
Abstract
As the predominant host defense against pathogens, neutrophil extracellular traps (NETs) have attracted increasing attention due to their vital roles in infectious inflammation in the past few years. Interestingly, NETs also play important roles in noninfectious conditions, such as rheumatism and cancer. The process of NETs formation can be regulated and the form of cell death accompanied by the formation of NETs is regarded as “NETosis”. A large amount of evidence has confirmed that many stimuli can facilitate the release of NETs from neutrophils. Furthermore, it has been illustrated that NETs promote tumor growth and progression via many molecular pathways. Meanwhile, NETs also can promote metastasis in many kinds of cancers based on multiple studies. In addition, some researchs have found that NETs can promote coagulation and cancer-associated thrombosis. In the present review, it will highlight how NETosis, which is stimulated by various stimuli and signaling pathways, affects cancer biological behaviors via NETs. Given their crucial roles in cancer, NETs will become possible therapeutic targets for inhibiting proliferation, metastasis and thrombosis in cancer patients.
Similar content being viewed by others
Introduction
Neutrophil extracellular traps (NETs) are net-like structures composed of granule proteins and nuclear components (such as DNA and histones) [1]. DNA is decorated with granule proteins, including neutrophil elastase (NE), myeloperoxidase (MPO), matrix metalloproteinase 9 (MMP-9) [1], calprotectin [2], cathepsin G (CG) and proteinase 3 (PR3) [3]. Furthermore, mitochondria can also serve as a source of DNA for NET formation [4]. NETs were first recognized as a novel host defense mechanism. NETs have been shown to trap diverse pathogens, including bacteria [1], fungi [5, 6], viruses [7] and protozoan parasites [8]. Apart from infectious inflammation, NETs have been found to be involved in cancer [9] and sterile inflammatory diseases, such as ventilator-induced lung injury [10], lupus nephritis [11] and acute pancreatitis [12]. Furthermore, NETs also play a significant role in atherosclerosis disease [13]. A study has confirmed that deficiency of EGF-like repeats and discoidin I-like domain 3 can improve adverse cardiac healing through polarization of pro-inflammatory macrophage which is mediated by NETs [14]. In particular, NETs are even associated with preeclampsia and central nervous system diseases [15, 16].
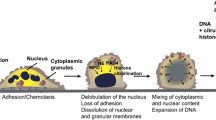
NETs are released into the extracellular environment in response to relevant stimuli. The formation of NETs involves a unique form of cell death that is dependent on the generation of reactive oxygen species (ROS) by NADPH oxidase, which is different from apoptosis and necrosis [17]. Steinberg and Grinstein regarded this form of cell death accompanied by the formation of NETs as “NETosis” [18]. When neutrophils encounter relevant stimuli, they undergo morphological changes. The most salient morphological differences seen in cells undergoing NETosis but not apoptotic and necrotic cells are disintegration of the nuclear envelope and mixing of nuclear and cytoplasmic material, loss of internal membranes, and disappearance of cytoplasmic organelles. Cell death is initiated by ROS [17]. Decondensation of chromatin is a critical event for NETosis. NE exits azurophilic granules and translocates to the nucleus. Then, MPO translocates to the nucleus. MPO cooperates with NE to induce nuclear decondensation [19]. Peptidylarginine deiminase 4 (PAD4), which can promote citrullination of histones, is highly expressed in neutrophils. The hypercitrullination of histones by PAD4 promotes chromatin decondensation [20]. Next, the cell membrane breaks, and NETs are released [17]. Various stimuli have been shown to contribute the production of NETs, then inhibitors of NADPH oxidase and NE prevent NET formation [17, 19]. Therefore, investigating the signaling pathways that regulate the formation of NETs and identifying specific inhibitors may provide possibility for the treatments of human cancers.
Due to its vital roles in infectious and sterile inflammation, NETs have attracted increasing attention in the last two decades [1, 10]. In the area of malignancy, NETs have been shown to promote thrombosis, proliferation and metastasis of cancer cells [21], cause organ damage in cancer patients [9] and even predict the prognosis of cancer patients [22, 23]. In this review, we will highlight the mechanisms by which NETosis, which is stimulated by various stimuli and signaling pathways, promotes tumor progression and metastasis and cancer-associated thrombosis via NETs.
Molecular pathway of NETosis stimulated by various agonists
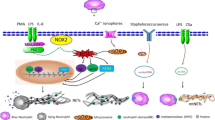
To date, many stimuli have been confirmed to induce the release NETs from neutrophils, such as lipopolysaccharide (LPS) [24] and phorbol 12-myristate 13-acetate (PMA) [1]. Some cytokines and proteins, including high mobility group 1 (HMGB1) protein [25], cathepsin C (CTSC) [26], granulocyte colony stimulating factor (G-CSF) [27] and interleukin-8 (IL-8) [28], can also stimulate the formation of NETs. A summary of the molecular pathways by which NETosis is activated by various agonists is shown in Fig. 1.

Molecular pathway of NETosis stimulated by various agonists. Many agonists which can induce the formation of NETs are described, including LPS, PMA, HMGB1, G-CSF, IL-8 and CTSC
IL-8
IL-8, which is also named C-X-C motif chemokine ligand 8 (CXCL8), is a member of the CXC subfamily of chemokines. IL-8 is released by not only malignant cells but also stromal cells in the tumor microenvironment [29]. The plasma levels of IL-8 are higher in cancer patients than in healthy individuals [30]. IL-8 is increased in breast cancer tissues compared to adjacent normal breast tissues [31]. IL-8 secreted from tumors promotes the release of NETs [32, 33]. Furthermore, IL-8 is positively correlated with NETs in non-small cell lung cancer (NSCLC) and melanoma [32]. IL-8 can easily bind with C-X-C motif chemokine receptor 1 (CXCR1) and C-X-C motif chemokine receptor 2 (CXCR2) [34]. Agonists of CXCR1 and CXCR2 are the main mediators of NETosis induced by cancer. Blocking CXCR1 and CXCR2 in mice with breast cancer with reparixin, a specific small-molecule inhibitor of CXCR1 and CXCR2, results in decreased levels of NETs [35, 36]. Podaza et al. [37] found that plasma from chronic lymphocytic leukemia patients could promote the formation of NETs through the plasmatic IL-8-CXCR2 axis. In diffuse large B cell lymphoma (DLBCL), the IL-8-CXCR2 axis induces NETosis via src, p38 and extracellular-signal-regulated kinase (ERK) rather than the phosphoinositide 3-kinase (PI3K) signaling pathway [38]. However, in another study, IL-8 was found to promote the phosphorylation of serine/threonine-protein kinase (AKT), a key mediator downstream of the PI3K signaling pathway. The IL-8-CXCR2 axis mediates NET formation via the PI3K/AKT/ROS axis in tumor-infiltrating neutrophils [39]. In our ongoing study, we also have found that IL-8 can influence the biological behavior of gastric cancer (GC) by promoting the release of NETs through CXCR1/2. These studies demonstrate formation of NETs regulated by IL-8 and its receptors, CXCR1 and CXCR2, via various signaling pathways.
G-CSF
G-CSF, a cytokine produced by leukocytes, endothelium, and tumors, is increased in the peripheral blood of cancer patients [40,41,42]. Some studies have shown increased generation of NETs in tumors induced by G-CSF [43, 44]. G-CSF can directly stimulate neutrophils to release NETs in cancer patients [45]. However, the molecular pathway by which G-CSF promotes NETosis has not been clearly identified.
