
Abstract
In mammals, a basal level of autophagy, a self-eating cellular process, degrades cytosolic proteins and subcellular organelles in lysosomes to provide energy, recycles the cytoplasmic components, and regenerates cellular building blocks; thus, autophagy maintains cellular and tissue homeostasis in all eukaryotic cells. In general, adaptive autophagy increases when cells confront stressful conditions to improve the survival rate of the cells, while destructive autophagy is activated when the cellular stress is not manageable and elicits the regenerative capacity. Hypoxia-reoxygenation (H/R) injury and ischemia-reperfusion (I/R) injury initiate excessive autophagy and endoplasmic reticulum (ER) stress and consequently induce a string of damage in mammalian tissues or organs. Mesenchymal stem cell (MSC)-based therapy has yielded promising results in repairing H/R- or I/R-induced injury in various tissues. However, MSC transplantation in vivo must overcome the barriers including the low survival rate of transplanted stem cells, limited targeting capacity, and low grafting potency; therefore, much effort is needed to increase the survival and activity of MSCs in vivo. Modulating autophagy regulates the stemness and the anti-oxidative stress, anti-apoptosis, and pro-survival capacity of MSCs and can be applied to MSC-based therapy for repairing H/R- or I/R-induced cellular or tissue injury.
Similar content being viewed by others


Introduction
Although timely restoration of blood flow or oxygen has long been appreciated to reduce the injury in ischemic tissue by replenishment of cellular ATP, hypoxia-reoxygenation (H/R) and ischemia-reperfusion (I/R) injury, which aggravate the progression of organ dysfunction by reintroducing oxygen-rich blood to the impaired tissue [1], initiate autophagy and endoplasmic reticulum (ER) stress in mammals; consequently, they induce a string of damage in tissues or organs [2, 3]. During the ischemic period, ischemia or hypoxia is able to break down the endothelial cell barrier and increase vascular permeability and leakage by downregulating acti-adenylate cyclase activity and intracellular cAMP levels [4, 5]. The mitochondria then produce less ATP, leading to anaerobic metabolism, impairment of sodium-potassium pumps, and detachment of ribosomes [6]. In the ischemic stage, reactive oxygen species (ROS) can be produced from the xanthine oxidase system, NADPH oxidase system, mitochondrial electron transport chain, and uncoupled nitric oxide synthases and lead to cytokine cascades and pathological conditions [7, 8]. Reperfusion then upregulates the release of mitochondria-derived ROS [9], consequently disrupting normal ATP generation [10] and upregulating the mitochondrial permeability transition level [11, 12]. Excessive ROS cause oxidative stress, which promotes endothelial dysfunction, DNA damage, and local inflammatory responses [13]. Reperfusion predominantly triggers the release of intracellular damage-associated molecular pattern molecules (DAMPs) including high-mobility group box 1 and ATP and extracellular DAMPs including adenosine, thus promoting the accumulation of inflammatory cells including monocytes, dendritic cells, and granulocytes and activating the complement system [14,15,16]. Moreover, I/R also activates innate and adaptive immune responses including pattern-recognition receptors such as Toll-like receptors (TLRs) and inflammatory cell trafficking into the diseased organ (innate and adaptive immune activation) [17]. Moderate I/R simultaneously activate the recovery system after activation of autophagy, although autophagy does not clear all dysfunctional mitochondria during the intracellular perfusion stage [18]. If damage is severe, cell death may be induced via apoptotic or necrotic pathways, while a shorter duration of I/R may activate cell survival programs to control ROS generation and cell damage [19]. In addition, long-term I/R injury induces the generation of fibrotic scar tissue along with altered structure and reduced function in the injured site [20]. The current mechanisms of I/R- and H/R-induced injury should be further clarified for injury prevention throughout the entire process (Fig. 1).

The potential mechanisms of I/R- and H/R-induced injury in organs
Recently, the transplantation of mesenchymal stem cells (MSCs) has emerged as an effective strategy in regenerative medicine to repair injured organ function via regulating autophagy [21]. MSCs are multipotent cells that retain plasticity under standard culture conditions; these cells express high levels of makers CD105, CD73, and CD90 but rarely express CD45, CD34, CD14 or CD11b, CD79a or CD19, and HLA class II [22]. MSCs with high stemness and multipotency exhibit a high level of constitutive autophagy, while autophagy inhibitors prohibit proteolytic degradation and reduce the survival and differentiation of MSCs [23]. On the other hand, MSCs can undergo angiogenesis and secrete angiogenic factors in ischemic sites, thus promoting the recovery of I/R-injured tissues [24]. In addition, the immunosuppressive and pro-angiogenic effects of MSCs serve as important mechanisms for repairing tissue functions. MSCs not only inhibit macrophage-mediated inflammation and monocyte differentiation into immature dendritic cells [25] but also suppress the proliferation of B cells, antigen-primed T cells, and inactive natural killer cells [25,26,27,28]. Intriguingly, upregulating autophagy remarkably enhances the secretion of transforming growth factor (TGF)-β1 from MSCs and suppresses the proliferation of CD4+ T lymphocytes [29], whereas inhibiting autophagy reduces the responsiveness of T cells to mitogen interleukin (IL)-2 and increases the production of immunosuppressive prostaglandin E2 (PGE2) [30]. With such fascinating properties, MSCs may potentially be used for clinical applications in vessel repair and ischemic diseases and may be able to successfully treat ischemic tissues.
Autophagy is a type II programmed cell death [31] that can be activated to promote cell survival [32] or result in cell death [33] by stimulating various physiological and pathological factors. A basal level of autophagy occurs as a self-eating cellular process to degrade cytosolic proteins and subcellular organelles in lysosomes, recycle the cytoplasmic components, and regenerate cellular building blocks and energy, thus maintaining cellular and tissue homeostasis in all eukaryotic cells [34, 35]. Modulating autophagy regulates the stemness, differentiation, survival, and apoptosis of MSCs (Fig. 2). Although MSC transplantation has yielded promising results in treating H/R- or I/R-induced injury, the application of MSCs is limited by their low survival and low grafting potency. Therefore, further efforts to increase the survival and activity of MSCs in vitro and in vivo will shed light on the treatment of H/R- or I/R-induced injury in multiple organs.

Modulating autophagy regulates the stemness, differentiation, survival, and apoptosis of MSCs
In this review, we have comprehensively summarized the modulation of autophagy for improving the therapeutic effects of MSC transplantation for repairing H/R- and I/R-induced injury. Further studies of strategies to control autophagic flux will augment the application of MSCs in tissue engineering and regenerative medicine.
