onlus and by Fondazione Frisiani-Santini.
Availability of data and materials
The raw data supporting our findings can be requested from the corresponding author.
Author information
Authors and Affiliations
Contributions
MP analyzed the data and wrote the paperaccording to the revisions of MRR, AT and FLC; TO, MD, VA and LC performed the assays and reported results. TO wrote methods and drew the figure too. MP, AF, EC, MPB, AC, GG, SP, APL and AT treated the patient. AT, FLC, MRR, SM, RL, TO approved the final version. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent was obtained from the patient for the publication of this case report and the accompanying images.
Competing interests
Authors declare no competing financial interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Piedimonte, M., Ottone, T., Alfonso, V. et al. A rare BCR-ABL1 transcript in Philadelphia-positive acute myeloid leukemia: case report and literature review. BMC Cancer 19, 50 (2019). https://doi.org/10.1186/s12885-019-5265-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12885-019-5265-5