PMA
PMA (also named TPA), a potent neutrophil activator, results in morphological changes in neutrophils that are quite different from those typical of apoptosis or necrosis [46, 47]. Some studies have confirmed that PMA can promote the release of NETs from neutrophils in both healthy donors and cancer patients [48,49,50]. Ermert et al. [51] found that PMA can promote the generation of ROS in neutrophils in a mouse model, and neutrophils from NADPH oxidase-deficient mice failed to produce ROS and did not die or release NETs upon stimulation. In chronic granulomatous disease patients, the neutrophils have NADPH oxidase mutations and are thus unable to generate ROS even upon PMA activation [17]. PMA promotes the formation of NETs by NADPH oxidase-mediated ROS generation. Furthermore, the nonphysiological agent PMA is an activator of protein kinase C (PKC) [52]. Hakkim et al. confirmed that the Raf-MAP kinase ERK kinase (MEK)-ERK pathway is upstream of NADPH oxidase and downstream of PKC. Blocking PKC with staurosporine, a PKC inhibitor, results in decreased formation of NETs. PMA stimulates the generation of NETs through the Raf-MEK-ERK signaling pathway via the activation of PKC [53]. However, in another study, ERK was found to be downstream of ROS generation and upstream of P38 mitogen-activated protein kinase (MAPK) signaling during PMA-induced NETosis [54]. Moreover, activation of Akt is dependent on NADPH oxidase-mediated ROS production, which was essential for the NETosis induced by PMA [55]. Spleen associated tyrosine kinase (Syk) and PI3K, which mediate the generation of ROS, are crucial for NETosis elicited by PMA [56, 57].
LPS
LPS, a main component of the gram-negative bacteria cell wall, has been identified as a potent stimulator of the formation of NETs [58, 59]. LPS can stimulate NETosis in tumors [60, 61]. Furthermore, LPS can upregulate NADPH oxidase in neutrophils [62]. Khan et al. [63] found that LPS induces activation of c-jun N-terminal kinase (JNK), which is upstream of NADPH oxidase, in a toll-like receptor 4 (TLR4)-dependent manner in neutrophils. Both JNK activation and TLR4 signaling are important for LPS-mediated ROS production and NETosis in neutrophils. However, LPS does not induce NET formation in purified neutrophils [64], which is consistent with another study reporting that LPS indirectly promotes NET generation [65]. Some research has shown that LPS-induced NETosis depends on the presence of platelets [66, 67]. The structure of LPS might result in these two different consequences [68]. LPS promotes the activation of platelets through platelet TLR4, inducing the interaction of platelets with neutrophils and the generation of NETs [69, 70]. NETosis induced by LPS-stimulated platelets is dependent on ROS. This process is mediated by interferon regulatory factor 1 (IRF-1) [71]. However, in another study, ROS were found to not be involved in the NETosis induced by platelets, which was instead activated by LPS and platelet-triggered NET release through the ERK, PI3K, and Src kinases [67]. It shows that PKC and generation of ROS are essential for PMA induced NETosis.
HMGB1
HMGB1 protein is both a nuclear factor and a secreted protein [72]. HMGB1 is released by living immune cells or passively released from dead, dying, and injured cells [73]. Several studies have demonstrated that HMGB1 is prone to binding with receptor for advanced glycation end products (RAGE), TLR2, TLR4 and TLR9 [74,75,76]. Tadie et al. found a role of HMGB1 in contributing to NET formation, and HMGB1 was found to induce the generation of NETs both in vitro and in vivo through a TLR4-dependent mechanism [25, 77]. This result is consistent with a study conducted by Zhou and colleagues in lung cancer. The researchers found that the downstream molecules of TLR4, p38 MAPK and ERK, were activated during the formation of NETs [78]. Moreover, HMGB1 was found to promote NET formation via tumor necrosis factor (TNF) and receptor-interacting-protein kinase-1 (RIPK1) kinase activity during tumorigenesis of skin [79].
CTSC
CTSC, also known as dipeptidyl peptidase I, is a lysosomal cysteine protease essential for many serine proteases, including CG, NE, PR3, granzymes A/B and mast cell chymases [80,81,82]. A study discovered that CTSC secreted by tumors promotes the formation of NETs in breast cancer cells. CTSC induces neutrophil ROS production and the formation of NETs by activating the neutrophil membrane-bound PR3-interleukin-1β (IL-1β)-p38 axis [83].
NETs and cancer
Neutrophils play an important role in cancer. Tumor-associated neutrophils are divided into two phenotypes according to their functions in the tumor microenvironment: the antitumor N1 versus the protumor N2 phenotype [84]. In a spontaneous intestinal tumorigenesis model, low-density neutrophils display clear features of N2 neutrophils and spontaneously undergo NETosis via complement 3a receptor signaling [85]. Several studies have confirmed the presence of increased levels of NETs in advanced cancer patients, including DLBCL and esophageal and lung adenocarcinoma patients [22, 38, 86]. NETs can also accelerate deterioration of colorectal cancer (CRC) [87]. Moreover, in our two previous studies, we have discovered that NETs and its key component MPO are prognostic factors affecting poor survival in patients with GC [22, 88]. As a serum biomarker, NETs have a better diagnostic value than carcinoembryonic antigen and carbohydrate antigen 19-9 in GC. The level of NETs is inversely correlated with short-term efficacy in GC patients who have received medical treatment [22]. NETs are also associated with tumor burden and have been shown to promote growth, progression and metastasis in cancer [89,90,91]. Furthermore, a relationship between NETs and cancer-associated thrombosis has been confirmed [27]. The next three parts will highlight the mechanisms of NETs in tumor growth, progression, metastasis and cancer-associated thrombosis.
NETs promote tumor growth and progression
NETs have been shown to promote tumor growth and progression. In a nonalcoholic steatohepatitis (NASH) model, the progression of NASH to hepatocellular carcinoma (HCC) can be reduced by blocking NETs [92]. In glioma, NETs promote the proliferation of cancer cells [39]. To date, some roles of NETs in tumor growth and progression have been identified (Fig. 2).

NETs promote tumor growth and progression via many molecular pathways. HMGB1, released by NETs, can promote tumor growth by binding with TLR9 or RAGE to activate STAT3, p38, MAP and NF-κB. NE directly alters metabolism and upregulates mitochondrial biogenesis in colon cancer cells via the TLR4-PGC1-α pathway and thus promotes tumor growth. NETs awaken cancer cells and cleave laminin via NE and MMP-9 through the activity of integrin α3β1 and FAK/ERK/MLCK/YAP signaling. NET DNA drives pancreatic tumor growth by activating pancreatic stellate cells through interaction with RAGE. In our ongoing study, we have illustrated that NETs can promote tumor growth by BRF1 in GC cells
NETs can directly increase TLR9 expression in DLBCL and promote tumor progression via the NF-κB, signal transducer and activator of transcription 3 (STAT3) and p38 pathways [38]. Furthermore, the mechanisms by which NETs promote tumor growth and progression have been confirmed.
HMGB1, a damage-associated molecular pattern, has been shown to participate in the growth and progression of cancer [93, 94]. HMGB1 is a constituent protein of NETs [21, 95]. In glioblastoma, HMGB1 derived from NETs promotes the proliferation of cancer cells by interacting with RAGE and activating the NF-κB pathway [39]. In another experiment performed by Tohme et al. [21], HMGB1 released from NETs interacted with TLR9 in MC38 CRC cells and then activated the MAP kinase pathway to exert a protumorigenic function.
DNA is a significant element in NETs. Miller-Ocuin et al. found that DNA released from NETs promotes the proliferation of Panc02 murine pancreatic cancer cells. NET DNA drives pancreatic tumor growth in a murine subcutaneous tumor model by activating pancreatic stellate cells through interaction with RAGE [96].
NE is also an important component of NETs. Yazdani and colleagues found that NETs and mitochondrial biogenesis factors, including peroxisomes proliferator-activated receptor gamma coactivator 1-alpha (PGC-1α), mitochondrial transcription factor A (TFAM) and nuclear respiratory factor 1 (NRF-1), are increased in HCC and CRC tissues compared to their nontumor counterparts. NE directly alters metabolism and upregulates mitochondrial biogenesis in MC38 cells via the TLR4-PGC1-α pathway and thus promotes tumor growth [91].
In our ongoing study, we have illustrated that NETs can promote transcription factor IIB-related factor 1 (BRF1) expression in GC cells. BRF1 is known to affect the transcription and expression of RNA polymerase-3 related genes. In previous results, we found that BRF1 is a key molecule affecting the proliferation of GC [88].