Autophagy process
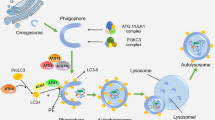
In mammals, three types of autophagy, including macroautophagy [36], microautophagy [37], and chaperone-mediated autophagy (CMA) [38], serve as a quality control mechanism for proteins and organelles [39, 40]. Multiple signaling pathways regulate autophagy; the mammalian target of rapamycin (mTOR) and AMP-activated protein kinase (AMPK) pathways are the two major pathways that regulate autophagy in mammals [41]. In addition, other pathways, such as the AKT/PKB, p52, and inositol pathways, also play critical roles in modulating autophagy [42,43,44]. Macroautophagy can be divided into five steps, including initiation, vesicle nucleation, vesicle elongation, fusion, and degradation [45]. mTOR complex 1 (mTORC1), which regulates the phosphorylation and inactivation of UNC51-like kinase (ULK) 1/2 and Atg-13, downregulates the assembly of the ULK protein complex, which consists of Ulk1, Atg-13, FIP200, and Atg-101. Stressful conditions induce the dissociation of mTORC1 from the ULK protein complex, thus initiating macroautophagy [46]. Then, the Beclin-1/class III phosphatidylinositol-3-kinase (PI3K) complex forms, which are coordinated by the interactions of multiple proteins such as Beclin-1, Atg14, p150, Ambra1, endophilin B1, vacuolar protein-sorting 34, B cell leukemia/lymphoma 2, and the UV irradiation resistance-associated tumor suppressor gene; this complex helps activate PI3K to produce phosphatidylinositol-3-phosphate for vesicle nucleation [47]. Several Atg proteins (Atg5, Atg8, Atg12, and Atg16) are assembled into two ubiquitin-like markers and are tethered to the membrane of the pre-autophagosomes to elongate the vesicle. At the final stage, the vesicle closes to generate a complete autophagosome that fuses with an endosome or lysosome to generate an autophagolysosome that recycles materials or organelles [48]. However, a feedback mechanism terminates macroautophagy, as reactivation of mTOR generates proto-lysosomal vesicles that extrude from the autophagolysosome and develop into mature lysosomes [49]. This feedback inhibits excessive activation of autophagy under stressful conditions, thereby providing the full complement of the autophagy machinery. Microautophagy is a process through which cytoplasmic contents enter the lysosome through an invagination or deformation of the lysosomal membrane; this process helps maintain the normal size of organelles and membrane homeostasis and enhances cellular survival under stressful conditions [50]. Finally, CMA is a uniquely selective form of autophagy that degrades a wide range of substrate proteins after transporting them one by one into the lysosome [51]. Then, the no-longer-useful cytoplasmic, long-lived proteins and organelles, including mitochondria, peroxisomes, Golgi apparatus, and ER, are degraded by activated lysosomal enzymes. Impaired lysosomal activity leads to the accumulation of autophagosomes, thus impairing cellular function and activating caspase-mediated cell death [52]. In general, we refer to macroautophagy as autophagy in this review because it is the major type of autophagy in most live cells and tissues (Fig. 3).

Macroautophagy serves as a quality control mechanism for proteins and organelles in mammals
Modulation of autophagy to resist against oxidative stress
Slight autophagy is rapidly activated to recycle cytoplasmic materials to initiate a survival mechanism by producing energy under stressful conditions, including irradiation, pathogen infection, hypoxia, and starvation [53]; thus, this process certainly inhibits apoptosis and prolongs the survival time of engrafted MSCs in vivo. However, excessive activation of autophagy results in increased cell death, while inhibition of excessive autophagy allows MSCs to withstand the stressful conditions [54].
Autophagic flux is activated as a self-defensive mechanism at the early stage of hydrogen peroxide (H2O2)-induced injury in MSCs and is abolished after long-term exposure (i.e., 6 h) to a stressful condition, which results in the upregulation of caspase-3 and caspase-6 [55]. Pretreatment or cotreatment with rapamycin significantly ameliorates H2O2-induced cell death, while 3-MA exacerbates H2O2-induced cell apoptosis [55]. Treatment with H2O2 upregulates autophagy and apoptosis of MSCs in a concentration- and time-dependent manner, and augmenting the autophagy JNK inhibitor reduces apoptosis and increases the MSC survival rate in response to oxidative stress [56]. Since SDF-1β activation consequently activates the stromal cell-derived factor-1 (SDF-1)/CXC chemokine receptor 4 (CXCR4) signaling axis to improve MSC survival under oxidative stress, Herberg et al. demonstrated that the overexpression of SDF-1β protects MSCs from H2O2-induced cell death via upregulation of autophagy and downregulation of caspase-3-mediated apoptosis, even though SDF-1β overexpression did not exert any effect on the proliferation rate of MSCs in vitro [57]. Furthermore, MSCs cultured on chitosan substrates show spheroid formation and upregulated expression of the autophagosomal marker LC3 II in a Ca2+-dependent manner, and these changes are accompanied by a higher survival rate in response to H2O2 than in cells grown as a monolayer culture. In addition, chitosan substrates also increase the expression levels of the upper autophagy signal pathway-related proteins, including Ulk1, Atg-13, and autophagy/Beclin-1 regulator 1, both before and after the addition of H2O2 compared with general culture conditions [58]. Jun et al. argued that melatonin increases the levels of manganese superoxide dismutase (MnSOD) and catalase while suppressing H2O2-induced ER stress and autophagy to reduce the apoptosis rate of MSCs via upregulation of prion protein (PrPC) [59].
Irradiation induces ROS accumulation-associated DNA damage in MSCs, while rapamycin attenuates the damage via inducing autophagy [60]. Long-term incubation with palmitate impairs adaptive autophagic flux and enhances the accumulation of p62 in MSCs, while rapamycin-pretreated MSCs resist apoptosis via the ROS-JNK/p38 MAPK signaling pathway [61]. MSCs confront an inflammatory microenvironment after incubation with TNF-α/cycloheximide and show a decreased survival rate; rapamycin significantly improves cell survival, and knockdown of Beclin-1 and 3-MA accelerates the apoptotic progress of MSCs [62]. Tetramethylpyrazine (a traditional Chinese medicine) and eicosapentaenoic acid (an omega-3 long-chain polyunsaturated fatty acid) protect MSCs against dexamethasone-induced cytotoxicity, autophagy, and apoptosis in an AMPK/mTOR pathway-dependent manner [63, 64]. Moreover, application of vitamin C dramatically protects MSCs from Sin-1-induced oxidative stress via downregulation of autophagy, thus increasing the survival rate and proliferation of MSCs exposed to oxidative stress [65].
Gene modification of MSCs may enable the overexpression or knockdown of the key candidate genes of autophagy at high efficiency and specificity, thus improving the resistance of MSCs against oxidative stress. Carbon monoxide protects MSCs from oxidative stress-induced death via improving autophagy and mitophagy in normal MSCs; however, depletion of LC3B and Beclin-1 in MSCs increases oxidative stress-induced injury, decreases intracellular ATP, reduces mitochondrial membrane potential, and increases mitochondrial ROS, and this deficiency cannot be reversed by preconditioning with carbon monoxide [66]. Overexpression of superoxide dismutase 3 markedly reduces ROS production and increases the survival rate of MSCs via upregulation of autophagy under serum-deprived conditions. Moreover, superoxide dismutase 3 protects MSCs against serum deprivation-induced injury via modulation of AMPK/sirtuin 1, activation of extracellular signal-regulated kinase, and promotion of Forkhead box O3a trafficking to the nucleus [67]. Overexpression of carnitine palmitoyltransferase 1 C (CPT1C) increases MSC survival under glucose deprivation via enhancement of autophagy, which promotes the accumulation of lipid droplets and ATP production [68]. In contrast, knockdown of Beclin-1 increases the survival rate of MSCs in a TNF-alpha- and IFN-gamma-induced in vitro inflammatory environment via inhibition of autophagy and enhancement of Bcl-2 expression via the ROS/MAPK1/3 pathway [69].
