
Abstract
Bacterial and archaeal diversity of two alkaline Indian hot springs, Jakrem (Meghalaya) and Yumthang (Sikkim), were studied. Thirteen major bacterial phyla were identified of which Firmicutes, Chloroflexi and Thermi were dominant in Jakrem and Proteobacteria in Yumthang. The dominant genera were Clostridium, Chloroflexus and Meiothermus at Jakrem (water temperature 46 °C, pH 9) and Thiobacillus, Sulfuritalea at Yumthang (water temperature 39 °C, pH 8) hot springs. The four Euryarchaeota taxa that were observed in both the hot springs were Methanoculleus, Methanosaeta, Methanosarcina and Methanocorposculum. Elstera litoralis, Thiovirga sp., Turneriella sp. were observed for the first time in association with hot springs along with Tepidibacter sp., Ignavibacterium sp., Teribacillus sp. and Dechloromonas sp. Individual bacterial phyla were found to be specifically correlated with certain physico-chemical factors such as temperature, dissolved SiO2, elemental S, total sulphide, calcium concentrations in hot spring water. Bacterial reads involved in sulfur cycle were identified in both16S rRNA gene library and sulfur metabolism may play key physiological functions in this hot spring. Members within Desulfobacterales and Thermodesulfovibrionaceae were identified and hypothesized their role in regulating sulfur cycle. The presence of many taxonomically unsolved sequences in the 16S rRNA gene tag datasets from these hot springs could be a sign of novel microbe richness in these less known hot water bodies of Northeastern India.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The Himalayas represent a unique area of geothermal system associated with continent- continent colliding zone and the Himalayan geothermal belt (HGB) extends from the north-western part to the north-eastern part of India over a length of 1500 sq km (Chauhan 2015). Geological survey of India has identified 340 hot-water springs in India and classified them on the basis of their geo-tectonic setup (Craig et al. 2013; Ghelani et al. 2015). Thermal springs located in Sikkim and Meghalaya are an integral part of HGB which is located within the Indo-Burma range and hot springs in HGB have alkaline pH and unique geochemistry i.e. elevated Na, Ca and SiO2 (Siangbood and Ramanujam 2011; Rakshak et al. 2013). Compared to many studies on hot springs at lower elevations such as Yellowstone National Park (Kan et al. 2011), Kamchatka in Russia (Reigstad et al. 2010), Iceland (Mirete et al. 2011) Indonesia (Aditiawati et al. 2009), Tunisia (Sayeh et al. 2010) and north-eastern Australia (Weidler et al. 2007), very little is known about the microbial diversity of high elevation Himalayan hot springs. Hot springs present in high elevation HGB are less explored in terms of biotic components (Ghosh et al. 2003). Northeast Himalayan geothermal sub-province harbors large number of thermal springs and is an important geothermal energy source in India (Razdan et al. 2008). Only a few studies have been performed on the microbial ecology of hot springs from North-eastern India (Rakshak et al. 2013; Sherpa et al. 2013) and still it is assumed that comprehensive understanding on the microbial community structure in these hot springs are less known.
Thermophilic microbial diversity is reported from many alkaline hot water springs previously (Pagaling et al. 2012; Coman et al. 2013) and it is assumed that it influenced the evolution of life on earth (Doolittle 1999). Metagenomics studies from extreme environments led to the discovery of biocatalysts, secondary metabolites and bioactive compounds (Wong 2010; Barone et al. 2014). For example sulphur-cycling genes from sulphidic deep sea hydrothermal vent communities (Cao et al. 2014), H2 oxidation genes from H2-rich serpentinite hydrothermal vent communities (Brazelton et al. 2011), lipid oxidation genes in DSHV communities (He et al. 2013) and genes for ammonia- oxidation (amoA) in the Guaymas Basin (Baker et al. 2012) were identified by community metagenome analysis.
Hot springs harbor rich bacterial diversity that could be the source of commercially important products specially enzymes, sugars, compatible solutes and antibiotics (Satyanarayana et al. 2005). Bacterial diversity analysis of such extreme environments by culture independent approaches has grown in significance because of their diverse, unusual chemistry and the opportunity they provide to identify rare compounds and genes (Kuddus and Ramtekke 2012). Hot springs of Indian subcontinent offer striking and demanding platform for researchers from the globe due to the existence of unknown and untapped microbial communities. Most of the hot springs present in Northeast of India are present in unexplored environments and their diversity studies could be of great interest to facilitate various industrial, agricultural and medicinal applications and offer potential solutions to environmental concerns including the demand for bio-fuels (Urbieta et al. 2015).
The objective of this research was to study the microbial community composition and diversity in hot springs of the HGB (Yumthang and Jakrem) located in Northeast India and to understand the influence of the hot spring physico-chemical properties on the microbial diversity. These analyses were based on the hypothesis that the alkaline hot springs of HGB will host important microbial species for bio-prospecting and that specific ecological parameters might favor the species diversity and richness.
Materials and methods
Sampling
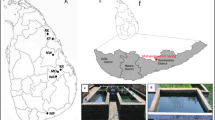
Microbial mat along with water and sediment was collected from Jakrem, Meghalaya (temp. 46 °C; elevation of 1450 m from MSL) and Yumthang, Sikkim (temp. 39 °C; elevation of 3564 m from MSL) hot springs of Northeast India. The geographical location of the sampling sites is shown in Fig. 1. The sample was collected from random sites using a hand trowel and pooled into sterile tubes, frozen in dry ice and transported to the laboratory for further analysis. The sediment/mat color, water temperature, pH and dissolved oxygen were recorded. XRD was performed to identify the mineralogy of collected solids (Huang et al. 2011). 500–1000 ml of the sample was filtered (0.22 µm) and split into several aliquots for analysis of various anion, cation and trace elements (sodium, calcium, potassium, magnesium, iron, arsenic, phosphorous, chloride, sulfur, nitrate, aluminium, silicon, dissolved silica and total sulphide) by Inductively Coupled Plasma Optical Emission Spectroscopy3r (ICP OES-7300, Perkin Elmer, USA).

Geographical location of the sampling sites
Community DNA extraction
Total DNA was extracted from 2 to 5 g of collected sample using Fast DNA™ Spin kit for soil (MP Biomedicals, USA). The DNA concentration was quantified using a microplate reader (SpectraMax 2E, Molecular Devices, USA). Agarose gel electrophoresis of the community DNA was carried out to check the quality of the DNA, stained with ethidium bromide and visualized under gel documentation system (G-Box, SynGene, USA).