Another relationship of note is that between NETs and tumor recurrence. Albrengues et al. induced sustained inflammation of the lung via tobacco smoke exposure or nasal instillation of LPS in Balb/c or nude mice that had been injected with MCF-7 breast cancer cells. They found an increase in NETosis and proliferation of awakened cancer cells in inflammatory lungs. NET DNA was prone to bind with the extracellular matrix (ECM) protein laminin. NETs awakened cancer cells and there was concomitant remodeling of laminin, which was sequentially cleaved by two NET-associated proteases, NE and MMP-9, through the activity of integrin α3β1 and focal adhesion kinase/ERK/myosin light chain kinase/yes-associated protein (FAK/ERK/MLCK/YAP) signaling [97].
NETs promote tumor metastasis
With a deeper focus on the relationship between NETs and cancer, many studies have confirmed the mechanisms of NETs in tumor metastasis (Fig. 3). Park et al. [98] found that NETs promote migration and invasion in mouse 4T1 murine breast cancer cells and human BT-549 breast cancer cells. Similarly, an in vitro experiment showed that NETs promote migration in the human DLBCL cell lines SU-DHL2, SU-DHL4, and SU-DHL6 and the mouse DLBCL cell line A20 [38].

Molecular mechanisms of NETs in tumor metastasis. HMGB1, which is released by NETs, can facilitate tumor metastasis by the increased expression of EMT-associated genes, ZEB1 and Snail. In addition to EMT, NETs can also regulate expression of COX-2 and MAP to induce tumor migration. In human breast cancer cells, NET DNA binds to CCDC25 and induces an ILK-β-PARVIN-RAC1-CDC42 cascade, resulting in the metastasis of cancer cells. TSP-1 can be degraded by NETs to eliminate its inhibitory effect on tumor migration
Many studies have reported the role of adhesion of NETs and cancer cells in tumor metastasis. Cools-Lartigue et al. [99] induced sepsis via cecal ligation and puncture and subsequently found that NETs promote cancer metastasis by trapping circulating tumor cells in circulation. This process is mediated by β1-integrin [100]. In an ovarian cancer model, NETs are located in the omentum, which is the premetastatic niche of ovarian cancer. NETs attach to ovarian cancer cells and promote metastasis [101].
Furthermore, another study found a novel pathway in breast cancer lung metastasis. NETs promote tumor metastasis by degrading thrombospondin-1 (TSP-1), a secreted extracellular matrix protein that inhibits tumor metastasis [26, 102].
An in vitro experiment showed that NET DNA significantly promotes the migration and adhesion of MDA-MB-231 human breast cancer cells. Proteins extracted from the cytoplasmic membrane of cancer cells and then inoculated with NET DNA were examined, and the transmembrane protein coiled-coil domain containing protein 25 (CCDC25) was found to function as a potential receptor for NETs to promote tumor metastasis. NET DNA binds to CCDC25 and induces an integrin linked kinase (ILK)-β-PARVIN-RAC1-CDC42 cascade, resulting in the metastasis of cancer cells [103].
HMGB1 played an important role in tumor metastasis [104, 105]. A study demonstrated that NETs can release HMGB1 and promote tumor metastasis [21]. Tohme et al. established an ischemia and reperfusion model to mimic surgical stress. They found that NETs induced by surgical stress promoted the development of gross metastases, which was mediated by HMGB1 from NETs through the TLR9-associated MAP kinase pathway in MC38 cells.
Endothelial to mesenchymal transition (EMT) is a key process by which cancer cells acquire migration ability [90]. Glomerular NETs have been confirmed to promote EMT in lupus nephritis patients and mouse models [11]. An in vitro experiment revealed that NETs can change the morphology of MCF-7 cells, accompanied by an increase in the transcription of EMT-related genes, such as zinc-finger E-box-binding homeobox 1 (ZEB1) and Snail. NETs promote migration ability in breast cancer through EMT [90]. This result is consistent with another study in GC [106]. It shows that process of EMT is significant for NETs induced tumor metastasis.
An interesting study assessed EMT, HMGB1 and NETs in cancer. NETs enhanced the migration and invasion of pancreatic cancer cells both in vivo and in vitro. Furthermore, the morphology of PANC-1 cells changed in the presence of NETs, and the transcription of genes involved in EMT, such as slug, ZEB1 and Snail, was upregulated. Immunofluorescence verified the colocalization of NETs and HMGB1. In this study, HMGB1 derived from NETs increased migration and invasion abilities through the EMT program [107].
In addition to the above pathways, the tumor microenvironment is also associated with tumor metastasis. The level of cyclooxygenase-2 (COX-2) is increased in MDA-MB-231 breast cancer cells that are treated with NETs. In addition, NETs induce a proinflammatory response [90]. An interesting experiment reported that the inflammatory response in the tumor microenvironment potentiates the metastatic potential of cancer via NETs. NETs promote metastasis of hepatocellular carcinoma by trapping HCC cells and promoting angiogenesis. In one study, RNA sequencing confirmed that a set of genes coding inflammatory mediators, for example, COX2, were upregulated. Related siRNAs were used to block the activity of TLR4/9, and in response, the NET-induced expression of COX2, which is downstream of TLR4/9, was obviously decreased [108]. These studies demonstrate that NETs promote potential metastasis of tumor via provoking tumorous inflammatory response.
NETs promote coagulation and cancer-associated thrombosis
Cancer patients commonly have a hypercoagulable condition, and thrombosis has been identified as an important cause of death in cancer patients [27]. NETs were first found to promote thrombosis in an infected wound [109]. Subsequently, various studies confirmed that NETs attributed to atherosclerosis and thrombosis [110]. Recently, a role of NETs in coagulation and cancer-associated thrombosis was identified (Fig. 4). NETs induced by cancer cell could promote not only cancer progression but also hypercoagulability [111]. Spontaneous NETosis was found to be associated with thrombosis at late stages of cancer in mammary tumor-bearing mice [27]. In colorectal cancer patients, the activated partial thromboplastin time is significantly shorter and d-dimer levels are obviously higher than those in healthy subjects. Treatment with DNase I decreases coagulation time in control plasma treated with NETs [112]. Similarly, administration of DNase I significantly reduces the procoagulant role of NETs released by neutrophils derived from patients with GC [113]. NETs are crucial for the generation of venous and arterial thrombi in both cancer patients and tumor-bearing mice [40, 44, 114].

NETs promote coagulation and cancer-associated thrombosis. NETs provide a scaffold for platelets and tumor-derived EVs. Exposure of PS on platelets and endothelial cells that are induced by NETs promotes coagulation and cancer-associated thrombosis. EVs, which recruited by NETs, lead to a prothrombotic state. TF, which is derived from EVs or NETs, can promote cancer-associated thrombosis by initiating extrinsic coagulation. The factor XII, which can trigger the contact system, is associated with the level of NETs. However, the mechanisms by which NETs drive factor XII- and cancer-associated thrombosis need next studies
NETs have been shown to promote thrombin formation by providing a scaffold for platelets. Studies have shown that NETs trap platelets to promote platelet activation and thrombus formation [109, 115]. NETs derived from CRC patients promote the procoagulant activity of platelets and phosphatidylserine (PS) exposure on platelets. PS provides the specific catalytic surface needed for the coagulation cascade. Moreover, the study also revealed an increase in PS exposure on human umbilical vein endothelial cells (HUVECs) that had been stimulated with NETs from CRC. CRC-derived NETs induced shortened coagulation time and massive fibrin release from endothelial cells. NETs promoted the coagulant activity of endothelial cells mostly via PS [112]. Another study confirmed that NETs derived from pancreatic cancer convert HUVECs towards the procoagulant phenotype via PS [116]. All above results implies that exposure of PS induced by NETs on platelets and endothelial cells plays an important role in procoagulant activity in tumor.
