
Abstract
Adult mesenchymal stem cells (MSCs) represent a subject of intense experimental and biomedical interest. Recently, trophic activities of MSCs have become the topic of a number of revealing studies that span both basic and clinical fields. In this review, we focus on recent investigations that have elucidated trophic mechanisms and shed light on MSC clinical efficacy relevant to musculoskeletal applications. Innate differences due to MSC sourcing may play a role in the clinical utility of isolated MSCs. Pain management, osteochondral, nerve, or blood vessel support by MSCs derived from both autologous and allogeneic sources have been examined. Recent mechanistic insights into the trophic activities of these cells point to ultimate regulation by nitric oxide, nuclear factor-kB, and indoleamine, among other signaling pathways. Classic growth factors and cytokines—such as VEGF, CNTF, GDNF, TGF-β, interleukins (IL-1β, IL-6, and IL-8), and C-C ligands (CCL-2, CCL-5, and CCL-23)—serve as paracrine control molecules secreted or packaged into extracellular vesicles, or exosomes, by MSCs. Recent studies have also implicated signaling by microRNAs contained in MSC-derived exosomes. The response of target cells is further regulated by their microenvironment, involving the extracellular matrix, which may be modified by MSC-produced matrix metalloproteinases (MMPs) and tissue inhibitor of MMPs. Trophic activities of MSCs, either resident or introduced exogenously, are thus intricately controlled, and may be further fine-tuned via implant material modifications. MSCs are actively being investigated for the repair and regeneration of both osteochondral and other musculoskeletal tissues, such as tendon/ligament and meniscus. Future rational and effective MSC-based musculoskeletal therapies will benefit from better mechanistic understanding of MSC trophic activities, for example using analytical “-omics” profiling approaches.
Similar content being viewed by others

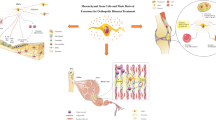
Background
From a research, medical, and business standpoint, mesenchymal stem cell (MSC)-based therapies are fascinating. Sales for stem cell products (e.g., as a subset of osteobiologics) were projected to top $600,000,000 by 2015 [1, 2], and a recent Scopus search for musculoskeletal and stem cells resulted in over 3000 documents, with more than a third being reviews. We have limited this review to highlighting noteworthy findings and concepts concerned with the understanding of and challenges with MSC musculoskeletal therapies.
MSCs were discovered in the 1960s [3], named in the early 1990s [4], and purportedly defined by the mid-2000s [5]. Despite the proposed criteria, the functional definition within the literature varies widely. MSCs can be defined by their ability to adhere to tissue culture plastic, their expression of several cell surface molecular epitopes—cluster of differentiation CD73, CD90, and CD105, and others—as well as their lack of several surface markers, including CD45 [6]. Some previously excluded markers are debated within certain circles, such as CD34 and CD146 [7–9]. MSCs can be isolated from a range of tissues, but the most commonly cited sources are bone marrow (BM), adipose tissue, muscle, bone, and perinatal tissues (e.g., Wharton’s Jelly, umbilical vein/cord blood (UV/UCB), and amnion).
While they were originally utilized clinically in hopes of harnessing their differentiation and proliferation potential, MSCs are increasingly thought to also influence, in addition to participating in, tissue function [10, 11], especially within osteochondral spaces [12]. These MSC influences can range from relatively rare activities that require cell contact, such as mitochondrial transfer and cell fusion, to relatively common paracrine MSC actions through extracellular microvesicles or secreted factors. MSCs may modulate the immune response, angiogenesis, apoptosis, oxidation level, migration, and/or differentiation/stimulation of surrounding cells [13]. Because of this alternative use of MSCs, Caplan and Sorell [14] suggested a renaming of MSCs to medicinal signaling cells to suggest a new era of MSC clinical relevance due to their immunomodulatory properties. While acknowledging progress in the other areas mentioned, this work will focus on the current debates concerning sourcing, MSC alterations of angiogenesis, cell differentiation/stimulation, and strategies to improve MSC differentiation.
Sourcing
Sourcing of MSCs has become an area of debate due to well-recognized potential differences in differentiation abilities and trophic activities of the derived MSCs. Alternatively sourced MSCs may have different differentiation potentials as BM-MSCs, and they may require additional supplementation to achieve robust or similar differentiation. However, while relative abundance and ease of isolation of MSCs may allow their use for successful musculoskeletal interventions [15], there are concerns that diminished numbers of MSCs may be present in BM as patients age or succumb to disease [16]. One study found that mouse MSCs from four common sources (BM, adipose tissue, skeletal muscle, and myocardium) equally supported endothelial cell (EC) network formation in vitro and blood vessel formation in vivo [17]. Muscle-derived stem cells (MDSCs) and satellite cells are thought to contribute to repair of skeletal muscle and bone [18–20]. Although the exact mechanisms remain to be elucidated, cartilage and muscle health has been provocatively linked with changes in or lack of multipotent cell activity, including diseases such as osteoarthritis (OA, cartilage), sarcopenia (muscle), and related muscle and motor neuron diseases [21].
In one study attempting to address the most useful source of MSCs for angiogenesis through a hindlimb ischemia model, Bortolotti et al. examined adipose-derived MSCs (AD-MSCs) and BM-MSCs (along with a subpopulation of CD11-depleted BM-MSCs). They found, as have many others, that MSCs were not incorporated into the healing wound but that wounds, particularly those in muscles, healed more rapidly when exposed to BM-MSCs (regardless of MSC sorting) [22]. Classic proangiogenic, chemotactic, and remodeling molecules were identified as being expressed by MSCs, with several factors appearing prominently in the also effective conditioned medium (CM) (platelet-derived growth factor-B (PDGFB), transforming growth factor beta (TGF-β), stromal-derived factor-1 (SDF1), angiopoietin-1 (Ang1), regulator of G-protein signaling-5 (Rgs5), matrix metalloproteinase-9 (MMP-9), chemokine (C-X-C) ligand-10 (CXCL10), chemokine (C-C) ligand (CCL5)) [22].
Work with 5–6-week-old human embryonic BM-MSCs and MDSCs suggests that MSCs have innate propensities for adipogenic and myogenic differentiation, respectively, which ultimately affects the organization of the engineered tissues [16, 23]. MSCs sourced from older tissue might overcome these propensities depending on the implantation culture environment [24]. Taking a cue from the successful use of stem cells from birth-associated tissues [25], one group recently investigated the angiogenic activity of endometrium/menstrual blood-sourced multipotent cells, showing that they support the recruitment of ECs, blood vessels, and, potentially, the proliferation of hematopoietic stem cells [26].
An encouraging finding is that the number of MSCs required to exert trophic action may be far less than originally calculated as necessary for tissue replacement, because a prospective study of BM-MSC vs mixed BM-MSC + lipoaspirate therapy for OA found no difference in patient-reported outcomes between the two groups [27]. Interestingly, increasing body mass index (BMI) appeared to correlate with patient-reported improvement of function, a link that should be explored in the future [28].
MSC musculoskeletal clinical use
Evidence for an altered view of MSC efficacy follows results from clinical trials, several of which have recently begun to yield data about long-term MSC efficacy in disease treatment. A large area of MSC-based musculoskeletal research has been directed towards the degenerative joint disease OA, which currently affects approximately 20 million Americans and is projected to affect 20 % of American adults by 2030 [29]. OA is characterized by the degeneration of articular cartilage and synovial inflammation, which alters associated soft tissue and subchondral bone, resulting in bony lesion and osteophyte formation. These degenerative events cause pain and loss of joint mobility and function. Because cartilage has a lower regenerative capacity than other, more vascularized tissues in the body, arthritis and joint degeneration are growing targets of MSC-based therapies.
Recent investigations into MSC treatment of OA have begun to include formal, controlled clinical trials [30] in addition to many uncontrolled trials by private entities. Several international companies, including Cartistem, Regenexx, Regeneus, BioHeart, and Mesoblast, are carrying out phase I and II clinical trials for the treatment of degenerative joint diseases with allogeneic or autologous, multipotent cell types that are capable of mesenchymal differentiation, usually derived from BM or adipose origin [15, 31, 32]. Several other companies have chosen to facilitate MSC isolation within the clinic by constructing machines that quickly sort stem cells from the mixed populations present in surgically isolated tissue [33]. Although published data have been relatively scarce for completed trials, adverse events such as tumors, infections, or premature trial closures have rarely been reported, suggesting safety of MSC-based therapies [34, 35]. The majority of reviewed studies utilize either dissociated cells injected into the joint space or cells delivered via seeding in various biocompatible and/or biodegradable materials.