Illumina sequencing
The V4 region of the 16S rRNA gene was amplified using 515F/806R (5′ GTGCCAGCMGCCGCGGTAA 3′; 5′ GGACTACHVGGGTWTCTAAT 3′) primers (Caporaso et al. 2012; Moonsamy et al. 2013). Cycling conditions for the PCR reaction were 98 °C for 30 s, followed by 30 cycles of 98 °C for 10 s and 72 °C for 30 s, with a 5 s elongation step at 72 °C followed by 4 °C hold. The paired-end sequencing (2 × 251 base pairs) was performed on an Illumina Mi-Seq platform at Scigenome India Pvt Ltd, Cochin, India.
Phylogenetic and statistical analysis
QIIME data analysis package was used for 16S rRNA data analysis. Quality check on raw sequences was performed, Chimeras were removed using UCHIME, pre-processed consensus V4 sequences were grouped into operational taxonomic units (OTUs) using the clustering program UCLUST at a similarity threshold of 0.97 (Edgar 2010). The representative sequence was finally aligned against Greengenes core set of sequences using PyNAST program and representative sequence for each OTU was classified using RDP classifier and Greengenes database. Sequences which are not classified were classified as unknown. The Shannon diversity indices were calculated and it represents OTU abundance, richness and evenness. The original sequencing output files of Jakrem and Yumthang hot spring have been deposited in the Sequence Read Archive (SRA) service of the National Centre for Biotechnology Information (NCBI) database under the accession numbers SRS932137 and SRS932073, respectively.
Canonical correlation analysis was performed to determine the correlation between microbial diversity and geochemical factors using PAST: Paleontological statistics software (Hammer et al. 2001). Pearson correlation between hot spring physico-chemical parameters and bacterial phyla were calculated using PASW statistics 18 (SPSS Inc, Chicago, USA).
Analysis of metabolic potential
The bioinformatics pipeline PICRUSt (Phylogenetic Investigation of Communities by Reconstruction of Unobserved States) (Langille et al. 2013) was used to address the functional potential of the microbes present in the hot springs. The closed-reference OTU picking protocol using QIIME 1.9.1 (Caporaso et al. 2012) was used and sequences were searched against the Greengenes database, version 13_05, taxonomically assigned using uclust with default parameters (Edgar 2010). The OTU table was created and analyzed by PICRUSt pipeline. The PICRUSt pipeline scans KEGG functional database and uses the OTU table of assigned taxa and their relative distribution to generate the relative abundance of functional categories. Data produced by the PICRUSt pipeline was statistically evaluated with the STAMP bioinformatics package (Parks and Beiko 2010).
Results
Geochemical analysis
Microbial mats in Jakrem were green in color whereas white microbial mats and grey macroscopic filaments observed at Yumthang. The temperature, pH, dissolved elemental composition and mineralogical data of the hot springs are described in Tables 1 and 2. The aqueous concentrations of cations such as sodium, calcium and potassium were highest in Jakrem hot spring, where as total sulphide concentration was high in Yumthang hot spring. XRD analysis shows that quartz is predominant in both hot spring sediments, whereas other minerals included are tridymite, wollastonite and kyanite (Table 2).
Analysis of bacterial diversity
Alpha diversity indices including Shannon, Chao1 and observed species metrices showed that sample YM1 is more diverse. The Shannon index was 2.10 and 1.96 for the Yumthang and Jakrem hot spring, respectively. Similarly the number of OTU and Chao index was higher in Yumthang as compared to Jakrem hot spring (Table 3). The metric calculation was performed using QIIME software. Rarefaction curve for Shannon metric indicated that the sample has reached near saturation for higher taxonomic levels (Fig. 2).

Rarefaction analysis of alpha diversity among JM1 and YM1. Shannon diversity matrix was used
Bacterial, archaeal diversity and community composition
The bacterial and archaeal diversity was assessed by illumina sequencing of V4 hyper-variable region of bacterial and archaeal small sub-unit rRNA genes with reference to these two unexplored hot springs of North- Eastern India. Jakrem 16S rRNA gene library comprised of 682,049 reads with 342.38 Mb data and average sequence length of 251 bp (Table 4). The G + C content was 57.87% and more than 90% sequence had a Phred score >=Q30. A total of 509,150 raw sequences (255. 59 Mb data) were obtained from Yumthang mat high throughput 16S rRNA library. These sequence reads clustered into Operational Taxonomic Units (OTUs) based on their sequence similarity using Uclust program (similarity cutoff = 0.97). A total of 881, 622 preprocessed reads were clustered into 1188 OTUs. Sample libraries ranged from 540,082 (JM1) to 341,540 (YM1) sequence reads (Table 4). Approximately, 561 OTUs were obtained from Jakrem, whereas 891 OTUs found in Yumthang library. Forty OTUs from Jakrem and 80 OTUs from Yumthang high throughput 16S metagenome library didn’t cluster with any of the previously known microbial classifications.
The archaeal community consisted of sequences closely related to Euryarchaeota and Crenarchaeota in both springs. Two identified order under the phyla Euryarchaeota were Methanomicrobiales and Methanosarcinales. Members under Crenarchaeota belonged to Marine Benthic Group (MBGA). Archaeal diversity did not show much variation between the two study sites.
The phylum level bacterial diversity identified in the high throughput 16S rRNA libraries from JM1 (Jakrem) and YM1 (Yumthang) hot springs is presented in Figs. 3 and 4. A total of 19 distinct phyla in the Yumthang 16S rRNA library dominated by Proteobacteria (83.68%), Bacteroidetes (10.93%) and Thermi (1.78%) whereas Jakrem 16S rRNA library accounted for 36.08% of Firmicutes, 34.18% of Chloroflexi and 25.44% of Thermi (Fig. 4).

Taxonomic classification of OTUs at phylum level (JM1—Jakrem samples, YM1—Yumthang samples)

Taxonomic classification of reads at phylum level (JM1—Jakrem samples, YM1—Yumthang samples)
Thermophilic OTUs belonging to the alpha-, beta-, delta- and gammaproteobacteria were detected in both the samples but with variable abundance. The four classes of Proteobacteria are represented by the families Rhodobacteraceae, Caulobacteraceae, Oxalobacteraceae, Comamonadaceae, Thiotrichaceae, Moraxellaceae, Halomonadaceae, Halothiobacillaceae, Desulfomicrobiaceae and Desulfobacteraceae. The most abundant taxonomic groups among Proteobacteria are Betaproteobacteria (82%) represented by dominated OTU 617 classified further under the order Rhodocyclales with other Proteobacterial sequences affiliated with Alphaproteobacteria (0.79%), Gammaproteobacteria (0.36%) and Deltaproteobacteria (0.33%) in the hot spring microbial mat of Yumthang. The other dominant OTUs within Betaproteobacteria were OTU 128, 1320 and 940 were classified under the order Burkholderiales, Thiobacterales and Hydrogenophilales respectively (Additional file 1: Table S1). Previous studies show the predominance Proteobacteria, particularly of Betaproteobacteria in a circumneutral hot spring from the Uzon Caldera, Kamchatka, Russia (Wemheuer et al. 2013; Chan et al. 2015) and other acidic thermal springs (Wilson et al. 2008). The information on distribution of genera is listed in Additional file 1: Table S2.