NETs also serve as the backbone for tumor-derived extracellular vesicles (EVs) [44, 117]. Exosomes and microparticles (MPs) are two common types of EVs [44]. Exosomes are the products of the endolysosomal pathway and range in size from 30 to 150 nm [118]. NETs promote both venous and arterial thrombosis in a murine 4T1 mammary carcinoma model. Exosomes derived from 4T1 cells exhibit a procoagulant effect in a dose-dependent manner and tend to adhere to NETs in vitro. These results suggest that NETs might lead to a prothrombotic state in 4T1-bearing mice via recruitment of tumor-derived exosomes [44]. Moreover, another study found pancreatic tumor-derived MPs expressing tissue factor (TF) bound to NETs in vitro [117]. MPs are cell-derived membrane fragments that range in size from 0.1 to 1 μm [119]. In a deep vein thrombosis model generated by inferior vena cava stenosis, PANC02-derived MPs were infused into the blood of mice. As a result, the MPs adhered to NETs and accumulated at the site of pathological thrombosis, which promoted the formation of cancer-associated deep vein thrombosis in vivo [117].
TF is a transmembrane protein that functions as a receptor and activator of factor VII, subsequently initiating extrinsic coagulation [120, 121]. Some studies have confirmed that TF derived from MPs is correlated with coagulation activation and thrombosis in cancer [120, 122]. Furthermore, TF is also derived from neutrophils and released during the formation of NETs [123]. NETs have been shown to express functional TF [124, 125]. Another study confirmed that NETs can promote cancer-associated thrombosis by trapping tumor-derived MPs expressing TF in vitro [117]. In specimens of colonic adenocarcinoma and respective metastatic lymph nodes, TF colocalized with NETs [126]. In a model of murine pancreatic adenocarcinoma, coagulation was reduced via a decrease in the levels of TF, which was induced by abolishment of NETs [123]. It shows that TF modulated by NETs is vital for coagulation.
Factor XII can trigger the contact system, which initiates the intrinsic coagulation pathway [127]. In patients with HCC, the plasma levels of NETs and factor XIIa are elevated [128]. Similarly, markers of the contact system in plasma, including activated factor XIIa and high-molecular-weight kininogen, are significantly increased in acute leukemia samples compared with normal controls. The factor XIIa level is significantly correlated with the level of histone-DNA complexes, which seem to be a marker of NETs. Thus, contact system activation is associated with NET formation [129]. NETs have been shown to bind with factor XII via their negatively charged surface and subsequently activate intravascular thrombus formation [130]. However, the mechanisms by which NETs drive factor XII- and cancer-associated thrombosis need further investigation.
The clinical value of NETs
In recent years, the value of NETs in cancer diagnosis, efficacy prediction and prognosis has been concerned. In our previous study, we discovered that NETs had novel diagnostic, therapeutic predictive, and prognostic value in GC patients [22]. Besides, elevated levels of NETs were associated with higher mortality in patients with cancer [131]. Recently, a study demonstrated that NETs were linked to a poor prognosis in patients with terminal cancer [132]. Thus, if we find the related way to inhibit the pathway of NETosis, it may control tumor invasion, evasion and metastasis, and ultimately improve the prognosis of cancer patients [133]. At present, NETs become a potential therapeutic target to inhibit cancer progression and metastasis [134, 135]. Some experiments in vivo have further proven this hypothesis. It has been confirmed that blocking NETosis via drugs against the components of NETs can effectively decrease ability of tumor growth and metastasis, such as DNase [21, 44], PAD4 inhibitors [97, 98, 136] and NE inhibitors [99]. Notably, a recombinant human DNase which named Pulmozyme was applied in a phase 1 trial in patients with head and neck cancer [134]. Danshen, the dried root of Salvia miltiorrhiza, which was traditional Chinese medicine, was found that it could exert antitumor effect via suppression of NETosis [137]. Moreover, inhibitors of CXCR1 and CXCR2, as the receptors of NETosis, were confirmed to block NETs [35, 36]. Meanwhile, the inhibitor of TLR9 which was a receptor of NETosis could retard tumor progression [38]. It provides a potential treatment about blocking key points of NETosis may control the progression of cancer effectively.
Conclusions and future perspectives
Shortly after their discovery, NETs were recognized for their function in host defense against pathogens. Once NETs were reported in cancer, the relationship between tumors and NETs received more attention. In this review, the regulatory mechanism of NETs in cancer biological behavior is discussed in detail. Some stimuli that are exogenous or secreted from tumors, such as IL-8, G-CSF, PMA, LPS, HMGB1, and CTSC, have been shown to promote the formation of NETs in cancer. NETs can promote cancer cell proliferation and cancer progression via many kinds of pathways, including the TLR and RAGE pathways. Moreover, NETs potentiate metastasis by changing the adhesion of cancer cells, promoting EMT, and enhancing inflammation in the microenvironment. In addition, NETs can promote cancer-associated thrombosis by providing scaffolds for activated platelets and mediating coagulant molecules via EVs. In particular, NETs regulate tumor biological behaviors through interaction with tumor microenvironment, which is different from traditional molecule targets. Because, NETs is a complex structure in tumor microenvironment, not a single molecule. It reflects the new function of infiltrated neutrophils in the tumor microenvironment. Analysis of the molecular mechanisms underlying NET formation has revealed that some key receptors and signaling pathways participate in both NETosis and tumor biological behaviors.
Interestingly, we found that NETs could promote tumor progression in a variety of tumor types, such as GC [22], head and neck cancer [23], DLBCL [38] and breast cancer [103]. On the other hand, the presence of HCC did not further increase the levels of NETs as compared to patients with cirrhosis only [138]. However, the presence of NETs accelerated transition of NASH to HCC [92]. It indicates that heterogenous presence of NETs in cancer progression may attribute to the complex causes of different cancers. In general, we still suggest that tumor-associated inflammation represented by NETs may be one of the common mechanisms to trigger cancer progression.
Given their crucial roles in cancer, NETs are possible therapeutic targets in cancer patients. Blocking NETosis through drugs against the components of NETs or the receptors of NETosis can effectively decrease ability of tumor growth and metastasis. Similarly, in recent years, immunotherapy by targeting T lymphocytes in the tumor microenvironment has changed the current state of medical antitumor therapy. Accordingly, we hope that NETs will become possible therapeutic targets for inhibiting proliferation, metastasis and thrombosis in cancer patients in the future.
Availability of data and materials
Not applicable.
Code availability
Not applicable.
Abbreviations
- NETs:
-
Neutrophil extracellular traps
- NE:
-
Neutrophil elastase
- MPO:
-
Myeloperoxidase
- MMP-9:
-
Matrix metalloproteinase 9
- CG:
-
Cathepsin G
- PR3:
-
Proteinase 3
- ROS:
-
Reactive oxygen species
- PAD4:
-
Peptidylarginine deiminase 4
- LPS:
-
Lipopolysaccharide
- PMA:
-
Phorbol 12-myristate 13-acetate
- HMGB1:
-
High mobility group 1
- CTSC:
-
Cathepsin C
- G-CSF:
-
Granulocyte colony-stimulating factor
- IL-8:
-
Interleukin-8
- CXCL8:
-
C-X-C motif chemokine ligand 8
- NSCLC:
-
Non-small cell lung cancer
- CXCR1:
-
C-X-C motif chemokine receptor 1
- CXCR2:
-
C-X-C motif chemokine receptor 2
- DLBCL:
-
Diffuse large B cell lymphoma
- ERK:
-
Extracellular-signal-regulated kinase
- PI3K:
-
Phosphoinositide 3-kinase
- AKT:
-
Serine/threonine-protein kinase
- GC:
-
Gastric cancer
- PKC:
-
Protein kinase C
- MEK:
-
MAP kinase ERK kinase
- MAPK:
-
Mitogen-activated protein kinase
- Syk:
-
Spleen associated tyrosine kinase
- JNK:
-
C-jun N-terminal kinase
- TLR4:
-
Toll-like receptor 4
- IRF-1:
-
Interferon regulatory factor 1
- RAGE:
-
Receptor for advanced glycation end products
- TNF:
-
Tumor necrosis factor
- RIPK1:
-
Receptor-interacting-protein kinase-1
- IL-1β:
-
Interleukin-1β
- NASH:
-
Nonalcoholic steatohepatitis
- HCC:
-
Hepatocellular carcinoma
- STAT3:
-
Signal transducer and activator of transcription 3
- CRC:
-
Colorectal cancer
- PGC-1α:
-
Peroxisomes proliferator-activated receptor gamma coactivator 1-alpha
- TFAM:
-
Mitochondrial transcription factor A
- NRF-1:
-
Nuclear respiratory factor 1
- ECM:
-
Extracellular matrix
- FAK:
-
Focal adhesion kinase
- MLCK:
-
Myosin light chain kinase
- YAP:
-
Yes-associated protein
- TSP-1:
-
Thrombospondin-1
- CCDC25:
-
Coiled-coil domain containing protein 25
- ILK:
-
Integrin linked kinase
- EMT:
-
Endothelial to mesenchymal transition
- ZEB1:
-
Zinc-finger E-box-binding homeobox 1
- COX-2:
-
Cyclooxygenase-2
- PS:
-
Phosphatidylserine
- HUVECs:
-
Human umbilical vein endothelial cells
- EVs:
-
Extracellular vesicles
- MPs:
-
Microparticles
- TF:
-
Tissue factor
References
Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303(5663):1532–5.