A recent review details results of nine OA articular cartilage (knee) clinical trials which utilized cultured BM-MSCs, noncultured BM concentrate, peripheral blood-derived stem cells, or cells from the adipose stromal vascular fraction [36]. Some cells were immobilized with hyaluronan, collagen, platelet gel, and/or fibrin glue. Others were injected into the joint or at a defect using only saline. Regardless of cell origin, intra-articular injection of cells (vs hydrogel or flap immobilization through open surgery) resulted in improved clinical function over untreated controls in some studies as long as 5 years post treatment [36]. Pain relief with minor return of function was noted in most studies.
Noting the ameliorative effect of MSCs on joint pain, chronic lower back pain has recently become a target of MSC therapy. Autologous, scaffold-less BM-MSC injection into patients with spinal cord injury in a Brazilian clinical trial suggested clinically meaningful pain relief and possible improvement in cartilage structure after 6 months and as long as 2 years post treatment, although the high number of MSCs utilized coupled with the high cost of the procedure were identified as potential areas for improvement [37]. In a Spanish study, 7 of 12 patients showed mild return of function after 6 months when enrolled in a phase I safety and efficacy study for BM-MSC injection for long-term (>6 months) spinal cord injury [38]; although positive in terms of apparent MSC effect, the study faced almost immediate criticism from other researchers due to the small sample size and lack of appropriate controls [39]. A study by Mesoblast reported decreased lower back pain in 48 % of allogeneic BM-MSC-treated patients vs 13 % in placebo controls up to 2 years post injection [40].
A portion of these analgesic effects could be due to the anti-inflammatory activity of MSCs. Evidence that a decrease in granulocyte macrophage colony-stimulating factor (GM-CSF) resulted from MSC treatment and may decrease disease severity after 4 months comes from a rheumatoid arthritis (RA) clinical trial that used MOR103 antibodies to deplete serum GM-CSF [41]. In an excellent and very recent review of MSC applications to RA, De Bari described how immunomodulation could play a role in RA-specific joint degeneration. Immunoregulators, including interferon gamma (IFN-γ) and tumor necrosis factor alpha (TNF-α), which are regulated through indoleamine 2,3-dioxygenase (IDO) or nitric oxide (NO), and MSC effects on forkhead box p3+ (Foxp3+) Tregs or CD4+ Th17 cells have been suggested [42]. Interestingly, the “transformation hypothesis” proposes that MSCs may become transformed by interplay with chronic inflammatory processes in the joint, resulting in a more aggressive cell type with abilities to either invade the articular cartilage and/or circulate, spreading arthritis to unaffected joints [43, 44]. UV-MSCs may help to relieve the severity of RA symptoms when combined with disease-modifying antirheumatic treatments [45].
Recent exploration of immunomodulation showed that AD-MSC surface-bound glycoprotein A repetitions predomain/leucine-rich repeat containing-32 (GARP/LRRC32), found on CD4+/Foxp3+ Tregs, megakaryocytes, and platelets, binds to membrane-bound TGF-β1, holding it in an inactivated but readily-accessible state. GARP silencing results in increased secretion and activation of TGF-β1 and impaired proliferation of AD-MSC as well as activation of T cells [46]. Immunosuppressive effects of membrane-bound TGF-β1, especially when bound to extracellular vesicles (EVs), have also been reported for other MSC types [47–49].
Mechanisms of MSC trophic activity
Insight into the mechanisms of MSC trophic activity is advancing across multiple fields (Table 1). Within the joint space, the MSC secretome is thought to influence the anabolic tendencies of chondrocytes, chondrocyte progenitor cells (CPCs), cartilage-derived stem/progenitor cells (CSPCs), synovium-resident multipotent progenitor cells, osteoblasts/osteoclasts/resident MSCs within the subchondral bone (especially after microfracture), and chondrogenic cells within the infrapatellar fat pad [36, 50, 51]. The MSC secretome can be modified through permanent or temporary alterations. Several studies have found that the exposure of MSCs to proinflammatory factors, sometimes for as little as a few hours, can alter the gene and protein expression of MSCs for days afterwards [52]. Factors known to be secreted or bound to MSC membranes with anti-inflammatory activities (activation of Tregs/tolerogenic dendritic cell phenotype; pro-resolving/M2 macrophage activation; inhibition or proapoptosis of T cells, B cells, NK cells, or dendritic cells; decreasing cytokine production) include: purines, bone morphogenetic proteins (BMPs, specifically BMP-4), CD274, CCL2, Connexin 43, cyclooxygenase (COX)/prostaglandin (PG), CD95/CD95 ligand, galectins, heme oxygenase-1, human leukocyte antigen-G (HLA-G), IDO/kynurenine, interleukin-6 (IL-6), leukemia inhibitory factor (LIF), NO, TGF-β, tumor necrosis factor-inducible gene-6 (TSG6), and vascular endothelial growth factor (VEGF) [53].
In addition to some of the classic chemotactic growth factors and molecules already mentioned (HGF, PDGF, and bFGF), MSCs are strongly influenced by the binding of CXCL12 (SDF1) to CXCR4 [54]. Embryonic muscle growth and adult muscle repair are thought to be heavily influenced by MMP-10-regulated CXCL12 stimulation of MSC migration [55, 56]. Additionally, MSCs express a variety of receptors, including various integrins and selectins, that allow extravasation at repair sites [57].
Clues to the mechanisms of MSC trophic activities (Fig. 1) can also be found in the extensive work done in other fields, particularly exploration of the stem cell secretome in the cardiac field [58]. Identified factors include adrenomedullin, angiogenin, fibroblast growth factor-2 (FGF2), CXCL12, cistatin C, cysteine-rich angiogenic inducer 61 (Cyr61), Dickkopf-related proteins (Dkk), hepatocyte growth factor (HGF), insulin-like growth factor (IGF), IL-1, IL-6, pigmented epithelium-derived factor (PEDF), placental growth factor (PLGF), SDF1, TSG6, VEGF, MMP-2, tissue inhibitor of metalloproteinase-1 (TIMP-1), TIMP-2, secreted frizzled related protein-2 (SFRP-2), thrombospondin-1, and tenascin C [58]. Belying their osseous origin, CM of BM-MSC appears enriched in molecules typically secreted by or influencing osteoblasts, including decorin, osteoprotegerin, Dkk-3, receptor activator of nuclear factor-kB (RANK), osteopontin, and CCL5; inflammatory factors maximally produced by BM-MSCs include CCL2, TIMP-2, IL-6, IL-7, IL-3, MMP-7, chemokine (C-X-C) receptor-16 (CXCR16), and MMP-10. CCL2 and CCL7 produced by BM-MSCs appear to strongly influence nascent bone formation [59, 60]. Recent work also suggests that AD-MSC, BM-MSC, and dental pulp stem cell-secreted CXCL14 and CCL2 help to recruit CXCR4+ cells and chemokine (C-C) receptor-2+ (CCR2+) vessel-associated cells, without inducing proliferation [61]. Besides these influential but potentially short-lived proteins, some MSCs secrete EVs which may contain any number of influential molecules, protected from systemic degradation by virtue of their natural, membrane-bound packaging [62–65].

MSC trophic mechanisms depend on MSC interactions with and modification of the local environment. MSC trophic functions can be both achieved and altered through dynamic ECM–cytoskeletal interactions, cell–cell contacts, and soluble and transcription factor signaling. ECM extracellular matrix, miRNA microRNA, MMP matrix metalloproteinase, MSC mesenchymal stem cell, TIMP tissue inhibitor of metalloproteinase
In many cell types, EVs of varying sizes, including ectosomes/exosomes and microvesicles/microparticles, were derived from either cytoplasmic protrusions or lipid raft internalization and subsequent endosomal fusion with the plasma membrane [66–69]. These 30 nm–1 μm vesicles may be studded with multiple proteins, usually tetraspanins, and filled with a combination of proteins, lipids, and copious amounts of mRNA and microRNA (miRNA), particularly miR22 and miR-19a [58]. EC and cancer cell-derived microparticles may potently increase MSC NF-kB activity, stimulating local trophic support [70]. Gap junctions, formed via connexins, are another avenue allowing direct cell–cell communication, with strong evidence for membrane and (to a lesser extent) cytoplasmic exchange between MSCs and ECs [71].