PICRUSt analysis
PICRUSt uses the OTU table of assigned taxa and their relative distribution to generate the relative abundance of functional categories based on sequenced genomes. On the basis of different KEGG functional gene ontology, the functions are arranged into five functional modules: metabolism, genetic information processing, environmental information processing, cellular processes, organismal systems. The fact that no differences were observed among both the samples via. PICRUSt analysis (Additional file 1: Figure S1) could be attributed to a low quantity and quality of annotated genomes that are related to the species observed in the hot spring samples. The increase of prevalence of genes encoding carbon fixation through photosynthesis in Jakrem can be explained by the high diversity of phototrophs (Additional file 1: Figure S1). Methane and Sulfur metabolism gene modules were also identified by PICRUSt pipeline accounting for the role of methane and sulfur in the regulation of geochemical cycle. The most abundant gene categories were purine, pyrimidine, arginine, proline, amino sugar and nucleotide sugar metabolites which reflects the basic requirements of microbial life.
Linking microbial community structure and hot spring geochemistry
Principal component analysis was used to analyze the major geochemical factors responsible for shaping the microbial community structure in the microbial mat of both the hot springs. Simultaneously, the microbial community and geochemical parameters from Manikaran hot spring, India (north western Himalayas) (Bhatia et al. 2015; Chandrasekharam et al. 2005) and Sungai Klah (SK) alkaline hot spring, Malaysia (Chan et al. 2015) were taken into account to overcome the problem of small sample size in the present study for two component analysis. PCA method showed that the community composition was significantly (p < 0.05) linked to temperature, dissolved SiO2, elemental S, total sulphide, calcium etc. (Fig. 5). Thermi and Chloroflexi were negatively correlated with phosphorous (p < 0.01). The correlation analysis showed that few dominant bacterial phyla were positively correlated with particular geochemical factor such as the Firmicutes with temperature, Ca, Cl, dissolved SiO2; Thermi and Chloroflexi with pH, Si and elemental S; Proteobacteria specifically correlates with total sulphide (Additional file 1: Table S3; Fig. 5).

Correlative relationship between dominant microbial phyla and geochemical factors (Temperature, pH, Na, Ca, Cl, dissolved SiO2, K, elemental S, total sulphide and Si)
Discussion
The taxonomic and metabolic compositions of microbial communities are both shaped and constrained by the characteristics of their local environment (Alsop et al. 2014). There are recent published studies focusing on few environmental parameters i.e. temperature and pH to determine the evolution of thermophilic microbial communities (Cowan et al. 2015). The present study is an attempt to investigate the factors determining the composition of thermophilic microbial communities by including multiple geochemical parameters. The present investigation characterized the archaeal and bacterial communities of two alkaline springs with similar temperature and pH, but different geochemical parameters. The archaeal and bacterial diversity varied across both the hot springs.
Tepidimonas taiwanensis and Tepidimonas fonticaldi are both thermophilic bacteria isolated from a hot spring of southern Thailand (Chen et al. 2006) and Antun hot spring from Taiwan (Chen et al. 2013). This study also identified few Tepidimonas OTUs in Jakrem 16S rRNA library. It is the third description worldwide in association with hot springs. Members of the genus Tepidimonas were shown to produce strong alkaline protease activity (Chen et al. 2006). One hundred thirty four V4 16S rDNA reads from the Yumthang library share more than 90% identity with members of Dechloromonas group those are poorly described from other thermal environments. There are only three type species reported from genus Dechloromonas i.e. Dechloromonas agitata (Achenbach et al. 2001); Dechloromonas denitrificans (Horn et al. 2005); Dechloromonas hortensis (Wolterink et al. 2005) till date. The microbes isolated from Gedongsongo hot spring (pH 6.0–7.0) Indonesia were closely related to Dechloromonas genera (Aminin et al. 2008). Dechloromonas represents a unique genus with a broad range of novel metabolic capabilities and bioremediative applicability. There are reports that Dechloromonas strains RCB and JJ can completely mineralize various mono-aromatic compounds including benzene to CO2 in the absence of O2 with nitrate as the electron acceptor (Coates et al. 2001). PICRUSt predicted the presence of certain xenobiotic metabolizing genes during the present investigations (Additional file 1: Figure S1).
Partial 16SrDNA sequences from Yumthang and Jakrem hot springs share more than 96% identity with the members of Thiobacillus sajanensis strain 4G. This is a new obligate autotrophic sulfur-oxidizing bacterium isolated from Khoito-Gol hydrogen-sulfide springs in Buryatia (Dul’tseva et al. 2006). The bacteria belonging to Thiobacillus group are mesophilic to moderately thermophilic and were found in thermal environments of Africa (Khavarpour et al. 2011), Italy (Pentecost et al. 2004), New Zealand (Jones and Renaut 2007) and Romania (Coman et al. 2013). V4 16S rDNA reads from Yumthang share identity with Sulfuritalea hydrogenivorans, a novel sulfur-oxidizing betaproteobacterium. Delta proteobacterial 16S rDNA sequences observed from Yumthang library are closely related to Desulfomicrobium baculatum, a gram-negative, motile, sulfate-reducing bacterium isolated from water-saturated manganese carbonate ore (Copeland et al. 2009).
The most dominant OTU within Chloroflexi was OTU 1166 classified under the genus Chloroflexus present in Jakrem were closely related to Chloroflexus aurantiacus strain J-10-fl which is also reported from microbial mats of various neutral to alkaline hot springs of the world viz. Romania (Coman et al. 2013); China (Lau and Pointing 2009; Lau et al. 2009); Thailand (Kanokratana et al. 2004; Portillo et al. 2009); Japan (Sekiguchi et al. 2003); USA (Costa et al. 2009); France (Gregoire et al. 2011a, b) or Russia (Bryanskaya et al. 2006). The Chloroflexus is a filamentous anoxygenic phototrophic bacterium. At taxonomic level, four Chloroflexi OTUs from Yumthang showed 87% similarity with Thermomarinilinea lacunofontalis strain SW7 isolated from the main hydrothermal vent of the Taketomi submarine hot spring field located on the southern part of Yaeyama Archipelago, Japan. These OTUs were encountered in similar hot springs from China (Lau and Pointing 2009; Lau et al. 2009), Thailand (Kanokratana et al. 2004; Portillo et al. 2009), Japan (Sekiguchi et al. 2003), USA (Costa et al. 2009), France (Gregoire et al. 2011a, b), Russia (Bryanskaya et al. 2006) and Romania (Coman et al. 2013).
