
Abstract
Termites constitute part of diverse and economically important termite fauna in Africa, but information on gut microbiota and their associated soil microbiome is still inadequate. In this study, we assessed and compared the bacterial diversity and community structure between termites’ gut, their mounds and surrounding soil using the 454 pyrosequencing-based analysis of 16S rRNA gene sequences. A wood-feeder termite (Microcerotermes sp.), three fungus-cultivating termites (Macrotermes michaelseni, Odontotermes sp. and Microtermes sp.), their associated mounds and corresponding savannah soil samples were analyzed. The pH of the gut homogenates and soil physico-chemical properties were determined. The results indicated significant difference in bacterial community composition and structure between the gut and corresponding soil samples. Soil samples (Chao1 index ranged from 1359 to 2619) had higher species richness than gut samples (Chao1 index ranged from 461 to 1527). The bacterial composition and community structure in the gut of Macrotermes michaelseni and Odontotermes sp. were almost identical but different from that of Microtermes and Microcerotermes species, which had unique community structures. The most predominant bacterial phyla in the gut were Bacteroidetes (40–58 %), Spirochaetes (10–70 %), Firmicutes (17–27 %) and Fibrobacteres (13 %) while in the soil samples were Acidobacteria (28–45 %), Actinobacteria (20–40 %) and Proteobacteria (18–24 %). Some termite gut-specific bacterial lineages belonging to the genera Dysgonomonas, Parabacteroides, Paludibacter, Tannerella, Alistipes, BCf9-17 termite group and Termite Treponema cluster were observed. The results not only demonstrated a high level of bacterial diversity in the gut and surrounding soil environments, but also presence of distinct bacterial communities that are yet to be cultivated. Therefore, combined efforts using both culture and culture-independent methods are suggested to comprehensively characterize the bacterial species and their specific roles in these environments.
Similar content being viewed by others
Background
Termites (Isoptera) are a large and diverse group of soil macrofauna comprising of >2600 species worldwide (Ahmed et al. 2011). The greatest termite diversity is in Africa (Eggleton 2000), where they play diverse roles in semi-arid and humid ecosystems: As soil engineers, termites have an impact on the soil structure (Holt and Lepage 2000), which modifies the soil environment thereby controlling diversity and activity of other soil organisms (Jones et al. 1997). Their influence on the soil microbial component is due to their major construction activities of complex galleries and mounds, which partly contribute to soil heterogeneity in the tropical regions (Holt and Lepage 2000). The termite mound is made from a mineral matrix mixed with feces or saliva, depending on the termite species and forms a specific habitat for soil microbes since the physical and chemical properties are different from the surrounding soil (Brauman 2000; Holt and Lepage 2000).
The type of a mound constructed depends on the feeding habit of the termite species (Holt and Lepage 2000): Soil-feeders (subfamily Termitinae) build their mounds with fecal matter mixed with coarse, inorganic particles (Noirot and Darlington 2000) and have a limited effect on the surrounding soil of about 20 cm in depth and within a range of a few meters (Harry et al. 2001). However, the fungus-growing termites (subfamily Macrotermitinae) build their mounds using soil and clay cemented by salivary secretions, which make the mounds enriched with clay particles but impoverished in carbon (Harry et al. 2001). The nest-walls consist of organo-mineral aggregates, characterized by a low stability and thus mineralize easily (Garnier-Sillam et al. 1988). They have a wider range of activity on the surrounding soil of 1–3 m in depth and within a range of a 2–8 m (Harry et al. 2001), which may influence the soil properties and fertility. The question is whether the fungus-feeding termites induce soil microbial changes as those observed in soil-feeders (Harry et al. 2001; Fall et al. 2004; Roose-Amsaleg et al. 2004; Fall et al. 2007). This forms the principle objective of this study with a focus on bacterial community structure in the different environments (termite gut, associated mound and surrounding soil ecosystems).
Previously, studies on microbial communities between termite guts and mounds (Roose-Amsaleg et al. 2004; Fall et al. 2007), mounds (Fall et al. 2004) and termite mounds and surrounding soils (Holt 1996; Harry et al. 2001) indicated differences in the microbial community abundance. Moreover, the gut bacterial communities have been assessed by using traditional molecular methods such as Sanger sequencing-based analysis of 16S rRNA gene libraries or fingerprinting techniques (Schmitt-Wagner et al. 2003; Shinzato et al. 2005, 2007; Fisher et al. 2007; Fall et al. 2007; Mackenzie et al. 2007; Mathew et al. 2012; Makonde et al. 2013a). These results not only indicated high bacterial diversity in the guts, but also termite-specific bacterial lineages (Shinzato et al. 2005). Such methods, however, were often limited to the analysis of a relative small number of clones.