Likely mediated through EC-stimulated VEGF production, CXCR4-enriched exosomes derived from MSCs generated from induced pluripotent stem cell (iPSC) lines via medium change (iMSCs) improved recovery from myocardial infarction [72]. In work addressing hindlimb ischemia in mice, iMSC exosomes were associated with a higher number of CD31+ and CD34+ cells in damaged muscle tissue as well as increased EC secretion of VEGF, TGF-β1, and angiogenin, suggesting enhanced vascular recruitment by vesicles alone [73]. Another group generated iMSCs through TGF-β-pathway inhibition and medium changes; those iMSCs, surprisingly, did less to promote cancer than BM-MSCs, appearing to express and produce lower amounts of several of the inflammatory and differentiation factors (particularly TGF-β receptor-2 and, interestingly, hyaluronan (HA)) when cultured with various types of cancer cells [74].
In a recent study, Baglio et al. [75] characterized the RNA contents of exosomes obtained through ultracentrifugation, and found that exosomes were enriched for tRNA, in particular tRNA CTC. Their work further suggested that the differentiation state of a MSC might be deduced by the content of its exosomes, particularly the presence of full-length tRNA and tRNA long fragments, consistent with a stem-like state of the cells [76]. miRNA loading within vesicles is not random, as dexamethasone treatment of both C2C12 cells and diabetic rats increased the concentration of miR-23a and miR-182 in collected microvesicles and urine, respectively [77], thus providing MSCs with a dynamic way to influence and respond to their microenvironment. Microvesicles may also suppress the infiltration of macrophages into damaged tissues [75]. However, the relevance of vesicles/microvesicles to classical and clinically approved MSCs is questionable, because a different group noted that MSCs produced much fewer vesicles than immortalized ESC-derived MSCs [78]. The group offered an immortalization strategy that would maximize the yield of MSC-produced vesicles, should they prove effective in the clinic.
Altering MSC activity prior to bone or cartilage implantation
Because “plain” MSC implantation has faced such varied success in the clinic, the next generation of MSC-based strategies seeks to harness and direct MSC trophic activities. It was observed that substrate composition and stiffness, sensed through various integrins, could influence the expression of myogenic factors [79]. Substrate stiffness, known to affect the differentiation of MSCs, acting possibly through regulation of alpha-smooth muscle actin (αSMA), could also play a role in the eventual differentiation capacity of culture-expanded MSCs [80]. Substrate stiffness, in turn, affects MSC trophic properties; stiff (40 kPA) polyacrylamide gels coated with fibronectin induced proangiogenic factor secretion by BM-MSCs [81]. Through internalization and recycling of focal adhesions and receptors, caveolins play an intriguing role in MSC sensing of both substrate stiffness and surrounding soluble signals, particularly in vascular, muscular, and osteogenic settings [82]. Alternatively, hydrogels made from autologous plasma may also help to temporarily concentrate either AD-MSCs or AD-MSC-CM at the injury site [83].
The ability of MSCs to self-generate abundant collagenous extracellular matrix (ECM) may partially explain some of the positive effects witnessed in some joint degeneration trials involving MSCs [27]. Medium supplements or additional modifications to the substrate to mimic other ECM molecules or bioactive factors, such as osteonectin, may further increase MSC secretion of bioactive molecules (FGF2, CCL5, and VEGF), supporting native cell migration and differentiation [84].
The milieu present in culture serum appears to strongly influence the fate of cultured cells, more so than any single exogenous supplement [85]. One method to both encourage MSC trophic activity as well as ease immunological concerns in the clinic could be to utilize autologously derived cell culture supplements such as platelet lysate for autologous MSC expansion and culture [86]. Platelet-rich plasma, for example, may protect cartilage from injury by enhancing collagen II (COLII)/aggrecan (AGN) expression and suppressing MMP-3, COX2, iNOS, and associated NO and PGE2 production [87]. Once established in vitro, however, the role of serum becomes less clear; while serum content affects MSC gene expression and growth rate, inherent multipotency and stem cell marker surface expression do not appear to be affected by the absence of serum [88]. For that reason, MSC-CM concentration and injection may sidestep issues of autophagy [89] or apoptosis of MSCs upon in-vitro expansion [13].
Culture under hypoxia is another attractive method to increase initial MSC production of trophic factors. Under hypoxia, hypoxia inducible factor-1 alpha (HIF1α) expression increases, driving VEGF and other proangiogenic, antiapoptotic, and antioxidant molecules [90]. Under certain circumstances, hypoxic MSCs may serve to prevent harmful fibrosis through HGF production, TGF-β1/COLII, and IL-1β downregulation, and fibronectin expression [91]. Work in vitro with chondrocytes has specifically identified HGF as an antifibrotic agent released by AD-MSCs [92]. Furthermore, FGF1 secreted by MSCs in contact with chondrocytes may stimulate the proliferation of and help to preserve the function of chondrocytes [93, 94].
In-vitro evidence suggests that the addition of BM-MSCs to OA cartilage may initially increase IL-1β and IL-8 production but ultimately reduce the amount of soluble glycosaminoglycan (GAG) released by the cartilage over time, making the exact influence of MSCs on cartilage structure unclear [95].
Reported pain relief associated with the introduction of MSCs may be the result of immunomodulation but was by no means universal [36]. In-vitro studies suggest that exposure of MSCs to chondrocytes may induce expression of MSC major histocompatibility complex (MHC) I/II and other costimulatory molecules [96].
Pretreatment of MSCs, especially with IFN-γ to prime their immunosuppressive activity, may result in decreased MSC-associated tissue degradation [97]. Possibly in response to IL-1α, MSCs exposed to platelet lysate have the capacity to encourage a proinflammatory/M1 or proresolving/M2 macrophage phenotype through GM-CSF and PGE2 activity, respectively [53, 98]. Growing evidence suggests that the native, inflamed cartilage environment may both trigger the release and limit the effectiveness of MSC-anti-inflammatory PGE2 [99]. MSCs may recruit CD4+ T cells, which can also play a role in increasing local osteogenic activity [100]. Care must be taken with extensive culture, because MSCs may gain genetic abnormalities and lose some ability to differentiate, promoting senescence [88].
Vascular/inflammation regulation
Several key targets of regenerative medicine therapies, including restored nerve and muscle function and the previously discussed bone repair, rely heavily on the influence of vascular cells [101]. Consequently, MSC effects on blood vessel cells have long fascinated researchers. Recent work has begun to elucidate mechanisms by which MSCs may affect blood vessel morphology and function. Chen et al. [102] reported that MSC-produced HGF, upon interaction with ECs in coculture and, to a lesser extent, via paracrine signaling, caused an increase in EC cadherin and F-actin remodeling, thereby decreasing EC permeability [90, 103, 104]. This and earlier work with ECs suggests that MSC–EC interactions may temporarily restrict both the physical clearance of MSCs and the invasion of inflammatory cells. Once in the bloodstream, MSC inhibition of NF-kB, perhaps through IL-10 or other factors, may decrease the binding of monocytes to the endothelium, further decreasing inflammation at wound/MSC injection sites [45]. This temporary pause in the battle with chronic inflammation may explain some of the positive results seen with MSCs.
Following a more classical approach focused on EC motility and activity, VEGF, ANG, and NF-kB pathways have all been implicated in regulating angiogenesis. Recent work suggests that, in the short term, the NF-kB pathway may control EC response through BM-MSC-produced IL-6 and IL-1β; following this NF-kB activation, ECs activate P-selectin, producing CCL23, CXCL2, and CXCL3. In turn, MSCs showed signs of early differentiation towards a smooth muscle phenotype in coculture, influenced by TGF-β1 and TGF-β3 [105]. BM-MSC production of VEGF may be stimulated by IL-8, either through paracrine or autocrine mechanisms [106]. In turn, BM-MSCs may stabilize ECs by upregulating ANG1, thereby downregulating EC proliferation [107]. Conversely, the interaction of MSCs with ECs, particularly through endothelin 1 (ET1) and PDGFB, may prime cells to survive transplantation and differentiate more easily upon reimplantation [108].