Urban CF, Ermert D, Schmid M, Abu-Abed U, Goosmann C, Nacken W, et al. Neutrophil extracellular traps contain calprotectin, a cytosolic protein complex involved in host defense against Candida albicans. PLoS Pathog. 2009;5(10):e1000639.
Averhoff P, Kolbe M, Zychlinsky A, Weinrauch Y. Single residue determines the specificity of neutrophil elastase for Shigella virulence factors. J Mol Biol. 2008;377(4):1053–66.
Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009;16(11):1438–44.
Urban CF, Reichard U, Brinkmann V, Zychlinsky A. Neutrophil extracellular traps capture and kill Candida albicans yeast and hyphal forms. Cell Microbiol. 2006;8(4):668–76.
Bruns S, Kniemeyer O, Hasenberg M, Aimanianda V, Nietzsche S, Thywissen A, et al. Production of extracellular traps against Aspergillus fumigatus in vitro and in infected lung tissue is dependent on invading neutrophils and influenced by hydrophobin RodA. PLoS Pathog. 2010;6(4):e1000873.
Guimaraes-Costa AB, Nascimento MT, Froment GS, Soares RP, Morgado FN, Conceicao-Silva F, et al. Leishmania amazonensis promastigotes induce and are killed by neutrophil extracellular traps. Proc Natl Acad Sci USA. 2009;106(16):6748–53.
Saitoh T, Komano J, Saitoh Y, Misawa T, Takahama M, Kozaki T, et al. Neutrophil extracellular traps mediate a host defense response to human immunodeficiency virus-1. Cell Host Microbe. 2012;12(1):109–16.
Cedervall J, Zhang Y, Huang H, Zhang L, Femel J, Dimberg A, et al. Neutrophil extracellular traps accumulate in peripheral blood vessels and compromise organ function in tumor-bearing animals. Cancer Res. 2015;75(13):2653–62.
Rossaint J, Herter JM, Van Aken H, Napirei M, Doring Y, Weber C, et al. Synchronized integrin engagement and chemokine activation is crucial in neutrophil extracellular trap-mediated sterile inflammation. Blood. 2014;123(16):2573–84.
Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. 2017;37(7):1371–9.
Linders J, Madhi R, Rahman M, Morgelin M, Regner S, Brenner M, et al. Extracellular cold-inducible RNA-binding protein regulates neutrophil extracellular trap formation and tissue damage in acute pancreatitis. Lab Invest. 2020;100(12):1618–30.
Perez-Olivares L, Soehnlein O. Contemporary lifestyle and neutrophil extracellular traps: an emerging link in atherosclerosis disease. Cells. 2021;10(8):1985.
Wei X, Zou S, Xie Z, Wang Z, Huang N, Cen Z, et al. EDIL3 deficiency ameliorates adverse cardiac remodeling by neutrophil extracellular traps (NET)-mediated macrophage polarization. Cardiovasc Res. 2021. https://doi.org/10.1093/cvr/cvab269.
Vokalova L, van Breda SV, Ye XL, Huhn EA, Than NG, Hasler P, et al. Excessive neutrophil activity in gestational diabetes mellitus: could it contribute to the development of preeclampsia? Front Endocrinol. 2018;9:542.
Guo Y, Zeng H, Gao C. The role of neutrophil extracellular traps in central nervous system diseases and prospects for clinical application. Oxid Med Cell Longev. 2021;2021:9931742.
Fuchs TA, Abed U, Goosmann C, Hurwitz R, Schulze I, Wahn V, et al. Novel cell death program leads to neutrophil extracellular traps. J Cell Biol. 2007;176(2):231–41.
Steinberg BE, and Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE. 2007;2007(379):pe11.
Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010;191(3):677–91.
Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184(2):205–13.
Tohme S, Yazdani HO, Al-Khafaji AB, Chidi AP, Loughran P, Mowen K, et al. Neutrophil extracellular traps promote the development and progression of liver metastases after surgical stress. Cancer Res. 2016;76(6):1367–80.
Zhang Y, Hu Y, Ma C, Sun H, Wei X, Li M, et al. Diagnostic, therapeutic predictive, and prognostic value of neutrophil extracellular traps in patients with gastric adenocarcinoma. Front Oncol. 2020;10:1036.
Decker AS, Pylaeva E, Brenzel A, Spyra I, Droege F, Hussain T, et al. Prognostic role of blood NETosis in the progression of head and neck cancer. Cells. 2019;8(9):946.
McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. 2012;12(3):324–33.
Tadie JM, Bae HB, Jiang S, Park DW, Bell CP, Yang H, et al. HMGB1 promotes neutrophil extracellular trap formation through interactions with Toll-like receptor 4. Am J Physiol Lung Cell Mol Physiol. 2013;304(5):L342–9.
Xiao Y, Cong M, Li J, He D, Wu Q, Tian P, et al. Cathepsin C promotes breast cancer lung metastasis by modulating neutrophil infiltration and neutrophil extracellular trap formation. Cancer Cell. 2021;39(3):423-437.e7.
Demers M, Krause DS, Schatzberg D, Martinod K, Voorhees JR, Fuchs TA, et al. Cancers predispose neutrophils to release extracellular DNA traps that contribute to cancer-associated thrombosis. Proc Natl Acad Sci USA. 2012;109(32):13076–81.
Gupta AK, Joshi MB, Philippova M, Erne P, Hasler P, Hahn S, et al. Activated endothelial cells induce neutrophil extracellular traps and are susceptible to NETosis-mediated cell death. FEBS Lett. 2010;584(14):3193–7.
Sanmamed MF, Perez-Gracia JL, Schalper KA, Fusco JP, Gonzalez A, Rodriguez-Ruiz ME, et al. Changes in serum interleukin-8 (IL-8) levels reflect and predict response to anti-PD-1 treatment in melanoma and non-small-cell lung cancer patients. Ann Oncol. 2017;28(8):1988–95.
Thalin C, Lundstrom S, Seignez C, Daleskog M, Lundstrom A, Henriksson P, et al. Citrullinated histone H3 as a novel prognostic blood marker in patients with advanced cancer. PLoS ONE. 2018;13(1):e0191231.
Cai Z, Zhang M, Boafo Kwantwi L, Bi X, Zhang C, Cheng Z, et al. Breast cancer cells promote self-migration by secreting interleukin 8 to induce NET formation. Gene. 2020;754:144902.
de Andrea CE, Ochoa MC, Villalba-Esparza M, Teijeira A, Schalper KA, Abengozar-Muela M, et al. Heterogenous presence of neutrophil extracellular traps in human solid tumours is partially dependent on interleukin-8. J Pathol. 2021;255(2):190–201.
Alfaro C, Teijeira A, Onate C, Perez G, Sanmamed MF, Andueza MP, et al. Tumor-produced interleukin-8 attracts human myeloid-derived suppressor cells and elicits extrusion of neutrophil extracellular traps (NETs). Clin Cancer Res. 2016;22(15):3924–36.
Raghuwanshi SK, Su Y, Singh V, Haynes K, Richmond A, Richardson RM. The chemokine receptors CXCR1 and CXCR2 couple to distinct G protein-coupled receptor kinases to mediate and regulate leukocyte functions. J Immunol. 2012;189(6):2824–32.