To compressively describe and compare the microbial community structure in different ecosystems, high-throughput methods (Droege and Hill 2008; Glenn 2011) are necessary. Recently, high resolution analyses from five genera of the Macrotermitinae revealed that community composition almost resembles host phylogeny and their gut microbiotas are distinct from those of other termites (Otani et al. 2014). Elsewhere, analysis of the gut environment and bacterial microbiota (Köhler et al. 2012) revealed functional compartmentation on wood-feeding higher termites (Nasutitermes spp.). In this study, we used 454 pyrosequencing-based analysis of the 16S rRNA gene region to assess and compare the bacterial diversity and community structure in the gut of termites, associated termite mounds and surrounding soil environments. This is the first study that attempts to comparatively assess the bacterial diversity and structure in termite gut and surrounding habitats using the high-throughput sequencing approach. The results indicated variation in bacterial diversity and structure in the different environments.
Results
Description of the samples
The pH of the gut homogenates was within the neutral range (pH 7–8). The soils were slightly acidic (pH range 5–7) with overall high sand (76 %) and a relative increase in clay content (30 and 20 %) in the two mounds compared to the corresponding savannah soil (27.5 and 2.5 %). Similarly, organic carbon (OC) and nitrogen (N) contents had overall slightly higher values in savannah soil (3.0 and 0.3 mg/g, respectively) compared to the mounds (2.0 and 0.2 mg/g, respectively). The C/N ratios ranged from 9 to 11 (see Additional file 1a).
Distribution of phyla across the samples
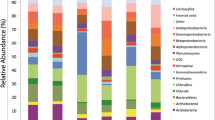
A total of 17, 528 reads were obtained for the bacterial samples. After quality filtering and chimera check 14, 301, the resulting sequences (≥300 bp) were clustered into 4, 157 operational taxonomic units [OTUs] (Table 1) at 3 % genetic distance according to the approach described by Huse et al. (2010). Taxonomic assignment of the resulting sequences against the SILVA database showed that a total of 21 phyla were represented and the major ones were: Bacteroidetes, Acidobacteria, Spirochaetes, Actinobacteria, Proteobacteria, Firmicutes, Fibrobacteres and Chloroflexi (Fig. 1a; Table 2). The other 13 phyla were represented at varying levels in one or more samples at <5 % of the effective sequences (Table 2).

a Relative abundances of phylogenetic groups in the samples. b Relative abundances of bacterial groups (at class level) in the samples. MCG8 Microcerotermes sp. gut homogenate, MIG7 Microtermes sp. gut homogenate, OTG1 Odontotermes sp. gut homogenate, MTG4 M. michaelseni gut homogenate, OTN2 soil from mound C of Odontotermes sp., MTN5 soil from mound D of M. michaelseni, MTS6 soil collected 3 m away from mound D, OTS3 soil collected 3 m away from mound C. Phylogenetic groups accounting for ≤0.4 % of the analyzed sequences were included in the artificial group ‘others’
Bacterial community structure across samples
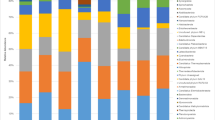
Bacterial composition at the phylum level differed between the termite guts, mounds, and soil environments (Fig. 1a). Each environment was dominated by a particular phylum/phyla (≥5 % of the effective sequences). In the termite guts (MTG4, OTG1 and MIG7 samples), Bacteroidetes (>40 %) was the most abundant phylum while sample MCG8 was dominated by the phylum Spirochaetes (>70 %). Within the mounds (samples OTN2 and MTN5), the most abundant phyla were Actinobacteria (34–40 %), followed by Acidobacteria (28–32 %), whereas the corresponding soils (samples OTS3 and MTS6) were predominated by Acidobacteria (36–45 %). Four major phyla (Bacteroidetes, Proteobacteria, Firmicutes and Actinobacteria) were shared by all samples, but in different relative abundances (Fig. 1a; Table 2). Two phyla (Deferribacteres and Synergistetes) were exclusively detected in the gut samples while Gemmatimonadetes, Nitrospirae, Armatimonadetes, Candidate division TM7, Candidate division WS3, SM2F11 and WCHB1-60 were only detected in the mounds and surrounding soil in one or more samples (Fig. 1a; Table 2). Moreover, differences in bacterial community members were observed at the class level (Fig. 1b). At the family level, members from different families Porphyromonadaceae, Rikenellaceae, Ruminococcaceae and Lachnospiraceae were more abundant within the guts than in the mounds and savannah soil samples. However, the wood-feeding termite (sample MCG8) was dominated by the family Spirochaetaceae and termite gut group, representing 70 and 12 % of the effective sequences, respectively (Additional file 2).
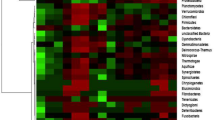
At high taxonomic resolution, there were salient differences in relative abundance of majority genera across the samples. Within the gut samples, there were 13 genera with known members that were represented with a value ≥2 % in one or more samples (Additional file 2). Notably, the genus Termite Treponema cluster was the most abundant in sample MCG8 (>50 %), while the genus Treponema (≥19 %) was more abundant in MCG8 and MIG7 than in samples OTG1 and MTG4, which were dominated by the genus Alistipes (>30 %) (Additional file 2). For the mounds and savannah soil samples, there were nine genera with known members that were represented by a value ≥2 % in one or more samples. They included; Bryobacter, Acidothermus, Frankia, Hamadaea, Rugosimonospora, Nocardioides, Streptomyces, Rhizomicrobium and Blastobacter (Additional file 2). Clustering of samples based on community similarity clustered the gut and soil samples separately (Figs. 2a, 3). The gut samples (MTG4 and OTG1) had identical communities, thus, clustered together compared to MCG8 and MIG7 samples. Likewise, the mound samples (MTS6 and OTS3) had more similar communities compared to surrounding soil samples (OTS3 and OTN2).

