
Abstract
Neutrophil extracellular traps (NETs) released by activated neutrophils typically consist of DNA-histone complexes and granule proteins. NETs were originally identified as a host defense system against foreign pathogens and are strongly associated with autoimmune diseases. However, a novel and predominant role of NETs in cancer is emerging. Increasing evidence has confirmed that many stimuli can facilitate NET formation in an NADPH oxidase (NOX)-dependent/NOX-independent manner. In cancer, NETs have been linked to cancer progression, metastasis, and cancer-associated thrombosis. In this review, we aimed to summarize the current available knowledge regarding NET formation and focused on the role of NETs in cancer biological behaviors. The potential target for cancer therapy will be further discussed.
Similar content being viewed by others
Background
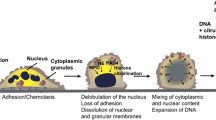
Cancer-related inflammation has long been recognized as a driving force of tumorigenesis development. Increasing evidence has shown that immune cells constitute the innate and adaptive immune system and enable the ability of tumor cells to escape immunosurveillance [1]. Neutrophils (innate immune cells) are the most abundant heterogeneous leukocytes in humans, and play a critical role in host defense against pathogens, including bacteria, fungi, and viruses. A number of mechanisms are involved, including reactive oxygen species (ROS) production, phagocytosis, and degranulation [2]. In 2004, Brinkmann et al. discovered a novel immune defense mechanism of neutrophils called neutrophil extracellular traps (NETs), a special form of degranulation [3]. NETs are composed of DNA fibers, histones, granular content, and antimicrobial proteins, which contribute to entrapping and killing invasive bacteria [4]. A unique type of cell death accompanied by the formation of NETs is known as neutrophil extracellular trap-osis (NETosis), which unlike apoptosis and necrosis, is dependent on the generation of ROS by NADPH oxidase [5]. Under relevant stimuli (microbial infection or foreign invasion), neutrophils are rapidly activated and accumulate, after which they undergo morphological changes. These events can be found in sequence in cells undergoing NETosis with nuclear envelope disintegration, mixing of nuclear and cytoplasmic material, cytoplasmic organelle disappearance, chromatin decondensation, cell membrane rupture, and NET release [6]. Apart from the primary advantageous role of protection against foreign bodies, and when neutrophils are dysregulated, NETs are implicated in many inflammatory diseases, including gout, rheumatoid arthritis, systemic lupus erythematosus, and others [7]. In the last two decades, several studies focused on investigating the role of NETs in malignant tumors because of their vital roles in infectious and immune-related diseases [8]. On the one hand, NETs can exert an antitumor effect by directly killing tumor cells and inhibiting tumor growth and metastasis. On the other hand, NETs have been shown to contribute to exerting protumor activity by inhibiting apoptosis and inducing tumor angiogenesis. Although many studies have shown that NETs may be more inclined to promote tumor proliferation and metastasis, their role has not yet been completely elucidated [9, 10].
Furthermore, interactions between neutrophils and other immune cells in the tumor microenvironment (TME) have been demonstrated in previous studies. However, the underlying mechanism of interaction between these cells in the TME is still inconclusive [11]. Studies in this field have become the focus and difficulty of current cancer research, which will bring an understanding of identifying cancer biomarkers and developing novel therapeutics. Until now, NETs have been found in animal models, peripheral blood, and tumor specimens from cancer patients. In this review, we will focus on the known steps of the biological characteristics of NETs, tumor progression, and metastasis, and cancer-associated thrombosis. Subsequently, the role of potential NET markers as prognostic biomarkers and their ability to serve as potential targets for cancer therapy will be discussed.
Mechanism of NET formation
In general, neutrophils are recognized as the core component of the innate immune system and play roles in endothelial adherence, chemotaxis, oxidization, phagocytosis, and the release of toxic granules, resulting in microbial killing [12]. The formation of NETs widely exists in neutrophils, which are considered a defense mechanism in response to harmful stimuli (bacteria, fungal hyphae, immune complexes, activated platelets, and biochemical stimuli) [7, 13]. NETs are mainly composed of DNA filaments with a diameter of 15–17 nm and many spherical protein substances with a diameter of approximately 25 nm. NET proteins have been identified as histones H1-H4, neutrophil elastase (NE), myeloperoxidase (MPO) and cathepsin G. Among NET proteins, histones H2-H4 account for approximately 70% and are core proteins [14]. In previous studies, it was widely believed that the formation of NETs is presumably a biologically conserved process. Two key mechanisms of NET formation have been discovered based on the death fate of neutrophils (NADPH oxidase-dependent/independent or NOX-dependent/independent suicidal NET formation) [15]. However, the underlying molecular mechanisms of NET formation are still not entirely understood.
NADPH oxidase-dependent NET formation
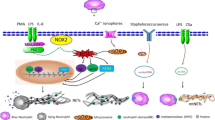
Under physiological conditions, histones are highly wrapped around most DNA strands in the nuclei of neutrophils, which results in transcriptional inactivity because of the constraint of DNA extension by protein‒DNA interactions. Under various stimuli conditions, such as pathogenic microorganisms in vivo or phorbol 12-myristate 13-acetate (PMA), interleukin-8 (IL-8), and lipopolysaccharide (LPS) in vitro, neutrophils can be activated [16, 17]. After 3 to 8 h of neutrophil activation, NADPH oxidase (NOX)-dependent NETs will form, which is the first discovered mechanism of NET formation. It is a type of suicidal approach because the formation of NETs is followed by neutrophil death [15]. Different from apoptosis and necrosis, this unique type of neutrophil death is considered NETosis, which is insensitive to caspase inhibition. In this period, neutrophils experience a series of biochemical and biological processes, which eventually lead to the release of potential energy along with chromatin and granulated protein expulsion [18]. The vital step during NETosis is the decondensed DNA strands into fibrous polymers in neutrophils. The critical step of chromatin decondensation is the generation of ROS, which is mediated by NADPH oxidase 2 (Nox2) [7]. Following PMA stimulation, the activity of protein kinase C was increased for the entry of endoplasmic calcium into the cytoplasm, which then phosphorylates Nox2, thereby driving the production of ROS [19]. Similar to PMA, phosphorylation of Nox2 was followed by LPS stimulation through the c-Jun N-terminal kinase (JNK) pathway [20]. Two key protein enzymes, MPO and NE, which are stored in the cytoplasmic granules of naïve neutrophils, can be released and activated by ROS. Once NE is translocated into the nucleus, chromatin decondensation starts. First, core histones (H2A, H2B, H3, and H4) that package nuclear DNA were disrupted and degraded by activated NE. Subsequently, MPO binds to nuclear chromatin for further decondensation with NE [21]. In addition to NE and MPO, another vital protease, peptidyl-arginine deiminase-4 (PAD4), catalyzes the conversion of arginine to citrullines in histones. In this process, citrullinated histones strongly weaken histone-DNA binding, which further boosts the chromatin decondensation of nuclear DNA [22]. Previous studies have demonstrated that PAD4 or NE deficiency affects NET formation in mouse models [23, 24]. However, the role of PAD4 remains controversial because in various studies, it has been shown that PAD4 is not always essential for NET formation [25]. After breakdown of the nuclear membrane, decondensed chromatin decorated with histones is released into the cytoplasm, mixes with granule proteins and extrudes throughout the cellular membrane after disintegration of the plasma membrane with the help of gasdermin D, which results in the release of NETs and neutrophil death [26, 27] (Fig. 1 and Table 1).

NADPH oxidase (NOX)-dependent NET formation pathways. Neutrophils are activated by extracellular microbes or PMA, LPS and IL-8, followed by activation of various pathways, including MEK/Erk, c-JNK, and PI3K/Akt signals. The endoplasmic calcium in the cytoplasm then phosphorylates NADPH Oxidase (Nox2), thereby driving the production of reactive oxygen species (ROS). Subsequently, neutrophil elastase (NE) and myeloperoxidase (MPO) stored in cytoplasmic granules translocate into the nucleus and contribute to chromatin decondensation with the assistance of calcium-dependent protein protein-arginine deiminase type 4 (PAD4), which citrullinates histones. Decondensed chromatin mixed with granule proteins is first released into the cytoplasm and then out of the cell membrane, and forms Neutrophil Extracellular Traps (NETs).