Bertini R, Allegretti M, Bizzarri C, Moriconi A, Locati M, Zampella G, et al. Noncompetitive allosteric inhibitors of the inflammatory chemokine receptors CXCR1 and CXCR2: prevention of reperfusion injury. Proc Natl Acad Sci USA. 2004;101(32):11791–6.
Teijeira A, Garasa S, Gato M, Alfaro C, Migueliz I, Cirella A, et al. CXCR1 and CXCR2 chemokine receptor agonists produced by tumors induce neutrophil extracellular traps that interfere with immune cytotoxicity. Immunity. 2020;52(5):856-871.e8.
Podaza E, Sabbione F, Risnik D, Borge M, Almejun MB, Colado A, et al. Neutrophils from chronic lymphocytic leukemia patients exhibit an increased capacity to release extracellular traps (NETs). Cancer Immunol Immunother. 2017;66(1):77–89.
Nie M, Yang L, Bi X, Wang Y, Sun P, Yang H, et al. Neutrophil extracellular traps induced by IL8 promote diffuse large B-cell lymphoma progression via the TLR9 signaling. Clin Cancer Res. 2019;25(6):1867–79.
Zha C, Meng X, Li L, Mi S, Qian D, Li Z, et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol Med. 2020;17(1):154–68.
Thalin C, Demers M, Blomgren B, Wong SL, von Arbin M, von Heijne A, et al. NETosis promotes cancer-associated arterial microthrombosis presenting as ischemic stroke with troponin elevation. Thromb Res. 2016;139:56–64.
Savarese TM, Mitchell K, McQuain C, Campbell CL, Guardiani R, Wuu J, et al. Coexpression of granulocyte colony stimulating factor and its receptor in primary ovarian carcinomas. Cancer Lett. 2001;162(1):105–15.
Kowanetz M, Wu X, Lee J, Tan M, Hagenbeek T, Qu X, et al. Granulocyte-colony stimulating factor promotes lung metastasis through mobilization of Ly6G+Ly6C+ granulocytes. Proc Natl Acad Sci USA. 2010;107(50):21248–55.
Guo L, Chen G, Zhang W, Zhou L, Xiao T, Di X, et al. A high-risk luminal A dominant breast cancer subtype with increased mobility. Breast Cancer Res Treat. 2019;175(2):459–72.
Leal AC, Mizurini DM, Gomes T, Rochael NC, Saraiva EM, Dias MS, et al. Tumor-derived exosomes induce the formation of neutrophil extracellular traps: implications for the establishment of cancer-associated thrombosis. Sci Rep. 2017;7(1):6438.
Arpinati L, Shaul ME, Kaisar-Iluz N, Mali S, Mahroum S, Fridlender ZG. NETosis in cancer: a critical analysis of the impact of cancer on neutrophil extracellular trap (NET) release in lung cancer patients vs. mice. Cancer Immunol Immunother. 2020;69(2):199–213.
Takei H, Araki A, Watanabe H, Ichinose A, Sendo F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J Leukoc Biol. 1996;59(2):229–40.
MacLeod KT, Harding SE. Effects of phorbol ester on contraction, intracellular pH and intracellular Ca2+ in isolated mammalian ventricular myocytes. J Physiol. 1991;444:481–98.
Najmeh S, Cools-Lartigue J, Giannias B, Spicer J, Ferri LE. Simplified human neutrophil extracellular traps (NETs) isolation and handling. J Vis Exp. 2015;16(98):e52687.
Healy LD, Puy C, Itakura A, Chu T, Robinson DK, Bylund A, et al. Colocalization of neutrophils, extracellular DNA and coagulation factors during NETosis: development and utility of an immunofluorescence-based microscopy platform. J Immunol Methods. 2016;435:77–84.
Lukasova E, Koristek Z, Klabusay M, Ondrej V, Grigoryev S, Bacikova A, et al. Granulocyte maturation determines ability to release chromatin NETs and loss of DNA damage response; these properties are absent in immature AML granulocytes. Biochim Biophys Acta. 2013;1833(3):767–79.
Ermert D, Urban CF, Laube B, Goosmann C, Zychlinsky A, Brinkmann V. Mouse neutrophil extracellular traps in microbial infections. J Innate Immun. 2009;1(3):181–93.
Wolfe PC, Chang EY, Rivera J, Fewtrell C. Differential effects of the protein kinase C activator phorbol 12-myristate 13-acetate on calcium responses and secretion in adherent and suspended RBL-2H3 mucosal mast cells. J Biol Chem. 1996;271(12):6658–65.
Hakkim A, Fuchs TA, Martinez NE, Hess S, Prinz H, Zychlinsky A, et al. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat Chem Biol. 2011;7(2):75–7.
Keshari RS, Verma A, Barthwal MK, Dikshit M. Reactive oxygen species-induced activation of ERK and p38 MAPK mediates PMA-induced NETs release from human neutrophils. J Cell Biochem. 2013;114(3):532–40.
Douda DN, Yip L, Khan MA, Grasemann H, Palaniyar N. Akt is essential to induce NADPH-dependent NETosis and to switch the neutrophil death to apoptosis. Blood. 2014;123(4):597–600.
Romao S, Tejera E, Nytko KJ, Siler U, Munz C, Reichenbach J. Defective nuclear entry of hydrolases prevents neutrophil extracellular trap formation in patients with chronic granulomatous disease. J Allergy Clin Immunol. 2015;136(6):1703–6.
Tatsiy O, McDonald PP. Physiological stimuli induce PAD4-dependent, ROS-independent NETosis, with early and late events controlled by discrete signaling pathways. Front Immunol. 2018;9:2036.
Shi Y, Liu T, Nieman DC, Cui Y, Li F, Yang L, et al. Aerobic exercise attenuates acute lung injury through NET inhibition. Front Immunol. 2020;11:409.
Okeke EB, Louttit C, Fry C, Najafabadi AH, Han K, Nemzek J, et al. Inhibition of neutrophil elastase prevents neutrophil extracellular trap formation and rescues mice from endotoxic shock. Biomaterials. 2020;238:119836.
Richardson JJR, Hendrickse C, Gao-Smith F, Thickett DR. Neutrophil extracellular trap production in patients with colorectal cancer in vitro. Int J Inflamm. 2017;2017:4915062.
Garley M, Dziemianczyk-Pakiela D, Grubczak K, Surazynski A, Dabrowska D, Ratajczak-Wrona W, et al. Differences and similarities in the phenomenon of NETs formation in oral inflammation and in oral squamous cell carcinoma. J Cancer. 2018;9(11):1958–65.
DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, et al. Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J Clin Invest. 1998;101(2):455–63.
Khan MA, Farahvash A, Douda DN, Licht JC, Grasemann H, Sweezey N, et al. JNK activation turns on LPS- and Gram-negative bacteria-induced NADPH oxidase-dependent suicidal NETosis. Sci Rep. 2017;7(1):3409.
Chapman EA, Lyon M, Simpson D, Mason D, Beynon RJ, Moots RJ, et al. Caught in a trap? Proteomic analysis of neutrophil extracellular traps in rheumatoid arthritis and systemic lupus erythematosus. Front Immunol. 2019;10:423.
Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H, et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep. 2016;6:37252.
McDonald B, Davis RP, Kim SJ, Tse M, Esmon CT, Kolaczkowska E, et al. Platelets and neutrophil extracellular traps collaborate to promote intravascular coagulation during sepsis in mice. Blood. 2017;129(10):1357–67.
Carestia A, Kaufman T, Rivadeneyra L, Landoni VI, Pozner RG, Negrotto S, et al. Mediators and molecular pathways involved in the regulation of neutrophil extracellular trap formation mediated by activated platelets. J Leukoc Biol. 2016;99(1):153–62.
Pieterse E, Rother N, Yanginlar C, Hilbrands LB, van der Vlag J. Neutrophils discriminate between lipopolysaccharides of different bacterial sources and selectively release neutrophil extracellular traps. Front Immunol. 2016;7:484.
Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13(4):463–9.