a Heatmap shows hierarchical clustering of taxa (relative abundance ≥1.0 % of the analyzed sequences). The scale bar represents color saturation gradient based on the relative abundances of the bacterial genera. The dendrogram at the top shows the weighted Euclidean distance analysis of community similarity. Classification is presented at the genus and phylum levels. b PCA of bacterial communities based on the relative abundances (≥2.3 %) of selected genera. The vectors indicate the direction and impact of each genus on the overall variance. R squared (r2) = 0.49. MCG8 Microcerotermes sp. gut homogenate, MIG7 Microtermes sp. gut homogenate, OTG1 Odontotermes sp. gut homogenate, MTG4 M. michaelseni gut homogenate, OTN2 soil from mound C of Odontotermes sp., MTN5 soil from mound D of M. michaelseni, MTS6 soil collected 3 m away from mound D, OTS3 soil collected 3 m away from mound C

A 3 dimensional PCoA plot showing the degree of similarity of bacterial communities on termite guts, mounds and soil samples. R squared (r2) = 0.69. MCG8 Microcerotermes sp. gut homogenate, MIG7 Microtermes sp. gut homogenate, OTG1 Odontotermes sp. gut homogenate, MTG4 M. michaelseni gut homogenate, OTN2 soil from mound C of Odontotermes sp., MTN5 soil from mound D of M. michaelseni, MTS6 soil collected 3 m away from mound D, OTS3 soil collected 3 m away from mound C
Bacterial diversity and richness
Bacterial diversity and richness for the selected sequences from each sample (Table 1) was evaluated by rarefaction as shown in Fig. 4 and Additional file 1b. At 3, 5 and 10 % sequence divergence, most rarefaction curves did not reach saturation, indicating that the surveying efforts did not fully cover the extent of taxonomic diversity at these genetic distances, but a substantial fraction of the bacterial diversity within individual samples was assessed. The diversity measures indicated that MTN5 had the most genus-level taxa (645, Table 1) and MCG8 the least (261, Table 1), that OTN2 was richest (Chao 1 index), while MCG8 was poorest. Despite variation in community composition, Simpson (1/D) and Shannon indices were similar across communities, ranging from 0.9 to 1.0 and 4.0 to 5.4, respectively. Comparison between any pair of bacterial communities using unweighted UniFrac PCoA (Fig. 3) showed a distinct clustering by environment but the p value of 0.283 and R value of 0.091 indicated that the grouping of samples is weak. For instance, samples OTG1 and MTG4 clustered together (Fig. 3), indicating similarities in the two guts. Sample MIG7 was slightly away from OTG1 and MTG4 meaning that its bacterial communities and community structure are different from the two. Sample MCG8 was very distinct and far away from the other gut samples, implying that its bacterial communities are unique to those of fungus-cultivating termites. Likewise, the mound samples (OTN2 and MTN5) and soils samples (OTS3 and MTS6) clustered together (Figs. 2a, 3), indicating that the bacterial community structure of the sample types was almost identical. The PCA (Fig. 2b), indicated that the relative abundances of Alistipes, Treponema, Bryobacter and Frankia are the major effect determining the overall variance of the genus compositions in the samples. Differences regarding the other genera detected in the samples are minimal. Alistipes abundance increases in the direction of the M. michaelseni (sample MTG4) and Odontotermes sp. (sample OTG1), while abundance of Treponema increases towards Microcerotermes sp. (sample MCG8) and Microtermes sp. (sample MIG7). Bryobacter and Frankia, however, increases towards the mounds (samples OTN2 and MTN5) and soil (samples OTS3 and MTS6).