Hypoxia- and/or serum deprivation-induced injury
The in vitro oxygen concentration (approximately 20%) is much higher than that of the in vivo MSC niche; thus, engrafted MSCs promptly lose their activity and senesce to an apoptotic state after transplantation due to the stressful hypoxic environment [70]. However, hypoxia or serum deprivation may exert destructive or protective effects on MSCs according to the specific conditions (Table 1). An environment containing 0% O2 increases the autophagic flux via activation of the ERK1/2 pathway [71], and 1% O2 increases apoptosis via activation of the AMPK/mTOR pathway in MSCs; in contrast, 3-MA and the AMPK inhibitor compound C abolish hypoxia-induced apoptosis [72]. Ischemia induced by sodium azide (NaN3) and 2-deoxygluocose (2DG) blocks glycolysis and oxidative phosphorylation and consequently induces cellular autophagy and apoptosis in MSCs [73]. However, preconditioning with serum deprivation for 48 h reduces the energetic demand of MSCs and sends the cells into a quiescent state; then, the preconditioned MSCs maintain their viability and ATP levels via suppression of mTOR, activation of autophagy and enhancement of glycolysis even under exposure to 0.1% O2, and total glucose depletion for up to 14 days in vitro [74].
Transplantation of hypoxia-pretreated MSCs has become a primary method to improve the prognosis of various diseases via modulating autophagy. Preconditioning with 1% O2 enhances the proliferation of MSCs and increases the expression of hypoxia-inducible factor-1α (HIF-1α), apelin, Beclin-1, and LC3II/LC3I in a time-dependent manner [75]. Liu et al. argued that hypoxic pretreatment did not influence the cellular activities of MSCs under normal conditions but significantly improved cell activity and decreased the apoptosis rate of MSCs treated with lipopolysaccharide via enhancing HIF-1α-mediated autophagy [76]. Preconditioning with 0.5% O2 significantly upregulates autophagy to protect MSCs from hypoxia and serum deprivation (H/SD)-induced injury, while 3-MA and leptin-shRNA eliminate the protective effects via downregulating AMPK/mTOR-mediated autophagy [77]. Pezzi et al. argued that MSCs exposed to hypoxia reduce autophagy flux, which is accompanied by a smaller size, greater cellular complexity, and lower mitochondrial activity without altering oxidative stress in MSCs [78].
In addition to hypoxic pretreatment, some clinical drugs and growth factors can regulate autophagic flux in MSCs under hypoxic conditions. Atorvastatin (ATV) significantly activates autophagy via the AMPK/mTOR pathway to enhance MSC survival and reduce the apoptosis of MSCs under H/SD [79]. Pretreatment with macrophage migration inhibitory factor (MIF) protects MSCs against H/SD-induced apoptosis via upregulating autophagy and increasing the AMPK/mTOR pathway [80]. Because HIF-1α is a major regulator in mammals under hypoxic conditions, HIF-1α has been implicated as a major regulator of autophagy [81]. Overexpression of HIF-1α improves cell activity and reduces the apoptosis rate of MSCs, thus protecting MSCs from oxygen-glucose deprivation (OGD)-induced injury via activation of autophagy and inactivation of the PI3K/AKT/mTOR pathway [82]. However, sitagliptin (a dipeptidyl peptidase-4 inhibitor) effectively attenuates the apoptosis and autophagy of MSCs via regulating the Bcl-2/Beclin-1 pathway under an H/SD environment [83].
I/R-induced injury
MSC-based therapy is widely used to treat I/R-induced injury to organs such as the heart, brain, liver, retina, and limb, but the transplanted MSCs often fail to engraft within the targeted tissue or organ, in part due to the poor survival of MSCs in response to hypoxia, nutrient starvation, inflammatory reactions, and oxidative stress. Autophagy may regulate MSC activities in vivo and repair I/R-induced injury in different tissues (Table 2).
Acute myocardial infarction
AMI is a life-threatening disease induced by disrupted coronary blood flow that affects myocardial function; prolonged ischemia triggers molecular and structural changes in myocytes and leads to myocardial dysfunction. The incidence of AMI is 935 per 100,000 persons and remains the leading cause of mortality in the USA [84]. I/R-induced injury leads to heart failure in up to 40% of patients, and any delay in restoring coronary flow causes extensive cardiac cell death and results in ventricular remodeling in the injured heart tissue [85]. Although pharmacological agents or angioplasty is immediately used to reopen the occluded coronary artery after the onset of ischemic insult [86], the remaining inflammatory factors induced by AMI continue to impair myocytes in vivo. In vitro experiments have demonstrated that exposure to H/R upregulates autophagic flux in cultured rat neonatal cardiomyocytes [87] and H9C2 cells [88, 89]. Moreover, ischemia of differing severity leads to varied effects on autophagy modulation in cardiomyocytes under ischemic conditions. In cultured H9C2 cells, 2-deoxy-d-glucose induces mild ischemia and upregulates both autophagic flux and apoptosis, while sodium dithionate alone or with 2-deoxy-d-glucose induces apoptosis and necrosis without initiating autophagy [90].
Enhancing autophagy by hypoxia before MSC transplantation significantly improves the survival rate and the therapeutic effects of MSCs in repairing the ischemic myocardium, thus preserving heart function and eliminating fibrosis [21]. Injecting MSC-derived exosomes into rats that had undergone AMI markedly decreased the apoptosis rate of MSCs and reduced the myocardial infarct size via upregulating myocardial LC3B expression and activating the AMPK/mTOR and AKT/mTOR pathways [91]. However, Xiao et al. argued that the transplantation of MSCs and MSC-derived exosomes reduced the infarct size and improved the cardiac function via reducing apoptosis and autophagic flux in a mouse AMI model [92]. In addition, overexpressing Let-7b significantly decreased the apoptosis rate and increased the survival rate of MSCs by downregulating autophagy under high ROS conditions. Intramyocardial injection of these Let-7b-overexpressing MSCs significantly improved left ventricular function and microvessel density in AMI rats by directly targeting the caspase-3 signaling pathway [93]. The regulation of autophagy during I/R-induced myocardial injury appears to depend on the severity and duration of the ischemia. MSC transplantation regulates resident myocyte regeneration and heart function by regulating autophagy and ROS.