NADPH oxidase-independent NET formation
In 2004, in the first few years since NETs were first identified, the term “NETosis” has been widely used. However, in 2018, it was strongly recommended that the term NETosis should be replaced with NETs because a great number of studies reported that the formation of NETs does not accompany neutrophil death [52, 53]. In fact, NADPH oxidase (Nox)-independent NET formation was described in 2012. This is a fast calcium-activated pathway. In recent years, the detailed molecular mechanisms related to it, have received great attention, and some breakthroughs have been made [54]. Although NADPH oxidase is not necessary for NET formation, ROS generation is required. Similar to PMA or LPS, some Nox-independent NET formation agonists, such as calcium ionophores, nicotine and A23187, have been suggested to trigger NETosis via mitochondrial ROS (mROS) that is generated by the activation of the calcium-activated small conductance potassium (SK) channel member SK3 [19, 48]. In the past few years, mROS production mediated by CK channels has been considered to be associated with apoptosis, but this process has been linked to several autoimmune diseases. Therefore, we propose a novel role for mitochondria in neutrophils as ROS generators to participate in Nox-independent NET formation, thereby playing a role in innate immune function [55, 56].
NETs and cancer
Neutrophils play a vital role in cancer. Acting as an arm of neutrophils, NETs in cancer were first identified by Demers et al. Based on the status of the immune system or TME, the role of NETs is variable [57]. On the one hand, NETs play an antitumor role in colorectal cancer (CRC) and head and neck squamous cell carcinoma by inducing apoptosis [58, 59]. Recently, in some studies, it was reported that NETs may have an antitumor function in ovarian cancer and melanoma by inducing necrosis[60, 61]. The most direct mechanism may be its direct killing of tumor cells or stimulation of the immune system to fight against the tumor [62]. In melanoma, MPO is a representative component of NETs, which can kill melanoma cells (cell line A375) and decline the ability of proliferation and metastasis after implementation [61]. Higher values of the S100A8/CRP ratio, the release of which is associated with NETs, were found to correlate with favorable survival of high grade serous ovarian cancer (HGSOC) patients [60]. Moreover, neutrophils secrete high levels of H2O2 when stimulated by PMA that could inhibit metastatic seeding in the mouse lung cancer models [63]. On the other hand, an increasing number of studies have focused on the protumor role of NETs in various types of malignant tumors [lung cancer (LC), breast cancer (BC), and myeloproliferative neoplasms] through the promotion of tumor proliferation and metastasis [64,65,66]. Furthermore, NETs are associated with tumor angiogenesis and cancer-associated thrombosis [57]. Subsequently, the underlying mechanisms of NETs in tumor proliferation, metastasis, angiogenesis, and cancer-associated thrombosis will be highlighted.
NETs and the tumor microenvironment
Neutrophils are leukocytes originating from the bone marrow and spleen and are considered the first line of defense against microorganism infections or injuries. Neutrophils are normally generated every day and can be further increased by proinflammatory factors during infection. NETs are also induced [67]. In recent years, in various studies, it has been demonstrated that NETs can also be stimulated by tumor cells in the absence of an infection and can act as important components of the TME, thereby playing a pivotal role in cancer [7]. Based on their function, there are two phenotypes of tumor-associated neutrophils (TANs) in the TME: the N1 (antitumor) phenotype and the N2 (protumor) phenotype [43]. N1 TANs have been demonstrated to enhance proinflammatory cytokines, including tumor necrosis factor-α (TNF-α) and intercellular adhesion molecule-1 (ICAM-1). On the other hand, C-X-C motif chemokine ligand 8 (CXCL8, also called interleukin-8, IL-8) and CXCL5 are upregulated in N2 TANs. In addition, N2 TANs in the TME are also associated with tumor angiogenesis by recruiting matrix metalloproteinase-9 (MMP-9) [68]. The upregulation of the expression of proinflammatory cytokines [including TNF-α, IL-8, and interleukin-6 (IL-6)] and neutrophil survival have been reported to promote protumorigenic (N2) phenotype TANs in a breast cancer model. In a lung cancer cell model, Shaul et al. showed that the immunosuppressive cytokine transforming growth factor-β (TGF-β) can polarize neutrophils into an N2 phenotype [69]. Granulocyte colony stimulating factor (G-CSF) can stimulate neutrophil production and maintain neutrophil survival in the bone marrow. The formation of NETs can be increased by the upregulation of G-CSF in the TME. As the upstream regulatory cytokine of G-CSF, interleukin-1 beta (IL-1β) was found to influence NET production in breast cancer [70]. Tumor-derived G-CSF can drive NET generation, and cancer-associated fibroblasts (CAFs) in the TME have recently been considered one of the key factors in NET formation [71]. Furthermore, the generation of IL-8 is mainly regulated by the upstream transcription factor nuclear factor kappa B (NF-κB). Interactions with neutrophils of IL-8 are exerted through C-X-C motif chemokine receptors 1 and 2 (CXCR1 and CXCR2). Tumor-derived IL-8 has been shown to induce NET generation in many malignant tumors, including BC, LC, hepatocellular carcinoma (HCC), and melanoma [72, 73]. From the above discussion, these findings demonstrate that NET formation is closely related to the TME and promotes pro-tumoral function (Table 1).
NETs promote proliferation
Recent evidence has confirmed that neutrophils are a significant component of the TME, and a high neutrophil infiltration or a higher neutrophil to lymphocyte ratio is associated with faster progression and poor prognosis in various malignant tumors [74]. NETs play a complex role in tumor progression (proliferation and growth) by different mechanisms. First, in nonsolid tumors, such as chronic lymphocytic leukemia and diffuse large B-cell lymphoma (DLBCL), NETs have been shown to enhance the proliferative ability by increasing activation markers and inhibiting apoptosis of tumor cells [75]. Moreover, activation of the NF-κB pathway and signal transducer and activator of transcription 3 (STAT3)/p38 signaling stimulated by NETs is another proposed mechanism of promoting tumor proliferation in DLBCL [76]. Second, in solid tumors, multiple underlying mechanisms have been elucidated. Circulating tumor cells (CTCs) are cancer cells that “have fallen off” a tumor to circulate in the bloodstream and undergo a state of dormancy when exposed to an adverse microenvironment (lack of adequate angiogenesis and nutrient supply) [77]. In a mouse model injected with dormant MCF-7 BC cells, Albruenges et al. reported that NET formation and awakened tumor cell proliferation increased in sustained inflammatory lungs after exposure to LPS [45]. Multiple cytokines and chemokines secreted by tumor cells, including IL-8, IL-17, G-CSF, and CXCL6, can recruit bone marrow-derived neutrophils to tumor sites and trigger NETosis. Subsequently, there is increased neutrophil infiltration and NET formation in the TME, which ultimately leads to increased tumor cell proliferation [72, 78]. As a key component of NETs, NE plays an important role in the tumorigenesis of digestive system tumors (CRC and HCC). Yazdani et al. demonstrated that NETs can activate toll-like receptor 4 (TLR4)-proliferator activated receptor gamma coactivator 1-α (PGC-1α) signaling in CRC MC38 cells. Subsequently, mitochondrial adenosine triphosphate (ATP) is produced through the NE-activated TLR4-PGC-1α pathway, which is involved in tumor cell proliferation and metastasis [47]. In another MC38 CRC cell study, high mobility group box 1 (HMGB1), a constituent part of NETs, interacted with toll-like receptor 9 (TLR9), followed by activation of MAP kinase signaling to perform tumorigenic functions [79]. In addition, neutrophil infiltration and NETs formation were previously reported in neurological cancers. HMGB1, which acts as a ligand of RAGE, has been shown to participate in glioma tumor cell proliferation by activating the NF-κB pathway and promoting IL-8 secretion [80]. Furthermore, NETs protect tumor cells from cytotoxicity by suppressing infiltrating CD8 + and natural killer (NK) cells in the TME, thereby promoting tumor cell survival and growth [81].
