
Abstract
Immunotherapy, especially anti-programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) treatment has significantly improved the survival of non-small cell lung cancer (NSCLC) patients. However, the overall response rate remains unsatisfactory. Many factors affect the outcome of anti-PD-1/PD-L1 treatment, such as PD-L1 expression level, tumor-infiltrating lymphocytes (TILs), tumor mutation burden (TMB), neoantigens, and driver gene mutations. Further exploration of biomarkers would be favorable for the best selection of patients and precisely predict the efficacy of anti-PD-1/PD-L1 treatment. In this review, we summarized the latest advances in this field, and discussed the potential applications of these laboratory findings in the clinic.
Similar content being viewed by others
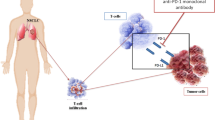


Background
Lung cancer has a high incidence rate worldwide and is the main cause of cancer deaths [1]. The 5-year survival rate varies in different regions [2]. Non-small cell lung cancer (NSCLC) accounts for approximately 80–85% of all lung cancers [3, 4]. Recently, the anti-programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) treatment has substantially changed the treatment patterns of NSCLC. The anti-PD-1/PD-L1 treatment with or without platinum-based chemotherapy has become the first-line strategy for NSCLC without driver gene mutations [5].
The immune system can specifically recognize the expression of tumor-specific antigens and eliminate tumor cells [6]. Alterations in effector cell signal transduction molecule (T cell receptor/CD3), the levels of tumor antigens, the maturation of antigen-presenting cells (APC), tumor-derived soluble factors such as vascular endothelial growth factor (VEGF), transforming growth factor-β (TGF-β), and IL-10 propel tumor immune escape [7,8,9,10,11]. PD-1 and PD-L1 are type I transmembrane proteins[12]. The interaction of PD-1 and PD-L1 leads to the phosphorylation of the cytoplasmic immunoreceptor tyrosine-based inhibitory motif (ITIM) and the immunoreceptor tyrosine-based switch motif (ITSM) and recruits Src homology 2 domain containing phosphatases 1/2 (SHP1/2) [13]. The recruitment of SHP1/2 inhibits the activation of T cells [14]. SHP1/2 and their downstream inhibitory signaling pathways suppress the activation of phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT) and mitogen-activated protein kinase (MAPK) [15, 16].
The anti-PD-1/PD-L1 treatment blocks the interaction of PD-1 and its ligands, interferes with inhibitory signal transduction, restores the vitality of T cells, and thereby restarts the anti-tumor immune effect [17, 18]. NSCLC has high level of heterogeneity. The heterogeneity of molecular immune subtypes and immune microenvironment results in the differences in the efficacy of PD-1/PD-L1 inhibitors [19]. The low response rate to PD-1/PD-L1 inhibitors hinders the clinical application [20]. Therefore, it is urgent to find reliable biomarkers to effectively predict the efficacy of PD-1/PD-L1 inhibitors. In this review, we summarized the latest advances in the predictive biomarkers of anti-PD-1/PD-L1 therapy in NSCLC.
Tumor feature related biomarkers
PD-L1 expression level
A known mechanism for PD-1/PD-L1 to promote tumor immune escape is adaptive immune resistance [21]. Multiple clinical trials have been performed to evaluate the relationship between the expression of PD-L1 on tumor cells and the response rate to PD-1/PD-L1 inhibitors (Fig. 1). The high level of PD-L1 expression heralds the potential benefit of anti-PD-1/PD-L1 treatment [22, 23]. In the phase I KEYNOTE-001 study, among patients who had previously treated with anti-PD-1 therapy, patients with PD-L1 tumor proportion score (TPS) ≥ 50% had a median overall survival (OS) of 15.4 months (95% CI: 10.6–18.8 months) (Table 1) and the 5-year OS rate was 25.0%; while in the PD-L1 TPS 1%-49% group and PD-L1 TPS ≤ 1% group, the median OS were 8.5 months (95% CI: 6.0–12.6 months) and 8.6 months (95% CI: 5.5–10.6 months), and the 5-year OS rates were 12.6% and 3.5%, respectively [22]. In the multicenter, single-arm, open-label phase II clinical trial (PePS2), the incidence of durable clinical benefit (DCB) in the PD-L1 TPS ≥ 50% group was 53% (95% CI: 30–75%) (Table 1), while the PD-L1 TPS 1–49% group and PD-L1 TPS ≤ 1% group were 47% (95% CI: 25–70%) and 22% (95% CI: 11–41%) [23]. In KEYNOTE-024 study, pembrolizumab treatment lengthened the survival time of NSCLC patients with PD-L1 TPS ≥ 50%, relative to platinum-based chemotherapy (HR = 0.63, 95% CI: 0.47–0.86, p = 0.002) [24] (Table 1). However, only evaluating PD-L1 level can’t accurately select patients. Other studies showed that regardless of the level of PD-L1 expression, renal cell cancer (RCC) or NSCLC patients with anti-PD-1/PD-L1 treatment had survival benefits [25, 26]. The outcome of PD-1/PD-L1 blockade therapy was also determined by other characteristics including the immune status, the activity of the tumor-infiltrating T cells and the sensitivity of cancer cells to T cells [27]. Therefore, clinical decisions should be made carefully based on the results of PD-L1 expression.

Predictive biomarkers of anti-PD-1/PD-L1 therapy in NSCLC. First, increased PD-L1 level is an indicator of the pre-existed anti-tumor immune response which is positively correlated to response rate to anti-PD-1/PD-L1 treatment. Second, TIL is the effectors of anti-tumor immune response, which could be boosted by PD-1/PD-L1 inhibitors. Besides, TMB and neoantigens determine cancer immunogenicity which is the basis of anti-tumor response. In addition, multiple other factors such as suppressive immune cells, driver gene mutations, gut microbiota, tumor metabolites such as IDO1 also participate in anti-tumor immunity and affect the efficacy of anti-PD-1/PD-L1 therapies
Tumor mutation burden (TMB)
Whole-exome sequencing (WES) and sequencing of cancer gene panels (CGPs) are used to measure deoxyribonucleic acid (DNA) mutations in tumor tissue [28]. The tumor tissue TMB (tTMB) is positively correlated with tumor neoantigen load (Fig. 1) [29]. Multiple retrospective studies showed that tTMB was closely associated with the efficacy of PD-1/PD-L1 inhibitors and patient’s prognosis. In KEYNOTE-158, for patients treated with pembrolizumab, the tTMB-High group had a higher objective response rate than the non-tTMB-High group (29 vs. 6%) (Table 1) [30]. In CHECKMATE-026, patients with high tTMB receiving nivolumab treatment had a longer progression-free survival (PFS) (9.7 vs. 5.8 months; HR = 0.62, 95% CI: 0.38–1.00) (Table 1) and higher response rate (47 vs. 28%) than patients receiving chemotherapy [31]. Similarly, the results of CHECKMATE-227 showed that in patients with high tTMB, nivolumab plus ipilimumab group had a longer PFS than chemotherapy group (7.2 vs. 5.5 months; HR = 0.58, 97.5% CI: 0.41–0.81, p < 0.001) [32] (Table 1).
Blood TMB (bTMB) is discovered as a new and less invasive alternative, which is measured by detecting plasma cell-free DNA (Fig. 1) [28]. bTMB is positively correlated to tTMB [28]. Compared with bTMB < 6 subgroup, the bTMB ≥ 6 subgroup had higher objective response rate (39.3 vs. 9.1%) and longer PFS (HR = 0.39, 95% CI: 0.18–0.84, p = 0.01) (Table 1) [28]. However, the relationship between bTMB and patient’s survival showed a non-linear correlation [33]. For patients treated with PD-L1 inhibitors, the bTMB-High (≥ 14 mutations/Mb) and bTMB-Low (≤ 7 mutations/Mb) subgroups had longer PFS and OS than bTMB-Medium (8–13 mutations/Mb) subgroup [33]. The positive correlation between baseline circulating tumor DNA (ctDNA) and bTMB score explained the better prognosis of the bTMB-low patients [33]. In addition, compared with patients of bTMB-Medium, bTMB-low patients had longer response duration and higher stable disease rate [33]. In general, hypermutation promoted the production of tumor neoantigens, enhanced tumor immunogenicity and improved the response rate to PD-L1 inhibitors [34].