Cerebral I/R injury
Even though the expression level of autophagosomes in the mature brain under normal conditions and starvation is always lower than in other organs, such as the heart, liver, pancreas, kidney, and skeletal muscle [94], modulating autophagy is vital for maintaining homeostasis and protein quality control in neurons. Autophagy enhances brain tissue damage at the early stage after focal brain ischemia but indispensably enhances the reconstruction of injured neurons and brain function by removing harmful protein aggregates and damaged organelles during the postischemic phase [95]. However, the authentic function of autophagy is still under debate, as autophagy plays different roles in ischemic brain injury according to brain maturity, the brain region, ischemia severity, ischemia stage and the timing of therapeutic interventions. In early studies, upregulated autophagy was widely accepted as a deteriorative factor for repairing brain function. Nitatori et al. first reported that cerebral ischemia increased the number of cathepsin B-immunopositive lysosomes in neurons via upregulating autophagy [96], and neonatal ischemia induced autophagy-mediated cell death in the absence of apoptotic markers during reperfusion [97, 98]. In contrast, human brain tissue from adult patients after traumatic brain injury (TBI) exhibits higher levels of autophagosomes and LC3-II than control tissue [99], and the levels of p62/sequestosome 1 and Beclin-1 are significantly higher in cerebrospinal fluid from children with severe TBI than in normal control cerebrospinal fluid [100]. Coculture with MSCs protected the cortical neurons from cell death via alleviating apoptosis-inducing factor-mediated parthanatos, attenuating receptor-interacting protein kinase (RIP) 1- and 3-mediated necroptosis, and downregulating caspase-3-mediated apoptosis, but coculture exerted no effects on the regulation of autophagy [101]. However, mild hypothermia enhanced autophagic flux and preserved the hippocampal neural function via downregulating lysosomal number and acidity, the ratio of autolysosomes to autophagosomes, and expression of LAMP2 in OGD-induced injury in hippocampal neurons [102]. Overexpression of HIF-1α protects MSCs from OGD-induced injury by enhancing cell viability, inhibiting apoptosis, and activating autophagy via activating the AMPK pathway and inhibiting the mTOR pathway [103].
To attenuate autophagy-induced cell death, Yin et al. transplanted MSCs via the retro-orbital route, which significantly promoted neurite growth and regeneration in rats with spinal cord I/R injury [104]. MSC transplantation also decreased the expression levels of LC3, Beclin-1, and connexin 43, thus decreasing autophagy and apoptosis-related neuronal cell death in the hippocampus after TBI [105]. Transplanting MSCs into a middle cerebral artery occlusion (MCAO) rat model significantly reduced the expression of the autophagy-associated proteins LC3 and Beclin-1 and consequently enhanced behavioral recovery, reduced the cerebral infarction volume, and downregulated the rate of apoptosis in rat neurons via activation of the PI3K/AKT/mTOR signaling pathway [106]. In contrast, other studies demonstrated that MSC transplantation protects the brain from I/R-induced injury by augmenting autophagy in vivo. MSC transplantation significantly increases the brain-derived neurotrophic factor level and reduces the activation of mTOR pathway; thus, MSC transplantation subsequently increases autophagy and exerts neuroprotection, attenuating the behavioral deficits after hypoxia-ischemia-induced injury in the brain [107]. Transplantation of HIF-1α-MSCs into MCAO rats significantly reduced brain infarct volume and improved neurobehavioral outcome via activating autophagy, inhibiting pro-inflammatory cytokine generation, and enhancing neurotrophin secretion [103]. In addition, transplantation of exosomes from pigment epithelium-derived factor (PEDF)-overexpressing MSCs at 3 days prior to MCAO surgery also ameliorated cerebral I/R injury by upregulating autophagy and inhibiting neuronal apoptosis [108].
I/R-induced liver injury
Liver transplantation, partial hepatectomy, abdominal trauma, and hemorrhagic shock completely or partially interrupt liver blood flow and oxygen supply, followed by reperfusion to re-establish a new supply of blood and oxygen, but the reperfusion process aggravates the liver injury [109, 110]. Liver transplantation is the most effective treatment for patients with end-stage liver disease or patients with irreversible liver tumors [111]. However, long-term stimulation of excessive autophagy induced by severe ischemia in the liver destroys the structural organelles and leads to hepatocellular death. Strategies to improve the prognosis of I/R-induced liver injury via modulating autophagy are a current hot topic in experimental and clinical tests. Ischemic preconditioning (IPC) is divided into two steps, including cutting off hepatic inflow around the portal triad for 10–15 min and removing the clamp for another 10–15 min of reperfusion before liver transplantation [112]. Liu et al. recently reported that IPC and intestinal I/R prior to hepatic I/R confers protection against liver I/R-induced injury as demonstrated by the downregulation of serum aminotransferase and inflammatory cytokines via enhancing heme oxygenase-1 (HO-1)-mediated autophagy [113, 114]. Rapamycin-preconditioned MSCs enhanced the migration into the injured site and anti-inflammatory properties via regulation of the CXCR4/CXCL12 axis, thus affording additional protection against liver I/R injury [115]. In contrast, overexpression of HO-1 obviously increased the levels of autophagy-related proteins, ERK and p-ERK, but decreased the levels of mTOR and p-mTOR to increase autophagy in MSCs; then, HO-1-MSCs protected against I/R-induced injury in liver grafts following reduced-size liver transplantation [116]. Administering heat shock protein (HSP)-overexpressing MSCs significantly decreased the levels of serum aminotransferases and Suzuki scores and improved cell survival and histopathology in hepatic I/R rats by increasing autophagy and reducing apoptosis [117]. Activating autophagy prior to or after I/R improves the survival rate of MSCs in the liver and provides protective effects against liver I/R injury.
I/R-induced retinal injury
Hypoxia or ischemia in the retina may induce sight-threatening disorders, including central retinal artery occlusion, diabetic retinopathy, and glaucoma, and result in the degeneration and loss of retinal ganglion cells. Retinal ischemia induced by elevated intraocular pressure upregulates the expression of LC3BII at the early phase of recovery, and posttreatment with rapamycin exerts no protection against the retinal lesion but increases the apoptosis of retinal cells [118]. However, Russo et al. demonstrated that rapamycin consistently upregulates autophagy and improves retinal ganglion cell survival in the hypoxic-ischemic retina, thus providing a potential therapy in retinas associated with ischemic stress [119]. Intravitreal transplantation of MSCs eliminated ischemic damage in the retina by suppressing apoptosis, attenuating the inflammatory response and vascular permeability, and preserving autophagy in a rat model [120]. Thus, MSC transplantation can rescue vision loss and serve as an effective treatment of ischemia-associated retinal degeneration.
I/R-induced limb injury
Peripheral artery disease is characterized by pathophysiologic arterial narrowing and consequent stenosis, and accumulating evidence indicates that MSC transplantation can effectively restore blood flow in the ischemic limb. Volarevic et al. demonstrated that MSC transplantation could potentially serve as an alternative revascularization therapy for treating diabetic lower limb ischemia via pro-angiogenic effects [121]. Moreover, hypoxia-pretreated MSCs exhibited activated AMPK/mTOR signaling, enhanced autophagy, and improved pro-angiogenic effects, which improved the therapeutic effects in repairing lower limb function in ischemic diabetic rats [76]. Apelin significantly enhanced MSC survival in ischemic hindlimbs, restored hindlimb blood perfusion, and repaired limb functions by promoting autophagy, activating AMPK, and inhibiting mTOR in peripheral arterial disease [122]. In contrast, melatonin-pretreated MSCs enhanced neovascularization in a murine hindlimb ischemia model and promoted functional limb recovery via enhancing PrPC expression and suppressing autophagy [59].
Conclusions
In general, transplanted MSCs are confronted with a harsh environment in vivo, and the therapeutic application of MSCs is limited by their poor survival after engraftment. However, autophagy serves as a strict control to enhance the survival, engraftment, and paracrine effects of MSCs in vivo. Furthermore, this highly networked process offers protective effects against H/R- or I/R-induced injury via clearing away specific, no-longer-useful sets of macromolecules and providing additional energy. Targeting the potential mechanisms that modulate autophagy and thus control the anti-oxidative stress, anti-inflammation, and survival rate of MSCs will help improve the therapeutic effects of the engrafted MSCs in various I/R-induced injured tissues. Preconditioning MSCs by modulating autophagy via gene modification and specific activators or inhibitors may shed light on improving the regenerative capacity of MSCs in vitro and in vivo.