Photosynthetic Cyanobacterial reads detected in Jakrem hot spring are closely related to filamentous Cyanobacterial Arthronema and Leptolyngbya genera. Some Oscillatoriales Cyanobacteria (especially Leptolyngbya sp.) were observed to dominate hot springs with temperature of about 55 °C (Roeselers et al. 2007; McGregor and Rasmussen 2008; Sompong et al. 2008). Their filamentous structures and polysaccharide matrices probably represent the backbone of the microbial mat (Van Gemerden 1993). The metabolic abilities of filamentous members of the Oscillatoria order can shed light on the role played by closely related clones encountered in the Jakrem spring mats. Oscillatoria species are known to dwell in anaerobic, sulfide-containing habitats (Castenholz 1989). At night they can grow by fermenting glycogen and other compounds produced during day time photosynthesis. Some species are also capable of growth in the dark via sulfur respiration (Richardson and Castenholz 1987; Teske and Stahl 2002). Elemental sulfur is produced as an intermediate of anoxygenic photosynthesis and is abundant in the Jakrem spring. Therefore, the anoxic conditions (3.0 mg/l dissolved oxygen along with high elemental sulfur concentrations) in the Jakrem spring represent an ideal habitat for members of the order Oscillatoriales.
The occurrence “Candidatus Xiphinematobacter,” in Jakrem 16S rRNA library is of interest as only few species reported till date. ‘Candidatus Xiphinematobacter’ gen. nov., along with three new candidate species, ‘Candidatus Xiphinematobacter americani’ sp. nov., ‘Candidatus Xiphinematobacter rivesi’ sp. nov. and ‘Candidatus Xiphinematobacter brevicolli’ sp. nov., were reported (Vandekerckhove et al. 2000). The non-Proteobacterial obligately methanotrophic bacterium Kam1 belonging to the Verrucomicrobia was recovered from an acidic hot spring in Kamchatka, Russia and is more thermoacidophilic than any other known methanotroph with optimal growth at 55 °C and pH 3.5 (Islam et al. 2008). The majority of the Firmicutes sequences in Jakrem 16S rRNA library were affiliated with the genus Clostridium. The most dominant OTU among the Firmicutes phyla is OTU 1235 (read = 154,229), having close sequence similarity with the Clostridium sp. TB10. Approximately, 25% of the total bacterial clone sequences in Jakrem library were closely related to Deinococcus—Meiothermus. The Bacteroidetes sequences observed in the Yumthang library and Flavobacterium represented the predominant genus of this phylum.
Other bacterial reads such as Elstera litoralis, Thiovirga sp., Turneriella sp. were observed for the first time in association with the hot spring. The presence of sequence reads from bacterial taxa Tepidibacter sp., Ignavibacterium sp., Teribacillus sp., Dechloromonas sp., could be the representatives of novel species within these genera. The genus Tepidibacter (Firmicutes) was proposed (Slobodkin et al. 2003) with three reported type species i.e. Tepidibacter thalassicus, Tepidibacter formicigenes and Tepidibacter mesophilus (Tan et al. 2012). Thus, the OTUs from Jakrem hot spring representing Tepidibacter (Firmicutes) possibly are the novel species with in this genus. The bacterium Ignavibacterium album was reported to be the only members of the bacterial phylum Chlorobi (Iino et al. 2010; Liu et al. 2012). Ignavibacterium was the only member of the phylum Chlorobi detected in these two hot springs. The presence of number of taxonomically unsolved sequence reads in both the hot spring high throughput 16S rRNA library is a sign of many novel microbes indigenous to these selected hot water springs. The findings of the molecular survey of these two so far not investigated sites showed that these hot springs are repository of unique bacterial and archaeal species in the biodiversity rich regions of the world.
Low diversity of archaea was found with genus-level OTUs corresponding to Methanocorposculum, Methanosaeta and Methanosarcina in both the springs and with an addition of Methanoculleus in Yumthang hot spring alone. Methanogenic microbes can use H2, acetate, formate, methanol, carbon monoxide and various methylamines as energy sources (Balch 1979). Methane could be playing a major role in geochemical cycling at Yumthang hot spring which is indicated by the presence of methanogenic archaeal sequence reads and methane metabolism genes in in silico analysis by PICRUSt (Additional file 1: Figure S1) in this environment. Newell et al. (2008) measured gas concentrations at various springs along the southern margin of the Tibetan plateau and observed variable CH4 concentrations from 110 to 500 ppm. The presence of methanogenic reads in Yumthang spring indicates methane may be derived from deeply buried carbon-bearing rocks or it could be produced in the near surface by organic matter fermentation (Whiticar 1986).
Thermophilic strains of Methanosarcina sp. have been reported with growth temperature of 55 °C (Zinder and Mah 1979) and Methanosaeta thermophila (Nozhevnikova and Yagodina 1983) from a hot spring (55 °C) of Kamchatka, Russia. However, the occurrence of Methanosarcina and Methanosaeta sequence reads in Yumthang hot spring 16S rRNA illumina library (water temperature 39 °C) suggests that there are members of both of these genera that grow at lower temperatures (similar to and below that found in Yumthang). There are reports on occurrence of methanogens including Methanomicrobiales and Methanosarcinales and relatives of Methanomassiliicoccus luminyensis from hot springs of Armenia (Hedlund et al. 2013). Our study revealed the dominance of Euryarchaeota over Crenarchaeota in these hot springs.
Thermi and Chloroflexi were negatively correlated with phosphorous (p < 0.01). The correlation analysis showed that few dominant bacterial phyla were positively correlated with particular geochemical factor such as the Firmicutes with temperature, Ca, Cl, dissolved SiO2; Thermi and Chloroflexi with pH, Si and elemental S; Proteobacteria specifically correlates with total sulphide (Additional file 1: Table S3; Fig. 5). Desulfomicrobiaceae and Desulfobacteraceae. Thermi and Chloroflexi were negatively correlated with phosphorous (p < 0.01). The correlation analysis showed that few dominant bacterial phyla were positively correlated with particular geochemical factor such as the Firmicutes with temperature, Ca, Cl, dissolved SiO2; Thermi and Chloroflexi with pH, Si and elemental S; Proteobacteria specifically correlates with total sulphide (Additional file 1: Table S3; Fig. 5). In the complete dataset, only few significant correlations were observed between hot spring geochemical factors and dominant phyla which may be attributed to small sample size in the present study.