Tamassia N, Calzetti F, Ear T, Cloutier A, Gasperini S, Bazzoni F, et al. Molecular mechanisms underlying the synergistic induction of CXCL10 by LPS and IFN-gamma in human neutrophils. Eur J Immunol. 2007;37(9):2627–34.
Liu S, Yue Y, Pan P, Zhang L, Su X, Li H, et al. IRF-1 intervention in the classical ROS-dependent release of NETs during LPS-induced acute lung injury in mice. Inflammation. 2019;42(1):387–403.
Scaffidi P, Misteli T, Bianchi ME, Erratum. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2010;467(7315):622.
Kang R, Xie Y, Zhang Q, Hou W, Jiang Q, Zhu S, et al. Intracellular HMGB1 as a novel tumor suppressor of pancreatic cancer. Cell Res. 2017;27(7):916–32.
Hori O, Brett J, Slattery T, Cao R, Zhang J, Chen JX, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270(43):25752–61.
Yang H, Hreggvidsdottir HS, Palmblad K, Wang H, Ochani M, Li J, et al. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc Natl Acad Sci USA. 2010;107(26):11942–7.
Tian J, Avalos AM, Mao SY, Chen B, Senthil K, Wu H, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8(5):487–96.
Shinde-Jadhav S, Mansure JJ, Rayes RF, Marcq G, Ayoub M, Skowronski R, et al. Role of neutrophil extracellular traps in radiation resistance of invasive bladder cancer. Nat Commun. 2021;12(1):2776.
Zhou J, Yang Y, Gan T, Li Y, Hu F, Hao N, et al. Lung cancer cells release high mobility group box 1 and promote the formation of neutrophil extracellular traps. Oncol Lett. 2019;18(1):181–8.
Hoste E, Maueroder C, van Hove L, Catrysse L, Vikkula HK, Sze M, et al. Epithelial HMGB1 delays skin wound healing and drives tumor initiation by priming neutrophils for NET formation. Cell Rep. 2019;29(9):2689–701.
Pham CTN, Ley TJ. Dipeptidyl peptidase I is required for the processing and activation of granzymes A and B in vivo. Proc Natl Acad Sci USA. 1999;96:8627–32.
Wolters PJ, Pham CT, Muilenburg DJ, Ley TJ, Caughey GH. Dipeptidyl peptidase I is essential for activation of mast cell chymases, but not tryptases, in mice. J Biol Chem. 2001;276(21):18551–6.
Adkison AM, Raptis SZ, Kelley DG, Pham CT. Dipeptidyl peptidase I activates neutrophil-derived serine proteases and regulates the development of acute experimental arthritis. J Clin Invest. 2002;109(3):363–71.
Li Y, Yuan R, Ren T, Yang B, Miao H, Liu L, et al. Role of Sciellin in gallbladder cancer proliferation and formation of neutrophil extracellular traps. Cell Death Dis. 2021;12(1):30.
Fridlender ZG, Sun J, Kim S, Kapoor V, Cheng G, Ling L, et al. Polarization of tumor-associated neutrophil phenotype by TGF-beta: “N1” versus “N2” TAN. Cancer Cell. 2009;16(3):183–94.
Guglietta S, Chiavelli A, Zagato E, Krieg C, Gandini S, Ravenda PS, et al. Coagulation induced by C3aR-dependent NETosis drives protumorigenic neutrophils during small intestinal tumorigenesis. Nat Commun. 2016;7(1):11037.
Rayes RF, Mouhanna JG, Nicolau I, Bourdeau F, Giannias B, Rousseau S, et al. Primary tumors induce neutrophil extracellular traps with targetable metastasis promoting effects. JCI Insight. 2019;5(16):e128008.
Shang A, Gu C, Zhou C, Yang Y, Chen C, Zeng B, et al. Exosomal KRAS mutation promotes the formation of tumor-associated neutrophil extracellular traps and causes deterioration of colorectal cancer by inducing IL-8 expression. Cell Commun Signal. 2020;18(1):52.
Zhang Y, Wu H, Yang F, Ning J, Li M, Zhao C, et al. Prognostic value of the expression of DNA repair-related biomarkers mediated by alcohol in gastric cancer patients. Am J Pathol. 2018;188(2):367–77.
Wolach O, Sellar RS, Martinod K, Cherpokova D, McConkey M, Chappell RJ, et al. Increased neutrophil extracellular trap formation promotes thrombosis in myeloproliferative neoplasms. Sci Transl Med. 2018;10(436):eaan8292.
Martins-Cardoso K, Almeida VH, Bagri KM, Rossi MID, Mermelstein CS, Konig S, et al. Neutrophil extracellular traps (NETs) promote pro-metastatic phenotype in human breast cancer cells through epithelial–mesenchymal transition. Cancers. 2020;12(6):1542.
Yazdani HO, Roy E, Comerci AJ, van der Windt DJ, Zhang H, Huang H, et al. Neutrophil extracellular traps drive mitochondrial homeostasis in tumors to augment growth. Cancer Res. 2019;79(21):5626–39.
van der Windt DJ, Sud V, Zhang H, Varley PR, Goswami J, Yazdani HO, et al. Neutrophil extracellular traps promote inflammation and development of hepatocellular carcinoma in nonalcoholic steatohepatitis. Hepatology. 2018;68(4):1347–60.
Hernandez C, Huebener P, Pradere JP, Antoine DJ, Friedman RA, Schwabe RF. HMGB1 links chronic liver injury to progenitor responses and hepatocarcinogenesis. J Clin Invest. 2018;128(6):2436–51.
Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, et al. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015;63(1):114–21.
Whittall-Garcia LP, Torres-Ruiz J, Zentella-Dehesa A, Tapia-Rodriguez M, Alcocer-Varela J, Mendez-Huerta N, et al. Neutrophil extracellular traps are a source of extracellular HMGB1 in lupus nephritis: associations with clinical and histopathological features. Lupus. 2019;28(13):1549–57.
Miller-Ocuin JL, Liang X, Boone BA, Doerfler WR, Singhi AD, Tang D, et al. DNA released from neutrophil extracellular traps (NETs) activates pancreatic stellate cells and enhances pancreatic tumor growth. Oncoimmunology. 2019;8(9):e1605822.
Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, et al. Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science. 2018;361(6409):eaao4227.
Park J, Wysocki RW, Amoozgar Z, Maiorino L, Fein MR, Jorns J, et al. Cancer cells induce metastasis-supporting neutrophil extracellular DNA traps. Sci Transl Med. 2016;8(361):361ra138.
Cools-Lartigue J, Spicer J, McDonald B, Gowing S, Chow S, Giannias B, et al. Neutrophil extracellular traps sequester circulating tumor cells and promote metastasis. J Clin Invest. 2013;123(8):3446–58.
Najmeh S, Cools-Lartigue J, Rayes RF, Gowing S, Vourtzoumis P, Bourdeau F, et al. Neutrophil extracellular traps sequester circulating tumor cells via beta1-integrin mediated interactions. Int J Cancer. 2017;140(10):2321–30.
Lee W, Ko SY, Mohamed MS, Kenny HA, Lengyel E, Naora H. Neutrophils facilitate ovarian cancer premetastatic niche formation in the omentum. J Exp Med. 2019;216(1):176–94.
Catena R, Bhattacharya N, El Rayes T, Wang S, Choi H, Gao D, et al. Bone marrow-derived Gr1+ cells can generate a metastasis-resistant microenvironment via induced secretion of thrombospondin-1. Cancer Discov. 2013;3(5):578–89.
Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, et al. DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature. 2020;583(7814):133–8.
Yan W, Chang Y, Liang X, Cardinal JS, Huang H, Thorne SH, et al. High-mobility group box 1 activates caspase-1 and promotes hepatocellular carcinoma invasiveness and metastases. Hepatology. 2012;55(6):1863–75.
Wang Z, Yang C, Li L, Jin X, Zhang Z, Zheng H, et al. Tumor-derived HMGB1 induces CD62L(dim) neutrophil polarization and promotes lung metastasis in triple-negative breast cancer. Oncogenesis. 2020;9(9):82.