Abbreviations
- AMI:
-
Acute myocardial infarction
- AMPK:
-
AMP-activated protein kinase
- Atg:
-
Autophagy-related gene
- ATV:
-
Atorvastatin
- CMA:
-
Chaperone-mediated autophagy
- CPT1C:
-
Carnitine palmitoyltransferase 1 C
- CXCR4:
-
CXC chemokine receptor 4
- DAMPs:
-
Damage-associated molecular pattern molecules
- ER:
-
Endoplasmic reticulum
- H/R:
-
Hypoxia-reoxygenation
- H/SD:
-
Hypoxia and serum deprivation
- H2O2 :
-
Hydrogen peroxide
- HIF-1α:
-
Hypoxia-inducible factor-1α
- HO-1:
-
Heme oxygenase-1
- HSP:
-
Heat shock protein
- I/R:
-
Ischemia-reperfusion
- IPC:
-
Ischemic preconditioning
- MCAO:
-
Middle cerebral artery occlusion
- MIF:
-
Migration inhibitory factor
- MnSOD:
-
Manganese superoxide dismutase
- MSCs:
-
Mesenchymal stem cells
- mTOR:
-
Mammalian target of rapamycin
- mTORC1:
-
mTOR complex 1
- OGD:
-
Oxygen-glucose deprivation
- PEDF:
-
Pigment epithelium-derived factor
- PGE2:
-
Prostaglandin E2
- PI3K:
-
Phosphatidylinositol-3-kinase
- PrPC :
-
Prion protein
- RIP1:
-
Receptor-interacting protein kinase 1
- ROS:
-
Reactive oxygen species
- SDF-1:
-
Stromal cell-derived factor-1
- TBI:
-
Traumatic brain injury
- TGF:
-
Transforming growth factor
- TLRs:
-
Toll-like receptors
- ULK:
-
UNC51-like kinase
References
Reimer KA, Lowe JE, Rasmussen MM, Jennings RB. The wavefront phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation. 1977;56(5):786–94.
Ulamek-Koziol M, Kocki J, Bogucka-Kocka A, Petniak A, Gil-Kulik P, Januszewski S, et al. Dysregulation of autophagy, mitophagy, and apoptotic genes in the medial temporal lobe cortex in an ischemic model of Alzheimer’s disease. J Alzheimers Dis. 2016;54(1):113–21.
Ulamek-Koziol M, Kocki J, Bogucka-Kocka A, Januszewski S, Bogucki J, Czuczwar SJ, et al. Autophagy, mitophagy and apoptotic gene changes in the hippocampal CA1 area in a rat ischemic model of Alzheimer’s disease. Pharmacol Rep. 2017;69(6):1289–94.
Ogawa S, Gerlach H, Esposito C, Pasagian-Macaulay A, Brett J, Stern D. Hypoxia modulates the barrier and coagulant function of cultured bovine endothelium. Increased monolayer permeability and induction of procoagulant properties. J Clin Invest. 1990;85(4):1090–8.
Ogawa S, Koga S, Kuwabara K, Brett J, Morrow B, Morris SA, et al. Hypoxia-induced increased permeability of endothelial monolayers occurs through lowering of cellular cAMP levels. Am J Phys. 1992;262(3 Pt 1):C546–54.
Wu MY, Yiang GT, Liao WT, Tsai AP, Cheng YL, Cheng PW, et al. Current mechanistic concepts in ischemia and reperfusion injury. Cell Physiol Biochem. 2018;46(4):1650–67.
Lee MC, Velayutham M, Komatsu T, Hille R, Zweier JL. Measurement and characterization of superoxide generation from xanthine dehydrogenase: a redox-regulated pathway of radical generation in ischemic tissues. Biochemistry. 2014;53(41):6615–23.
Ma MW, Wang J, Zhang Q, Wang R, Dhandapani KM, Vadlamudi RK, et al. NADPH oxidase in brain injury and neurodegenerative disorders. Mol Neurodegener. 2017;12(1):7.
Loor G, Kondapalli J, Iwase H, Chandel NS, Waypa GB, Guzy RD, et al. Mitochondrial oxidant stress triggers cell death in simulated ischemia-reperfusion. Biochim Biophys Acta. 2011;1813(7):1382–94.
Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88(2):581–609.
Bernardi P, Rasola A, Forte M, Lippe G. The mitochondrial permeability transition pore: channel formation by F-ATP synthase, integration in signal transduction, and role in pathophysiology. Physiol Rev. 2015;95(4):1111–55.
Di Lisa F, Bernardi P. Modulation of mitochondrial permeability transition in ischemia-reperfusion injury of the heart. Advantages and limitations. Curr Med Chem. 2015;22(20):2480–7.
Ornellas FM, Ornellas DS, Martini SV, Castiglione RC, Ventura GM, Rocco PR, et al. Bone marrow-derived mononuclear cell therapy accelerates renal ischemia-reperfusion injury recovery by modulating inflammatory, antioxidant and apoptotic related molecules. Cell Physiol Biochem. 2017;41(5):1736–52.
Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–42.
Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11(9):785–97.
Atkinson C, He S, Morris K, Qiao F, Casey S, Goddard M, et al. Targeted complement inhibitors protect against posttransplant cardiac ischemia and reperfusion injury and reveal an important role for the alternative pathway of complement activation. J Immunol. 2010;185(11):7007–13.
Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. 2010;10(12):826–37.
Qin J, Zhou J, Dai X, Zhou H, Pan X, Wang X, et al. Short-term starvation attenuates liver ischemia-reperfusion injury (IRI) by Sirt1-autophagy signaling in mice. Am J Transl Res. 2016;8(8):3364–75.
McCully JD, Wakiyama H, Hsieh YJ, Jones M, Levitsky S. Differential contribution of necrosis and apoptosis in myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol. 2004;286(5):H1923–35.
White HD, Norris RM, Brown MA, Brandt PW, Whitlock RM, Wild CJ. Left ventricular end-systolic volume as the major determinant of survival after recovery from myocardial infarction. Circulation. 1987;76(1):44–51.
Zhang Z, Yang C, Shen M, Yang M, Jin Z, Ding L, et al. Autophagy mediates the beneficial effect of hypoxic preconditioning on bone marrow mesenchymal stem cells for the therapy of myocardial infarction. Stem Cell Res Ther. 2017;8(1):89.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8(4):315–7.
Oliver L, Hue E, Priault M, Vallette FM. Basal autophagy decreased during the differentiation of human adult mesenchymal stem cells. Stem Cells Dev. 2012;21(15):2779–88.
Hsiao ST, Lokmic Z, Peshavariya H, Abberton KM, Dusting GJ, Lim SY, et al. Hypoxic conditioning enhances the angiogenic paracrine activity of human adipose-derived stem cells. Stem Cells Dev. 2013;22(10):1614–23.
Gazdic M, Volarevic V, Arsenijevic N, Stojkovic M. Mesenchymal stem cells: a friend or foe in immune-mediated diseases. Stem Cell Rev. 2015;11(2):280–7.