NETs promote metastasis
Tumor metastasis, the process by which tumor cells spread from the primary lesion to a distant site (tissues or organs), is the leading cause of cancer-related death [82]. With a deeper understanding on the function of NETs in cancer, the relationship between NETs and cancer metastasis has become an emerging topic of interest. Recent studies have confirmed the underlying mechanisms of NETs in the tumor invasion-metastasis process. The epithelial to mesenchymal transition (EMT) is a crucial mechanism by which tumor cells acquire motility and invasiveness [83]. The degradation of VE-cadherin (CD144) accompanied by the activation of the Wnt/β-catenin pathway is an important process of EMT formation induced by NETs, which was first reported by Pieterse et al. [84]. NETs have been shown to change the morphology from MCF7 cells from an epithelial to a mesenchymal phenotype (EMT) in a BC mouse model, thereby promoting the tumor migration ability. The underlying mechanism of action involved upregulation of the expression of EMT-related genes, including ZEB1 and Snail [66]. Another in vitro study confirmed that the DNA component of NETs (NETs-DNA) significantly enhanced the migration and adhesion ability of BC cells (MDA-MB-231) with the help of coiled-coil domain containing protein 25 (CCDC25), which functions as a NET receptor and binds NETs-DNA [85]. Jin et al. revealed that the migration, invasion, and EMT in pancreatic ductal adenocarcinoma (PDAC) cells was promoted via IL-1b/epidermal growth factor receptor (EGFR)/extracellular signal regulated kinase (ERK) signaling when introduced to NET supernatant [46]. As mentioned earlier, CTCs undergo a state of dormancy under adverse conditions. However, dormant cells eventually break out of the dormant state and metastasize. Albrengues et al. demonstrated that NETs participate in the awakening of dormant tumor cells in mouse models of metastatic lung cancer. Lung inflammation stimulated by NETs (nasal instillation of LPS) can awaken dormant lung cancer cells and facilitate metastasis. Mechanistically, NET-derived NE and MMP-9 proteases were required for reactivating dormant cells through extracellular matrix (ECM) remodeling rather than through direct contact between NETs and dormant tumor cells. NE and MMP-9 are necessary to cleave and remodel laminin, which activates downstream integrin α3β1 and FAK/MEK/ERK signaling, subsequently allowing dormant tumor cells to reenter the cell cycle, leading to the resumption of aggressive metastatic growth of tumor cells [45]. In addition to its role in tumor proliferation, HMGB1 plays a nonnegligible role in tumor metastasis. HMGB1 is a NET-related component protein that can activate the TLR9 pathway and subsequently stimulate the p38 and JNK pathways to promote CRC cell migration and metastasis [79]. Furthermore, HMGB1 can enhance tumor migration and invasion abilities through EMT formation [83] (Fig. 2).

Molecular mechanisms of NETs in tumor metastasis. HMGB1, released by NETs, promotes tumor metastasis by binding to TLR9 to activate p38/MAPK signaling. HMGB1 also facilitates metastasis by binding to TLR4 or by regulating the degradation of VE-cadherin (CD144), followed by increased expression of EMT-associated genes, ZEB1 and Snail. The NETs component, NE, directly regulates mitochondrial metabolism via the TLR4-p38-PGC1-α pathway and promotes tumor proliferation and metastasis. NETs-derived NE and MMP-9 proteases are required for reactivating dormant cancer cells through degradation of the extracellular matrix (ECM) through the cleavage of laminin. In several cancer cells, NET-DNA directly binds to CCDC25, resulting in tumor metastasis.
NETs promote angiogenesis
Angiogenesis is a hallmark of malignant tumors and can provide sufficient oxygen and nutrients for tumor proliferation and metastasis [86]. The vascular endothelial growth factor (VEGF) family, including VEGF-A-D and placental growth factor (PlGF), are major angiogenic molecules [87]. VEGF levels were found to be higher in peripheral blood neutrophils of BC patients compared to that in neutrophils from healthy control [88]. Neutrophil-derived MMP-9 has been shown to be linked to VEGF activation and angiogenesis. In summary, neutrophils have higher levels of VEGF and MMP-9, which, in general, are connected to angiogenesis [36]. The angiopoietin (ANGPT) family includes ANGPT1 and ANGPT2, which are another group of key proangiogenic factors. ANGPT1 is produced by pericytes and some types of immune cells and acts as an agonist of the tyrosine kinase receptor TIE2 on endothelial cells (ECs). ANGPT2 is an antagonist of TIE2 [89]. Both ANGPT1 and ANGPT2 can induce neutrophil adhesion into ECs. Subsequently, NET formation increases by prolonging the incubation of neutrophils with ANGPT1/2 [90]. To investigate the relationship between NETs and angiogenesis, a series of in vivo experiments were conducted. Aldabbous and collaborators first revealed that NET-DNA can enhance vascularization by subcutaneous injection in a mouse model and that NETs can induce angiogenesis in human pulmonary artery endothelial cells (HPAECs) [90]. This evidence suggests that ANGPT1/2 can promote the formation of NETs, which exert proangiogenic functions.
NETs and cancer-associated thrombosis
Thrombosis is caused by damage to ECs and blood clot formation, and blocks normal blood flow in arteries or veins. This pathological condition can lead to a variety of fatal diseases, including ischemic stroke and venous thromboembolism (VTE), which contribute to the global burden of disease [91]. Cancer-associated thrombosis has been identified as the second leading cause of death in cancer patients with hypercoagulable conditions. In previous studies, NETs have been found to contribute to thrombosis in infected wounds. In the last decade, NETs have significantly changed our view of cancer-associated thrombosis [92]. This may result from the interaction of various mechanisms. Recent data provide strong evidence that NETs enhance not only platelet adhesion, activation, and aggregation but also erythrocyte adhesion, which directly leads to fibrin deposition and clotting processes, thereby accelerating cancer-related thrombosis [93]. Functional release of platelets is directly caused by neutrophil-derived histone proteasomes in a TLR2- and TLR4-dependent manner [94]. In an in vivo model of pancreatic cancer, Norbaini et al. recently demonstrated that AsPC-1 pancreatic cancer cells can activate rapid NET formation. In addition, when platelets were incubated with neutrophils, they were preincubated with AsPC-1 cells, which can promote the release of NETs, thereby promoting thrombosis formation [95]. The same phenomenon was observed in gastric cancer patients, and NETs promoted the generation of thrombin and fibrin [96]. In myeloproliferative neoplasms (MPNs), NETs have been reported to contribute to thrombogenesis via platelet activation, which is a major cause of mortality in patients [97].
NETs as potential therapeutic targets
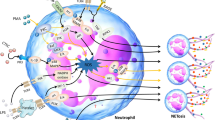
Considering that NETs are closely related to a higher mortality and poor prognosis of cancer patients, inhibiting the related pathway of NET formation can be a potential therapeutic target to control cancer progression and metastasis. Given the above knowledge, blocking NET formation via small molecule drugs against NET constituents, such as DNase I [98, 99], NE inhibitors [100], MPO inhibitors [47], and PAD4 inhibitors [101, 102] may have great potential. As the core component of NETs, DNA can be targeted using DNase I. In an in vitro assay, DNase I treatment suppressed pancreatic cell growth and decreased gastric cancer cell adhesion [103]. Moreover, after DNase I treatment, gastric cancer cells exhibited an epithelial phenotype rather than a mesenchymal phenotype (invasive and migratory phenotype) [104]. In animal models, the tumor growth of human CRC and HCC was suppressed under DNase I treatment. Therefore, the metastatic potential of BC and LC was significantly reduced after DNase I therapy [105, 106]. Chromatin densification is the most critical step in NET formation and is dependent on the presence of PAD4 via histone citrullination. In global PAD4 gene-deleted mice, it was shown that NET production was decreased and tumor cell proliferation was mitigated compared to wild-type (WT) mice [47]. PAD4 deficiency can also promote tumor cell apoptosis and reduce metastatic burden [107]. Decades ago, the FDA approved DNase for cystic fibrosis patients, thereby demonstrating its safety profile as a drug [108]. Unfortunately, only a handful of clinical trials are being conducted to validate the effectiveness of DNase in cancer patients. Pulmozyme, a recombinant human DNase, is currently being tested in a phase 1 trial (NCT00536952) in patients with head and neck cancer who undergo radiotherapy and chemotherapy. In a phase 2 clinical trial in acute myeloid/lymphoid leukemia, Oshadi D and Oshadi R (DNase in an Oshadi carrier) were evaluated (NCT02462265). NETs have recently been linked to cancer resistance and immunotherapy. Previous studies have shown that deoxyribonuclease I (DNase I) disruption of NETs is promising in efforts to improve CAR-T efficiency [109]. In a CRC mouse model, light-regulated release of DNase I sensitized immune checkpoint inhibitors (ICIs) treatment [110].