Neural support
MSCs have long been known to support nerve growth through the support of Schwann cells, secretion of neurovascular factors (including FGF2 and VEGF-A), and, possibly, transdifferentiation into Schwann-like cells. Combined with varying types of biocompatible and bioactive materials, such as poly-lactic acid (PLA), polycaprolactone (PCL), polyurethane (PU), polyethylene (PE), and silicone (for strength) and COLI, HA, and so forth (for bioactivity), several groups have observed enhanced nerve extension and functional improvements in a range of animal models [109, 110]. The most recent work refines previous findings that guidance fibers of particular spacing and architecture may aid MSCs in further accelerating the nerve healing process [111–113]. AD-MSCs, at sufficient density, secrete brain-derived neurotrophic factor (BDNF) in response to autocrine IFN-β [114]. Stimulating cocktails that increase cyclic-adenosine monophosphate (cAMP) and include retinoic acid (pretreatment), FGF2, PDGFAA, and different forms of neuregulin have been shown to increase neurite outgrowth in vitro and nerve extension after injury in vivo [115]. In addition to neurotrophic BDNF, nerve growth factor (NGF), and glial cell line-derived neurotrophic factor (GDNF), as well as angiogenic VEGF and ANG1 identified in many other experiments, recent work identified the antiapoptotic activity of AD-MSCs, possibly by decreasing neuronal c-jun [116]. As mentioned in previous sections, such pretreatment is relatively common in nonclinical work, and may become de rigeur as new progenitor cell sources are explored for musculoskeletal therapies [117]. Crucially, it appears that MSCs should not be directly injected intrathecally for early spinal cord repair, as the subsequent inflammation seemed to prevent MSC migration to neuronal injury sites [118]; later injection may prove beneficial [119].
Ciliary neurotrophic factor (CNTF) is a particularly well known neuroprotective factor produced by MSCs. While the factor has potent therapeutic effects on nerve apoptosis, neuroinflammation, and neuronal proliferation, it has been linked with altered metabolism (due to neurogenesis in the hypothalamus as well as direct action on adipocytes) when administered systemically and may negatively affect osteoblast differentiation and mineralization [120–122]. GDNF, another potent neurotrophic molecule often produced by MSCs, may help to ease allodynia and hyperalgesia experienced in dorsal root ganglia sensory nerves [123]. Amniotic membrane-derived MSCs expressed more ANG1, FGF1, IGF1, and VEGFA (but not FGF2) than AD-MSCs in a mouse sciatic nerve injury trial [124].
While CM from cells treated under hypoxic and normoxic conditions both increased the observed number of differentiating neurons in vitro, hypoxia-cultured Wharton’s Jelly (WJ)-derived MSCs upregulated thymosin B and eukaryotic elongation factor (EF2) and may have contributed to a slight increase in total neuron maturity [125]. WJ-MSCs under normoxic conditions were shown to produce PDGFAA, HGF, TGF-β2, IL-6, IL-8, IL-1ra, CCL5, CCL2, and CXCL10 at much larger concentrations than BM-MSCs and AD-MSCs [126]. Whatever the mechanism for neural support, tissue response to neurological directives is critical to the ultimate utility of the repaired nerve.
Muscles and miscellany
Intriguing results suggest that MSCs derived from less traditional sources could one day be utilized therapeutically. One readily available source for MSCs could be skeletal muscles. Our research group has worked extensively with blast-traumatized muscle-derived multipotent cells [127–129]. This particular type of muscle-derived multipotent cells is especially attractive therapeutically due to its relative abundance and ease of isolation [130] as well as neurotrophic activity [131]. MDSCs should be used cautiously when attempting to rebuild musculoskeletal tissues, because several groups have identified populations that seem predisposed to mineralize ectopically [132–134], especially in the presence of muscular genetic abnormalities [135]. Growth factor coinjection might attenuate this ectopic bone formation, as growth hormone–insulin-like growth factor-1 (GH-IGF1) activity promotes muscle cell proliferation, regulates muscle fiber size and type, controls osteoblast proliferation and differentiation, inhibits osteoclast activity, stimulates renal conversion of 25-OH-vitamin D3, and controls phosphate reabsorption [136]. By contrast, this matrix-modifying MSC activity may help to attenuate disease severity and ultimately contribute to useful muscle mass [137]. Harmful proliferation and contraction of myofibroblasts, as occurs in Dupuytren's contracture, may be attenuated in the presence of the CM of both AD-MSCs or BM-MSCs as well as the physical presence of AD-MSCs (but not BM-MSCs) [138]. BM-MSCs appear to contribute to pathological myofibroblast proliferation while AD-MSCs appear to inhibit the activity slightly [138]. MMP-2 and MMP-9 are required for efficient skeletal muscle regeneration and are enhanced by mouse BM-MSCs/MSC-CM along with reduced TIMP-1/2 levels. Muscle cell motility may also be encouraged by BM-MSC-secreted MMP-2 [139].
Aside from their ECM-modifying properties, the immunomodulatory properties of MSCs are intriguing from a therapeutic standpoint but must be used carefully, because MSC treatment, concurrent with a Staphylococcus aureus infection, was shown to increase the severity of bone loss, despite increased MSC proinflammatory cytokine expression, in an osteomyelitis model [140]. Conversely, encouraging results were recently published from a small idiopathic osteonecrosis trial in Japan, where BM-MSCs were isolated, cultured for 2 weeks, and returned to osteonecrotic patients along with tricalcium phosphate chips (Osferion) and tricortical iliac crest bone [141]; after a 12-week rehabilitation program, all patients reported reduced pain and increased physical function with no serious adverse events reported in the study [142]. The likelihood of MSC engraftment being the cause for the recovery is low, however, as MSCs have been found to migrate towards apoptotic cells, via HGF signaling, but not HGF produced in the presence of necrotic cells [143].
Evidence of MSC trophic efficacy has generated intense excitement in clinically focused research. This excitement is evident in the increasing number of reviews examining MSC trophic properties. Marked therapeutic successes will likely hinge on technological and computational advancements that allow dynamic, high-resolution, and quantitative observation of MSC–ECM, MSC–paracrine, and MSC–cellular interactions to better define the appropriate perspective on the true activity of MSCs.
Conclusions
The application of allogeneic and autologous MSC therapies for the treatment of diseases and dysfunctions of multiple musculoskeletal tissues has received increasing attention. Exciting in-vitro and in-vivo investigations on tendon [117, 144, 145], meniscus [146–148], and ligaments [149, 150] have been reported, along with the use of autologous products such as platelet-rich plasma/plasma lysate [151]. Studies using larger, clinically relevant animal models are both underway and necessary before human clinical trials can be developed [152].
This review has primarily explored secreted trophic factors produced by MSCs. A whole host of therapies are dedicated to engineering or modifying the physical environment and ECM of MSCs to affect their therapeutic potential. A recently developed approach attempts to anchor cells to the collagenous tissue matrix by engineering collagen anchors [153], to promote local action of MSCs and minimize their systemic loss to the lungs, liver, and spleen. Changes in substrate composition (especially the presence of collagen) and stiffness may expand the potential applications of MSC therapies to include muscle volume loss through stimulation of muscle-resident progenitor cells [134, 154, 155]. Local ECM modifications are known to affect MSC differentiation potential [156, 157] and are beyond the scope of this review. Through continuing advancements in genetic engineering, MSCs may eventually be used to treat genetic musculoskeletal conditions, including osteogenesis imperfect [158] and Duchenne’s muscular dystrophy [159, 160]. Careful selection of the therapeutic cells, taking into account subtle tissue source-related differences, may be the key to successful clinical dystrophy therapies [35]. To prove their efficacy in the clinic, these potential treatments will need to be tested in well-controlled studies to assess physical functions for an extended period of time [27].