As the two hot springs from Jakrem and Yumthang showed small differences in temperature and pH, the difference in bacterial community may be due to differences in aqueous geochemistry. Sulfur metabolism involves sulfur oxidation and sulfur reduction both. Because reads detected in 16S rRNA gene library were closest to the sulfate reducing microbes, we conclude that both hot spring communities preferably generate the reductive form of sulfur compounds (Fig. 6). The Yumthang hot spring had low dissolved oxygen (4 mg/l) and alkaline pH and these conditions appear to favour sulfate-reducing microorganisms. For example, the reads of Deltaproteobacterial order Desulfobacterales (Widdel 1987), Thermodesulfovibrionaceae family from Nitrospirae (Haouari et al. 2008) capable of reducing sulfates to sulfides were identified. The presence of large amount of total sulphide (0.16 mg/l) (Table 1) in Yumthang may be possible due to the presence of sulfate-reducing micro organisms.

KEGG based analysis of sulfur metabolism
Sulfate-reducing microorganisms are important in degrading organic matter under anoxic environments. In the Jakrem community, the organisms related to sulfate reduction identified from library those having closest hits to Desulfomicrobium apsheronum (Rozanova et al. 1988), Desulfomicrobium thermophilum (Thevenieau et al. 2007) and Desulfomicrobium sp. B1 (Chen et al. 2008). These sulfate-reducing microorganisms play an important role in energy production as well as the maintenance of the microbial community (Elshahed et al. 2003; Douglas and Douglas 2001). They depend on sulfate and elemental sulfur as the terminal electron acceptor during anaerobic metabolism. Reduction of sulfate to sulfite was activated by the formation of adenosine-5′-phosphosulfate (APS) and 3′-phosphoadenosine-5′-phosphosulfate (PAPS) which was further reduced to sulfite and hydrogen sulfide using the enzyme of phosphoadenosine phosphosulfate reductase and sulfite reductase. The abundance of δ-Proteobacteria and purple sulfur γ-Proteobacteria (80–83%) in the microbial mat bacterial diversity of the studied North east Indian hot spring samples was consistent with previous observations in mesophilic sulfide-rich springs (Elshahed et al. 2003).
Conclusions
This culture-independent study has provided an important insight into the potentially novel microbial diversity and community composition of two alkaline hot springs of Himalayan Geothermal Belt. Jakrem hot spring (39 °C) was abundant with the bacterial genera Clostridium, Chloroflexus and Meiothermus whereas Thiobacillus, Sulfuritalea was abundant in Yumthang (45 °C) hot springs. Bacterial phyla were found to be specifically correlated with physico-chemical factors of the hot spring water such as the Firmicutes with temperature, Ca, Cl, dissolved SiO2; Thermi and Chloroflexi with pH, Si and elemental S; Proteobacteria specifically correlates with total sulphide. Several bacterial genera with known industrial importance were identified from the hot spring metagenomes. The presence of high sulphide concentration as well as sulfate-reducing micro organisms in Yumthang indicates an active sulphur cycle in this hot spring. Many sequence reads not closely similar to any of the known species identified in the present study indicates the possibility of novel microbes in these habitats. Further studies with cultivation followed by physiological analysis of these important microbes would be required to determine their precise functional roles within these communities.
References
Achenbach LA, Michaelidou U, Bruce RA, Fryman J, Coates JD (2001) Dechloromonas agitata gen. nov., sp. nov. and Dechlorosoma suillum gen. nov., sp. nov., two novel environmentally dominant (per)chlorate-reducing bacteria and their phylogenetic position. Int J Syst Evol Microbiol 51:527–533
Aditiawati P, Yohandini H, Madayanti F, Akhmaloka (2009) Microbial diversity of acidic hot spring (kawah hujan B) in geothermal field of kamojang area, West Java-Indonesia. Open Microbiol J 3:58–66
Alsop EB, Boyd ES, Raymond J (2014) Merging metagenomics and geochemistry reveals environmental controls on biological diversity and evolution. BMC Ecol 14:16. doi:10.1186/1472-6785-14-16
Aminin ALN, Warganegara FM, Aditiawati P, Akhmaloka (2008) Simple enrichment and independent cultures to expand bacterial community analysis from Gedongsongo hot spring. J Biosci Bioeng 106(2):211–214
Baker BJ, Lesniewski RA, Dick GJ (2012) Genome-enabled transcriptomics reveals archaeal populations that drive nitrification in a deep-sea hydrothermal plume. ISME J 6:2269–2279
Balch WE, Fox GE, Margum LJ, Woese CR, Wolfe Wolfe RS (1979) Methanogens: reevaluation of a unique biological group. Microbiol Rev 43:260–296
Barone R, De Santi C, Esposito FP, Tedesco P, Galati F, Visone M, Scala AD, Pascale DD (2014) Marine metagenomics, a valuable tool for enzymes and bioactive compounds discovery. Front Mar Sci 1:1–6
Bhatia S, Batra N, Pathak A, Green SJ, Joshi A, Chauhan A (2015) Metagenomic evaluation of bacterial and archaeal diversity in the geothermal hot springs of Manikaran, India. Genome Announc 3(1):e01544-14
Brazelton WJ, Nelson B, Schrenk MO (2011) Metagenomic evidence for H2 oxidation and H2 production by serpentinite-hosted subsurface microbial communities. Front Microbiol 2:268. doi:10.3389/fmicb.2011.00268
Bryanskaya AV, Namsaraev ZB, Kalashnikova OM, Barkhutova DD, Namsaraev BB, Gorlenko VM (2006) Biogeochemical processes in the algal–bacterial mats of the Urinskii alkaline hot spring. Microbiology 75:611–620
Cao H, Wang Y, Lee OO, Zeng X, Shao Z, Qian P-Y (2014) Microbial sulfur cycle in two hydrothermal chimneys on the southwest Indian ridge. MBio 5:e00980-13
Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R (2012) Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6:1621–1624