Neoantigens
Neoantigens are derived from somatic mutation [35], which bind to major histocompatibility class I (MHCI) and are expressed on the surface of cancer cells. Neoantigens endow the tumor with high immunogenicity and induce anti-tumor immune response (Fig. 1) [36]. Neoantigens are released by tumor cells and captured by professional APC, and then the effector T cells targeting cancer specific antigens are activated [37]. Activated T cells migrate and infiltrate into tumor bed, specifically recognize the antigens on tumor cells and kill cancer cells [37]. The tumor clones with potent immunogenicity are eliminated, and the cancer cells with weak immunogenicity escape immune surveillance [38]. Many studies proved that anti-PD-1/PD-L1 therapy combined with radiotherapy or oncolytic virus increased the release of neoantigens and amplified the specific immune response [39,40,41]. Compared with no durable clinical benefit (NDB) patients, DCB patients had higher burden of candidate neoantigens. High candidate neoantigen burden was associated with improvement in PFS (HR = 0.23, 95%CI: 0.09–0.58, p = 0.002) [42] (Table 1). The efficacy of immunotherapy was not only related to the quantity of neoantigens, but also related to the quality of neoantigens [43]. High-quality neoantigens especially clonal neoantigens, could bind to multiple HLA alleles [43]. The clonal neoantigens promoted the activation and infiltration of neoantigen reactive T cells expressing high level of PD-1, and tumors enriched clonal neoantigens were more sensitive to PD-1 blockers [34]. The incidence rate of DCB in patients with high mutation burden and low neoantigen subclonal fraction was higher than patients with high subclonal neoantigen fraction or low clonal neoantigen burden (92 vs. 11%) [34] (Table 1). Immune elimination of neoantigen-containing tumor cell subpopulations and genetic events such as chromosomal deletions or loss of heterozygosity in tumor cells lead to the loss of neoantigens, which contribute to the emergence of acquired resistance to anti-PD-1/PD-L1 treatment [44].
Driver gene mutations
Next-generation sequencing (NGS) is widely used for tumor genome analysis [45]. The gene alterations detected by targeting NGS may herald the response rate to PD-1/PD-L1 inhibitors (Fig. 1) [45]. Kirsten rat sarcoma 2 viral oncogene homolog (KRAS) mutation status was positively correlated with PD-L1 expression [46]. In addition, KRAS mutant-type tumors had more TILs and higher TMB, which presented the inflammatory phenotype of adaptive immune resistance and increased immunogenicity [46]. Compared with KRAS wild subgroup, KRAS mutated subgroup had a higher objective response rate (odds ratio = 1.51, 95% CI: 1.17–1.96, p = 0.002) (Table 1) [46]. TP53-mutated tumors had high PD-L1 expression and CD8+ T cell density [47]. Patients with TP53 mutations and no serine/threonine kinase 11 (STK11) or epidermal growth factor receptor (EGFR) co-mutations had higher response rate and longer PFS to anti-PD-1 therapy (HR = 0.32, 95%CI: 0.16–0.63, p < 0.001). Pathways related to immune cell cytotoxicity, T cell chemotaxis, antigen processing were upregulated in this tumor subtype [47]. EGFR with exon 19 deletion, L858R mutation and T790M mutation upregulated the expression of PD-L1, which attenuated cytotoxicity of lymphocytes and induced T-cell exhaustion through PD-1/PD-L1 axis [48,49,50]. Among patients who treated with anti-PD-1 therapy, patients with EGFR mutations had worse prognosis (median PFS: 5.3 months, 95% CI: 1.3–12.4 months) [51]. The anaplastic lymphoma kinase (ALK)-rearranged upregulated PD-L1 expression and promoted tumor immune escape [52]. However, ALK-mutated patients who treated with anti-PD-1 therapy presented worse PFS than patients with EGFR mutations (ALK: 0.6 (95% CI: 0.2–2.1) months, EGFR: 1.8 (95% CI: 1.2–2.1) months), suggesting that PD-L1 expression was not a reliable biomarker for immunotherapy for patients with ALK rearrangement [53]. The mesenchymal epithelial transition (MET) exon 14 skipping alterations occur in 3%-4% of lung cancers [54]. A large proportion of lung cancer cells with MET exon 14 alterations expressed PD-L1 [55]. Lung cancer patients with MET exon 14 mutations responded modestly to single-agent or combination immune checkpoint inhibitors (objective response rate: 17%, 95% CI: 6%-36%) [55], and didn’t seem to benefit from immunotherapy [56]. Kelch-like ECH-associated protein 1 (KEAP1) somatic mutations promoted tumorigenesis and reduced therapeutic sensitivity by activating the KEAP1/nuclear factor erythroid-2-related factor-2 (NFE2L2) stress response pathway [57,58,59,60]. NFE2L2/KEAP1 mutations were associated with high TMB and PD-L1 expression, and the efficacy of immunotherapy was better in patients with NFE2L2/KEAP1 mutations than other treatments (median OS: 22.52 months vs. 12.89 months, p = 0.0034) [61]. The mutation status of other rare driver genes such as ROS1, HER2, RET may also affect the response to PD-1/PD-L1 inhibitors [62, 63].
Inflammation related genes
Some expression signatures reflect the inflammatory state of tumors, such as genes related to T cell activation, chemokine expression, and adaptive immune resistance (Fig. 1) [64, 65]. Patients with significantly elevated inflammatory profile scores tended to be sensitive to PD-1/PD-L1 inhibitors. Compared with non-responders, responders had significantly higher inflammation signature scores [65]. In addition, inflammation scores was correlated with epithelial-mesenchymal transition (EMT) scores. Thompson’s study showed that the combination of EMT phenotypic feature scores and inflammation gene scores increased the accuracy of prediction [65]. Therefore, it is predicted that reversal of EMT may improve the resistance to anti-PD-1/PD-L1 therapy [65]. Further study found that in the same NSCLC cohort, the eight genes associated with antigen processing machinery (APM) scores could more effectively predict the efficacy than inflammation scores [66]. Also, our previous study indicated that some immune response-related signatures related to the efficacy of immune checkpoint inhibitor in lung adenocarcinoma [4].
microRNA(miRNA)
MiRNA modifies the expression of target genes by regulating protein translation [67]. miRNA dysregulation is closely associated with carcinogenesis and can promote or suppress cancer by targeting a group of genes (Fig. 1) [68]. In addition, miRNA regulates anti-tumor immunity. Some miRNAs interfere with antigen processing and presentation, upregulate human leukocyte antigen (HLA)-G expression and downregulate natural killer group 2, member D (NKG2D) ligand to form immune escape [69]. Circular RNA circ-CPA4 upregulated PD-L1 expression in NSCLC cells by downregulating let-7 miRNA [70]. 10-high expressed miRNAs (miR-93, miR-138-5p, miR-200, miR-27a, miR-424, miR-34a, miR-28, miR-106b, miR-193a-3p, miR-181a) were found in responders treated with anti-PD-1 treatment, and associated with significantly improved PFS and OS (median PFS: 6.25 months vs. 3.21 months, HR = 0.45, 95% CI: 0.25–0.76; median OS: 7.65 months vs. 3.2 months, HR = 0.39, 95% CI: 0.15–0.68) (Table 1) [71].