Rarefaction curves indicating the observed number of operational taxonomic units (OTUs). The samples are marked by different colors. MCG8 Microcerotermes sp. gut homogenate, MIG7 Microtermes sp. gut homogenate, OTG1 Odontotermes sp. gut homogenate, MTG4 M. michaelseni gut homogenate, OTN2 soil from mound C of Odontotermes sp., MTN5 soil from mound D of M. michaelseni, MTS6 soil collected 3 m away from mound D, OTS3 soil collected 3 m away from mound C
Discussion
The profiling of bacterial communities indicated that the termite gut environment harbor bacterial communities that are unique and different to those of soil environment. There was a significant difference in the bacterial composition and community structure between the guts and savannah soils as indicated by the distribution of the major bacterial phyla (Bacteroidetes, Actinobacteria, Proteobacteria, Spirochaetes, Firmicutes, Fibrobacters and Acidobacteria) and genera (Fig. 1a; Additional file 2) across the samples. The gut samples of the fungus-feeders were dominated by members of Bacteroidetes, which were detected to a lesser extent in the mound and surrounding soil samples. Interestingly, the gut Bacteroidetes were mainly from members of the class Bacteroidia while those from soil samples belonged primarily to the Flavobacteria, Cytophagia, and Sphingobacteria classes. The genera Dysgonomonas, Parabacteroides, Paludibacter, Tannerella, Alistipes and BCf9-17 termite group were the major genera, but were rarely detected in the mounds and savannah soil (Additional file 2). These genera may represent the termite-specific bacterial lineages reported in other termites (Shinzato et al. 2007; Otani et al. 2014). The relatively low abundance of Bacteroidetes in the gut of the Microcerotermes sp. was evident. Members of Bacteroidetes are thought to be specialized in degradation of complex organic matter in the biosphere (Church 2008), implicating their roles in plant biomass degradation.
Moreover, the dominancy of Spirochaetes in the gut of Microcerotermes species was more pronounced than in the gut of fungus-feeders and soil samples. This demonstrates that members of Spirochaetes are host-associated and form an abundant group in the gut of wood-feeding termites (Köhler et al. 2012), where they are thought to be responsible for H2 production (Graber et al. 2004). Notably, Spirochaetes are rarely detected in fungus-feeding termites, ranging from almost absence in Macrotermes species (Otani et al. 2014), to approximate 10 % in Odontotermes species (Liu et al. 2013; Makonde et al. 2013a; Otani et al. 2014) and 22–36 % in Microtermes species (Makonde et al. 2013a; Otani et al. 2014). Again, Spirochaetes have been insignificantly detected within the mounds (Fall et al. 2007) and soils (Nacke et al. 2011).
The abundance of members of Actinobacteria within the mounds is noteworthy since the savannah soil was dominated by members of the group Acidobacteria. Importantly, are the genera Arthrobacter, Nocardioides, Streptomyces and Solirubrobacter that were not only relatively abundant in the mounds (Additional file 2), but are potential candidates for bioremediation (Shi et al. 2011). Besides, Actinobacteria have been demonstrated to produce antimicrobial compounds, which partly help prevent contamination in the farming of fungus gardens (Moriya et al. 2005), by inhibiting growth of some Pseudoxylaria and Termitomyces species (Visser et al. 2012).
The members of Proteobacteria were relative abundant in the mounds and savannah soil, however, they did not form the dominant group. This may be due to changes in soil properties, especially pH that has been shown to negatively influence the abundance of some Proteobacteria subdivisions (Nacke et al. 2011). Contrarily, members of the phylum Proteobacteria were represented by relative low abundances in the termite guts despite being known to have crucial role. For instance, members of δ-Proteobacteria such as Desulfovibrio spp. isolated from termite guts display high rates of H2-dependent oxygen reduction (Kuhnigk et al. 1996). The relative abundance of members belonging to the phylum Firmicutes, especially the class Clostridia was higher in the gut compared to the mounds and savannah soil, which contradicts the results of Fall et al. (2007) who reported relatively high abundance of Firmicutes in the mound belonging to soil-feeding termites. The relative high abundance of members of the phylum Fibrobacteres in Microcerotermes sp. may be associated with the degradation of plant-based cellulose (Qi et al. 2005), which is the main diet for these termites.
The bacterial composition between the mounds and savannah soil showed low variation, as demonstrated by weak grouping of samples (Fig. 3). Studies indicate that discrepancies in community structures between termite mounds and surrounding soil could be attributed by the trophic and mound construction behavior of the termites (Harry et al. 2001; Fall et al. 2007). The use of feces in building materials by termites may create an environment conducive to the development or the sustenance of particular microorganisms (Harry et al. 2001). Besides, the presence of clay may offer protection to some microorganisms (Harry et al. 2001) thereby increasing their survival. The tendency of increase in clay content in the mounds than the surrounding soil samples may demonstrate that soils in the mounds are enriched with clay particles due to the preferred selection of clay particles by termites (Manuwa 2009; Muwawa et al. 2014).
Conclusions
The findings from this study have revealed diverse bacterial communities in the gut and surrounding soil environments, the majority of which are uncharacterized. We also note tendency of increase in clay particles in the mounds, which may be one of the factor influencing the prevalence of bacterial communities in the investigated environments. The bacterial community composition and structure in gut and soil environments were different but that of mound and surrounding soil were negligible. Although the methodology applied in this study cannot help infer physiological roles for the uncultured bacteria, the data obtained contribute to understanding the bacterial diversity and community structure in the gut and surrounding soil environments.
Methods
Research authorization
The Research Authorization was obtained from National Commission for Science, Technology and Innovation (NACOSTI) in Kenya. Kenya Wildlife Services (KWS) and the National Environmental Management Authority (NEMA) of Kenya approved the research and provided permits and other necessary documents for sample collection in Kenya.