Corcione A, Benvenuto F, Ferretti E, Giunti D, Cappiello V, Cazzanti F, et al. Human mesenchymal stem cells modulate B-cell functions. Blood. 2006;107(1):367–72.
Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005;105(7):2821–7.
Gazdic M, Simovic Markovic B, Vucicevic L, Nikolic T, Djonov V, Arsenijevic N, et al. Mesenchymal stem cells protect from acute liver injury by attenuating hepatotoxicity of liver natural killer T cells in an inducible nitric oxide synthase- and indoleamine 2,3-dioxygenase-dependent manner. J Tissue Eng Regen Med. 2018;12(2):e1173–85.
Gao L, Cen S, Wang P, Xie Z, Liu Z, Deng W, et al. Autophagy improves the immunosuppression of CD4+ T cells by mesenchymal stem cells through transforming growth factor-beta1. Stem Cells Transl Med. 2016;5(11):1496–505.
Volarevic V, Gazdic M, Simovic Markovic B, Jovicic N, Djonov V, Arsenijevic N. Mesenchymal stem cell-derived factors: Immuno-modulatory effects and therapeutic potential. Biofactors. 2017;43(5):633–44.
Uchiyama Y. Autophagic cell death and its execution by lysosomal cathepsins. Arch Histol Cytol. 2001;64(3):233–46.
Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26(7):1749–60.
Bursch W. The autophagosomal-lysosomal compartment in programmed cell death. Cell Death Differ. 2001;8(6):569–81.
Cuervo AM. Autophagy: many paths to the same end. Mol Cell Biochem. 2004;263(1):55–72.
Finn PF, Dice JF. Proteolytic and lipolytic responses to starvation. Nutrition. 2006;22(7–8):830–44.
Gomes LC, Scorrano L. Mitochondrial morphology in mitophagy and macroautophagy. Biochim Biophys Acta. 2013;1833(1):205–12.
Dubouloz F, Deloche O, Wanke V, Cameroni E, De Virgilio C. The TOR and EGO protein complexes orchestrate microautophagy in yeast. Mol Cell. 2005;19(1):15–26.
Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004;36(12):2435–44.
Mizushima N, Levine B. Autophagy in mammalian development and differentiation. Nat Cell Biol. 2010;12(9):823–30.
Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–41.
Srinivas V, Bohensky J, Shapiro IM. Autophagy: a new phase in the maturation of growth plate chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells Tissues Organs. 2009;189(1–4):88–92.
Yoo YM, Park YC. Streptozotocin-induced autophagy reduces intracellular insulin in insulinoma INS-1E cells. DNA Cell Biol. 2018;37(3):160–7.
Tan HY, Wang N, Man K, Tsao SW, Che CM, Feng Y. Autophagy-induced RelB/p52 activation mediates tumour-associated macrophage repolarisation and suppression of hepatocellular carcinoma by natural compound baicalin. Cell Death Dis. 2015;6:e1942.
Frej AD, Otto GP, Williams RS. Tipping the scales: lessons from simple model systems on inositol imbalance in neurological disorders. Eur J Cell Biol. 2017;96(2):154–63.
Kim KH, Lee MS. Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol. 2014;10(6):322–37.
Wong AS, Cheung ZH, Ip NY. Molecular machinery of macroautophagy and its deregulation in diseases. Biochim Biophys Acta. 2011;1812(11):1490–7.
Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. 2007;9(10):1102–9.
Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Res. 2010;20(7):748–62.
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010;465(7300):942–6.
Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012;69(7):1125–36.
Cuervo AM. Chaperone-mediated autophagy: selectivity pays off. Trends Endocrinol Metab. 2010;21(3):142–50.
Walls KC, Ghosh AP, Franklin AV, Klocke BJ, Ballestas M, Shacka JJ, et al. Lysosome dysfunction triggers Atg7-dependent neural apoptosis. J Biol Chem. 2010;285(14):10497–507.
Denton D, Xu T, Kumar S. Autophagy as a pro-death pathway. Immunol Cell Biol. 2015;93(1):35–42.
Molaei S, Roudkenar MH, Amiri F, Harati MD, Bahadori M, Jaleh F, et al. Down-regulation of the autophagy gene, ATG7, protects bone marrow-derived mesenchymal stem cells from stressful conditions. Blood Res. 2015;50(2):80–6.
Song C, Song C, Tong F. Autophagy induction is a survival response against oxidative stress in bone marrow-derived mesenchymal stromal cells. Cytotherapy. 2014;16(10):1361–70.
Liu GY, Jiang XX, Zhu X, He WY, Kuang YL, Ren K, et al. ROS activates JNK-mediated autophagy to counteract apoptosis in mouse mesenchymal stem cells in vitro. Acta Pharmacol Sin. 2015;36(12):1473–9.
Herberg S, Shi X, Johnson MH, Hamrick MW, Isales CM, Hill WD. Stromal cell-derived factor-1beta mediates cell survival through enhancing autophagy in bone marrow-derived mesenchymal stem cells. PLoS One. 2013;8(3):e58207.
Yang CM, Huang YJ, Hsu SH. Enhanced autophagy of adipose-derived stem cells grown on chitosan substrates. Biores Open Access. 2015;4(1):89–96.
Lee JH, Yoon YM, Han YS, Jung SK, Lee SH. Melatonin protects mesenchymal stem cells from autophagy-mediated death under ischaemic ER-stress conditions by increasing prion protein expression. Cell Prolif. 2019;52(2):e12545.
Hou J, Han ZP, Jing YY, Yang X, Zhang SS, Sun K, et al. Autophagy prevents irradiation injury and maintains stemness through decreasing ROS generation in mesenchymal stem cells. Cell Death Dis. 2013;4:e844.
Liu Y, Wang N, Zhang S, Liang Q. Autophagy protects bone marrow mesenchymal stem cells from palmitate-induced apoptosis through the ROSJNK/p38 MAPK signaling pathways. Mol Med Rep. 2018;18(2):1485–94.
Yang R, Ouyang Y, Li W, Wang P, Deng H, Song B, et al. Autophagy plays a protective role in tumor necrosis factor-alpha-induced apoptosis of bone marrow-derived mesenchymal stem cells. Stem Cells Dev. 2016;25(10):788–97.
Wang L, Zhang HY, Gao B, Shi J, Huang Q, Han YH, et al. Tetramethylpyrazine protects against glucocorticoid-induced apoptosis by promoting autophagy in mesenchymal stem cells and improves bone mass in glucocorticoid-induced osteoporosis rats. Stem Cells Dev. 2017;26(6):419–30.
Gao B, Han YH, Wang L, Lin YJ, Sun Z, Lu WG, et al. Eicosapentaenoic acid attenuates dexamethasome-induced apoptosis by inducing adaptive autophagy via GPR120 in murine bone marrow-derived mesenchymal stem cells. Cell Death Dis. 2016;7:e2235.
Sangani R, Periyasamy-Thandavan S, Pathania R, Ahmad S, Kutiyanawalla A, Kolhe R, et al. The crucial role of vitamin C and its transporter (SVCT2) in bone marrow stromal cell autophagy and apoptosis. Stem Cell Res. 2015;15(2):312–21.
Ghanta S, Tsoyi K, Liu X, Nakahira K, Ith B, Coronata AA, et al. Mesenchymal stromal cells deficient in autophagy proteins are susceptible to oxidative injury and mitochondrial dysfunction. Am J Respir Cell Mol Biol. 2017;56(3):300–9.