Castenholz RW (1989) Order oscillatoriales. In: Williams ST, Sharpe ME, Holt JG (eds) Bergey’s manual of systematic bacteriology, vol 3. Williams & Wilkins, Baltimore, p 1771
Chan CS, Chan KG, Tay YL, Chua YH, Goh KM (2015) Diversity of thermophiles in a Malaysian hot spring determined using 16S rRNA and shotgun metagenome sequencing. Front Microbiol 6:177. doi:10.3389/fmicb.2015.00177
Chandrasekharam D, Alam MA, Minissale A (2005) Thermal discharges at Manikaran, Himachal Pradesh, India. Proceedings World Geothermal Congress. Antalya, Turkey 1–4
Chauhan BC (2015) Geothermal energy and earthquakes in Western Himalayas. IJSRSET 1(1):170–175
Chen TL, Chou YJ, Chen WM, Arun B, Young CC (2006) Tepidimonas taiwanensis sp. nov., a novel alkaline-protease-producing bacterium isolated from a hot spring. Extremophiles 10(1):35–40
Chen C, Ren N, Wang A, Yu Z, Lee DJ (2008) Microbial community of granules in expanded granular sludge bed reactor for simultaneous biological removal of sulfate, nitrate and lactate. Appl Microbiol Biotechnol 79(6):1071. doi:10.1007/s00253-008-1503-5
Chen WM, Huang HW, Chang JS, Han YL, Guo TR, Sheu SY (2013) Tepidimonas fonticaldi sp. nov., a slightly thermophilic betaproteobacterium isolated from a hot spring. Int J Syst Evol Microbiol 63(5):1810–1816
Coates JD, Chakraborty R, Lack JG, O’Connor SM, Cole KA, Bender KS, Achenbach LA (2001) Anaerobic benzene oxidation coupled to nitrate reduction in pure culture by two novel organisms. Nature 411:1039–1043
Coman C, Druga B, Hegedus A, Sicora C, Dragos N (2013) Archaeal and bacterial diversity in two hot spring microbial mats from a geothermal region in Romania. Extremophiles 17:523–534
Copeland A, Spring S, Goker M, Schneider S, Lapidus A, Del Rio TG, Tice H, Cheng JF, Chen F, Nolan M, Bruce D, Goodwin L, Pitluck S, Lvanova N, Mavrommatis K, Ovchinnikova G, Pati A, Chen A, Palaniappan K, Land M, Hauser L, Chang YJ, Jeffries CC, Meincke L, Sims D, Brettin T, Detter J, Han C, Chain P, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Kypides NC, Klenk HP, Lucas S (2009) Complete genome sequence of Desulfomicrobium baculatum type strain (XT). Stand Genomic Sci 1(1):29–37. doi:10.4056/sigs.13134
Costa KC, Navarro JB, Shock EL, Zhang CL, Soukup D, Hedlund BP (2009) Microbiology and geochemistry of great boiling and mud hot springs in the United States Great Basin. Extremophiles 13(3):447–459
Cowan DA, Ramond JB, Makhalanyane TP, De Maayer P (2015) Metagenomics of extreme environments. Curr Opin Microbiol 25:97–102
Craig J, Absar A, Bhat G, Cadel G, Hafiz M, Hakhoo N, Kashkari R, Moore J, Ricchiuto TE, Thurow J, Thusu B (2013) Hot springs and the geothermal energy potential of Jammu and Kashmir State, N.W. Himalaya, India. Earth Sci Rev 126:156–177
Doolittle WF (1999) Phylogenetic classification and the universal tree. Science 284:2124–2128
Douglas S, Douglas DD (2001) Structural and geomicrobiological characteristics of a microbial community from a cold sulfide spring. Geomicrobiology 18:401–422
Dul’tseva NM, Turova TP, Spiridonova EM, Kolganova TV, Osipov GA, Gorlenko VM (2006) Thiobacillus sajanensis sp. nov., a new obligately autotrophic sulfur-oxidizing bacterium isolated from Khoito-Gol hydrogen-sulfide springs, Buryatia. Mikrobiologiia 75(5):670–681
Edgar RC (2010) Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461
Elshahed MS, Senko JM, Najar FZ, Kenton SM, Roe BA, Dewers TA, Spear JR, Krumholz LR (2003) Bacterial diversity and sulfur cycling in a mesophilic sulfide-rich spring. Appl Environ Microbiol 69(9):5609–5621
Ghelani A, Patel R, Mangrola A, Dudhagara P (2015) Cultivation-independent comprehensive survey of bacterial diversity in Tulsi Shyam hot springs, India. Genom Data 4:54–56
Ghosh D, Bal B, Kashyap VK, Pal S (2003) Molecular phylogenetic exploration of bacterial diversity in a Bakreshwar (India) hot spring and culture of Shewanella-related thermophiles. Appl Environ Microbiol 69:4332–4336
Gregoire P, Bohli M, Cayol JL, Joseph M, Guasco S, Dubourg K, Cambar J, Michotey V, Bonin P, Fardeau ML, Ollivier B (2011a) Caldilinea tarbellica sp. nov., a filamentous, thermophilic, anaerobic bacterium isolated from a deep hot aquifer in the Aquitaine Basin. Int J Syst Evol Microbiol 61(6):1436–1441
Gregoire P, Fardeau ML, Joseph M, Guasco S, Hamaide F, Biasutti S, Michotey V, Bonin P, Ollivier B (2011b) Isolation and characterization of Thermanaerothrix daxensis gen nov., sp. nov., a thermophilic anaerobic bacterium pertaining to the phylum ‘‘Chloroflexi’’, isolated from a deep hot aquifer in the Aquitaine Basin. Syst Appl Microbiol 34(7):494–497
Hammer O, Harper DAT, Ryan PD (2001) PAST: Paleontological statistics software package for education and data analysis. Palaeontol Electron 4(1):9
Haouari O, Fardeau M, Cayol JL, Fauque G, Casiot C, Elbaz-Poulichet F, Hamdi M, Ollivier B (2008) Thermodesulfovibrio hydrogeniphilus sp. nov., a new thermophilic sulphate-reducing bacterium isolated from a Tunisian hot spring. Syst Appl Microbiol 31(1):38–42
He Y, Xiao X, Wang F (2013) Metagenome reveals potential microbial degradation of hydrocarbon coupled with sulfate reduction in an oil-immersed chimney from guaymas basin. Front Microbiol 4:148
Hedlund BP, Dodsworth JA, Cole JK, Panosyan HH (2013) An integrated study reveals diverse methanogens, Thaumarchaeota, and yet-uncultivated archaeal lineages in Armenian hot springs. Antonie Van Leeuwenhoek 104(1):71–82
Horn MA, Ihssen J, Matthies C, Schramm A, Acker G, Drake HL (2005) Dechloromonas denitrificans sp. nov., Flavobacterium denitrificans sp. nov., Paenibacillus anaericanus sp. nov. and Paenibacillus terrae strain MH72, N2O-producing bacteria isolated from the gut of the earthworm Aporrectodea caliginosa. Int J Syst Evol Microbiol 55:1255–1265