Zhu T, Zou X, Yang C, Li L, Wang B, Li R, et al. Neutrophil extracellular traps promote gastric cancer metastasis by inducing epithelial–mesenchymal transition. Int J Mol Med. 2021;48(1):127.
Kajioka H, Kagawa S, Ito A, Yoshimoto M, Sakamoto S, Kikuchi S, et al. Targeting neutrophil extracellular traps with thrombomodulin prevents pancreatic cancer metastasis. Cancer Lett. 2021;497:1–13.
Yang LY, Luo Q, Lu L, Zhu WW, Sun HT, Wei R, et al. Increased neutrophil extracellular traps promote metastasis potential of hepatocellular carcinoma via provoking tumorous inflammatory response. J Hematol Oncol. 2020;13(1):3.
Fuchs TA, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers DD Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. 2010;107(36):15880–5.
Moschonas IC, Tselepis AD. The pathway of neutrophil extracellular traps towards atherosclerosis and thrombosis. Atherosclerosis. 2019;288:9–16.
Jung Hye Soo, Gu JaYoon, Kim Ji-Eun, Nam Youngwon, Song Jae Woo, Kim HK. Cancer cell-induced neutrophil extracellular traps promote both hypercoagulability and cancer progression. PLoS ONE. 2019;14(4):e0216055.
Zhang Y, Wang C, Yu M, Zhao X, Du J, Li Y, et al. Neutrophil extracellular traps induced by activated platelets contribute to procoagulant activity in patients with colorectal cancer. Thromb Res. 2019;180:87–97.
Yang C, Sun W, Cui W, Li X, Yao J, Jia X, et al. Procoagulant role of neutrophil extracellular traps in patients with gastric cancer. Int J Clin Exp Pathol. 2015;8(11):14075–86.
Mauracher LM, Posch F, Martinod K, Grilz E, Daullary T, Hell L, et al. Citrullinated histone H3, a biomarker of neutrophil extracellular trap formation, predicts the risk of venous thromboembolism in cancer patients. J Thromb Haemost. 2018;16(3):508–18.
Abdol Razak N, Elaskalani O. Pancreatic cancer-induced neutrophil extracellular traps: a potential contributor to cancer-associated thrombosis. Int J Mol Sci. 2017;18(3):487.
Yu M, Li T, Li B, Liu Y, Wang L, Zhang J, et al. Phosphatidylserine-exposing blood cells, microparticles and neutrophil extracellular traps increase procoagulant activity in patients with pancreatic cancer. Thromb Res. 2020;188:5–16.
Thomas GM, Brill A, Mezouar S, Crescence L, Gallant M, Dubois C, et al. Tissue factor expressed by circulating cancer cell-derived microparticles drastically increases the incidence of deep vein thrombosis in mice. J Thromb Haemost. 2015;13(7):1310–9.
Jiang M, Fang H, Shao S, Dang E, Zhang J, Qiao P, et al. Keratinocyte exosomes activate neutrophils and enhance skin inflammation in psoriasis. FASEB J. 2019;33(12):13241–53.
Thomas GM, Panicot-Dubois L, Lacroix R, Dignat-George F, Lombardo D, Dubois C. Cancer cell-derived microparticles bearing P-selectin glycoprotein ligand 1 accelerate thrombus formation in vivo. J Exp Med. 2009;206(9):1913–27.
Manly DA, Wang J, Glover SL, Kasthuri R, Liebman HA, Key NS, et al. Increased microparticle tissue factor activity in cancer patients with venous thromboembolism. Thromb Res. 2010;125(6):511–2.
Kambas K, Chrysanthopoulou A, Vassilopoulos D, Apostolidou E, Skendros P, Girod A, et al. Tissue factor expression in neutrophil extracellular traps and neutrophil derived microparticles in antineutrophil cytoplasmic antibody associated vasculitis may promote thromboinflammation and the thrombophilic state associated with the disease. Ann Rheum Dis. 2014;73(10):1854–63.
Kyrle P, Eichinger S, Quehenberger P, Sagaster V, Weber H, Kollars M, et al. Tissue factor-positive microparticles: cellular origin and association with coagulation activation in patients with colorectal cancer. Thromb Haemost. 2017;97(01):119–23.
Boone BA, Murthy P, Miller-Ocuin J, Doerfler WR, Ellis JT, Liang X, et al. Chloroquine reduces hypercoagulability in pancreatic cancer through inhibition of neutrophil extracellular traps. BMC Cancer. 2018;18(1):678.
Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS ONE. 2012;7(9):e45427.
Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, et al. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. 2015;36(22):1405–14.
Arelaki S, Arampatzioglou A, Kambas K, Papagoras C, Miltiades P, Angelidou I, et al. Gradient infiltration of neutrophil extracellular traps in colon cancer and evidence for their involvement in tumour growth. PLoS ONE. 2016;11(5):e0154484.
Park HS, Gu J, You HJ, Kim JE, Kim HK. Factor XII-mediated contact activation related to poor prognosis in disseminated intravascular coagulation. Thromb Res. 2016;138:103–7.
Seo JD, Gu JY, Jung HS, Kim YJ, Kim HK. Contact system activation and neutrophil extracellular trap markers: risk factors for portal vein thrombosis in patients with hepatocellular carcinoma. Clin Appl Thromb Hemost. 2019;25:1076029618825310.
Kim TY, Gu JY, Jung HS, Koh Y, Kim I, Kim HK. Elevated extracellular trap formation and contact system activation in acute leukemia. J Thromb Thrombolysis. 2018;46(3):379–85.
von Bruhl ML, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. 2012;209(4):819–35.
Grilz E, Mauracher LM, Posch F, Konigsbrugge O, Zochbauer-Muller S, Marosi C, et al. Citrullinated histone H3, a biomarker for neutrophil extracellular trap formation, predicts the risk of mortality in patients with cancer. Br J Haematol. 2019;186(2):311–20.
Rosell A, Aguilera K, Hisada Y, Schmedes C, Mackman N, Wallen H, et al. Prognostic value of circulating markers of neutrophil activation, neutrophil extracellular traps, coagulation and fibrinolysis in patients with terminal cancer. Sci Rep. 2021;11(1):5074.
Demkow U. Neutrophil extracellular traps (NETs) in cancer invasion, evasion and metastasis. Cancers. 2021;13(17):4495.
Chen Q, Zhang L, Li X, Zhuo W. Neutrophil extracellular traps in tumor metastasis: pathological functions and clinical applications. Cancers. 2021;13(11):2832.
Khan U, Chowdhury S, Billah MM, Islam KMD, Thorlacius H. Neutrophil extracellular traps in colorectal cancer progression and metastasis. Int J Mol Sci. 2021;22(14):7260.
Li M, Lin C, Deng H, Strnad J, Bernabei L, Vogl DT, et al. A novel peptidylarginine deiminase 4 (PAD4) inhibitor BMS-P5 blocks formation of neutrophil extracellular traps and delays progression of multiple myeloma. Mol Cancer Ther. 2020;19(7):1530–8.
Tao L, Xu M, Dai X, Ni T, Li D, Jin F, et al. Polypharmacological profiles underlying the antitumor property of Salvia miltiorrhiza root (Danshen) interfering with NOX-dependent neutrophil extracellular traps. Oxid Med Cell Longev. 2018;2018:4908328.
Zenlander R, Havervall S, Magnusson M, Engstrand J, Agren A, Thalin C, et al. Neutrophil extracellular traps in patients with liver cirrhosis and hepatocellular carcinoma. Sci Rep. 2021;11(1):18025.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Natural Science Foundation of Anhui Province (1908085QH333) and the Key Research and Development Project of Anhui Province (202004j07020044).
Author information
Authors and Affiliations
Contributions
KG and HW designed the study. YZ and HW drafted the manuscript. HW, QW, and XW drew pictures. All authors are personally accountable for their contributions to the review and approved the final manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wang, H., Zhang, Y., Wang, Q. et al. The regulatory mechanism of neutrophil extracellular traps in cancer biological behavior. Cell Biosci 11, 193 (2021). https://doi.org/10.1186/s13578-021-00708-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13578-021-00708-z