Agrahari G, Sah SK, Kim TY. Superoxide dismutase 3 protects mesenchymal stem cells through enhanced autophagy and regulation of FoxO3a trafficking. BMB Rep. 2018;51(7):344–9.
Roa-Mansergas X, Fado R, Atari M, Mir JF, Muley H, Serra D, et al. CPT1C promotes human mesenchymal stem cells survival under glucose deprivation through the modulation of autophagy. Sci Rep. 2018;8(1):6997.
Dang S, Yu ZM, Zhang CY, Zheng J, Li KL, Wu Y, et al. Autophagy promotes apoptosis of mesenchymal stem cells under inflammatory microenvironment. Stem Cell Res Ther. 2015;6:247.
Mastri M, Lin H, Lee T. Enhancing the efficacy of mesenchymal stem cell therapy. World J Stem Cells. 2014;6(2):82–93.
Wu J, Niu J, Li X, Li Y, Wang X, Lin J, et al. Hypoxia induces autophagy of bone marrow-derived mesenchymal stem cells via activation of ERK1/2. Cell Physiol Biochem. 2014;33(5):1467–74.
Zhang Z, Yang M, Wang Y, Wang L, Jin Z, Ding L, et al. Autophagy regulates the apoptosis of bone marrow-derived mesenchymal stem cells under hypoxic condition via AMP-activated protein kinase/mammalian target of rapamycin pathway. Cell Biol Int. 2016;40(6):671–85.
Tse KH, Kingham PJ, Novikov LN, Wiberg M. Adipose tissue and bone marrow-derived stem cells react similarly in an ischaemia-like microenvironment. J Tissue Eng Regen Med. 2012;6(6):473–85.
Moya A, Larochette N, Paquet J, Deschepper M, Bensidhoum M, Izzo V, et al. Quiescence preconditioned human multipotent stromal cells adopt a metabolic profile favorable for enhanced survival under ischemia. Stem Cells. 2017;35(1):181–96.
Li L, Li L, Zhang Z, Jiang Z. Hypoxia promotes bone marrow-derived mesenchymal stem cell proliferation through apelin/APJ/autophagy pathway. Acta Biochim Biophys Sin Shanghai. 2015;47(5):362–7.
Liu J, Hao H, Huang H, Tong C, Ti D, Dong L, et al. Hypoxia regulates the therapeutic potential of mesenchymal stem cells through enhanced autophagy. Int J Low Extrem Wounds. 2015;14(1):63–72.
Wang L, Hu X, Zhu W, Jiang Z, Zhou Y, Chen P, et al. Increased leptin by hypoxic-preconditioning promotes autophagy of mesenchymal stem cells and protects them from apoptosis. Sci China Life Sci. 2014;57(2):171–80.
Pezzi A, Amorin B, Laureano A, Valim V, Dahmer A, Zambonato B, et al. Effects of hypoxia in long-term in vitro expansion of human bone marrow derived mesenchymal stem cells. J Cell Biochem. 2017;118(10):3072–9.
Zhang Q, Yang YJ, Wang H, Dong QT, Wang TJ, Qian HY, et al. Autophagy activation: a novel mechanism of atorvastatin to protect mesenchymal stem cells from hypoxia and serum deprivation via AMP-activated protein kinase/mammalian target of rapamycin pathway. Stem Cells Dev. 2012;21(8):1321–32.
Xia W, Hou M. Macrophage migration inhibitory factor induces autophagy to resist hypoxia/serum deprivation-induced apoptosis via the AMP-activated protein kinase/mammalian target of rapamycin signaling pathway. Mol Med Rep. 2016;13(3):2619–26.
Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283(16):10892–903.
Lv B, Hua T, Li F, Han J, Fang J, Xu L, et al. Hypoxia-inducible factor 1 alpha protects mesenchymal stem cells against oxygen-glucose deprivation-induced injury via autophagy induction and PI3K/AKT/mTOR signaling pathway. Am J Transl Res. 2017;9(5):2492–9.
Wang XM, Yang YJ, Wu YJ, Zhang Q, Qian HY. Attenuating hypoxia-induced apoptosis and autophagy of mesenchymal stem cells: the potential of sitagliptin in stem cell-based therapy. Cell Physiol Biochem. 2015;37(5):1914–26.
Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, Ferguson TB, Ford E, Furie K, Gillespie C, Go A, Greenlund K, Haase N, Hailpern S, Ho PM, Howard V, Kissela B, Kittner S, Lackland D, Lisabeth L, Marelli A, MM MD, Meigs J, Mozaffarian D, Mussolino M, Nichol G, Roger VL, Rosamond W, Sacco R, Sorlie P, Thom T, Wasserthiel-Smoller S, Wong ND, Wylie-Rosett J. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010;121(7):e46–e215.
Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101(25):2981–8.
Hochman JS, Choo H. Limitation of myocardial infarct expansion by reperfusion independent of myocardial salvage. Circulation. 1987;75(1):299–306.
Dosenko VE, Nagibin VS, Tumanovskaya LV, Moibenko AA, Vaage J. Postconditioning prevents apoptotic necrotic and autophagic cardiomyocyte cell death in culture. Fiziol Zh. 2005;51(3):12–7.
Gurusamy N, Lekli I, Gorbunov NV, Gherghiceanu M, Popescu LM, Das DK. Cardioprotection by adaptation to ischaemia augments autophagy in association with BAG-1 protein. J Cell Mol Med. 2009;13(2):373–87.
Gurusamy N, Lekli I, Mukherjee S, Ray D, Ahsan MK, Gherghiceanu M, et al. Cardioprotection by resveratrol: a novel mechanism via autophagy involving the mTORC2 pathway. Cardiovasc Res. 2010;86(1):103–12.
Loos B, Genade S, Ellis B, Lochner A, Engelbrecht AM. At the core of survival: autophagy delays the onset of both apoptotic and necrotic cell death in a model of ischemic cell injury. Exp Cell Res. 2011;317(10):1437–53.
Liu L, Jin X, Hu CF, Li R, Zhou Z, Shen CX. Exosomes derived from mesenchymal stem cells rescue myocardial ischaemia/reperfusion injury by inducing cardiomyocyte autophagy via AMPK and Akt pathways. Cell Physiol Biochem. 2017;43(1):52–68.
Xiao C, Wang K, Xu Y, Hu H, Zhang N, Wang Y, et al. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of mir-125b. Circ Res. 2018.
Ham O, Lee SY, Lee CY, Park JH, Lee J, Seo HH, et al. let-7b suppresses apoptosis and autophagy of human mesenchymal stem cells transplanted into ischemia/reperfusion injured heart 7by targeting caspase-3. Stem Cell Res Ther. 2015;6:147.
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15(3):1101–11.
Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115(1):68–78.
Xiao C, Wang K, Xu Y, Hu H, Zhang N, Wang Y, et al. Transplanted mesenchymal stem cells reduce autophagic flux in infarcted hearts via the exosomal transfer of mir-125b. Circ Res. 2018;123(5):564-78.
Puyal J, Clarke PG. Targeting autophagy to prevent neonatal stroke damage. Autophagy. 2009;5(7):1060–1.