Huang Q, Dong CZ, Dong RM, Jiang H, Wang S, Wang G, Fanq B, Ding X, Niu L, Li X, Zhang C, Dong H (2011) Archaeal and bacterial diversity in hot springs on the Tibetan Plateau, China. Extremophiles 15(5):549–563
Iino T, Mori K, Uchino Y, Nakagawa T, Harayama S, Suzuki K (2010) Ignavibacterium album gen. nov., sp. nov., a moderately thermophilic anaerobic bacterium isolated from microbial mats at a terrestrial hot spring and proposal of Ignavibacteria classis nov., for a novel lineage at the periphery of green sulfur bacteria. Int J Syst Evol Microbiol 60(6):1376–1382
Islam T, Jensen S, Reigstad LJ, Larsen O, Nils-Kare Birkeland (2008) Methane oxidation at 55 °C and pH 2 by a thermoacidophilic bacterium belonging to the Verrucomicrobia phylum. PNAS 105(1):300–304
Jones B, Renaut RW (2007) Selective mineralization of microbes in Fe-rich precipitate (jarosite, hydrous ferric oxides) from acid hot springs in the Waiotapu geothermal area, North Island, New Zealand. Sed Geol 194:77–98
Kan J, Clingenpeel S, Macur RE, Inskeep WP, Lovalvo D, Varley J (2011) Archaea in Yellowstone Lake. ISME 5:1784–1795
Kanokratana P, Chanapan S, Pootanakit K, Eurwilaichitr L (2004) Diversity and abundance of bacteria and archaea in the Bor Khlueng hot spring in Thailand. J Basic Microbiol 44(6):430–444
Khavarpour M, Najafpour GD, Ghoreyshi AA, Jahanshahi M, Bambai B (2011) Enhanced Fe2+ oxidation by mixed culture originated from hot spring: application of response surface method. Afr J Biotechnol 10(19):3769–3783
Kuddus M, Ramtekke PW (2012) Recent developments in production and biotechnological applications of cold-active microbial proteases. Crit Rev Microbiol 38:380–388. doi:10.3109/1040841X.678477
Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Thurber RLV, Knight R, Beiko RG, Huttenhower C (2013) Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 31:814–821
Lau MCY, Pointing SB (2009) Vertical partitioning and expression of primary metabolic genes in a thermophilic microbial mat. Extremophiles 13(3):533–540
Lau MCY, Aitchison JC, Pointing SB (2009) Bacterial community composition in thermophilic microbial mats from five hot springs in central Tibet. Extremophiles 13(1):139–149
Liu Z, Frigaard NU, Vogl K, Iino T, Ohkuma M, Overmann J, Bryant DA (2012) Complete genome of Ignavibacterium album, a metabolically versatile, flagellated, facultative anaerobe from the phylum Chlorobi. Front Microbiol 29(3):185–199
McGregor GB, Rasmussen JP (2008) Cyanobacterial composition of microbial mats from an Australian thermal spring: a polyphasic evaluation. FEMS Microbiol Ecol 63(1):23–35
Mirete S, de Figueras CG, Gonzalez-Pastor JE (2011) Diversity of Archaea in Icelandic hot springs based on 16S rRNA and chaperonin genes. FEMS Microbiol Ecol 77:165–175
Moonsamy PV, Williams T, Bonella P, Holcomb CL, Höglund BN, Hillman G, Goodridge D, Turenchalk GS, Blake LA, Daigle DA, Simen BB, Hamilton A, May AP, Erlich HA (2013) High throughput HLA genotyping using 454 sequencing and the Fluidigm access array system for simplified amplicon library preparation. Tissue Antigens 81:141–149. doi:10.1111/tan.12071
Newell DL, Jessup MJ, Cottle JM, Hilton DR, Sharp ZD, Fischer TP (2008) Aqueous and isotope geochemistry of mineral springs along the southern margin of the Tibetan plateau: implications for fluid sources and regional degassing of CO2. Geochem Geophys Geosyst 9(8):1–20
Nozhevnikova AN, Yagodina TG (1983) A thermophilic acetate methane-producing bacterium. Microbiology 51:534–541
Pagaling E, Grant WD, Cowan DA, Jones BE, Ma Y, Ventosa A, Heaphy S (2012) Bacterial and archaeal diversity in two hot spring microbial mats from the geothermal region of Tengchong, China. Extremophiles 16(4):607–618
Parks DH, Beiko RG (2010) Identifying biologically relevant differences between metagenomic communities. Bioinformatics 26:715–721
Pentecost A, Coletta P (2004) A note on the travertines of Suio, Roccamonfina, with reference to their microbial communities and geochemical origins. Geol Rom 37:109–112
Portillo MC, Sririn V, Kanoksilapatham W, Gonzalez JM (2009) Differential microbial communities in hot spring mats from Western Thailand. Extremophiles 13:321–331
Rakshak K, Ravinder K, Nupur Srinivas TNR, Anil Kumar P (2013) Caldimonas meghalayensis sp. nov., a novel thermophilic betaproteobacterium isolated from a hot spring of Meghalaya in northeast India. Antonie van Leeuwenhoek 104(6):1217–1225
Razdan PN, Agarwal RK, Singh R (2008) Geothermal energy resources and its potential in India. E J Earth Sci India 1(1):30–42
Reigstad LJ, Jorgensen SL, Schleper C (2010) Diversity and abundance of Korarchaeota in terrestrial hot springs of Iceland and Kamchatka. ISME J 4:346–356
Richardson LL, Castenholz RW (1987) Enhanced survival of the cyanobacterium Oscillatoria terebriformis in darkness under anaerobic conditions. Appl Environ Microbiol 53:2151–2158
Roeselers G, Norris TB, Castenholz RW, Rysgaard S, Glud RN, Kuhl M, Muyzer G (2007) Diversity of phototrophic bacteria in microbial mats from Arctic hot springs (Greenland). Environ Microbiol 9(1):26–38
Rozanova EP, Nazina TN, Galushko AS (1988) Isolation of a new genus of sulfate-reducing bacteria and description of a new species of this genus, Desulfomicrobium apsheronum gen. nov., sp. nov. Mikrobiologiya 57:634–641
Satyanarayana T, Raghukumar C, Shivaji S (2005) Extremophilic microbes: diversity and perspectives. Curr Sci 89:78–90
Sayeh R, Birrien JL, Alain K, Barbier G, Hamdi M, Prieur D (2010) Microbial diversity in Tunisian geothermal springs as detected by molecular and culture-based approaches. Extremophiles 14(6):501–514
Sekiguchi Y, Yamada T, Hanada S, Ohashi A, Harada H, Kamagata Y (2003) Anaerolinea thermophila gen. nov., sp. nov. and Caldilinea aerophila gen. nov., sp. nov., novel filamentous thermophiles that represent a previously uncultured lineage of the domain Bacteria at the subphylum level. Int J Syst Evol Microbiol 53(6):1843–1851