New or more precise modes of MSC trophic activity may be discovered by adopting contemporary analytical technologies to evaluate and compare genomic, transcriptomic, proteomic, metabolomic, and secretomic profiles, exemplified by the great strides that have been made in genetic and metabolic diseases [161–164]. Lessons learned from previous iterations of MSC therapies and clinical drug trials should overcome some of the regulatory and therapeutic hurdles to MSC use [2, 32, 33]. It is also noteworthy that while genetic engineering of iPSCs may hold the answer to unlimited numbers of perfectly-tuned stem cells, managing the safety concerns of iPSCs and negotiating the patent landscape of this saturated market will be highly challenging [165]. Another major challenge is the uncertainty in terms of biological responsiveness of the diseased tissue, because evidence in several fields suggests that ischemic tissue may be incapable of responding to MSCs [166]. Finally, each of these avenues should be explored while juggling the needs for rigorous science, proven therapeutic efficacy, regulatory approval (e.g., by the Food and Drug Agency) (and thus reproducibility) of a final therapy, and cost/benefit for the patient.
Abbreviations
- AD-MSC:
-
Adipose-derived mesenchymal stem cell
- AGN:
-
Aggrecan
- Ang:
-
Angiopoietin
- BDNF:
-
Brain-derived neurotrophic factor
- BM:
-
Bone marrow
- BMI:
-
Body mass index
- BMP:
-
Bone morphogenetic protein
- cAMP:
-
Cyclic adenosine monophosphate
- CCR/L:
-
Chemokine (C-C) receptor/ligand
- CD:
-
Cluster of differentiation
- CM:
-
Conditioned medium
- CNTF:
-
Ciliary neurotrophic factor
- COL:
-
Collagen
- COX:
-
Cyclooxygenase
- CPC:
-
Chondrocyte progenitor cell
- CSPC:
-
Cartilage-derived stem/progenitor cell
- CXCR/L:
-
Chemokine (C-X-C) receptor/ligand
- Cyr61:
-
Cysteine-rich angiogenic inducer 61
- Dkk:
-
Dickkopf-related proteins
- EC:
-
Endothelial cell
- ECM:
-
Extracellular matrix
- EF2:
-
Eukaryotic elongation factor
- ET:
-
Endothelin
- EV:
-
Extracellular vesicle
- FGF:
-
Fibroblast growth factor
- Foxp3:
-
Forkhead box p3
- GAG:
-
Glycosaminoglycan
- GARP:
-
Glycoprotein A repetitions predomain
- GDNF:
-
Glial cell line-derived neurotrophic factor
- GH-IGF:
-
Growth hormone-insulin-like growth factor
- GM-CSF:
-
Granulocyte macrophage colony-stimulating factor
- HA:
-
Hyaluronan
- HGF:
-
Hepatocyte growth factor
- HIF:
-
Hypoxia inducible factor
- HLA:
-
Human leukocyte antigen
- IDO:
-
Indoleamine 2,3-dioxygenase
- IFN:
-
Interferon
- IGF:
-
Insulin-like growth factor
- IL:
-
Interleukin
- iMSC:
-
MSC generated from induced pluripotent stem cell (iPSC) lines via medium change
- iNOS:
-
Nitric oxide synthase
- LIF:
-
Leukemia inhibitory factor
- LRRC32:
-
Leucine-rich repeat containing-32
- MDSC:
-
Muscle-derived stem cell
- MHC:
-
Major histocompatibility complex
- miRNA:
-
MicroRNA
- MMP:
-
Matrix metalloproteinase
- mRNA:
-
Messenger RNA
- MSC:
-
Mesenchymal stem cell
- NF-κB:
-
Nuclear factor-kappa B
- NGF:
-
Nerve growth factor
- NO:
-
Nitric oxide
- OA:
-
Osteoarthritis
- PCL:
-
Polycaprolactone
- PDGF:
-
Platelet-derived growth factor
- PE:
-
Polyethylene
- PEDF:
-
Pigment epithelium-derived factor
- PG:
-
Prostaglandin
- PLA:
-
Poly-lactic acid
- PLGF:
-
Placental growth factor
- PU:
-
Polyurethane
- RA:
-
Rheumatoid arthritis
- RANK:
-
Receptor activator of nuclear factor-κB
- Rgs:
-
Regulator of G-protein signaling
- SDF:
-
Stromal-derived factor
- SFRP:
-
Secreted frizzled related protein
- TGF:
-
Transforming growth factor
- TIMP:
-
Tissue inhibitor of metalloproteinase
- TNF:
-
Tumor necrosis factor
- TSG:
-
Tumor necrosis factor-inducible gene
- UCB:
-
Umbilical cord blood
- UV:
-
Umbilical vein
- VEGF:
-
Vascular endothelial growth factor
- WJ:
-
Wharton’s jelly
- αSMA:
-
Alpha smooth muscle actin
References
Wei C, Lin AB, Hung S. Mesenchymal stem cells in regenerative medicine for musculoskeletal diseases: bench, bedside, and industry. Cell Transplant. 2014;23:505–12.
Heathman TR, Nienow AW, McCall MJ, Coopman K, Kara B, Hewitt CJ. The translation of cell-based therapies: clinical landscape and manufacturing challenges. Regen Med. 2015;10:49–64.
Friedenstein AJ, Piatetzky-Shapiro II, Petrakova KV. Osteogenesis in transplants of bone marrow cells. J Embryol Exp Morphol. 1966;16:381–90.
Caplan AI. Mesenchymal stem cells. J Orthop Res. 1991;9:641–50.
Dominici M, Le Blanc K, Mueller I, Slaper-Cortenbach I, Marini F, Krause D, et al. Minimal criteria for defining multipotent mesenchymal stromal cells The International Society for Cellular Therapy position statement. Cytotherapy. 2006;8:315–7.
Salem HK, Thiemermann C. Mesenchymal stromal cells: current understanding and clinical status. Stem Cells. 2010;28:585–96.
Dmitrieva RI, Minullina R, Bilibina AA, Tarasova OV, Anisimov SV, Zaritskey AY. Bone marrow- and subcutaneous adipose tissue-derived mesenchymal stem cells: differences and similarities. Cell Cycle. 2012;11:377–83.
Sidney LE, Branch MJ, Dunphy SE, Dua HS, Hopkinson A. Evidence for CD34 as a common marker for diverse progenitors. Stem Cells. 2014;32:1380–9.
Lv F-J, Tuan RS, Cheung KMC, Leung VYL. The surface markers and identity of human mesenchymal stem cells. Stem Cells. 2014;32:1408–19.
Caplan AI, Correa D. The MSC: an injury drugstore. Cell Stem Cell. 2011;9:11–5.
Vonk LA, de Windt TS, Slaper-Cortenbach ICM, Saris DBF. Autologous, allogeneic, induced pluripotent stem cell or a combination stem cell therapy? Where are we headed in cartilage repair and why: a concise review. Stem Cell Res Ther. 2015;6:1–11.
Ruetze M, Richter W. Adipose-derived stromal cells for osteoarticular repair: trophic function versus stem cell activity. Expert Rev Mol Med. 2014;16:e9.
Liang X, Ding Y, Zhang Y, Tse H, Lian Q. Paracrine mechanisms of mesenchymal stem cell-based therapy: current status and perspectives. Cell Transplant. 2014;23:1045–59.
Caplan AI, Sorrell JM. The MSC curtain that stops the immune system. Immunol Lett. 2015;168:136–9.
Diederichs S, Shine KM, Tuan RS. The promise and challenges of stem cell-based therapies for skeletal diseases. Bioessays. 2013;35:220–30.
Hass R, Kasper C, Böhm S, Jacobs R. Different populations and sources of human mesenchymal stem cells (MSC): a comparison of adult and neonatal tissue-derived MSC. Cell Commun Signal. 2011;9:1–14.
Mahdi NS, Rahbarghazi R. Interactions of mesenchymal stem cells with endothelial cells. Stem Cells Dev. 2014;23:319–32.
Tamaki T, Okada Y, Uchiyama Y, Tono K, Masuda M, Wada M, et al. Clonal multipotency of skeletal muscle-derived stem cells between mesodermal and ectodermal lineage. Stem Cells. 2007;25:2283–90.
Zou J, Yuan C, Wu C, Cao C, Shi Q, Yang H. Isolation and osteogenic differentiation of skeletal muscle-derived stem cells for bone tissue engineering. Mol Med Rep. 2013;9:185–91.