Site description and Sampling
The samples used in this study were collected from Thika district, Kenya (latitude 1°5′54.68″N, longitude 37°1′1.10″W) as described elsewhere (Makonde et al. 2013b). Termite mounds (C and D, approximately 2 km far apart were colonized by Odontotermes sp. [JQ247986] (OTG1) and Macrotermes michaelseni [JQ247993] (MTG4) together with Microtermes sp. [JQ247990] (MIG7), respectively) were excavated to a depth of 0.5–1.0 m. Next to mound D [about 2 m a way, was a colony of a wood feeding termite species (Microcerotermes sp. (MCG8)] that was also collected and analyzed. Termites (n = 200 workers and 50 soldiers) were sampled into sterile plastic boxes. Worker-caste termites were used in the experiments due to their foraging behaviour. The identity of the termites was confirmed by sequencing the mitochondrial cytochrome oxidase II gene in DNA extracted from the heads of soldiers (Makonde et al. 2013b) and comparing it to the sequences of previously identified specimens (Inward et al. 2007). In addition, soil samples (~40 g collected at ~5 cm depth) from the termite mounds C (OTN2) and D (MTN5) and their surrounding soils samples (OTS3 and MTS6, collected 3 m away from each mound, respectively), were included in the analyses.
Physico-chemical analyses
Standard physical soil analyses, which involved texture and bulk density analyses of the soil samples, were performed according to Ackerman et al. (2007). Particle size distribution was determined by the hydrometer method for determining the silt and clay fraction as described by Manuwa (2009). Determination of pH and inorganic nitrogen of the samples were performed according to the methods described by Muwawa (2014). Carbon content was determined by the WalkleyBlack method (Walkley and Black 1934) while nitrate concentrations was determined by colorimetric method (Muwawa 2014).
DNA extraction
The exterior surfaces of the termites were washed with 70 % ethanol and then rinsed with sterile distilled water. The guts were aseptically removed with forceps (Schmitt-Wagner et al. 2003). A total of 165 guts (approximately 1 g) of the Odontotermes sp. (OTG1) and Macrotermes michaelseni (MTG4) and 198 guts (approximately 1 g) of Microtermes sp. (MIG7) and 176 guts (approximately 1 g) of Microcerotermes sp. (MCG8) were put separately into sterile micro tubes containing 0.5 ml of TE buffer (10 mM Tris–HCl, 1 mM EDTA, pH 8.0). They were then homogenized using a sterile glass rod. The corresponding homogenates were then transferred into sterile tubes and used for total DNA extraction. The soil samples were homogenized separately and coarse stones and roots were removed. Subsequently, soil samples (~4 g) were used for total microbial DNA extraction. Total DNA extraction for all samples was performed using MoBio PowerMax Soil DNA isolation kit (MoBio Laboratories, Inc. CA, USA) according to the manufacturer’s protocol. DNA concentration was quantified by using a NanoDrop Spectrophotometer (NanoDrop Technologies, USA) as recommended by the manufacturer.
Amplification of 16S rRNA genes and 454-pyrosequencing
The V3–V5 region of the bacterial DNA was PCR amplified using the universal 16S rRNA primers 357F (5′-TACGGRAGGCAGCAG-3′) (Wilson et al. 1990) and 926R (5′-CCGTCAATTCMTTTGAGT-3′) (Muyzer et al. 1996). The bacterial primers were modified for 454 pyrosequencing by attaching an Adaptor sequence, a key and a unique 12 Nucleotide MID for multiplexing purposes (Caporaso et al. 2010). Each PCR reaction (50 μL) contained forward and reverse primers (10 μM, each), dNTP’s (10 mM each), Phusion GC buffer (Finzymes), Phusion high fidelity polymerase (0.5 U μL−1) and 25 ng of template DNA. Cycling conditions were as described by Nacke et al. (2011); however for the bacterial DNA annealing was at 66 °C for 45 s. Amplification was confirmed by separating 2 µL of the PCR product on a 1 % TAE agarose gel (40 mM Tris base, 20 mM glacial acetic acid, 1 mM EDTA, 1.5 % (w/v) agarose run for 1 h at 100 V. Later three independent PCR products per sample were pooled in equal amounts, separated on a gel and extracted using the peqGOLD gel extraction kit (PeqLab Biotechnologie GmbH, Erlangen, Germany). Quantification of the PCR products was performed by using the Nanodrop (NanoDrop Technologies, USA) method and a Qubit fluorometer mbH, (Invitrogen GmbH Karlsruhe, Germany) as recommended by the manufacturer. Sequencing of the PCR amplicons was done at the Göttingen Genomics Laboratory using Roche GS-FLX 454 pyrosequencer (Roche, Mannheim, Germany) as recommended in the instructions of the manufacturer for amplicon sequencing.