Tumor microenvironment related biomarkers
Tumor-infiltrating lymphocyte (TIL)
Previous reports shown that PD-L1 expression was significantly associated with intratumoral T cells infiltration in NSCLC [72]. The transcription factor thymocyte selection-associated high mobility group box gene (TOX) in tumor-infiltrating CD8+ T cells promotes T cell exhaustion by upregulating the expression of immune checkpoint proteins PD-1, T cell immunoglobulin and mucin-domain containing-3 (TIM-3) [73], T cell immunoglobulin and ITIM domain (TIGIT) [74], and cytotoxic T lymphocyte antigen 4 (CTLA-4), thereby attenuates the outcome of anti-PD-1 therapy (Fig. 1) [75]. Based on PD-L1/TIL status, NSCLC tumor immune microenvironments were divided into type I (PD-L1+, TIL+), type II (PD-L1−, TIL−), type III (PD-L1+, TIL−) and type IV (PD-L1−, TIL+) [76]. The difference in clinical factors related to different tumor immune microenvironment types determines the patient selection for combination immunotherapies [76]. Type I tumors benefit greatly from anti-PD-1/PD-L1 therapy. However, Type III tumors are resistant to anti-PD-1/PD-L1 monotherapy, which could be reversed by the combining adjuvant therapy to recruit T cells into tumor bed [77]. The proportion of CD8+ cells among the overall population of CD3+ TILs has a close relationship with anti-PD-1/PD-L1 treatment outcomes. It has been shown that High CD8-to-CD3 ratio was positively correlated with disease-free survival (DFS) and OS (DFS: HR = 0.954, 95%CI: 0.965–0.983, p = 0.002; OS: HR = 0.965, 95%CI: 0.931–1.001, p = 0.057) (Table 1) [78]. The early proliferation of CD8+ T cells after anti-PD-1 therapy heralded a good clinical response to anti-PD-1 therapy [79]. T cell receptor (TCR) is expressed on the surface of T cells and composed of α chains and β chains, which form diversity and specificity through somatic DNA rearrangement [80]. TCR binds to MHC/antigen short peptide complex and triggers immune response (Fig. 1) [81]. The TCR β chain complementarity determining region 3 of PD-1+ CD8+ T cells was sequenced by multiplex PCR. The diversity of TCR before anti-PD-1/PD-L1 treatment heralded a better survival outcome (6.4 vs. 2.5 months, HR = 0.39, 95% CI: 0.17–0.94, p = 0.021), and the clonality of TCR after treatment also heralded clinical benefit (7.3 vs. 2.6 months, HR = 0.26, 95% CI: 0.08–0.86, p = 0.002) [82].
Consolidation therapy with durvalumab after concurrent chemo-radiotherapy (cCRT) could significantly improve the overall survival and median progression-free survival of patients as compared with placebo group [83]. Radiotherapy stimulated anti-tumor immunity by promoting the release of tumor neoantigens and driving the immune attack of CD8 + TILs [84]. Post-cCRT PD-L1 upregulation might be in response to radiotherapy-related immune attack, which provided theoretical basis for the application of PD-L1 blockers following cCRT [85]. In addition, increased CD8 + TIL density after cCRT was associated with favorable survival [85].
Suppressive immune cell
Tumor-infiltrating regulatory T lymphocytes (Tregs) express PD-L1, PD-L2 on the surface, which highly inhibit the activity of tumor-specific effector T cells [86]. Indoleamine 2,3-dioxygenase 1 (IDO1) induces T cells immune suppression and Treg hyperactivation by l-tryptophan (Trp) depletion and kynurenine (Kyn) accumulation in the tumor microenvironment (Fig. 1) [87]. Serum kyn/trp ratio may reflect the anti-PD-1 immune resistance mechanism [88]. Myeloid-derived suppressor cells (MDSCs) mainly play an immunosuppressive role in the tumor microenvironment [89]. Some inflammatory factors such as TGF-β, IFN-γ, and IL-6 drive the activation of MDSCs [90]. Chemokines such as C–C motif chemokine ligand 2 (CCL2) [91] and C-X-C motif chemokine ligand 12 (CXCL12) [92] recruit MDSCs to tumor sites. MDSCs inhibit the immune response of tumor-specific T cells by upregulated PD-L1 expression (Fig. 1) [93].
Extracellular vesicles (EVs)
EVs are a collection of membrane-bound carriers, which carry lipids, proteins, and nucleic acids [94]. Budding inward through endosomal pathways to form exosomes and sprouting out of the plasma membrane to form microvesicles [95]. EVs bind to target cells and initiate signal transduction through receptor-ligand interactions or internalize through endocytosis [96]. EVs mediate cancer cell sensitivity to chemotherapy and radiotherapy, and are promising strategy in liquid biopsy for cancer diagnosis and predictive markers [97, 98]. The exchange of EVs between immune cells affects innate immunity and adaptive immunity [99]. Local dendritic cells (DCs) secreted-EVs could induce T cell activation [95]. EVs are key components in the microenvironment that bridge the communication between tumor cells and stromal cells [100]. By extracting EVs miRNAs from advanced NSCLC patients receiving anti-PD-1/PD-L1 therapy for sequencing analysis, a remarkable difference in the concentration of specific miRNAs between responders and non-responders was found [101]. As a non-invasive liquid biopsy, early detection of tumor-derived EVs may help to predict the efficacy of anti-PD-1/PD-L1 therapy [102,103,104].
Biomarkers in peripheral blood
Circulating cancer-associated macrophage-like cells (CAMLs)
Tumor associated macrophage (TAM) promotes the invasion characteristics of malignant cells by secreting growth factors and cytokines such as VEGF, MMP, TNF-α [105]. TAM and circulating tumor cells (CTC) migrate to the blood circulation through lymphatic or capillary barrier, which enhance tumor invasion and distant metastasis [106]. As a diffuse TAM (Fig. 1), the isolation of CAMLs from peripheral blood of various cancer patients may be evidence of tumor metastasis and neovascularization [107]. CAMLs were quantified by the CellSieve system using multiplex immunostaining [108]. CAMLs ≥ 50 μm was defined as giant CAMLs. The size of CAMLs after completion of CRT was related to disease progression and patient’s survival [109]. The presence of giant CAMLs before anti-PD-L1 maintenance therapy indicated a poor prognosis (median PFS: 8 months, HR = 2.5, 95% CI: 1.1–5.8, p = 0.025; median OS: 25 months, HR = 3.5, 95% CI: 1.3–9.6, p = 0.034) (Table 1). The tumor-stimulating effect of CAMLs may limit the efficacy of anti- PD-L1 therapy [109].
PD-L1+ aneuploid circulating tumor endothelial cells (CTECs)
The aneuploidy of chromosome influences gene expression and determines tumor heterogeneity, which is closely related to the evolution of tumor [110,111,112]. CTECs, aneuploid CD31+ circulating tumor endothelial cells [113], are derived from aneuploid CD31+ tumor endothelial cells in tumor tissue and promote tumor angiogenesis [114, 115]. The PD-L1+ CTECs had morphological and karyotype changes after immunotherapy [116]. Anti-PD-1 could effectively eliminate haploid small CTECs, while relatively increase polyploid large PD-L1+ CTECs [116]. Patients with PD-L1+ CTECs subtype were resistant to anti-PD-1 treatment. The median PFS of patients with PD-L1+ CTECs was 5 months (95% CI: 3.9–6.1 months) (Table 1), which was shorter than that of patients without PD-L1+ CTECs (8 months, 95% CI: 4.9–11 months). It was speculated that the interaction of PD-L1 on CTECs with PD-1 on T cells inhibited the tumor-specific immune attack of CD8+ T cells and affected the efficacy of immunotherapy (Fig. 1) [116].
Other peripheral blood cells
Among many indicators that reflect inflammation, the high neutrophil to lymphocyte ratio (NLR) heralded a poor prognosis in many malignant tumors [117, 118]. Multiple studies found that NSCLC patients with high NLR had low response rate to immune checkpoint inhibitors (ICIs) [119, 120]. A meta-analysis showed that patients with high NLR before ICIs therapy had poor prognosis (PFS: HR = 1.44, 95%CI: 1.26–1.65, p < 0.001; OS: HR = 2.86, 95%CI: 2.11–3.87, p < 0.001) (Table 1) [119]. Similarly, another retrospective study also verified the predictive value of NLR for anti-PD-1 treatment [120]. Lactate dehydrogenase (LDH) is an indicator of cancer-related inflammation [121]. According to the values of LDH and NLR, lung cancer patients were divided into 3 groups (good, 0 factors; intermediate, 1 factor; poor, 2 factors). Compared with the good group, the intermediate group and poor group were more easily resist to anti-PD-1/PD-L1 treatment [121]. In addition, NLR and LDH might be useful indicators for predicting irAEs [122]. Neutrophils were highly correlated with myeloid phenotype, which promoted lymphocyte depletion [123]. Tumor-infiltrating CD8+ T cells to neutrophils (CD8/PMN) ratio could distinguish responders treated with anti-PD-1 therapy [123]. Combining neutrophil antagonists improved immunotherapy outcomes [123]. Besides, the amount and activity of NK cells in responders were highly elevated [124].