Another way to inhibit NETs is by blocking CXCR1 and CXCR2, which are key mediators of neutrophil chemotactic recruitment. The CXCR1/2-IL8 axis plays an important role in neutrophil chemotaxis as well as in NET formation [75]. Based on the protumor functions of the axis, CXCR1/2 and IL-8 have attracted much attention as therapeutic targets. IL-8 production may be triggered by IL-17, and inhibition of IL-17/IL-17RA signaling increases immune checkpoint blockade (anti-PD-1, anti-CTLA4) sensitivity in transplanted orthotopic PDAC mouse models [111]. Currently, in a phase1/2 clinical trial (NCT03400332) the combined treatment safety and efficacy of IL-8 inhibitor with nivolumab (anti-PD-1 mAb) or nivolumab plus ipilimumab (anti-CTLA4 mAb) is explored [112]. Several CXCR1/2 inhibitors combined with ICIs are currently being tested in clinical trials. SX-682, a CXCR1/2 inhibitor, is currently being tested in a phase 1 trial (NCT03161431) in advanced melanoma patients in combination with pembrolizumab (anti-PD-1 mAb). Navarixin, which targets both CXCR1/2, has been evaluated in combination with pembrolizumab in an ongoing phase 2 trial (NCT03473925) in advanced solid tumors. In addition, combination treatment with reparixin (another molecular drug against CXCR1/2) and paclitaxel showed antitumor activity along with a great safety profile in a phase 1b study in HER2-negative metastatic BC patients. A phase 2 clinical study is ongoing in patients with metastatic triple-negative BC (NCT02370238) [113] (Fig. 3). Table 2 summarizes potential therapeutics used to target NETs in cancer.

NETs as potential therapeutic targets. Inhibition of NETs formation via targeting crucial players, such as NE, MPO, or PAD4 in the formation pathway. Another way to inhibit NETs is by blocking CXCR1/2 and IL-8, key mediators of neutrophil chemotactic recruitment. Blockage of NETs-cancer cells interaction via targeting the TLR4/9 and CCDC25 can prevent the effect of NETs on cancer cells. As the core component of NETs, DNA can cause targeted destruction using DNAse. In addition, N2 neutrophils (pro-tumorigenic) can be converted to N1 neutrophils (anti-tumorigenic) by TGF-β inhibitors. Therapeutics used to target NETs may be a potential beneficial approach in combination with immunotherapy.
These findings support the potential treatment of blocking NETs to effectively control tumor progression and metastasis. However, the current study has some limitations. Firstly, current studies on anti-NETs therapy mainly rely on xenograft mouse models, which do not reflect the complex microenvironment seen in tumor patients. Secondly, in current clinical trials, injection of these NET inhibitors may have off-target effects, especially in elderly cancer patients with compromised immunity. Finally, targeting NETs is quickly becoming an optimistic treatment option in the cancer field, but it is clear to see that unwanted effects on the immune system have been found [126].
Conclusion
In recent years, increased attention has been given to tumor-associated neutrophils and their role in the TME. In addition, NETs from neutrophils play a complex and key role in cancer progression, metastasis, angiogenesis, cancer-associated thrombosis, and therapy. This review elaborates on the underlying mechanism of NET formation and its role in tumors. Most studies on the involvement of NETs in cancer biological behavior were based on animal or cellular models. Further studies are needed to understand the molecular mechanisms that regulate NET formation in tumors. Given their crucial roles in cancer, NETs have an important clinical application value. NET inhibitors against the components or receptors of NETs have great potential in the prevention and treatment of tumors. In previous studies, the combination of NET-interfering drugs and ICIs in the treatment of tumors has achieved some efficacy.
In conclusion, NETs will become possible therapeutic targets in cancer patients in the future, and clinical trials to verify the efficacy of NET-interfering drugs in cancers will be further explored.
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Abbreviations
- ATP:
-
Adenosine triphosphate
- BC:
-
Breast cancer
- CRC:
-
Colorectal cancer
- CAFs:
-
Cancer-associated fibroblasts
- CXCR1:
-
C-X-C motif chemokine receptors 1
- CXCR2:
-
C-X-C motif chemokine receptors 2
- CXCL8:
-
C-X-C motif chemokine ligand 8
- CXCL5:
-
C-X-C motif chemokine ligand 5
- CTCs:
-
Circulating tumor cells
- CCDC25:
-
Coiled-coil domain containing protein 25
- DLBCL:
-
Diffuse large B-cell lymphoma
- ECs:
-
Endothelial cells
- EMT:
-
Epithelial to mesenchymal transition
- EGFR:
-
Epidermal growth factor receptor
- ERK:
-
Extracellular signal regulated kinase
- ECM:
-
Extracellular matrix
- G-CSF:
-
Granulocyte colony stimulating factor
- G-CSFR:
-
Granulocyte colony stimulating factor receptor
- HGSOC:
-
High grade serous ovarian cancer
- HNSCC:
-
Head and neck squamous cell carcinoma
- HMGB1:
-
High mobility group box 1
- HPAECs:
-
Human pulmonary artery endothelial cells
- HCC:
-
Hepatocellular carcinoma
- IL-1β:
-
Interleukin-1 beta
- IL-6:
-
Interleukin-6
- IL-8:
-
Interleukin-8
- ICAM-1:
-
Intercellular adhesion molecule-1
- ICIs:
-
Immune checkpoint inhibitors
- JNK:
-
C-Jun N-terminal kinase
- LC:
-
Lung cancer
- LPS:
-
Lipopolysaccharide
- MPNs:
-
Myeloproliferative neoplasms
- MMP-9:
-
Matrix metalloproteinase-9
- MPO:
-
Myeloperoxidase
- NETs:
-
Neutrophil extracellular traps
- NOX:
-
NADPH oxidase
- NF-κB:
-
Nuclear factor kappa B
- NE:
-
Neutrophil elastase
- NK:
-
Natural killer
- PMA:
-
Phorbol 12-myristate 13-acetate
- PKC:
-
Protein kinase C
- PGC-1α:
-
Proliferator activated receptor gamma coactivator 1-α
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PlGF:
-
Placental growth factor
- PAD4:
-
Peptidyl-arginine deiminase-4
- ROS:
-
Reactive oxygen species
- RAGE:
-
Receptor for advanced glycation endproducts
- STAT3:
-
Signal transducer and activator of transcription 3
- TME:
-
Tumor microenvironment
- TANs:
-
Tumor-associated neutrophils
- TNF-α:
-
Tumor necrosis factor-α
- TGF-β:
-
Transforming growth factor-β
- TLR4:
-
Toll-like receptor 4
- TLR9:
-
Toll-like receptor 9
- VEGF:
-
Vascular endothelial growth factor
- VTE:
-
Venous thromboembolism
- WT:
-
Wild-type
References
Cristinziano L, Modestino L, Antonelli A, Marone G, Simon HU, Varricchi G, et al. Neutrophil extracellular traps in cancer. Semin Cancer Biol. 2022;79:91–104. https://doi.org/10.1016/j.semcancer.2021.07.011 (Epub 2021/07/20).
Shao BZ, Yao Y, Li JP, Chai NL, Linghu EQ. The role of neutrophil extracellular traps in cancer. Front Oncol. 2021;11:714357. https://doi.org/10.3389/fonc.2021.714357 (Epub 2021/09/04).
Chen Q, Zhang L, Li X, Zhuo W. Neutrophil extracellular traps in tumor metastasis: pathological functions and clinical applications. Cancers. 2021;13(11):2832. https://doi.org/10.3390/cancers13112832 (Epub 2021/07/03).
Snoderly HT, Boone BA, Bennewitz MF. Neutrophil extracellular traps in breast cancer and beyond: current perspectives on net stimuli, thrombosis and metastasis, and clinical utility for diagnosis and treatment. Breast Cancer Res. 2019;21(1):145. https://doi.org/10.1186/s13058-019-1237-6 (Epub 2019/12/20).
Wang H, Zhang Y, Wang Q, Wei X, Wang H, Gu K. The regulatory mechanism of neutrophil extracellular traps in cancer biological behavior. Cell Biosci. 2021;11(1):193. https://doi.org/10.1186/s13578-021-00708-z (Epub 2021/11/12).
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–91. https://doi.org/10.1083/jcb.201006052 (Epub 2010/10/27).
Yang D, Liu J. Neutrophil extracellular traps: a new player in cancer metastasis and therapeutic target. J Exp Clin Cancer Res. 2021;40(1):233. https://doi.org/10.1186/s13046-021-02013-6 (Epub 2021/07/18).
Chen Y, Han L, Qiu X, Wang G, Zheng J. Neutrophil extracellular traps in digestive cancers: warrior or accomplice. Front Oncol. 2021;11:766636. https://doi.org/10.3389/fonc.2021.766636 (Epub 2021/12/07).
Cedervall J, Hamidi A, Olsson AK. Platelets, nets and cancer. Thromb Res. 2018;164(Suppl 1):S148–52. https://doi.org/10.1016/j.thromres.2018.01.049 (Epub 2018/04/29).