Pyrosequencing data analysis
Raw sequence reads were quality filtered according to the published recommendations (Huse et al. 2007) using the QIIME release 1.5.0 (Caporaso et al. 2010). The denoised sequences (≥300 bp) were then evaluated for potential chimeric sequences using UCHIME within the USEARCH package v.4.2.66 (Edgar 2010). A sequence identity cutoff of 97 % was used to pick OTUs from the quality filtered non-chimeric sequences. Representative OTUs were picked using the de novo OUT clustering (Rideout et al. 2014) with standard UCLUST method using the default settings as implemented in QIIME at 97 % similarity level. OTU alignment was done using the python implementation of the NAST algorithm, PyNAST (Caporaso et al. 2010). Taxonomy was assigned to the representative sequences from each cluster using BLASTn against the SILVA SSU Reference 119 database at default e-value threshold of 0.001 in QIIME (Quast et al. 2013) at dissimilarity levels of 3, 5 and 10 %. Rarefied datasets were generated with the multiple_rarefaction function in QIIME in order to remove sample heterogeneity before diversity assessment. Rarefaction curves and diversity indices were calculated and plotted for each sample using QIIME (Caporaso et al. 2010). To determine the amount of dissimilarity (distance) between any pair of bacterial communities, we used the UniFrac metric (Lozupone and Knight 2005; Lozupone et al. 2007) that incorporates the degree of divergence in the phylogenetic tree of OTUs into Principal coordinates analysis (PCoA). UniFrac distances are based on the fraction of branch length shared between two communities within a phylogenetic tree constructed from the 16S rRNA gene sequences from all communities being compared. A relatively small UniFrac distance implies that two communities are compositionally similar, harboring lineages sharing a common evolutionary history. In unweighted UniFrac, only the presence or absence of lineages is considered. We used the analysis of similarities (ANOSIM) (Clarke 1993; Fierer et al. 2010) through 1000 permutations to test for differences in community composition among the groups of samples. Additionally, the relative abundance of the genera were used in hierarchical clustering using the pearson correlation distance metric implemented in MultiExperimentViewer version 4.9.0 (MeV 4.9.0). The relative abundances (≥2.2 % at least in one sample) of the cultivated genera were also used for correlation analysis using Principal Components Analysis (PCA) as implemented in XLSTAT version 2015.4.01. All pyrosequencing-derived 16S rRNA gene sequences datasets were deposited in the GenBank under accession number SRP019764.
References
Ackerman IL, Wenceslau GT, Susan JR, Johannes L, Erick CM (2007) The impact of mound-building termites on surface soil properties in a secondary forest of Central Amazonia. Appl Soil Ecol 37:267–276
Ahmed BM, Nkunika POY, Sileshi WG, French JRJ, Nyeko P, Jain S (2011) Potential impact of climate change on termite distribution in Africa. Brit J Environ Climate Change 1:172–189
Brauman A (2000) Effect of gut transit and mound deposit on soil organic matter transformations in the soil feeding termite: a review. Eur J Soil Biol 36:117–125
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Meth 7:335–336
Church MJ (2008) Resource control of bacterial dynamics in the sea. In: Kirchman DL, Hoboken NJ (eds) Microbial ecology of the oceans, 2nd edn. Wiley, New York
Droege M, Hill B (2008) The Genome Sequencer FLX (TM) System longer reads, more applications, straight forward bioinformatics and more complete data sets. J Biotechnol 136:3–10
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Eggleton P (2000) Global patterns of termite diversity. In: Abe T, Bignell DE, Higashi M (eds) Termites: evolution, sociality, symbioses, ecology, vol 1. Kluwer Academic, Dordrecht, The Nethaerlands
Fall S, Nazaret S, Chotte JL, Brauman A (2004) Bacterial density and community structure associated with aggregate size fractions of soil-feeding termite mounds. Microb Ecol 28:191–199
Fall S, Hamelin J, Ndiaye F, Assigbetse K, Aragno M, Chotte JL, Brauman A (2007) Differences between bacterial communities in the gut of a soil-feeding termite (Cubitermes niokoloensis) and its mounds. Appl Environ Microbiol 73:5199–5208
Fisher M, Miller D, Brewster C, Husseneder C, Dickerman A (2007) Diversity of gut bacteria of Reticulitermes flavipes as examined by 16S rRNA gene sequencing and amplified rDNA restriction analysis. Curr Microbiol 55:254–259
Garnier-Sillam E, Toutain F, Renoux J (1988) Comparaison de I’influence de deux termitie`res (humivore et champignonniste) surla stabilite´ structurale des sols forestiers tropicaux. Pedobiology 32:89–97
Glenn TC (2011) Field guide to next generation DNA sequencers. Mol Ecol Resour 15:759–769