Gut microbiota
Gut microbiota has a symbiotic relationship with the host [125]. In addition to playing a barrier role in the gastrointestinal tract, microorganisms are related to the immune function of the plora [126]. Immune cells are activated through cross-reactivity between microbial proteins and tumor antigens [127]. DCs induce activated T cells outside the intestine, recognize tumor antigens and exert anti-tumor effect [127]. In addition, the microbial proteins translocate from the intestine to the blood circulation, trigger initial immunity in secondary lymphoid organs and induce the activation of T cells. T cells migrate to the tumor site and participate in immune surveillance (Fig. 1) [127]. The composition of microorganisms may affect the efficacy of PD-1 inhibitors [128]. A study showed that the fecal Akkermansia muciniphila could be detected in 69% (11/16) and 58% (23/40) of patients exhibiting partial response or stable disease, whereas it was detectable in 34% (15/44) of patients who progressed or died [129]. Gut microbiota profiles of fecal specimens could be assessed by 16S ribosome RNA gene sequencing. Alipis putredinis, Prevotella copri and Bifidobacterium longum were enriched in the responders, and Ruminococcus_unclassified was enriched in non-responders. Patients with higher microbiota diversity had significantly longer PFS (HR = 4.2, 95%CI: 1.42–12.3, p = 0.009) (Table 1) [130]. The microbiota associated with clinical benefit varies in different studies, which implied that the difference between diet, host genetics, lifestyle factors, and human species may contribute to the diversity of gut microbiome and further affect the efficacy of ICIs [131, 132].
The application of cumulative antibiotics (ATB) could reduce the diversity of gut microbiota and disrupt the microbial balance [133, 134], which significantly weakened the efficacy of PD-L1 inhibitors and affected survival outcomes (median PFS: 1.9 months, HR = 1.5, 95%CI: 1.0–2.2, p = 0.03; median OS: 7.9 months, HR = 4.4, 95%CI: 2.6–7.7, p < 0.01) [135]. A study indicated that proton pump inhibitor (PPI) affected the diversity of gut microbiota through gastric acid [136]. The data of the phase II POPLAR and phase III OAK trial showed that in the population of anti-PD-L1 therapy, patients treated with ATB or PPI had shorter OS (HR = 1.20, 95%CI: 1.04–1.39) (Table 1), and the application of PPI was significantly related to shorter PFS (HR = 1.26, 95%CI: 1.10–1.44) [137]. As a promising treatment method, fecal microbiome transplantation (FMT) could improve the diversity of gut microbiota and the efficacy of immunotherapy [138, 139].
Patient's clinical characteristics
Factors such as genes, hormones contribute to the differences in immune response between males and females [140]. The differences may affect the efficacy of immunotherapy for male and female malignant tumors [140]. In a meta-analysis, by comparing the effects of anti-PD-1/PD-L1 plus chemotherapy and chemotherapy alone in men and women, it was found that the pooled OS-HRs were 0.76 (95% CI: 0.66–0.87) for men and 0.48 (95% CI: 0.35–0.67) for women [141]. Another meta-analysis showed that the pooled OS-HRs were 0.78 (95% CI: 0.60–1.00) in men and 0.97 (95% CI: 0.79–1.19) in women for anti-PD-1 alone, compared with 0.76 (95% CI: 0.64–0.91) in men and 0.44 (95% CI: 0.25–0.76) in women for anti-PD-1/PD-L1 plus chemotherapy [141] (Table 1). This implied that anti-PD-1 monotherapy may have a greater impact on men, and women may obtain greater survival benefits from the combination of anti-PD-1/PD-L1 and chemotherapy [141]. Nearly 80% of lung cancers are related to smoking. Exploratory analysis showed that among patients treated with anti-PD-1 treatment, current and former smokers had significantly higher overall response rate than non-smokers (36 vs. 26 vs. 14%) (Table 1) [142]. In addition, the increase of smoking years was associated with positive anti-PD-1 therapy response [143]. The patras immunotherapy score (PIOS) including the patient’s performance status (PS), body mass index (BMI), lines of treatment (LOT) and age was calculated through the formula (PS × BMI/LOT × age). Patients with high PIOS score had the best response to anti-PD-1 treatment (median PFS: 15 months vs. 5 months, HR = 0.469, 95% CI: 0.295–0.747; median OS: 32 months vs. 14 months, HR = 0.539, 95% CI: 0.317–0.918) (Table 1) [144].
Conclusion
Anti-PD-1/PD-L1 treatment is a promising treatment strategy for NSCLC. However, there are still numerous patients who are difficult to benefit from anti-PD-1/PD-L1 treatment. Various biomarkers for predicting efficacy are being explored. In the present stage, PD-L1 expression is the most widely adopted biomarker in clinical practice. TMB, TIL and neoantigen are significantly correlated with the efficacy of anti-PD-1/PD-L1 therapy. Gut microbiota, inflammatory genes, and dysregulated miRNA play an important role in anti-tumor immune regulation. Combining of multiple biomarkers may increase the predictive robustness and guide the implementation of cancer precision medicine.
Availability of data and materials
Not applicable.
Abbreviations
- PD-1:
-
Programmed cell death protein 1
- PD-L1:
-
Programmed cell death ligand 1
- NSCLC:
-
Non-small cell lung cancer
- TILs:
-
Tumor-infiltrating lymphocytes
- TMB:
-
Tumor mutational burden
- TPS:
-
Tumor proportion score
- OS:
-
Overall survival
- PFS:
-
Progression-free survival
- KRAS:
-
Kirsten rat sarcoma 2 viral oncogene homolog
- EGFR:
-
Epidermal growth factor receptor
- ALK:
-
Anaplastic lymphoma kinase
- MET:
-
Mesenchymal epithelial transition
- KEAP1:
-
Kelch-like ECH-associated protein 1
- NFE2L2:
-
Nuclear factor erythroid-2-related factor-2
- EMT:
-
Epithelial-mesenchymal transition
- TCR:
-
T cell receptor
- cCRT:
-
Concurrent chemo-radiotherapy
- MDSCs:
-
Myeloid-derived suppressor cells
- EVs:
-
Extracellular vesicles
- DCs:
-
Dendritic cells
- CAMLs:
-
Circulating cancer-associated macrophage-like cells
- TAM:
-
Tumor associated macrophage
- CTECs:
-
Circulating tumor endothelial cells
- NLR:
-
Neutrophil to lymphocyte ratio
- ATB:
-
Antibiotics
References
Deng Y, Zhao P, Zhou L, Xiang D, Hu J, Liu Y, Ruan J, Ye X, Zheng Y, Yao J, et al. Epidemiological trends of tracheal, bronchus, and lung cancer at the global, regional, and national levels: a population-based study. J Hematol Oncol. 2020;13:98.
Hirsch FR, Scagliotti GV, Mulshine JL, Kwon R, Curran WJ, Wu YL, Paz-Ares L. Lung cancer: current therapies and new targeted treatments. Lancet. 2017;389:299–311.
Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30.
Yi M, Li A, Zhou L, Chu Q, Luo S, Wu K. Immune signature-based risk stratification and prediction of immune checkpoint inhibitor’s efficacy for lung adenocarcinoma. Cancer Immunol Immunother. 2021. https://doi.org/10.1007/s00262-020-02817-z.
Hanna NH, Schneider BJ, Temin S, Baker S, Brahmer J, Ellis PM, Gaspar LE, Haddad RY, Hesketh PJ, Jain D, et al. Therapy for stage IV non-small-cell lung cancer without driver alterations: ASCO and OH (CCO) joint guideline update. J Clin Oncol. 2020;38:1608–32.
Swann JB, Smyth MJ. Immune surveillance of tumors. J Clin Invest. 2007;117:1137–46.
Kim R, Emi M, Tanabe K. Cancer immunoediting from immune surveillance to immune escape. Immunology. 2007; 1211–1214.