Meszaros LB, Usas A, Cooper GM, Huard J. Effect of host sex and sex hormones on muscle-derived stem cell-mediated bone formation and defect healing. Tissue Eng Part A. 2012;18:1751–9.
De Ceuninck F, Fradin A, Pastoureau P. Bearing arms against osteoarthritis and sarcopenia: when cartilage and skeletal muscle find common interest in talking together. Drug Discov Today. 2014;19:305–11.
Bortolotti F, Ukovich L, Razban V, Martinelli V, Ruozi G, Pelos B, et al. In vivo therapeutic potential of mesenchymal stromal cells depends on the source and the isolation procedure. Stem Cell Reports. 2015;4:332–9.
Krylova TA, Musorina AS, Zenin VV, Yakovleva TK, Poljanskaya GG. A comparative analysis of mesenchymal stem-cell lines derived from bone marrow and limb muscle of early human embryos. Cell Tissue Biol. 2014;8:441–53.
Baker N, Boyette LB, Tuan RS. Characterization of bone marrow-derived mesenchymal stem cells in aging. Bone. 2015;70:37–47.
Verdi J, Tan A, Shoae-Hassani A, Seifalian AM. Endometrial stem cells in regenerative medicine. J Biol Eng. 2014;8:20.
Alcayaga-Miranda F, Cuenca J, Luz-Crawford P, Aguila-Díaz C, Fernandez A, Figueroa FE, et al. Characterization of menstrual stem cells: angiogenic effect, migration and hematopoietic stem cell support in comparison with bone marrow mesenchymal stem cells. Stem Cell Res Ther. 2015;6:1–14.
Grässel S, Lorenz J. Tissue-engineering strategies to repair chondral and osteochondral tissue in osteoarthritis: use of mesenchymal stem cells. Curr Rheumatol Rep. 2014;16:1–16.
Centeno C, Pitts J, Al-Sayegh H, Freeman M. Efficacy of autologous bone marrow concentrate for knee osteoarthritis with and without adipose graft. Biomed Res Int. 2014;2014:370621.
Mobasheri A, Kalamegam G, Musumeci G, Batt ME. Chondrocyte and mesenchymal stem cell-based therapies for cartilage repair in osteoarthritis and related orthopaedic conditions. Maturitas. 2014;78:188–98.
Baugé C, Boumédiene K. Use of adult stem cells for cartilage tissue engineering: current status and future developments. Stem Cells Int. 2015;2015:438026.
Boregowda SV, Phinney DG. Therapeutic applications of mesenchymal stem cells: current outlook. BioDrugs. 2012;26:201–8.
Sharma RR, Pollock K, Hubel A, McKenna D. Mesenchymal stem or stromal cells: a review of clinical applications and manufacturing practices. Transfusion. 2014;54:1418–37.
Srijaya TC, Ramasamy TS, Kasim NHA. Advancing stem cell therapy from bench to bedside: lessons from drug therapies. J Transl Med. 2014;12:243.
Wang R, Rao MS. Application of mesenchymal stem cells in joint diseases. OA Musculoskelet Med. 2013;1:26.
Farini A, Sitzia C, Erratico S, Meregalli M, Torrente Y. Clinical applications of mesenchymal stem cells in chronic diseases. Stem Cells Int. 2014;2014:306573.
Counsel PD, Bates D, Boyd R, Connell DA. Cell therapy in joint disorders. Sports Health. 2014;7:27–37.
Mendonça MV, Larocca T, de Freitas SB, Villarreal C, Silva LF, Matos A, et al. Safety and neurological assessments after autologous transplantation of bone marrow mesenchymal stem cells in subjects with chronic spinal cord injury. Stem Cell Res Ther. 2014;5:126.
Orozco L, Soler R, Morera C, Alberca M, Sánchez A, García-Sancho J. Intervertebral disc repair by autologous mesenchymal bone marrow cells: a pilot study. Transplantation. 2011;92:822–8.
Kovacs FM, Abraira V, Gérvas J, Arana E, Peul WC, Schoene ML, et al. Overenthusiastic interpretations of a nonetheless promising study. Transplantation. 2012;93:e6–7.
Itescu S. Mesoblast—a global leader in cell based medicines. In: 34th Annual J.P. Morgan Healthcare Conference; San Francisco, CA; January 2016.
Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2014;74:1058–64.
De Bari C. Are mesenchymal stem cells in rheumatoid arthritis the good or bad guys? Arthritis Res Ther. 2015;17:113.
Lefèvre S, Knedla A, Tennie C, Kampmann A, Wunrau C, Dinser R, et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat Med. 2009;15:1414–20.
El-Jawhari JJ, El-Sherbiny YM, Jones EA, McGonagle D. Mesenchymal stem cells, autoimmunity and rheumatoid arthritis. QJM. 2014;107:505–14.
Letourneau PA, Menge TD, Wataha KA, Wade CE, S Cox C, Holcomb JB, et al. Human bone marrow derived mesenchymal stem cells regulate leukocyte-endothelial interactions and activation of transcription factor NF-kappa B. J Tissue Sci Eng. 2011;Suppl 3:001.
Carrillo-Galvez AB, Cobo M, Cuevas-Ocaña S, Gutiérrez-Guerrero A, Sánchez-Gilabert A, Bongarzone P, et al. Mesenchymal stromal cells express GARP/LRRC32 on their surface: effects on their biology and immunomodulatory capacity. Stem Cells. 2015;33:183–95.
Maumus M, Jorgensen C, Noël D. Mesenchymal stem cells in regenerative medicine applied to rheumatic diseases: role of secretome and exosomes. Biochimie. 2013;95:2229–34.
Mokarizadeh A, Delirezh N, Morshedi A, Mosayebi G, Farshid AA, Mardani K. Microvesicles derived from mesenchymal stem cells: potent organelles for induction of tolerogenic signaling. Immunol Lett. 2012;147:47–54.
Ottoboni L, De Feo D, Merlini A, Martino G. Commonalities in immune modulation between mesenchymal stem cells (MSCs) and neural stem/precursor cells (NPCs). Immunol Lett. 2015;168:228–39.
Jiang Y, Tuan RS. Origin and function of cartilage stem/progenitor cells in osteoarthritis. Nat Rev Rheumatol. 2014;11:206–12.
Uth K, Trifonov D. Stem cell application for osteoarthritis in the knee joint: a minireview. World J Stem Cells. 2014;6:629–36.
Czekanska EM, Czekanska EM, Ralphs JR, Alini M, Stoddart MJ. Enhancing inflammatory and chemotactic signals to regulate bone regeneration. Eur Cells Mater. 2014;28:320–34.
Ulivi V, Tasso R, Cancedda R, Descalzi F. Mesenchymal stem cell paracrine activity is modulated by platelet lysate: induction of an inflammatory response and secretion of factors maintaining macrophages in a proinflammatory phenotype. Stem Cells Dev. 2014;23:1858–69.
Maijenburg MW, van der Schoot CE, Voermans C. Mesenchymal stromal cell migration: possibilities to improve cellular therapy. Stem Cells Dev. 2012;21:19–29.
Gomez-Rodriguez V, Orbe J, Martinez-Aguilar E, Rodriguez JA, Fernandez-Alonso L, Serneels J, et al. Functional MMP-10 is required for efficient tissue repair after experimental hind limb ischemia. FASEB J. 2015;29:960–72.
Bobadilla M, Sainz N, Abizanda G, Orbe J, Rodriguez JA, Páramo JA, et al. The CXCR4/SDF1 axis improves muscle regeneration through MMP-10 activity. Stem Cells Dev. 2014;23:1417-27.
Chamberlain G, Fox J, Ashton B, Middleton J. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25:2739–49.
Gallina C, Turinetto V, Giachino C. A new paradigm in cardiac regeneration: the mesenchymal stem cell secretome. Stem Cells Int. 2015;2015:765846.
Ando Y, Matsubara K, Ishikawa J, Fujio M, Shohara R, Hibi H, et al. Stem cell-conditioned medium accelerates distraction osteogenesis through multiple regenerative mechanisms. Bone. 2014;61:82–90.