Masucci MT, Minopoli M, Del Vecchio S, Carriero MV. The emerging role of neutrophil extracellular traps (nets) in tumor progression and metastasis. Front Immunol. 2020;11:1749. https://doi.org/10.3389/fimmu.2020.01749 (Epub 2020/10/13).
Governa V, Trella E, Mele V, Tornillo L, Amicarella F, Cremonesi E, et al. The interplay between neutrophils and Cd8(+) T cells improves survival in human colorectal cancer. Clin Cancer Res. 2017;23(14):3847–58. https://doi.org/10.1158/1078-0432.CCR-16-2047 (Epub 2017/01/22).
Yipp BG, Kubes P. Netosis: How Vital Is It? Blood. 2013;122(16):2784–94. https://doi.org/10.1182/blood-2013-04-457671 (Epub 2013/09/07).
Yousefi S, Morshed M, Amini P, Stojkov D, Simon D, von Gunten S, et al. Basophils exhibit antibacterial activity through extracellular trap formation. Allergy. 2015;70(9):1184–8. https://doi.org/10.1111/all.12662 (Epub 2015/06/05).
Li T, Zhang Z, Li X, Dong G, Zhang M, Xu Z, et al. Neutrophil extracellular traps: signaling properties and disease relevance. Mediators Inflamm. 2020;2020:9254087. https://doi.org/10.1155/2020/9254087 (Epub 2020/08/11).
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–41. https://doi.org/10.1083/jcb.200606027 (Epub 2007/01/11).
Hamam HJ, Khan MA, Palaniyar N. Histone acetylation promotes neutrophil extracellular trap formation. Biomolecules. 2019;9(1):32. https://doi.org/10.3390/biom9010032 (Epub 2019/01/24).
Tsourouktsoglou TD, Warnatsch A, Ioannou M, Hoving D, Wang Q, Papayannopoulos V. Histones, DNA, and citrullination promote neutrophil extracellular trap inflammation by regulating the localization and activation of Tlr4. Cell Rep. 2020;31(5):107602. https://doi.org/10.1016/j.celrep.2020.107602 (Epub 2020/05/07).
Boeltz S, Amini P, Anders HJ, Andrade F, Bilyy R, Chatfield S, et al. To net or not to net: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019;26(3):395–408. https://doi.org/10.1038/s41418-018-0261-x (Epub 2019/01/10).
Ravindran M, Khan MA, Palaniyar N. Neutrophil extracellular trap formation: physiology, pathology, and pharmacology. Biomolecules. 2019;9(8):365. https://doi.org/10.3390/biom9080365 (Epub 2019/08/17).
Khan MA, Farahvash A, Douda DN, Licht JC, Grasemann H, Sweezey N, et al. Jnk activation turns on lps- and gram-negative bacteria-induced nadph oxidase-dependent suicidal netosis. Sci Rep. 2017;7(1):3409. https://doi.org/10.1038/s41598-017-03257-z (Epub 2017/06/15).
Efrimescu CI, Buggy PM, Buggy DJ. Neutrophil extracellular trapping role in cancer, metastases, and cancer-related thrombosis: a narrative review of the current evidence base. Curr Oncol Rep. 2021;23(10):118. https://doi.org/10.1007/s11912-021-01103-0 (Epub 2021/08/04).
Leshner M, Wang S, Lewis C, Zheng H, Chen XA, Santy L, et al. Pad4 mediated histone hypercitrullination induces heterochromatin decondensation and chromatin unfolding to form neutrophil extracellular trap-like structures. Front Immunol. 2012;3:307. https://doi.org/10.3389/fimmu.2012.00307 (Epub 2012/10/13).
Raup-Konsavage WM, Wang Y, Wang WW, Feliers D, Ruan H, Reeves WB. Neutrophil peptidyl arginine deiminase-4 has a pivotal role in ischemia/reperfusion-induced acute kidney injury. Kidney Int. 2018;93(2):365–74. https://doi.org/10.1016/j.kint.2017.08.014 (Epub 2017/10/25).
Rabadi M, Kim M, D’Agati V, Lee HT. Peptidyl arginine deiminase-4-deficient mice are protected against kidney and liver injury after renal ischemia and reperfusion. Am J Physiol Renal Physiol. 2016;311(2):F437–49. https://doi.org/10.1152/ajprenal.00254.2016 (Epub 2016/06/24).
Kenny EF, Herzig A, Kruger R, Muth A, Mondal S, Thompson PR, et al. Diverse stimuli engage different neutrophil extracellular trap pathways. Elife. 2017;6:e24437. https://doi.org/10.7554/eLife.24437 (Epub 2017/06/03).
Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3(26):eaar6689. https://doi.org/10.1126/sciimmunol.aar6689 (Epub 2018/08/26).
Tall AR, Westerterp M. Inflammasomes, neutrophil extracellular traps, and cholesterol. J Lipid Res. 2019;60(4):721–7. https://doi.org/10.1194/jlr.S091280 (Epub 2019/02/21).
Zhou SL, Dai Z, Zhou ZJ, Wang XY, Yang GH, Wang Z, et al. Overexpression of Cxcl5 Mediates Neutrophil Infiltration and Indicates Poor Prognosis for Hepatocellular Carcinoma. Hepatology. 2012;56(6):2242–54. https://doi.org/10.1002/hep.25907 (Epub 2012/06/20).
Yu PF, Huang Y, Han YY, Lin LY, Sun WH, Rabson AB, et al. Tnfalpha-activated mesenchymal stromal cells promote breast cancer metastasis by recruiting Cxcr2(+) Neutrophils. Oncogene. 2017;36(4):482–90. https://doi.org/10.1038/onc.2016.217 (Epub 2016/07/05).
De Larco JE, Wuertz BR, Furcht LT. The potential role of neutrophils in promoting the metastatic phenotype of tumors releasing interleukin-8. Clin Cancer Res. 2004;10(15):4895–900. https://doi.org/10.1158/1078-0432.CCR-03-0760 (Epub 2004/08/07).
Yang L, Liu L, Zhang R, Hong J, Wang Y, Wang J, et al. Il-8 mediates a positive loop connecting increased neutrophil extracellular traps (nets) and colorectal cancer liver metastasis. J Cancer. 2020;11(15):4384–96. https://doi.org/10.7150/jca.44215 (Epub 2020/06/04).
Peng ZP, Jiang ZZ, Guo HF, Zhou MM, Huang YF, Ning WR, et al. Glycolytic activation of monocytes regulates the accumulation and function of neutrophils in human hepatocellular carcinoma. J Hepatol. 2020;73(4):906–17. https://doi.org/10.1016/j.jhep.2020.05.004 (Epub 2020/05/15).
Yan B, Wei JJ, Yuan Y, Sun R, Li D, Luo J, et al. Il-6 Cooperates with G-Csf to induce protumor function of neutrophils in bone marrow by enhancing Stat3 activation. J Immunol. 2013;190(11):5882–93. https://doi.org/10.4049/jimmunol.1201881 (Epub 2013/05/01).
Zhu Q, Zhang X, Zhang L, Li W, Wu H, Yuan X, et al. The Il-6-Stat3 axis mediates a reciprocal crosstalk between cancer-derived mesenchymal stem cells and neutrophils to synergistically prompt gastric cancer progression. Cell Death Dis. 2014;5:e1295. https://doi.org/10.1038/cddis.2014.263 (Epub 2014/06/20).
Coffelt SB, Kersten K, Doornebal CW, Weiden J, Vrijland K, Hau CS, et al. Il-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis. Nature. 2015;522(7556):345–8. https://doi.org/10.1038/nature14282 (Epub 2015/03/31).
Kuang DM, Zhao Q, Wu Y, Peng C, Wang J, Xu Z, et al. Peritumoral neutrophils link inflammatory response to disease progression by fostering angiogenesis in hepatocellular carcinoma. J Hepatol. 2011;54(5):948–55. https://doi.org/10.1016/j.jhep.2010.08.041 (Epub 2010/12/15).
Wu P, Wu D, Ni C, Ye J, Chen W, Hu G, et al. Gammadeltat17 cells promote the accumulation and expansion of myeloid-derived suppressor cells in human colorectal cancer. Immunity. 2014;40(5):785–800. https://doi.org/10.1016/j.immuni.2014.03.013 (Epub 2014/05/13).
Wang Y, Wang K, Han GC, Wang RX, Xiao H, Hou CM, et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (Il-1)/Il-6 axis. Mucosal Immunol. 2014;7(5):1106–15. https://doi.org/10.1038/mi.2013.126 (Epub 2014/01/16).