Bai X, Yi M, Jiao Y, Chu Q, Wu K. Blocking TGF-β signaling to enhance the efficacy of immune checkpoint inhibitor. Onco Targets Ther. 2019;12:9527–38.
Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol. 2021;14:10.
Yi M, Jiao D, Xu H, Liu Q, Zhao W, Han X, Wu K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Mol Cancer. 2018;17:129.
Qin S, Li A, Yi M, Yu S, Zhang M, Wu K. Recent advances on anti-angiogenesis receptor tyrosine kinase inhibitors in cancer therapy. J Hematol Oncol. 2019;12:27.
Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10:727–42.
Sun C, Mezzadra R, Schumacher TN. Regulation and function of the PD-L1 checkpoint. Immunity. 2018;48:434–52.
Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–54.
Patsoukis N, Li L, Sari D, Petkova V, Boussiotis VA. PD-1 increases PTEN phosphatase activity while decreasing PTEN protein stability by inhibiting casein kinase 2. Mol Cell Biol. 2013;33:3091–8.
Karachaliou N, Cardona AF, Bracht JWP, Aldeguer E, Drozdowskyj A, Fernandez-Bruno M, Chaib I, Berenguer J, Santarpia M, Ito M, et al. Integrin-linked kinase (ILK) and src homology 2 domain-containing phosphatase 2 (SHP2): Novel targets in EGFR-mutation positive non-small cell lung cancer (NSCLC). EBioMedicine. 2019;39:207–14.
Seliger B. Basis of PD1/PD-L1 therapies. J Clin Med. 2019;8:2168.
Xie F, Xu M, Lu J, Mao L, Wang S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol Cancer. 2019;18:146.
Seo J-S, Kim A, Shin JY, Kim YT. Comprehensive analysis of the tumor immune micro-environment in non-small cell lung cancer for efficacy of checkpoint inhibitor. Sci Rep. 2018;8:14576.
Xu-Monette ZY, Zhang M, Li J, Young KH. PD-1/PD-L1 blockade: have we found the key to unleash the antitumor immune response? Front Immunol. 2017;8:1597.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568–71.
Garon EB, Hellmann MD, Rizvi NA, Carcereny E, Leighl NB, Ahn MJ, Eder JP, Balmanoukian AS, Aggarwal C, Horn L, et al. Five-year overall survival for patients with advanced non-small-cell lung cancer treated with pembrolizumab: results from the phase I KEYNOTE-001 study. J Clin Oncol. 2019;37:2518–27.
Middleton G, Brock K, Savage J, Mant R, Summers Y, Connibear J, Shah R, Ottensmeier C, Shaw P, Lee SM, et al. Pembrolizumab in patients with non-small-cell lung cancer of performance status 2 (PePS2): a single arm, phase 2 trial. Lancet Respir Med. 2020;8:895–904.
Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, Gottfried M, Peled N, Tafreshi A, Cuffe S, et al. Updated analysis of KEYNOTE-024: pembrolizumab versus platinum-based chemotherapy for advanced non-small-cell lung cancer with PD-L1 tumor proportion score of 50% or greater. J Clin Oncol. 2019;37:537–46.
Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, Tykodi SS, Sosman JA, Procopio G, Plimack ER, et al. Nivolumab versus everolimus in advanced renal-cell carcinoma. N Engl J Med. 2015;373:1803–13.
Rittmeyer A, Barlesi F, Waterkamp D, Park K, Ciardiello F, von Pawel J, Gadgeel SM, Hida T, Kowalski DM, Dols MC, et al. Atezolizumab versus docetaxel in patients with previously treated non-small-cell lung cancer (OAK): a phase 3, open-label, multicentre randomised controlled trial. Lancet. 2017;389:255–65.
Shen X, Zhao B. Efficacy of PD-1 or PD-L1 inhibitors and PD-L1 expression status in cancer: meta-analysis. BMJ. 2018;362:k3529.
Killock D. bTMB is a promising predictive biomarker. Nat Rev Clin Oncol. 2019;16:403.
Chan TA, Yarchoan M, Jaffee E, Swanton C, Quezada SA, Stenzinger A, Peters S. Development of tumor mutation burden as an immunotherapy biomarker: utility for the oncology clinic. Ann Oncol. 2019;30:44–56.
Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, Chung HC, Kindler HL, Lopez-Martin JA, Miller WH, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. 2020;21:1353–65.
Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, Felip E, van den Heuvel MM, Ciuleanu TE, Badin F, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med. 2017;376:2415–26.
Hellmann MD, Ciuleanu TE, Pluzanski A, Lee JS, Otterson GA, Audigier-Valette C, Minenza E, Linardou H, Burgers S, Salman P, et al. Nivolumab plus Ipilimumab in lung cancer with a high tumor mutational burden. N Engl J Med. 2018;378:2093–104.
Nie W, Qian J, Xu MD, Gu K, Qian FF, Hu MJ, Lu J, Gan L, Zhang XY, Cao SH, et al. A non-linear association between blood tumor mutation burden and prognosis in NSCLC patients receiving atezolizumab. Oncoimmunology. 2020;9:1731072.
McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 2016;351:1463–9.
Yi M, Dong B, Chu Q, Wu K. Immune pressures drive the promoter hypermethylation of neoantigen genes. Exp Hematol Oncol. 2019;8:32.
Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69–74.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10.
Jiang T, Shi T, Zhang H, Hu J, Song Y, Wei J, Ren S, Zhou C. Tumor neoantigens: from basic research to clinical applications. J Hematol Oncol. 2019;12:93.
Yi M, Qin S, Zhao W, Yu S, Chu Q, Wu K. The role of neoantigen in immune checkpoint blockade therapy. Exp Hematol Oncol. 2018;7:28.
Demaria S, Golden EB, Formenti SC. Role of local radiation therapy in cancer immunotherapy. JAMA Oncol. 2015;1:1325–32.
Kaufman HL, Kohlhapp FJ, Zloza A. Oncolytic viruses: a new class of immunotherapy drugs. Nat Rev Drug Discov. 2015;14:642–62.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
McGranahan N, Swanton C. Neoantigen quality, not quantity. Sci Transl Med. 2019;11:eaax7918.
Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, Zhang T, Adleff V, Phallen J, Wali N, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discov. 2017;7:264–76.
Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, Plodkowski A, Long N, Sauter JL, Rekhtman N, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol. 2018;36:633–41.
Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, Xu H, Lu Z, Huang J, Lei Y, et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Lett. 2020;470:95–105.
Biton J, Mansuet-Lupo A, Pécuchet N, Alifano M, Ouakrim H, Arrondeau J, Boudou-Rouquette P, Goldwasser F, Leroy K, Goc J, et al. TP53, STK11, and EGFR mutations predict tumor immune profile and the response to anti-PD-1 in lung adenocarcinoma. Clin Cancer Res. 2018;24:5710–23.
Chen N, Fang W, Zhan J, Hong S, Tang Y, Kang S, Zhang Y, He X, Zhou T, Qin T, et al. Upregulation of PD-L1 by EGFR activation mediates the immune escape in EGFR-driven NSCLC: implication for optional immune targeted therapy for NSCLC patients with EGFR mutation. J Thorac Oncol. 2015;10:910–23.
Peng S, Wang R, Zhang X, Ma Y, Zhong L, Li K, Nishiyama A, Arai S, Yano S, Wang W. EGFR-TKI resistance promotes immune escape in lung cancer via increased PD-L1 expression. Mol Cancer. 2019;18:165.
Akbay EA, Koyama S, Carretero J, Altabef A, Tchaicha JH, Christensen CL, Mikse OR, Cherniack AD, Beauchamp EM, Pugh TJ, et al. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013;3:1355–63.
Masuda K, Horinouchi H, Tanaka M, Higashiyama R, Shinno Y, Sato J, Matsumoto Y, Okuma Y, Yoshida T, Goto Y, et al. Efficacy of anti-PD-1 antibodies in NSCLC patients with an EGFR mutation and high PD-L1 expression. J Cancer Res Clin Oncol. 2020;147:245–51.