Ma L, Aijima R, Hoshino Y, Yamaza H, Tomoda E, Tanaka Y, et al. Transplantation of mesenchymal stem cells ameliorates secondary osteoporosis through interleukin-17-impaired functions of recipient bone marrow mesenchymal stem cells in MRL/lpr mice. Stem Cell Res Ther. 2015;6:104.
Hayashi Y, Murakami M, Kawamura R, Ishizaka R, Fukuta O, Nakashima M. CXCL14 and MCP1 are potent trophic factors associated with cell migration and angiogenesis leading to higher regenerative potential of dental pulp side population cells. Stem Cell Res Ther. 2015;6:111.
Koga Y, Yasunaga M, Moriya Y, Akasu T, Fujita S, Yamamoto S, et al. Exosome can prevent RNase from degrading microRNA in feces. J Gastrointest Oncol. 2011;2:215–22.
Subra C, Grand D, Laulagnier K, Stella A, Lambeau G, Paillasse M, et al. Exosomes account for vesicle-mediated transcellular transport of activatable phospholipases and prostaglandins. J Lipid Res. 2010;51:2105–20.
Borges FT, Reis LA, Schor N. Extracellular vesicles: structure, function, and potential clinical uses in renal diseases. Braz J Med Biol Res. 2013;46:824–30.
Katsuda T, Kosaka N, Takeshita F, Ochiya T. The therapeutic potential of mesenchymal stem cell-derived extracellular vesicles. Proteomics. 2013;13:1637–53.
Cocucci E, Meldolesi J. Ectosomes and exosomes: shedding the confusion between extracellular vesicles. Trends Cell Biol. 2015;25:364–72.
Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–83.
György B, Szabó TG, Pásztói M, Pál Z, Misják P, Aradi B, et al. Membrane vesicles, current state-of-the-art: emerging role of extracellular vesicles. Cell Mol Life Sci. 2011;68:2667–88.
Tan SS, Yin Y, Lee T, Lai RC, Yeo RWY, Zhang B, et al. Therapeutic MSC exosomes are derived from lipid raft microdomains in the plasma membrane. J Extracell Vesicles. 2013;2:22614.
Lozito TP, Tuan RS. Endothelial and cancer cells interact with mesenchymal stem cells via both microparticles and secreted factors. J Cell Mol Med. 2014;18:2372–84.
Boomsma RA, Geenen DL. Evidence for transfer of membranes from mesenchymal stem cells to HL-1 cardiac cells. Stem Cells Int. 2014;2014:653734.
Kang K, Ma R, Cai W, Huang W, Paul C, Liang J, et al. Exosomes secreted from CXCR4 overexpressing mesenchymal stem cells promote cardioprotection via Akt signaling pathway following myocardial infarction. Stem Cells Int. 2015. doi:10.1155/2015/659890.
Hu G, Li Q, Niu X, Hu B, Liu J, Zhou S, et al. Exosomes secreted by human-induced pluripotent stem cell-derived mesenchymal stem cells attenuate limb ischemia by promoting angiogenesis in mice. Stem Cell Res Ther. 2015;6:10.
Zhao Q, Gregory CA, Lee RH, Reger RL, Qin L, Hai B, et al. MSCs derived from iPSCs with a modified protocol are tumor-tropic but have much less potential to promote tumors than bone marrow MSCs. Proc Natl Acad Sci U S A. 2015;112:530–5.
Baglio SR, Rooijers K, Koppers-Lalic D, Verweij FJ, Pérez Lanzón M, Zini N, et al. Human bone marrow- and adipose-mesenchymal stem cells secrete exosomes enriched in distinctive miRNA and tRNA species. Stem Cell Res Ther. 2015. doi:10.1186/s13287-015-0116-z.
Hudson M, Woodworth-Hobbs M, Rahnert J, Zheng B, Price S. Glucocorticoids reduce muscle atrophy-related microRNAs via exosomal microRNA packaging (11634). FASEB J. 2014;28 Suppl 1:1163.4.
Müller G. Microvesicles/exosomes as potential novel biomarkers of metabolic diseases. Diabetes Metab Syndr Obes. 2012;5:247–82.
Chen TS, Arslan F, Yin Y, Tan SS, Lai RC, Choo ABH, et al. Enabling a robust scalable manufacturing process for therapeutic exosomes through oncogenic immortalization of human ESC-derived MSCs. J Transl Med. 2011;9:47.
Lv H, Li L, Sun M, Zhang Y, Chen L, Rong Y, et al. Mechanism of regulation of stem cell differentiation by matrix stiffness. Stem Cell Res Ther. 2015;6:103.
Talele NP, Fradette J, Davies JE, Kapus A, Hinz B. Expression of α-smooth muscle actin determines the fate of mesenchymal stromal cells. Stem Cell Reports. 2015;4:1016–30.
Abdeen AA, Weiss JB, Lee J, Kilian KA. Matrix composition and mechanics direct proangiogenic signaling from mesenchymal stem cells. Tissue Eng Part A. 2014;20:2737–45.
Baker N, Tuan RS. The less-often-traveled surface of stem cells: caveolin-1 and caveolae in stem cells, tissue repair and regeneration. Stem Cell Res Ther. 2013;4:90.
Linero I, Chaparro O. Paracrine effect of mesenchymal stem cells derived from human adipose tissue in bone regeneration. PLoS One. 2014;9:e107001.
Jose S, Hughbanks ML, Binder BYK, Ingavle GC, Leach JK. Enhanced trophic factor secretion by mesenchymal stem/stromal cells with Glycine-Histidine-Lysine (GHK)-modified alginate hydrogels. Acta Biomater. 2014;10:1955–64.
Tratwal J, Mathiasen AB, Juhl M, Brorsen SK, Kastrup J, Ekblond A. Influence of vascular endothelial growth factor stimulation and serum deprivation on gene activation patterns of human adipose tissue-derived stromal cells. Stem Cell Res Ther. 2015;6:62.
Li C, Wu X, Tong J, Yang X, Zhao J, Zheng Q, et al. Comparative analysis of human mesenchymal stem cells from bone marrow and adipose tissue under xeno-free conditions for cell therapy. Stem Cell Res Ther. 2015;6:55.
Xie X, Ulici V, Alexander PG, Jiang Y, Zhang C, Tuan RS. Platelet-rich plasma inhibits mechanically induced injury in chondrocytes. Arthrosc J Arthrosc Relat Surg. 2015;31:1142–50.
Bellayr IH, Catalano JG, Lababidi S, Yang AX, Lo Surdo JL, Bauer SR, et al. Gene markers of cellular aging in human multipotent stromal cells in culture. Stem Cell Res Ther. 2014;5:59.
Nuschke A, Rodrigues M, Stolz DB, Chu CT, Griffith L, Wells A. Human mesenchymal stem cells/multipotent stromal cells consume accumulated autophagosomes early in differentiation. Stem Cell Res Ther. 2014;5:140.
Liew A, O’Brien T. Therapeutic potential for mesenchymal stem cell transplantation in critical limb ischemia. Stem Cell Res Ther. 2012;3:28.
Lan YW, Choo KB, Chen CM, Hung TH, Chen YB, Hsieh CH, et al. Hypoxia-preconditioned mesenchymal stem cells attenuate bleomycin-induced pulmonary fibrosis. Stem Cell Res Ther. 2015;6:97.
Maumus M, Manferdini C, Toupet K, Peyrafitte JA, Ferreira R, Facchini A, et al. Adipose mesenchymal stem cells protect chondrocytes from degeneration associated with osteoarthritis. Stem Cell Res. 2013;11:834–44.
Wu L, Leijten J, van Blitterswijk CA, Karperien M. Fibroblast growth factor-1 is a mesenchymal stromal cell-secreted factor stimulating proliferation of osteoarthritic chondrocytes in co-culture. Stem Cells Dev. 2013;22:2356–67.
Song X, Xie Y, Liu Y, Shao M, Wang W. Beneficial effects of coculturing synovial derived mesenchymal stem cells with meniscus fibrochondrocytes are mediated by fibroblast growth factor 1: increased proliferation and collagen synthesis. Stem Cells Int. 2015;2015:926325.