Sherpa MT, Das S, Thakur N (2013) Physicochemical analysis of hot water springs of Sikkim—Polok Tatopani, Borong Tatopani and Reshi Tatopani. Recent Res Sci Technol 5(1):63–67
Siangbood H, Ramanujam P (2011) A report on thermophilic cyanophyta (cyanobacteria) from Jakrem Hotspring, Meghalaya. Int J Algae 13:178–185
Slobodkin AI, Tourova TP, Kostrikina NA, Chernyh NA, Bonch-Osmolovskaya EA, Jeanthon C, Jones BE (2003) Tepidibacter thalassicus gen. nov., sp. nov., a novel moderately thermophilic, anaerobic, fermentative bacterium from a deep-sea hydrothermal vent. Int J Syst Evol Microbiol 53:1131–1134
Sompong U, Anuntalabhochai S, Cutler RW, Castenholz RW, Peerapornpisal Y (2008) Morphological and phylogenetic diversity of cyanobacterial populations in six hot springs of Thailand. Sci Asia 34:153–162
Tan HQ, Wu XY, Zhang XQ, Wu M, Zhu XF (2012) Tepidibacter mesophilus sp. nov., a mesophilic fermentative anaerobe isolated from soil polluted by crude oil, and emended description of the genus Tepidibacter. Int J Syst Evol Microbiol 62:66–70
Teske A, Stahl DA (2002) Microbial mats and biofilms: evolution, structure, and function of fixed microbial communities. In: Stanley JT, Reysenbach AL (eds) Biodiversity of microbial life. Wiley, New York
Thevenieau F, Fardeau ML, Ollivier B, Joulian C, Baena S (2007) Desulfomicrobium thermophilum sp. nov., a novel thermophilic sulphate-reducing bacterium isolated from a terrestrial hot spring in Colombia. Extremophiles 11:295–303. doi:10.1007/s00792-006-0039-9
Urbieta MS, Donati ER, Chan KG, Shahar S, Sin LL, Gon KM (2015) Thermophiles in the genomic era: biodiversity, science, and applications. Biotechnol Adv. doi:10.1016/j.biotechadv.2015.04.007
Van Gemerden H (1993) Microbial mats: a joint venture. Mar Geol 113:3–25
Vandekerckhove TT, Willems A, Gillis M, Coomans A (2000) Occurrence of novel Verrucomicrobial species, endosymbiotic and associated with parthenogenesis in Xiphinema americanum-group species (Nematoda, Longidoridae). Int J Syst Evol Microbiol 50(6):2197–2205
Weidler GW, Dornmayr-Pfaffenhuemer M, Gerbl FW, Heinen W, Stan-Lotter H (2007) Communities of archaea and bacteria in a subsurface radioactive thermal spring in the Austrian Central Alps, and evidence of ammonia-oxidizing Crenarchaeota. Appl Environ Microbiol 73:259–270
Wemheuer B, Taube R, Akyol P, Wemheuer F, Daniel R (2013) Microbial diversity and biochemical potential encoded by thermal spring metagenomes derived from the Kamchatka Peninsula. Archaea. doi:10.1155/2013/136714
Whiticar MJ, Faber E, Schoell M (1986) Biogenic methane formation in marine and freshwater environments: CO2 reduction vs. acetate fermentation—Isotope evidence. Geochim Cosmochim Acta 50:693–709. doi:10.1016/0016-7037(86)90346-7
Widdel F (1987) New types of acetate-oxidizing, sulfate-reducing Desulfobacter species, D. hydrogenophilus sp. nov., D. latus sp. nov., and D. curvatus sp. nov. Arch Microbiol 148:286–291
Wilson MS, Siering PL, White CL, Hauser ME, Bartles AN (2008) Novel archaea and bacteria dominate stable microbial communities in North America’s Largest Hot Spring. Microb Ecol 56:292–305
Wolterink A, Kim S, Muusse M, Kim IS, Roholl PJ, van Ginkel CG, Stams AJ, Kengen SW (2005) Dechloromonas hortensis sp. nov. and strain ASK-1, two novel (per)chlorate-reducing bacteria, and taxonomic description of strain GR-1. Int J Syst Evol Microbiol 55:2063–2068
Wong D (2010) Applications of metagenomics for industrial bioproducts. In: Marco D (ed) Metagenomics: theory, methods and applications, 1st edn. Horizontal press, Norwich
Zinder SH, Mah RA (1979) Isolation and characterization of a thermophilic strain of Methanosarcina unable to use H2-CO2 for methanogenesis. Appl Environ Microbiol 38:996–1008
Authors’ contributions
AKP collected the samples, performed the experiments, analyzed the data, and wrote the paper. SDM helped in performing the experiments and analyzed the data. SPS and NSK critically analyzed the study and helped in drafting the article as well as edited the manuscript. All authors read and approved the final manuscript.
Acknowledgements
The authors are thankful to Department of Biotechnology, New Delhi for the infrastructural support in the form of State Biotech Hub to Mizoram University. The authors gratefully acknowledge Prof. Anil Gupta and his research group, Department of Molecular Biology and Genetic Engineering, GBPUAT, Pantnagar, Uttarakhand, India for their valuable inputs. The authors are grateful to Honorable Vice-Chancellor Kumaun University, Nainital for all administrative support.
Competing interests
The authors declare that they have no competing interests.
Declaration
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential competing interests.
Funding
This research was funded by a grant from SERB, Govt. of India, New Delhi vide Project No. SB/FT/LS-335/2012.
Author information
Authors and Affiliations
Corresponding author
Additional file
13568_2016_284_MOESM1_ESM.docx
Additional file 1: Table S1. Top fifteen OTU’s based on total read count number among the hot spring samples. Table S2. The distribution of genera in two samples. Table S3. Correlation matrix showing r values for Pearson’s correlation. Figure S1. Relative abundance of genes in the two hot spring samples (orange color: Yumthang hot spring, blue color: Jakrem hot spring) for selected functional KEGG pathways inferred from 16S rRNA gene data using PICRUSt.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Panda, A.K., Bisht, S.S., De Mandal, S. et al. Bacterial and archeal community composition in hot springs from Indo-Burma region, North-east India. AMB Expr 6, 111 (2016). https://doi.org/10.1186/s13568-016-0284-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13568-016-0284-y