Wislez M, Fleury-Feith J, Rabbe N, Moreau J, Cesari D, Milleron B, et al. Tumor-derived granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor prolong the survival of neutrophils infiltrating bronchoalveolar subtype pulmonary adenocarcinoma. Am J Pathol. 2001;159(4):1423–33. https://doi.org/10.1016/S0002-9440(10)62529-1 (Epub 2001/10/05).
Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6g+Ly6c+ granulocytes. Proc Natl Acad Sci USA. 2010;107(50):21248–55. https://doi.org/10.1073/pnas.1015855107 (Epub 2010/11/18).
Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. Hmgb1 promotes neutrophil extracellular trap formation through Interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L342–9. https://doi.org/10.1152/ajplung.00151.2012 (Epub 2013/01/15).
Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature. 2014;507(7490):109–13. https://doi.org/10.1038/nature13111 (Epub 2014/02/28).
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by Tgf-Beta: “N1” Versus “N2” Tan. Cancer Cell. 2009;16(3):183–94. https://doi.org/10.1016/j.ccr.2009.06.017 (Epub 2009/09/08).
SenGupta S, Hein LE, Xu Y, Zhang J, Konwerski JR, Li Y, et al. Triple-negative breast cancer cells recruit neutrophils by secreting Tgf-beta and Cxcr2 ligands. Front Immunol. 2021;12:659996. https://doi.org/10.3389/fimmu.2021.659996 (Epub 2021/04/30).
Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018. https://doi.org/10.1126/science.aao4227 (Epub 2018/09/29).
Jin L, Kim HS, Shi J. Neutrophil in the pancreatic tumor microenvironment. Biomolecules. 2021;11(8):1170. https://doi.org/10.3390/biom11081170 (Epub 2021/08/28).
Yazdani HO, Roy E, Comerci AJ, van der Windt DJ, Zhang H, Huang H, et al. Neutrophil extracellular traps drive mitochondrial homeostasis in tumors to augment growth. Cancer Res. 2019;79(21):5626–39. https://doi.org/10.1158/0008-5472.CAN-19-0800 (Epub 2019/09/15).
Douda DN, Khan MA, Grasemann H, Palaniyar N. Sk3 Channel and Mitochondrial ros mediate nadph oxidase-independent netosis induced by calcium influx. Proc Natl Acad Sci USA. 2015;112(9):2817–22. https://doi.org/10.1073/pnas.1414055112 (Epub 2015/03/03).
Pilsczek FH, Salina D, Poon KK, Fahey C, Yipp BG, Sibley CD, et al. A novel mechanism of rapid nuclear neutrophil extracellular trap formation in response to staphylococcus aureus. J Immunol. 2010;185(12):7413–25. https://doi.org/10.4049/jimmunol.1000675 (Epub 2010/11/26).
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. 2012;12(1):109–16. https://doi.org/10.1016/j.chom.2012.05.015 (Epub 2012/07/24).
Byrd AS, O’Brien XM, Johnson CM, Lavigne LM, Reichner JS. An extracellular matrix-based mechanism of rapid neutrophil extracellular trap formation in response to candida albicans. J Immunol. 2013;190(8):4136–48. https://doi.org/10.4049/jimmunol.1202671 (Epub 2013/03/20).
Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the nomenclature committee on cell death 2018. Cell Death Differ. 2018;25(3):486–541. https://doi.org/10.1038/s41418-017-0012-4 (Epub 2018/01/25).
White PC, Chicca IJ, Cooper PR, Milward MR, Chapple IL. Neutrophil extracellular traps in periodontitis: a web of intrigue. J Dent Res. 2016;95(1):26–34. https://doi.org/10.1177/0022034515609097 (Epub 2015/10/08).
Parker H, Dragunow M, Hampton MB, Kettle AJ, Winterbourn CC. Requirements for nadph oxidase and myeloperoxidase in neutrophil extracellular trap formation differ depending on the stimulus. J Leukoc Biol. 2012;92(4):841–9. https://doi.org/10.1189/jlb.1211601 (Epub 2012/07/18).
Fay AJ, Qian X, Jan YN, Jan LY. Sk channels mediate nadph oxidase-independent reactive oxygen species production and apoptosis in granulocytes. Proc Natl Acad Sci USA. 2006;103(46):17548–53. https://doi.org/10.1073/pnas.0607914103 (Epub 2006/11/07).
Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK, et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nat Med. 2016;22(2):146–53. https://doi.org/10.1038/nm.4027 (Epub 2016/01/19).
Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA. 2012;109(32):13076–81. https://doi.org/10.1073/pnas.1200419109 (Epub 2012/07/25).
Arelaki S, Arampatzioglou A, Kambas K, Papagoras C, Miltiades P, Angelidou I, et al. Gradient infiltration of neutrophil extracellular traps in colon cancer and evidence for their involvement in tumour growth. PLoS ONE. 2016;11(5):e0154484. https://doi.org/10.1371/journal.pone.0154484 (Epub 2016/05/03).
Millrud CR, Kagedal A, Kumlien Georen S, Winqvist O, Uddman R, Razavi R, et al. Net-producing Cd16(High) Cd62l(Dim) neutrophils migrate to tumor sites and predict improved survival in patients with Hnscc. Int J Cancer. 2017;140(11):2557–67. https://doi.org/10.1002/ijc.30671 (Epub 2017/03/02).
Muqaku B, Pils D, Mader JC, Aust S, Mangold A, Muqaku L, et al. Neutrophil extracellular trap formation correlates with favorable overall survival in high grade ovarian cancer. Cancers. 2020;12(2):505. https://doi.org/10.3390/cancers12020505 (Epub 2020/02/27).
Schedel F, Mayer-Hain S, Pappelbaum KI, Metze D, Stock M, Goerge T, et al. Evidence and impact of neutrophil extracellular traps in malignant melanoma. Pigment Cell Melanoma Res. 2020;33(1):63–73. https://doi.org/10.1111/pcmr.12818 (Epub 2019/08/14).
Liu Y, Liu L. The pro-tumor effect and the anti-tumor effect of neutrophils extracellular traps. Biosci Trends. 2020;13(6):469–75. https://doi.org/10.5582/bst.2019.01326.
Granot Z, Henke E, Comen EA, King TA, Norton L, Benezra R. Tumor entrained neutrophils inhibit seeding in the premetastatic lung. Cancer Cell. 2011;20(3):300–14. https://doi.org/10.1016/j.ccr.2011.08.012 (Epub 2011/09/13).
Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10(436):8292. https://doi.org/10.1126/scitranslmed.aan8292 (Epub 2018/04/13).
Houghton AM, Rzymkiewicz DM, Ji H, Gregory AD, Egea EE, Metz HE, et al. Neutrophil elastase-mediated degradation of Irs-1 accelerates lung tumor growth. Nat Med. 2010;16(2):219–23. https://doi.org/10.1038/nm.2084 (Epub 2010/01/19).
Martins-Cardoso K, Almeida VH, Bagri KM, Rossi MID, Mermelstein CS, Konig S, et al. Neutrophil extracellular traps (nets) promote pro-metastatic phenotype in human breast cancer cells through epithelial-mesenchymal transition. Cancers (Basel). 2020;12(6):1542. https://doi.org/10.3390/cancers12061542 (Epub 2020/06/18).
Mahmud Z, Rahman A, Mishu ID, Kabir Y. Mechanistic insights into the interplays between neutrophils and other immune cells in cancer development and progression. Cancer Metastasis Rev. 2022;41(2):405–32. https://doi.org/10.1007/s10555-022-10024-8 (Epub 2022/03/23).
Kessenbrock K, Plaks V, Werb Z. Matrix Metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67. https://doi.org/10.1016/j.cell.2010.03.015 (Epub 2010/04/08).
Shaul ME, Levy L, Sun J, Mishalian I, Singhal S, Kapoor V, et al. Tumor-associated neutrophils display a distinct N1 profile following Tgfbeta modulation: a transcriptomics analysis of pro- Vs. Antitumor Tans Oncoimmunology. 2016;5(11):e1232221. https://doi.org/10.1080/2162402X.2016.1232221 (Epub 2016/12/22).
Gomes T, Varady CBS, Lourenco AL, Mizurini DM, Rondon AMR, Leal AC, et al. Il-1beta blockade attenuates thrombosis in a neutrophil extracellular trap-dependent breast cancer model. Front Immunol. 2019;10:2088. https://doi.org/10.3389/fimmu.2019.02088 (Epub 2019/09/26).
Munir H, Jones JO, Janowitz T, Hoffmann M, Euler M, Martins CP, et al. Stromal-driven and amyloid beta-dependent induction of neutrophil extracellular traps modulates tumor growth. Nat Commun. 2021;12(1):683. https://doi.org/10.1038/s41467-021-20982-2 (Epub 2021/01/31).