Hong S, Chen N, Fang W, Zhan J, Liu Q, Kang S, He X, Liu L, Zhou T, Huang J, et al. Upregulation of PD-L1 by EML4-ALK fusion protein mediates the immune escape in ALK positive NSCLC: Implication for optional anti-PD-1/PD-L1 immune therapy for ALK-TKIs sensitive and resistant NSCLC patients. Oncoimmunology. 2016;5:e1094598.
Oya Y, Kuroda H, Nakada T, Takahashi Y, Sakakura N, Hida T. Efficacy of immune checkpoint inhibitor monotherapy for advanced non-small-cell lung cancer with ALK rearrangement. Int J Mol Sci. 2020;21:2623.
Frampton GM, Ali SM, Rosenzweig M, Chmielecki J, Lu X, Bauer TM, Akimov M, Bufill JA, Lee C, Jentz D, et al. Activation of MET via diverse exon 14 splicing alterations occurs in multiple tumor types and confers clinical sensitivity to MET inhibitors. Cancer Discov. 2015;5:850–9.
Sabari JK, Leonardi GC, Shu CA, Umeton R, Montecalvo J, Ni A, Chen R, Dienstag J, Mrad C, Bergagnini I, et al. PD-L1 expression, tumor mutational burden, and response to immunotherapy in patients with MET exon 14 altered lung cancers. Ann Oncol. 2018;29:2085–91.
Kron A, Scheffler M, Heydt C, Ruge L, Schaepers C, Eisert A, Merkelbach-Bruse S, Riedel R, Nogova L, Fischer RN, et al. Genetic heterogeneity of MET-aberrant non-small cell lung cancer and its impact on the outcome of immunotherapy. J Thorac Oncol. 2020. https://doi.org/10.1016/j.jtho.2020.11.017.
Gong M, Li Y, Ye X, Zhang L, Wang Z, Xu X, Shen Y, Zheng C. Loss-of-function mutations in KEAP1 drive lung cancer progression via KEAP1/NRF2 pathway activation. Cell Commun Signal. 2020;18:98.
Singh A, Daemen A, Nickles D, Jeon SM, Foreman O, Sudini K, Gnad F, Lajoie S, Gour N, Mitzner W, et al. NRF2 activation promotes aggressive lung cancer and associates with poor clinical outcomes. Clin Cancer Res. 2021;27:877–88.
Tian Y, Liu Q, Yu S, Chu Q, Chen Y, Wu K, Wang L. NRF2-driven KEAP1 transcription in human lung cancer. Mol Cancer Res. 2020;18:1465–76.
Tian Y, Wu K, Liu Q, Han N, Zhang L, Chu Q, Chen Y. Modification of platinum sensitivity by KEAP1/NRF2 signals in non-small cell lung cancer. J Hematol Oncol. 2016;9:83.
Xu X, Yang Y, Liu X, Cao N, Zhang P, Zhao S, Chen D, Li L, He Y, Dong X, et al. NFE2L2/KEAP1 mutations correlate with higher tumor mutational burden value/PD-L1 expression and potentiate improved clinical outcome with immunotherapy. Oncologist. 2020;25:e955–63.
Guisier F, Dubos-Arvis C, Viñas F, Doubre H, Ricordel C, Ropert S, Janicot H, Bernardi M, Fournel P, Lamy R, et al. Efficacy and safety of anti-PD-1 immunotherapy in patients with advanced NSCLC With BRAF, HER2, or MET mutations or RET translocation: GFPC 01–2018. J Thorac Oncol. 2020;15:628–36.
Mazieres J, Drilon A, Lusque A, Mhanna L, Cortot AB, Mezquita L, Thai AA, Mascaux C, Couraud S, Veillon R, et al. Immune checkpoint inhibitors for patients with advanced lung cancer and oncogenic driver alterations: results from the IMMUNOTARGET registry. Ann Oncol. 2019;30:1321–8.
Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127:2930–40.
Thompson JC, Hwang W-T, Davis C, Deshpande C, Jeffries S, Rajpurohit Y, Krishna V, Smirnov D, Verona R, Lorenzi MV, et al. Gene signatures of tumor inflammation and epithelial-to-mesenchymal transition (EMT) predict responses to immune checkpoint blockade in lung cancer with high accuracy. Lung Cancer. 2020;139:1–8.
Thompson JC, Davis C, Deshpande C, Hwang W-T, Jeffries S, Huang A, Mitchell TC, Langer CJ, Albelda SM. Gene signature of antigen processing and presentation machinery predicts response to checkpoint blockade in non-small cell lung cancer (NSCLC) and melanoma. J Immunother Cancer. 2020;8:e000974.
Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20:460–9.
Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. 2017;16:203–22.
Yi M, Xu L, Jiao Y, Luo S, Li A, Wu K. The role of cancer-derived microRNAs in cancer immune escape. J Hematol Oncol. 2020;13:25.
Hong W, Xue M, Jiang J, Zhang Y, Gao X. Circular RNA circ-CPA4/ let-7 miRNA/PD-L1 axis regulates cell growth, stemness, drug resistance and immune evasion in non-small cell lung cancer (NSCLC). J Exp Clin Cancer Res. 2020;39:149.
Fan J, Yin Z, Xu J, Wu F, Huang Q, Yang L, Jin Y, Yang G. Circulating microRNAs predict the response to anti-PD-1 therapy in non-small cell lung cancer. Genomics. 2020;112:2063–71.
Velcheti V, Schalper KA, Carvajal DE, Anagnostou VK, Syrigos KN, Sznol M, Herbst RS, Gettinger SN, Chen L, Rimm DL. Programmed death ligand-1 expression in non-small cell lung cancer. Lab Invest. 2014;94:107–16.
Qin S, Dong B, Yi M, Chu Q, Wu K. Prognostic Values of TIM-3 expression in patients with solid tumors: a meta-analysis and database evaluation. Front Oncol. 2020;10:1288.
Qin S, Xu L, Yi M, Yu S, Wu K, Luo S. Novel immune checkpoint targets: moving beyond PD-1 and CTLA-4. Mol Cancer. 2019;18:155.
Kim K, Park S, Park SY, Kim G, Park SM, Cho JW, Kim DH, Park YM, Koh YW, Kim HR, et al. Single-cell transcriptome analysis reveals TOX as a promoting factor for T cell exhaustion and a predictor for anti-PD-1 responses in human cancer. Genome Med. 2020;12:22.
Cui S, Dong L, Qian J, Ye L, Jiang L. Classifying non-small cell lung cancer by status of programmed cell death ligand 1 and tumor-infiltrating lymphocytes on tumor cells. J Cancer. 2018;9:129–34.
Teng MWL, Ngiow SF, Ribas A, Smyth MJ. Classifying cancers based on T-cell infiltration and PD-L1. Cancer Res. 2015;75:2139–45.
Mazzaschi G, Madeddu D, Falco A, Bocchialini G, Goldoni M, Sogni F, Armani G, Lagrasta CA, Lorusso B, Mangiaracina C, et al. Low PD-1 expression in cytotoxic CD8+ tumor-infiltrating lymphocytes confers an immune-privileged tissue microenvironment in NSCLC with a prognostic and predictive value. Clin Cancer Res. 2018;24:407–19.
Kamphorst AO, Pillai RN, Yang S, Nasti TH, Akondy RS, Wieland A, Sica GL, Yu K, Koenig L, Patel NT, et al. Proliferation of PD-1+ CD8 T cells in peripheral blood after PD-1-targeted therapy in lung cancer patients. Proc Natl Acad Sci U S A. 2017;114:4993–8.
Howie B, Sherwood AM, Berkebile AD, Berka J, Emerson RO, Williamson DW, Kirsch I, Vignali M, Rieder MJ, Carlson CS, et al. High-throughput pairing of T cell receptor α and β sequences. Sci Transl Med. 2015;7:301ra131.
Hennecke J, Wiley DC. T cell receptor-MHC interactions up close. Cell. 2001;104:1–4.
Han J, Duan J, Bai H, Wang Y, Wan R, Wang X, Chen S, Tian Y, Wang D, Fei K, et al. TCR repertoire diversity of peripheral PD-1+CD8+ T cells predicts clinical outcomes after immunotherapy in patients with non-small cell lung cancer. Cancer Immunol Res. 2020;8:146–54.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, Kurata T, Chiappori A, Lee KH, de Wit M, et al. Overall survival with durvalumab after chemoradiotherapy in stage III NSCLC. N Engl J Med. 2018;379:2342–50.