Graber JR, Leadbetter JR, Breznak JA (2004) Description of Treponema azotonutricium sp. nov. and Treponema primitia sp. nov., the first spirochetes isolated from termite guts. Appl Environ Microbiol 70:1315–1320
Harry M, Jusseaume N, Gambier B, Garnier-Sillam E (2001) Use of RAPD markers for the study of microbial community similarity from termite mounds and tropical soils. Soil Biol Biochem 33:417–427
Holt JA (1996) Mound-building termites and soil microbial biomass: an interaction influencing termite abundance. Insectes Soc 43:427–434
Holt JA, Lepage M (2000) Termites and soil properties. In: Abe T, Bignell DE, Higashi M (eds) Termites: evolution, sociality, symbioses, ecology, vol 2. Kluwer Academic, Dordrecht
Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM (2007) Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 8(7):R143
Huse SM, Welch DM, Morrison HG, Sogin ML (2010) Ironing out the wrinkles in the rare biosphere through improved OTU clustering. Environ Microbiol 12(7):1889–1898
Inward DJG, Vogler AP, Eggleton P (2007) A comprehensive phylogenetic analysis of termites (Isoptera) illuminates key aspects of their evolutionary biology. Mol Phylogenet Evol 44:953–967
Jones CG, Lawton JH, Shachak M (1997) Positive and negative effects of organisms as physical ecosystem engineers. Ecology 78:1946–1957
Köhler T, Dietrich C, Scheffrahn RH, Brune A (2012) High-resolution analysis of gut environment and bacterial microbiota reveals functional compartmentation in the gut of wood-feeding higher termites (Nasutitermes spp.). Appl Environ Microbiol 78:4691
Kuhnigk T, Branke J, Krekeler D, Cypionka H, König H (1996) A feasilble role of sulfate-reducing bacteria in the termite gut system. Appl Microbil 19:139–149
Liu N, Zhang L, Zhou H, Zhang M, Yan X, Wang Q, Long Y, Xie L, Wang S, Huang Y, Zhou Z (2013) Metagenomic insights into metabolic capacities of the gut microbiota in a fungus-cultivating termite (Odontotermes yunnanensis). PLoS One 8:e69184
Lozupone C, Knight R (2005) UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235
Lozupone CA, Hamady M, Kelley ST, Knight R (2007) Quantitative and qualitative (beta) diversity measures lead to different insights into factors that structure microbial communities. Appl Environ Microbiol 73(5):1576–1585
Mackenzie LM, Muigai AT, Osir EO, Lwande W, Keller M, Toledo G, Boga HI (2007) Bacterial diversity in intestinal tract of fungus-cultivating termite Macrotermes michaelseni (Sjöestedt). Afr J Biotechnol 6:658–667
Makonde HM, Boga HI, Osiemo Z, Mwirichia R, Mackenzie LM, Göker M, Klenk H-P (2013a) 16S-rRNA-based analysis of bacterial diversity in the gut of fungus-cultivating termites (Microtermes and Odontotermes species). Antonie Van Leeuwenhoek 104:869–883
Makonde HM, Boga HI, Osiemo Z, Mwirichia R, Stielow JB, Göker M, Klenk H-P (2013b) Diversity of Termitomyces associated with fungus-farming termites assessed by cultural and culture-independent methods. PLoS One 8(2):e56464
Manuwa SI (2009) Physico-chemical and dynamic properties of termite mound soil Relevant in sustainable food production. Afr Crop Sci Soc 9:356–369
Mathew GM, Ju Y-M, Lai C-Y, Mathew DC, Huang CC (2012) Microbial community analysis in the termite gut and fungus comb of Odontotermes formosanus: the implication of Bacillus as mutualists. FEMS Microbiol Ecol 79:504–517
Moriya S, Inoue T, Ohkuma M, Yaovapa T, Johjima T, Suwanarit P, Sangwani U, Vongkaluang C, Noparatnaraporn N, Kudo T (2005) Fungal community analysis of fungus gardens in termite nests. Microbes Environ 20:243–252
Muwawa EM (2014) Dynamics of nitrogen cycle microsymbionts and inorganic nutrients in Guts, Mounds and Soils of Odontotermes and Macrotermes species. Thesis, Jomo Kenyatta University of Agriculture and Technology
Muwawa EM, Makonde HM, Budambula NLM, Osiemo LZ, Boga HI (2014) Chemical properties associated with guts, soil and nest materials of Odontotermes and Macrotermes species from Kenya. J Biodiv Environ Sci 4(2):253–263
Muyzer G, Hottentrager S, Teske A, Wawer C (1996) Denaturing gradient gel electrophoresis of PCR-amplified 16S rDNA. A new molecular approach to analyze the genetic diversity of mixed microbial communities. In: Akkermans ADL, van Elsas JD, de Bruijn FJ (eds) Molecular microbial ecology manual. Kluwer Academic Publishing, Dordrecht
Nacke H, Thürmer A, Wollherr A, Will C, Hodac L, Herold N, Schöning I, Schrumpf M, Daniel R (2011) Pyrosequencing-based assessment of bacterial community structure along different management types in German Forest and Grassland Soils. PLoS One 6(2):e17000. doi:10.1371/journal.pone.0017000
Noirot C, Darlington JPEC (2000) Termite nest: architecture, regulation and defence. In: Abe T, Bignell DE, Higashi M (eds) Termites: evolution, sociality, symbioses, ecology, vol 2. Kluwer Academic, Dordrecht