Puyal J, Vaslin A, Mottier V, Clarke PG. Postischemic treatment of neonatal cerebral ischemia should target autophagy. Ann Neurol. 2009;66(3):378–89.
Clark RS, Bayir H, Chu CT, Alber SM, Kochanek PM, Watkins SC. Autophagy is increased in mice after traumatic brain injury and is detectable in human brain after trauma and critical illness. Autophagy. 2008;4(1):88–90.
Au AK, Aneja RK, Bayir H, Bell MJ, Janesko-Feldman K, Kochanek PM, et al. Autophagy biomarkers Beclin 1 and p62 are increased in cerebrospinal fluid after traumatic brain injury. Neurocrit Care. 2017;26(3):348–55.
Kong D, Zhu J, Liu Q, Jiang Y, Xu L, Luo N, et al. Mesenchymal stem cells protect neurons against hypoxic-ischemic injury via inhibiting parthanatos, necroptosis, and apoptosis, but not autophagy. Cell Mol Neurobiol. 2017;37(2):303–13.
Zhou T, Liang L, Liang Y, Yu T, Zeng C, Jiang L. Mild hypothermia protects hippocampal neurons against oxygen-glucose deprivation/reperfusion-induced injury by improving lysosomal function and autophagic flux. Exp Cell Res. 2017;358(2):147–60.
Lv B, Li F, Han J, Fang J, Xu L, Sun C, et al. Hif-1alpha overexpression improves transplanted bone mesenchymal stem cells survival in rat MCAO stroke model. Front Mol Neurosci. 2017;10:80.
Yin F, Meng C, Lu R, Li L, Zhang Y, Chen H, et al. Bone marrow mesenchymal stem cells repair spinal cord ischemia/reperfusion injury by promoting axonal growth and anti-autophagy. Neural Regen Res. 2014;9(18):1665–71.
Sun L, Gao J, Zhao M, Jing X, Cui Y, Xu X, et al. The effects of BMSCs transplantation on autophagy by CX43 in the hippocampus following traumatic brain injury in rats. Neurol Sci. 2014;35(5):677–82.
He H, Zeng Q, Huang G, Lin Y, Lin H, Liu W, et al. Bone marrow mesenchymal stem cell transplantation exerts neuroprotective effects following cerebral ischemia/reperfusion injury by inhibiting autophagy via the PI3K/Akt pathway. Brain Res. 2019;1707:124-32.
Zheng Z, Zhang L, Qu Y, Xiao G, Li S, Bao S, et al. Mesenchymal stem cells protect against hypoxia-ischemia brain damage by enhancing autophagy through brain derived neurotrophic factor/mammalin target of rapamycin signaling pathway. Stem Cells. 2018;36(7):1109–21.
Huang X, Ding J, Li Y, Liu W, Ji J, Wang H, et al. Exosomes derived from PEDF modified adipose-derived mesenchymal stem cells ameliorate cerebral ischemia-reperfusion injury by regulation of autophagy and apoptosis. Exp Cell Res. 2018;371(1):269–77.
Velmahos GC, Toutouzas K, Radin R, Chan L, Rhee P, Tillou A, et al. High success with nonoperative management of blunt hepatic trauma: the liver is a sturdy organ. Arch Surg. 2003;138(5):475–80 discussion 480-471.
Hassoun HT, Kone BC, Mercer DW, Moody FG, Weisbrodt NW, Moore FA. Post-injury multiple organ failure: the role of the gut. Shock. 2001;15(1):1–10.
Wertheim JA, Petrowsky H, Saab S, Kupiec-Weglinski JW, Busuttil RW. Major challenges limiting liver transplantation in the United States. Am J Transplant. 2011;11(9):1773–84.
Selzner N, Rudiger H, Graf R, Clavien PA. Protective strategies against ischemic injury of the liver. Gastroenterology. 2003;125(3):917–36.
Liu A, Fang H, Wei W, Dirsch O, Dahmen U. Ischemic preconditioning protects against liver ischemia/reperfusion injury via heme oxygenase-1-mediated autophagy. Crit Care Med. 2014;42(12):e762–71.
Kageyama S, Hata K, Tanaka H, Hirao H, Kubota T, Okamura Y, et al. Intestinal ischemic preconditioning ameliorates hepatic ischemia/reperfusion injury in rats: role of heme oxygenase 1 in the second window of protection. Liver Transpl. 2015;21(1):112–22.
Zheng J, Li H, He L, Huang Y, Cai J, Chen L, et al. Preconditioning of umbilical cord-derived mesenchymal stem cells by rapamycin increases cell migration and ameliorates liver ischaemia/reperfusion injury in mice via the CXCR4/CXCL12 axis. Cell Prolif. 2019;52(2):e12546.
Wang R, Shen Z, Yang L, Yin M, Zheng W, Wu B, et al. Protective effects of heme oxygenase-1-transduced bone marrow-derived mesenchymal stem cells on reduced-size liver transplantation: role of autophagy regulated by the ERK/mTOR signaling pathway. Int J Mol Med. 2017;40(5):1537–48.
Qiao PF, Yao L, Zhang XC, Li GD, Wu DQ. Heat shock pretreatment improves stem cell repair following ischemia-reperfusion injury via autophagy. World J Gastroenterol. 2015;21(45):12822–34.
Produit-Zengaffinen N, Pournaras CJ, Schorderet DF. Autophagy induction does not protect retina against apoptosis in ischemia/reperfusion model. Adv Exp Med Biol. 2014;801:677–83.
Russo R, Varano GP, Adornetto A, Nazio F, Tettamanti G, Girardello R, et al. Rapamycin and fasting sustain autophagy response activated by ischemia/reperfusion injury and promote retinal ganglion cell survival. Cell Death Dis. 2018;9(10):981.
Mathew B, Poston JN, Dreixler JC, Torres L, Lopez J, Zelkha R, et al. Bone-marrow mesenchymal stem-cell administration significantly improves outcome after retinal ischemia in rats. Graefes Arch Clin Exp Ophthalmol. 2017;255(8):1581–92.
Volarevic V, Arsenijevic N, Lukic ML, Stojkovic M. Concise review: mesenchymal stem cell treatment of the complications of diabetes mellitus. Stem Cells. 2011;29(1):5–10.
Liang D, Han D, Fan W, Zhang R, Qiao H, Fan M, et al. Therapeutic efficacy of apelin on transplanted mesenchymal stem cells in hindlimb ischemic mice via regulation of autophagy. Sci Rep. 2016;6:21914.
Acknowledgements
Not applicable.
Funding
This work was supported by National Natural Science Foundation of China (No.: 81700553), the Stem Cell and Translational Research, the National Key Research and Development Program of China (No. 2016YFA0101001) and the Postdoctoral Research Foundation of China (No. 2017M183789).
Availability of data and materials
All data are included in this published article.
Author information
Authors and Affiliations
Contributions
CH drafted the manuscript. LZ completed the data collection. DW revised the manuscript. LL contributed to the manuscript conception. CH and LL provided financial support. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hu, C., Zhao, L., Wu, D. et al. Modulating autophagy in mesenchymal stem cells effectively protects against hypoxia- or ischemia-induced injury. Stem Cell Res Ther 10, 120 (2019). https://doi.org/10.1186/s13287-019-1225-x
Published:
DOI: https://doi.org/10.1186/s13287-019-1225-x