Takeshima T, Chamoto K, Wakita D, Ohkuri T, Togashi Y, Shirato H, Kitamura H, Nishimura T. Local radiation therapy inhibits tumor growth through the generation of tumor-specific CTL: its potentiation by combination with Th1 cell therapy. Cancer Res. 2010;70:2697–706.
Yoneda K, Kuwata T, Kanayama M, Mori M, Kawanami T, Yatera K, Ohguri T, Hisaoka M, Nakayama T, Tanaka F. Alteration in tumoural PD-L1 expression and stromal CD8-positive tumour-infiltrating lymphocytes after concurrent chemo-radiotherapy for non-small cell lung cancer. Br J Cancer. 2019;121:490–6.
Briere D, Sudhakar N, Woods DM, Hallin J, Engstrom LD, Aranda R, Chiang H, Sodré AL, Olson P, Weber JS, et al. The class I/IV HDAC inhibitor mocetinostat increases tumor antigen presentation, decreases immune suppressive cell types and augments checkpoint inhibitor therapy. Cancer Immunol Immunother. 2018;67:381–92.
Munn DH, Mellor AL. Indoleamine 2,3 dioxygenase and metabolic control of immune responses. Trends Immunol. 2013;34:137–43.
Botticelli A, Mezi S, Pomati G, Cerbelli B, Cerbelli E, Roberto M, Giusti R, Cortellini A, Lionetto L, Scagnoli S, et al. Tryptophan catabolism as immune mechanism of primary resistance to Anti-PD-1. Front Immunol. 2020;11:1243.
Umansky V, Blattner C, Gebhardt C, Utikal J. The Role of myeloid-derived suppressor cells (MDSC) in cancer progression. Vaccines (Basel). 2016;4:36.
Condamine T, Ramachandran I, Youn JI, Gabrilovich DI. Regulation of tumor metastasis by myeloid-derived suppressor cells. Annu Rev Med. 2015;66:97–110.
Lesokhin AM, Hohl TM, Kitano S, Cortez C, Hirschhorn-Cymerman D, Avogadri F, Rizzuto GA, Lazarus JJ, Pamer EG, Houghton AN, et al. Monocytic CCR2(+) myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012;72:876–86.
Obermajer N, Muthuswamy R, Odunsi K, Edwards RP, Kalinski P. PGE(2)-induced CXCL12 production and CXCR4 expression controls the accumulation of human MDSCs in ovarian cancer environment. Cancer Res. 2011;71:7463–70.
Yamauchi Y, Safi S, Blattner C, Rathinasamy A, Umansky L, Juenger S, Warth A, Eichhorn M, Muley T, Herth FJF, et al. Circulating and tumor myeloid-derived suppressor cells in resectable non-small cell lung cancer. Am J Respir Crit Care Med. 2018;198:777–87.
Robbins PD, Dorronsoro A, Booker CN. Regulation of chronic inflammatory and immune processes by extracellular vesicles. J Clin Invest. 2016;126:1173–80.
Maas SLN, Breakefield XO, Weaver AM. Extracellular vesicles: unique intercellular delivery vehicles. Trends Cell Biol. 2017;27:172–88.
Tkach M, Théry C. Communication by extracellular vesicles: where we are and where we need to go. Cell. 2016;164:1226–32.
Li S, Yi M, Dong B, Jiao Y, Luo S, Wu K. The roles of exosomes in cancer drug resistance and its therapeutic application. Clin Transl Med. 2020;10:e257.
Li S, Yi M, Dong B, Tan X, Luo S, Wu K. The role of exosomes in liquid biopsy for cancer diagnosis and prognosis prediction. Int J Cancer. 2020. https://doi.org/10.1002/ijc.33386.
Lindenbergh MFS, Stoorvogel W. Antigen presentation by extracellular vesicles from professional antigen-presenting cells. Annu Rev Immunol. 2018;36:435–59.
Becker A, Thakur BK, Weiss JM, Kim HS, Peinado H, Lyden D. Extracellular vesicles in cancer: cell-to-cell mediators of metastasis. Cancer Cell. 2016;30:836–48.
Shukuya T, Ghai V, Amann JM, Okimoto T, Shilo K, Kim TK, Wang K, Carbone DP. Circulating MicroRNAs and extracellular vesicle-containing MicroRNAs as response biomarkers of anti-programmed cell death protein 1 or programmed death-ligand 1 therapy in NSCLC. J Thorac Oncol. 2020;15:1773–81.
Xie F, Zhou X, Fang M, Li H, Su P, Tu Y, Zhang L, Zhou F. Extracellular vesicles in cancer immune microenvironment and cancer immunotherapy. Adv Sci (Weinh). 2019;6:1901779.
Mathew M, Zade M, Mezghani N, Patel R, Wang Y, Momen-Heravi F. Extracellular vesicles as biomarkers in cancer immunotherapy. Cancers (Basel). 2020;12:2825.
Vasconcelos MH, Caires HR, Ābols A, Xavier CPR, Linē A. Extracellular vesicles as a novel source of biomarkers in liquid biopsies for monitoring cancer progression and drug resistance. Drug Resist Updat. 2019;47:100647.
Solinas G, Germano G, Mantovani A, Allavena P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol. 2009;86:1065–73.
Condeelis J, Pollard JW. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell. 2006;124:263–6.
Adams DL, Martin SS, Alpaugh RK, Charpentier M, Tsai S, Bergan RC, Ogden IM, Catalona W, Chumsri S, Tang CM, et al. Circulating giant macrophages as a potential biomarker of solid tumors. Proc Natl Acad Sci USA. 2014;111:3514–9.
Tang CM, Zhu P, Li S, Makarova OV, Amstutz PT, Adams DL. Filtration and analysis of circulating cancer associated cells from the blood of cancer patients. Methods Mol Biol. 2017;1572:511–24.
Augustyn A, Adams DL, He J, Qiao Y, Verma V, Liao Z, Tang CM, Heymach JV, Tsao AS, Lin SH. Giant circulating cancer-associated macrophage-like cells are associated with disease recurrence and survival in non-small-cell lung cancer treated with chemoradiation and atezolizumab. Clin Lung Cancer. 2020;S1525–7304:30210–2.
Kronenwett U, Huwendiek S, Ostring C, Portwood N, Roblick UJ, Pawitan Y, Alaiya A, Sennerstam R, Zetterberg A, Auer G. Improved grading of breast adenocarcinomas based on genomic instability. Cancer Res. 2004;64:904–9.
Dürrbaum M, Storchová Z. Effects of aneuploidy on gene expression: implications for cancer. FEBS J. 2016;283:791–802.
Sansregret L, Swanton C. The role of aneuploidy in cancer evolution. Cold Spring Harb Perspect Med. 2017;7:a028373.
Lin PP. Aneuploid CTC and CEC. Diagnostics (Basel). 2018;8(2):26.
Akino T, Hida K, Hida Y, Tsuchiya K, Freedman D, Muraki C, Ohga N, Matsuda K, Akiyama K, Harabayashi T, et al. Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am J Pathol. 2009;175:2657–67.
Cima I, Kong SL, Sengupta D, Tan IB, Phyo WM, Lee D, Hu M, Iliescu C, Alexander I, Goh WL, et al. Tumor-derived circulating endothelial cell clusters in colorectal cancer. Sci Transl Med. 2016;8:345ra389.
Zhang L, Zhang X, Liu Y, Zhang T, Wang Z, Gu M, Li Y, Wang DD, Li W, Lin PP. PD-L1 aneuploid circulating tumor endothelial cells (CTECs) exhibit resistance to the checkpoint blockade immunotherapy in advanced NSCLC patients. Cancer Lett. 2020; 469:355–366.
Templeton AJ, McNamara MG, Šeruga B, Vera-Badillo FE, Aneja P, Ocaña A, Leibowitz-Amit R, Sonpavde G, Knox JJ, Tran B, et al. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J Natl Cancer Inst. 2014;106:dju124.