Gonzalez-Aparicio M, Alfaro C. Influence of interleukin-8 and neutrophil extracellular trap (Net) formation in the tumor microenvironment: is there a pathogenic role? J Immunol Res. 2019;2019:6252138. https://doi.org/10.1155/2019/6252138 (Epub 2019/05/17).
De Meo ML, Spicer JD. The Role of Neutrophil Extracellular Traps in Cancer Progression and Metastasis. Semin Immunol. 2021;57:101595. https://doi.org/10.1016/j.smim.2022.101595.
Gago-Dominguez M, Matabuena M, Redondo CM, Patel SP, Carracedo A, Ponte SM, et al. Neutrophil to lymphocyte ratio and breast cancer risk: analysis by subtype and potential interactions. Sci Rep. 2020;10(1):13203. https://doi.org/10.1038/s41598-020-70077-z (Epub 2020/08/09).
Podaza E, Sabbione F, Risnik D, Borge M, Almejun MB, Colado A, et al. Neutrophils from chronic lymphocytic leukemia patients exhibit an increased capacity to release extracellular traps (Nets). Cancer Immunol Immunother. 2017;66(1):77–89. https://doi.org/10.1007/s00262-016-1921-7 (Epub 2016/11/01).
Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, et al. Neutrophil extracellular traps induced by Il8 promote diffuse large B-cell lymphoma progression via the Tlr9 signaling. Clin Cancer Res. 2019;25(6):1867–79. https://doi.org/10.1158/1078-0432.CCR-18-1226 (Epub 2018/11/18).
Sun N, Li X, Wang Z, Zhang R, Wang J, Wang K, et al. A Multiscale Tio2 nanorod array for ultrasensitive capture of circulating tumor cells. ACS Appl Mater Interfaces. 2016;8(20):12638–43. https://doi.org/10.1021/acsami.6b02178 (Epub 2016/05/14).
Mao Z, Zhang J, Shi Y, Li W, Shi H, Ji R, et al. Cxcl5 Promotes gastric cancer metastasis by inducing epithelial-mesenchymal transition and activating neutrophils. Oncogenesis. 2020;9(7):63. https://doi.org/10.1038/s41389-020-00249-z (Epub 2020/07/08).
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016;76(6):1367–80. https://doi.org/10.1158/0008-5472.CAN-15-1591 (Epub 2016/01/14).
Zha C, Meng X, Li L, Mi S, Qian D, Li Z, et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the Hmgb1/Rage/Il-8 axis. Cancer Biol Med. 2020;17(1):154–68. https://doi.org/10.20892/j.issn.2095-3941.2019.0353 (Epub 2020/04/17).
Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. Cxcr1 and Cxcr2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52(5):856-71 e8. https://doi.org/10.1016/j.immuni.2020.03.001 (Epub 2020/04/15).
Massague J, Obenauf AC. Metastatic colonization by circulating tumour cells. Nature. 2016;529(7586):298–306. https://doi.org/10.1038/nature17038 (Epub 2016/01/23).
Kajioka H, Kagawa S, Ito A, Yoshimoto M, Sakamoto S, Kikuchi S, et al. Targeting neutrophil extracellular traps with thrombomodulin prevents pancreatic cancer metastasis. Cancer Lett. 2021;497:1–13. https://doi.org/10.1016/j.canlet.2020.10.015 (Epub 2020/10/17).
Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. 2017;37(7):1371–9. https://doi.org/10.1161/ATVBAHA.117.309002 (Epub 2017/05/13).
Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via Ccdc25. Nature. 2020;583(7814):133–8. https://doi.org/10.1038/s41586-020-2394-6 (Epub 2020/06/13).
Bekes EM, Schweighofer B, Kupriyanova TA, Zajac E, Ardi VC, Quigley JP, et al. Tumor-recruited neutrophils and neutrophil timp-free Mmp-9 regulate coordinately the levels of tumor angiogenesis and efficiency of malignant cell intravasation. Am J Pathol. 2011;179(3):1455–70. https://doi.org/10.1016/j.ajpath.2011.05.031 (Epub 2011/07/12).
Marone G, Varricchi G, Loffredo S, Granata F. Mast cells and basophils in inflammatory and tumor angiogenesis and lymphangiogenesis. Eur J Pharmacol. 2016;778:146–51. https://doi.org/10.1016/j.ejphar.2015.03.088 (Epub 2015/05/06).
Kusumanto YH, Dam WA, Hospers GA, Meijer C, Mulder NH. Platelets and granulocytes, in particular the neutrophils, form important compartments for circulating vascular endothelial growth factor. Angiogenesis. 2003;6(4):283–7. https://doi.org/10.1023/B:AGEN.0000029415.62384.ba (Epub 2004/05/29).
Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, et al. Requisite role of angiopoietin-1, a ligand for the Tie2 receptor. During Embryonic Angiogenesis Cell. 1996;87(7):1171–80. https://doi.org/10.1016/s0092-8674(00)81813-9 (Epub 1996/12/27).
Lavoie SS, Dumas E, Vulesevic B, Neagoe PE, White M, Sirois MG. Synthesis of human neutrophil extracellular traps contributes to angiopoietin-mediated in vitro proinflammatory and proangiogenic activities. J Immunol. 2018;200(11):3801–13. https://doi.org/10.4049/jimmunol.1701203 (Epub 2018/04/25).
Laridan E, Martinod K, De Meyer SF. Neutrophil extracellular traps in arterial and venous thrombosis. Semin Thromb Hemost. 2019;45(1):86–93. https://doi.org/10.1055/s-0038-1677040 (Epub 2019/01/12).
Moschonas IC, Tselepis AD. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis. 2019;288:9–16. https://doi.org/10.1016/j.atherosclerosis.2019.06.919 (Epub 2019/07/08).
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr, et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107(36):15880–5. https://doi.org/10.1073/pnas.1005743107 (Epub 2010/08/28).
Zhou Y, Tao W, Shen F, Du W, Xu Z, Liu Z. The emerging role of neutrophil extracellular traps in arterial, venous and cancer-associated thrombosis. Front Cardiovasc Med. 2021;8:786387. https://doi.org/10.3389/fcvm.2021.786387.
Abdol Razak N, Elaskalani O, Metharom P. Pancreatic cancer-induced neutrophil extracellular traps: a potential contributor to cancer-associated thrombosis. Int J Mol Sci. 2017;18(3):487. https://doi.org/10.3390/ijms18030487 (Epub 2017/03/02).
Thalin C, Hisada Y, Lundstrom S, Mackman N, Wallen H. Neutrophil extracellular traps: villains and targets in arterial, venous, and cancer-associated thrombosis. Arterioscler Thromb Vasc Biol. 2019;39(9):1724–38. https://doi.org/10.1161/ATVBAHA.119.312463 (Epub 2019/07/19).
Craver BM, Ramanathan G, Hoang S, Chang X, Mendez Luque LF, Brooks S, et al. N-acetylcysteine inhibits thrombosis in a murine model of myeloproliferative neoplasm. Blood Adv. 2020;4(2):312–21. https://doi.org/10.1182/bloodadvances.2019000967 (Epub 2020/01/25).
Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123(8):3446–58. https://doi.org/10.1172/JCI67484 (Epub 2013/07/19).
Leal AC, Mizurini DM, Gomes T, Rochael NC, Saraiva EM, Dias MS, et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci Rep. 2017;7(1):6438. https://doi.org/10.1038/s41598-017-06893-7 (Epub 2017/07/27).
Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight. 2019;5(16):e128008. https://doi.org/10.1172/jci.insight.128008 (Epub 2019/07/26).
Cedervall J, Dragomir A, Saupe F, Zhang Y, Arnlov J, Larsson E, et al. Pharmacological targeting of peptidylarginine deiminase 4 prevents cancer-associated kidney injury in mice. Oncoimmunology. 2017;6(8):e1320009. https://doi.org/10.1080/2162402X.2017.1320009 (Epub 2017/09/19).
Doyle K, Lonn H, Kack H, Van de Poel A, Swallow S, Gardiner P, et al. Discovery of second generation reversible covalent Dpp1 inhibitors leading to an oxazepane amidoacetonitrile based clinical candidate (Azd7986). J Med Chem. 2016;59(20):9457–72. https://doi.org/10.1021/acs.jmedchem.6b01127 (Epub 2016/10/04).