Otani S, Mikaelyan A, Nobre T, Hansen LH, Kone N, Sørensen SJ, Aanen DK, Boomsma JJ, Brune A, Poulsen M (2014) Identifying the core microbial community in the gut of fungus-growing termites. Mol Ecol 23:4631–4644
Qi M, Nelson KE, Daugherty SC, Nelson WC, Hance IR, Morrison M, Forsberg CW (2005) Novel molecular features of the fibrolytic intestinal bacterium Fibrobacter intestinalis not shared with Fibrobacter succinogenes as determined by suppressive subtractive hybridization. J Bacterial 187:3739–3751
Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO (2013) The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucl Acids Res 41(DI):D590–D596
Rideout JR, He Y, Navas-Molina JA, Walters WA, Ursell LK, Gibbons SM, Chase J, McDonald D, Gonzalez A, Robbins-Pianka A, Clemente JC, Gilbert JA, Huse SM, Zhou H, Knight R, Caporaso JG (2014) Subsampled open-reference clustering creates consistent, comprehensive OTU definitions and scales to billions of sequences. Peer J 2:e545
Roose-Amsaleg C, Brygoo Y, Harry M (2004) Ascomycete diversity in soil-feeding termite nests and soils from a tropical rainforest. Environ Microbiol 6:462–469
Schmitt-Wagner D, Friedrich MW, Wagner B, Brune A (2003) Phylogenetic diversity, abundance, and axial distribution of bacteria in the intestinal tracts of two soil-feeding termites (Cubitermes spp). Appl Environ Microbiol 69:6007–6017
Shi W, Shukla G, Varma A (2011) Agricultural and ecological significance of soil enzymes: soil carbon sequestration and nutrient cycling soil enzymology. In: Varma A (ed) Springer Berlin Heidelberg
Shinzato N, Muramatsu M, Matsui T, Watanabe Y (2005) Molecular phylogenetic diversity of the bacterial community in the gut of the termite Coptotermes formosanus. Biosci Biotech Biochem 69:1145–1155
Shinzato N, Muramatsu M, Matsui T, Watanabe Y (2007) Phylogenetic analysis of the gut bacterial microflora of the fungus-growing termite Odontotermes formosanus. Biosci Biotech Biochem 71:906–915
Visser AA, Nobre T, Currie CR, Aanen DK, Poulsen M (2012) Exploring the potential for actinobacteria as defensive symbionts in fungus-growing termites. Microb Ecol 63(4):975–985
Walkley A, Black IA (1934) An examination of the Degtjareff method for determining soil organic matter and proposed modification of the chromic acid titration method. Soil Sci 37:29–38
Wilson KH, Blitchington RB, Greene RC (1990) Amplification of bacterial 16S ribosomal DNA with polymerase chain reaction. J Clin Microbiol 28:1942–1946
Authors’ contributions
HMM—conceived the idea, designed the study, performed the experiments and data analysis and drafted and wrote the manuscript. RM—participated in the data analysis, revised and critiqued the manuscript. HIB—designed the study and reviewed the manuscript. ZO—designed the study and critically revised the manuscript. HPK provided laboratory materials, participated in data analysis and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgements
This work was supported by a PhD scholarship from the Deutscher Akademischer Austauschdienst (DAAD) and a Grant (NCST/5/003/PG/224) from the Kenya National Commission for Science, Technology and Innovation (NACOSTI). Kenya Wildlife Services (KWS) and National Environmental Management Act (NEMA) provided permits for sample collection in Kenya.
Compliance with ethical guidelines
Competing interests The authors declare that they have no competing interest.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
a. Physical and chemical characteristics of the analyzed soil samples. b. Rarefaction curves indicating the observed number of operational taxonomic units (OTUs). (I) Indicates observed number of OTUs at 5 % genetic distance. (II) Indicates observed number of OTUs at 10 % genetic distance. Key: The samples are marked by different colors. MCG8, Microcerotermes sp. gut homogenate; MIG7, Microtermes sp. gut homogenate; OTG1, Odontotermes sp. gut homogenate; MTG4, M. michaelseni gut homogenate; OTN2, Soil from mound C of Odontotermes sp.; MTN5, Soil from mound D of M. michaelseni; MTS6, Soil collected 3m away from mound D; OTS3, Soil collected 3m away from mound C.
Additional file 2.
Relative abundances of bacterial groups across the samples. Key: MCG8, Microcerotermes sp. gut homogenate; MIG7, Microtermes sp. gut homogenate; OTG1, Odontotermes sp. gut homogenate; MTG4, M. michaelseni gut homogenate; OTN2, Soil from mound C of Odontotermes sp.; MTN5, Soil from mound D of M. michaelseni; MTS6, Soil collected 3m away from mound D; OTS3, Soil collected 3m away from mound C.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Makonde, H.M., Mwirichia, R., Osiemo, Z. et al. 454 Pyrosequencing-based assessment of bacterial diversity and community structure in termite guts, mounds and surrounding soils. SpringerPlus 4, 471 (2015). https://doi.org/10.1186/s40064-015-1262-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40064-015-1262-6