Mandaliya H, Jones M, Oldmeadow C, Nordman II. Prognostic biomarkers in stage IV non-small cell lung cancer (NSCLC): neutrophil to lymphocyte ratio (NLR), lymphocyte to monocyte ratio (LMR), platelet to lymphocyte ratio (PLR) and advanced lung cancer inflammation index (ALI). Transl Lung Cancer Res. 2019;8:886–94.
Li Y, Zhang Z, Hu Y, Yan X, Song Q, Wang G, Chen R, Jiao S, Wang J. Pretreatment neutrophil-to-lymphocyte ratio (NLR) may predict the outcomes of advanced non-small-cell lung cancer (NSCLC) patients treated with immune checkpoint inhibitors (ICIs). Front Oncol. 2020;10:654.
Simonaggio A, Elaidi R, Fournier L, Fabre E, Ferrari V, Borchiellini D, Thouvenin J, Barthelemy P, Thibault C, Tartour E, et al. Variation in neutrophil to lymphocyte ratio (NLR) as predictor of outcomes in metastatic renal cell carcinoma (mRCC) and non-small cell lung cancer (mNSCLC) patients treated with nivolumab. Cancer Immunol Immunother. 2020;69:2513–22.
Mezquita L, Auclin E, Ferrara R, Charrier M, Remon J, Planchard D, Ponce S, Ares LP, Leroy L, Audigier-Valette C, et al. Association of the lung immune prognostic index with immune checkpoint inhibitor outcomes in patients with advanced non-small cell lung cancer. JAMA Oncol. 2018;4:351–7.
Peng L, Wang Y, Liu F, Qiu X, Zhang X, Fang C, Qian X, Li Y. Peripheral blood markers predictive of outcome and immune-related adverse events in advanced non-small cell lung cancer treated with PD-1 inhibitors. Cancer Immunol Immunother. 2020;69:1813–22.
Kargl J, Zhu X, Zhang H, Yang GHY, Friesen TJ, Shipley M, Maeda DY, Zebala JA, McKay-Fleisch J, Meredith G, et al. Neutrophil content predicts lymphocyte depletion and anti-PD1 treatment failure in NSCLC. JCI Insight. 2019;4:e1308.
Cho Y-H, Choi MG, Kim DH, Choi YJ, Kim SY, Sung KJ, Lee JC, Kim SY, Rho JK, Choi CM. Natural killer cells as a potential biomarker for predicting immunotherapy efficacy in patients with non-small cell lung cancer. Target Oncol. 2020;15:241–7.
Yi M, Yu S, Qin S, Liu Q, Xu H, Zhao W, Chu Q, Wu K. Gut microbiome modulates efficacy of immune checkpoint inhibitors. J Hematol Oncol. 2018;11:47.
Nagasaka M, Sexton R, Alhasan R, Rahman S, Azmi AS, Sukari A. Gut microbiome and response to checkpoint inhibitors in non-small cell lung cancer-a review. Crit Rev Oncol Hematol. 2020;145:102841.
Zitvogel L, Ayyoub M, Routy B, Kroemer G. Microbiome and Anticancer Immunosurveillance. Cell. 2016;165:276–87.
Yi M, Jiao D, Qin S, Chu Q, Li A, Wu K. Manipulating gut microbiota composition to enhance the therapeutic effect of cancer immunotherapy. Integr Cancer Ther. 2019;18:1534735419876351.
Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillère R, Fluckiger A, Messaoudene M, Rauber C, Roberti MP, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–7.
Jin Y, Dong H, Xia L, Yang Y, Zhu Y, Shen Y, Zheng H, Yao C, Wang Y, Lu S. The diversity of gut microbiome is associated with favorable responses to anti-programmed death 1 immunotherapy in chinese patients with NSCLC. J Thorac Oncol. 2019;14:1378–89.
Blekhman R, Goodrich JK, Huang K, Sun Q, Bukowski R, Bell JT, Spector TD, Keinan A, Ley RE, Gevers D, et al. Host genetic variation impacts microbiome composition across human body sites. Genome Biol. 2015;16:191.
Gupta VK, Paul S, Dutta C. Geography, ethnicity or subsistence-specific variations in human microbiome composition and diversity. Front Microbiol. 2017;8:1162.
Tinsley N, Zhou C, Tan G, Rack S, Lorigan P, Blackhall F, Krebs M, Carter L, Thistlethwaite F, Graham D, et al. Cumulative antibiotic use significantly decreases efficacy of checkpoint inhibitors in patients with advanced cancer. Oncologist. 2020;25:55–63.
Yi M, Qin S, Chu Q, Wu K. The role of gut microbiota in immune checkpoint inhibitor therapy. Hepatobiliary Surg Nutr. 2018;7:481–3.
Derosa L, Hellmann MD, Spaziano M, Halpenny D, Fidelle M, Rizvi H, Long N, Plodkowski AJ, Arbour KC, Chaft JE, et al. Negative association of antibiotics on clinical activity of immune checkpoint inhibitors in patients with advanced renal cell and non-small-cell lung cancer. Ann Oncol. 2018;29:1437–44.
Imhann F, Bonder MJ, Vich Vila A, Fu J, Mujagic Z, Vork L, Tigchelaar EF, Jankipersadsing SA, Cenit MC, Harmsen HJM, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016;65:740–8.
Chalabi M, Cardona A, Nagarkar DR, Dhawahir Scala A, Gandara DR, Rittmeyer A, Albert ML, Powles T, Kok M, Herrera FG. Efficacy of chemotherapy and atezolizumab in patients with non-small-cell lung cancer receiving antibiotics and proton pump inhibitors: pooled post hoc analyses of the OAK and POPLAR trials. Ann Oncol. 2020;31:525–31.
McQuade JL, Ologun GO, Arora R, Wargo JA. Gut microbiome modulation via fecal microbiota transplant to augment immunotherapy in patients with melanoma or other cancers. Curr Oncol Rep. 2020;22:74.
Kaźmierczak-Siedlecka K, Daca A, Fic M, van de Wetering T, Folwarski M, Makarewicz W. Therapeutic methods of gut microbiota modification in colorectal cancer management—fecal microbiota transplantation, prebiotics, probiotics, and synbiotics. Gut Microbes. 2020;11:1518–30.
Klein SL, Flanagan KL. Sex differences in immune responses. Nat Rev Immunol. 2016;16:626–38.
Conforti F, Pala L, Bagnardi V, Viale G, De Pas T, Pagan E, Pennacchioli E, Cocorocchio E, Ferrucci PF, De Marinis F, et al. Sex-based heterogeneity in response to lung cancer immunotherapy: a systematic review and meta-analysis. J Natl Cancer Inst. 2019;111:772–81.
Li JJN, Karim K, Sung M, Le LW, Lau SCM, Sacher A, Leighl NB. Tobacco exposure and immunotherapy response in PD-L1 positive lung cancer patients. Lung Cancer. 2020;150:159–63.
Chiu M, Lipka MB, Bhateja P, Fu P, Dowlati A. A detailed smoking history and determination of status predict response to checkpoint inhibitors in advanced non-small cell lung cancer. Transl Lung Cancer Res. 2020;9:55–60.
Dimitrakopoulos FI, Nikolakopoulos A, Kottorou A, Kalofonou F, Liolis E, Frantzi T, Pyrousis I, Koutras A, Makatsoris T, Kalofonos H. PIOS (patras immunotherapy score) score is associated with best overall response, progression-free survival, and post-immunotherapy overall survival in patients with advanced non-small-cell lung cancer (NSCLC) treated with anti-program cell death-1 (PD-1) inhibitors. Cancers (Basel). 2020;12:1257.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (No.81874120, 82073370).
Author information
Authors and Affiliations
Contributions
MN drafted the manuscript and prepared the figures. MY and NL collected the related references and participated in discussion. SL and KW designed this review and revised the manuscript. All authors contributed to this manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Niu, M., Yi, M., Li, N. et al. Predictive biomarkers of anti-PD-1/PD-L1 therapy in NSCLC. Exp Hematol Oncol 10, 18 (2021). https://doi.org/10.1186/s40164-021-00211-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40164-021-00211-8