Hisada Y, Grover SP, Maqsood A, Houston R, Ay C, Noubouossie DF, et al. Neutrophils and neutrophil extracellular traps enhance venous thrombosis in mice bearing human pancreatic tumors. Haematologica. 2020;105(1):218–25. https://doi.org/10.3324/haematol.2019.217083 (Epub 2019/05/03).
Zhu T, Zou X, Yang C, Li L, Wang B, Li R, et al. Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelialmesenchymal transition. Int J Mol Med. 2021;48(1):127. https://doi.org/10.3892/ijmm.2021.4960 (Epub 2021/05/21).
Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138. https://doi.org/10.1126/scitranslmed.aag1711 (Epub 2016/11/01).
Shi L, Yao H, Liu Z, Xu M, Tsung A, Wang Y. Endogenous Pad4 in breast cancer cells mediates cancer extracellular chromatin network formation and promotes lung metastasis. Mol Cancer Res. 2020;18(5):735–47. https://doi.org/10.1158/1541-7786.MCR-19-0018.
Zhu B, Zhang X, Sun S, Fu Y, Xie L, Ai P. Nf-kappab and neutrophil extracellular traps cooperate to promote breast cancer progression and metastasis. Exp Cell Res. 2021;405(2):112707. https://doi.org/10.1016/j.yexcr.2021.112707 (Epub 2021/06/22).
Gray RD, McCullagh BN, McCray PB. Nets and Cf lung disease: current status and future prospects. Antibiotics. 2015;4(1):62–75. https://doi.org/10.3390/antibiotics4010062 (Epub 2015/01/01).
Volkov DV, Tetz GV, Rubtsov YP, Stepanov AV, Gabibov AG. Neutrophil extracellular traps (nets): opportunities for targeted therapy. Acta Naturae. 2021;13(3):15–23. https://doi.org/10.32607/actanaturae.11503 (Epub 2021/10/29).
Chen J, Hou S, Liang Q, He W, Li R, Wang H, et al. Localized degradation of neutrophil extracellular traps by photoregulated enzyme delivery for cancer immunotherapy and metastasis suppression. ACS Nano. 2022;16(2):2585–97. https://doi.org/10.1021/acsnano.1c09318 (Epub 2022/01/27).
Zhang Y, Chandra V, Riquelme Sanchez E, Dutta P, Quesada PR, Rakoski A, et al. Interleukin-17-Induced Neutrophil Extracellular Traps Mediate Resistance to Checkpoint Blockade in Pancreatic Cancer. J Exp Med. 2020;217(12):e20190354. https://doi.org/10.1084/jem.20190354 (Epub 2020/08/30).
Schalper KA, Carleton M, Zhou M, Chen T, Feng Y, Huang SP, et al. Elevated serum interleukin-8 is associated with enhanced intratumor neutrophils and reduced clinical benefit of immune-checkpoint inhibitors. Nat Med. 2020;26(5):688–92. https://doi.org/10.1038/s41591-020-0856-x (Epub 2020/05/15).
Schott AF, Goldstein LJ, Cristofanilli M, Ruffini PA, McCanna S, Reuben JM, et al. Phase Ib pilot study to evaluate reparixin in combination with weekly paclitaxel in patients with Her-2-negative metastatic breast cancer. Clin Cancer Res. 2017;23(18):5358–65. https://doi.org/10.1158/1078-0432.CCR-16-2748 (Epub 2017/05/26).
Xia Y, He J, Zhang H, Wang H, Tetz G, Maguire CA, et al. Aav-mediated gene transfer of dnase I in the liver of mice with colorectal cancer reduces liver metastasis and restores local innate and adaptive immune response. Mol Oncol. 2020;14(11):2920–35. https://doi.org/10.1002/1878-0261.12787 (Epub 2020/08/20).
Rayes RF, Vourtzoumis P, Bou Rjeily M, Seth R, Bourdeau F, Giannias B, et al. Neutrophil extracellular trap-associated ceacam1 as a putative therapeutic target to prevent metastatic progression of colon carcinoma. J Immunol. 2020;204(8):2285–94. https://doi.org/10.4049/jimmunol.1900240 (Epub 2020/03/15).
Jiang KL, Ma PP, Yang XQ, Zhong L, Wang H, Zhu XY, et al. Neutrophil elastase and its therapeutic effect on leukemia cells. Mol Med Rep. 2015;12(3):4165–72. https://doi.org/10.3892/mmr.2015.3946 (Epub 2015/06/18).
Zheng W, Warner R, Ruggeri R, Su C, Cortes C, Skoura A, et al. Pf-1355, a mechanism-based myeloperoxidase inhibitor, prevents immune complex vasculitis and anti-glomerular basement membrane glomerulonephritis. J Pharmacol Exp Ther. 2015;353(2):288–98. https://doi.org/10.1124/jpet.114.221788 (Epub 2015/02/24).
Knight JS, Subramanian V, O’Dell AA, Yalavarthi S, Zhao W, Smith CK, et al. Peptidylarginine deiminase inhibition disrupts net formation and protects against kidney, skin and vascular disease in Lupus-Prone Mrl/Lpr mice. Ann Rheum Dis. 2015;74(12):2199–206. https://doi.org/10.1136/annrheumdis-2014-205365 (Epub 2014/08/12).
Greene S, Robbins Y, Mydlarz WK, Huynh AP, Schmitt NC, Friedman J, et al. Inhibition of Mdsc trafficking with Sx-682, a Cxcr1/2 inhibitor, enhances Nk-cell immunotherapy in head and neck cancer models. Clin Cancer Res. 2020;26(6):1420–31. https://doi.org/10.1158/1078-0432.CCR-19-2625 (Epub 2019/12/19).
Nywening TM, Belt BA, Cullinan DR, Panni RZ, Han BJ, Sanford DE, et al. Targeting both tumour-associated Cxcr2(+) neutrophils and Ccr2(+) macrophages disrupts myeloid recruitment and improves chemotherapeutic responses in pancreatic ductal adenocarcinoma. Gut. 2018;67(6):1112–23. https://doi.org/10.1136/gutjnl-2017-313738 (Epub 2017/12/03).
Steele CW, Karim SA, Leach JDG, Bailey P, Upstill-Goddard R, Rishi L, et al. Cxcr2 inhibition profoundly suppresses metastases and augments immunotherapy in pancreatic ductal adenocarcinoma. Cancer Cell. 2016;29(6):832–45. https://doi.org/10.1016/j.ccell.2016.04.014 (Epub 2016/06/07).
Skov L, Beurskens FJ, Zachariae CO, Reitamo S, Teeling J, Satijn D, et al. Il-8 as antibody therapeutic target in inflammatory diseases: reduction of clinical activity in palmoplantar pustulosis. J Immunol. 2008;181(1):669–79. https://doi.org/10.4049/jimmunol.181.1.669 (Epub 2008/06/21).
Pastille E, Fassnacht T, Adamczyk A, Ngo TPN, Buer J, Westendorf AM. Inhibition of Tlr4 signaling impedes tumor growth in colitis-associated colon cancer. Front Immunol. 2021;12:66. https://doi.org/10.3389/fimmu.2021.669747 (Epub 2021/05/25).
Zhang S, Zhang Q, Wang F, Guo X, Liu T, Zhao Y, et al. Hydroxychloroquine inhibiting neutrophil extracellular trap formation alleviates hepatic ischemia/reperfusion injury by blocking Tlr9 in mice. Clin Immunol. 2020;216:108461. https://doi.org/10.1016/j.clim.2020.108461 (Epub 2020/05/22).
Peng H, Shen J, Long X, Zhou X, Zhang J, Xu X, et al. Local release of Tgf-beta inhibitor modulates tumor-associated neutrophils and enhances pancreatic cancer response to combined irreversible electroporation and immunotherapy. Adv Sci. 2022;9(10):e2105240. https://doi.org/10.1002/advs.202105240 (Epub 2022/02/08).
Mutua V, Gershwin LJ. A review of neutrophil extracellular traps (nets) in disease: potential anti-nets therapeutics. Clin Rev Allergy Immunol. 2021;61(2):194–211. https://doi.org/10.1007/s12016-020-08804-7 (Epub 2020/08/03).
Acknowledgements
No.
Funding
No.
Author information
Authors and Affiliations
Contributions
JH contributed to the conception of the study. YC, HH and ST were responsible for the draft of the manuscript and the data collection. YW and XF worked for the figures. QD and HZ worked for the Tables. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, Y., Hu, H., Tan, S. et al. The role of neutrophil extracellular traps in cancer progression, metastasis and therapy. Exp Hematol Oncol 11, 99 (2022). https://doi.org/10.1186/s40164-022-00345-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-022-00345-3