Leyh M, Seitz A, Dürselen L, Springorum HR, Angele P, Ignatius A, et al. Osteoarthritic cartilage explants affect extracellular matrix production and composition in cocultured bone marrow-derived mesenchymal stem cells and articular chondrocytes. Stem Cell Res Ther. 2014;5:77.
Lohan P, Coleman CM, Murphy JM, Griffin MD, Ritter T, Ryan AE. Changes in immunological profile of allogeneic mesenchymal stem cells after differentiation: should we be concerned? Stem Cell Res Ther. 2014;5:99.
Crop MJ, Baan CC, Korevaar SS, IJzermans JNM, Pescatori M, Stubbs AP, et al. Inflammatory conditions affect gene expression and function of human adipose tissue-derived mesenchymal stem cells. Clin Exp Immunol. 2010;162(I):474–86.
Waterman RS, Tomchuck SL, Henkle SL, Betancourt AM. A new mesenchymal stem cell (MSC) paradigm: polarization into a pro-inflammatory MSC1 or an immunosuppressive MSC2 phenotype. PLoS One. 2010;5:e10088.
Manferdini C, Maumus M, Gabusi E, Piacentini A, Filardo G, Peyrafitte JA, et al. Adipose-derived mesenchymal stem cells exert antiinflammatory effects on chondrocytes and synoviocytes from osteoarthritis patients through prostaglandin E2. Arthritis Rheum. 2013;65:1271–81.
Grassi F, Cattini L, Gambari L, Manferdini C, Piacentini A, Gabusi E, et al. T cell subsets differently regulate osteogenic differentiation of human mesenchymal stromal cells in vitro. J Tissue Eng Regen Med. 2016;10:305–14.
Faroni A, Mobasseri SA, Kingham PJ, Reid AJ. Peripheral nerve regeneration: experimental strategies and future perspectives. Adv Drug Deliv Rev. 2014. doi:10.1016/j.addr.2014.11.010.
Chen QH, Liu AR, Qiu HB, Yang Y. Interaction between mesenchymal stem cells and endothelial cells restores endothelial permeability via paracrine hepatocyte growth factor in vitro. Stem Cell Res Ther. 2015;6:44.
Bronckaers A, Hilkens P, Martens W, Gervois P, Ratajczak J, Struys T, et al. Mesenchymal stem/stromal cells as a pharmacological and therapeutic approach to accelerate angiogenesis. Pharmacol Ther. 2014;143:181–96.
Pati S, Khakoo AY, Zhao J, Jimenez F, Gerber MH, Harting M, et al. Human mesenchymal stem cells inhibit vascular permeability by modulating vascular endothelial cadherin/β-catenin signaling. Stem Cells Dev. 2011;20:89–101.
Li J, Ma Y, Teng R, Guan Q, Lang J, Fang J, et al. Transcriptional profiling reveals crosstalk between mesenchymal stem cells and endothelial cells promoting prevascularization by reciprocal mechanisms. Stem Cells Dev. 2015;24:610–23.
Hou Y, Ryu CH, Jun JA, Kim SM, Jeong CH, Jeun SS. IL-8 enhances the angiogenic potential of human bone marrow mesenchymal stem cells by increasing vascular endothelial growth factor. Cell Biol Int. 2014;38:1050–9.
Pedersen TO, Blois AL, Xue Y, Xing Z, Sun Y, Finne-Wistrand A, et al. Mesenchymal stem cells induce endothelial cell quiescence and promote capillary formation. Stem Cell Res Ther. 2014;5:23.
Lin RZ, Moreno-Luna R, Zhou B, Pu WT, Melero-Martin JM. Equal modulation of endothelial cell function by four distinct tissue-specific mesenchymal stem cells. Angiogenesis. 2012;15:443–55.
Kehoe S, Zhang XF, Boyd D. FDA approved guidance conduits and wraps for peripheral nerve injury: a review of materials and efficacy. Injury. 2012;43:553–72.
Tamaki T. Bridging long gap peripheral nerve injury using skeletal muscle-derived multipotent stem cells. Neural Regen Res. 2014;9:1333–6.
Oliveira JT, Bittencourt-Navarrete RE, de Almeida FM, Tonda-Turo C, Martinez AMB, Franca JG. Enhancement of median nerve regeneration by mesenchymal stem cells engraftment in an absorbable conduit: improvement of peripheral nerve morphology with enlargement of somatosensory cortical representation. Front Neuroanat. 2014;8:111.
Frattini F, Pereira Lopes FR, Almeida FM, Rodrigues RF, Boldrini LC, Tomaz MA, et al. Mesenchymal stem cells in a polycaprolactone conduit promote sciatic nerve regeneration and sensory neuron survival after nerve injury. Tissue Eng Part A. 2012;18:2030–9.
Carrier-Ruiz A, Evaristo-Mendonça F, Mendez-Otero R, Ribeiro-Resende V. Biological behavior of mesenchymal stem cells on poly-epsilon-caprolactone filaments and a strategy for tissue engineering of segments of the peripheral nerves. Stem Cell Res Ther. 2015;6:128.
Ryu H, Oh JE, Rhee KJ, Baik SK, Kim J, Kang SJ, et al. Adipose tissue-derived mesenchymal stem cells cultured at high density express IFN-β and suppress the growth of MCF-7 human breast cancer cells. Cancer Lett. 2015;37:213–21.
Maltman DJ, Hardy SA, Przyborski SA. Role of mesenchymal stem cells in neurogenesis and nervous system repair. Neurochem Int. 2011;59:347–56.
Kingham PJ, Kolar MK, Novikova LN, Novikov LN, Wiberg M. Stimulating the neurotrophic and angiogenic properties of human adipose-derived stem cells enhances nerve repair. Stem Cells Dev. 2014;23:741–54.
Bashir J, Sherman A, Lee H, Kaplan L, Hare JM. Mesenchymal stem cell therapies in the treatment of musculoskeletal diseases. PM R. 2014;6:61–9.
Schäfer S, Berger JV, Deumens R, Goursaud S, Hanisch UK, Hermans E. Influence of intrathecal delivery of bone marrow-derived mesenchymal stem cells on spinal inflammation and pain hypersensitivity in a rat model of peripheral nerve injury. J Neuroinflammation. 2014;11:157.
Jones J, Estirado A, Redondo C, Pacheco-Torres J, Sirerol-Piquer MS, Garcia-Verdugo JM, et al. Mesenchymal stem cells improve motor functions and decrease neurodegeneration in ataxic mice. Mol Ther. 2014;23:130–8.
Fargali S, Sadahiro M, Jiang C, Frick AL, Indall T, Cogliani V, et al. Role of neurotrophins in the development and function of neural circuits that regulate energy homeostasis. J Mol Neurosci. 2012;48:654–9.
McGregor NE, Poulton IJ, Walker EC, Pompolo S, Quinn JMW, Martin TJ, et al. Ciliary neurotrophic factor inhibits bone formation and plays a sex-specific role in bone growth and remodeling. Calcif Tissue Int. 2010;86:261–70.
Pasquin S, Sharma M, Gauchat JF. Ciliary neurotrophic factor (CNTF): new facets of an old molecule for treating neurodegenerative and metabolic syndrome pathologies. Cytokine Growth Factor Rev. 2015. doi:10.1016/j.cytogfr.2015.07.007.
Yu H, Fischer G, Ebert AD, Wu HE, Bai X, Hogan QH. Analgesia for neuropathic pain by dorsal root ganglion transplantation of genetically engineered mesenchymal stem cells: initial results. Mol Pain. 2015;11:1–13.
Li Y, Guo L, Ahn HS, Kim MH, Kim SW. Amniotic mesenchymal stem cells display neurovascular tropism and aid in the recovery of injured peripheral nerves. J Cell Mol Med. 2014;18:1028–34.
Teixeira FG, Panchalingam KM, Anjo SI, Manadas B, Pereira R, Sousa N, et al. Do hypoxia/normoxia culturing conditions change the neuroregulatory profile of Wharton Jelly mesenchymal stem cell secretome? Stem Cell Res Ther. 2015;6:133.
Amable PR, Teixeira MV, Carias RB, Granjeiro JM, Borojevic R. Protein synthesis and secretion in human mesenchymal cells derived from bone marrow, adipose tissue and Wharton’s jelly. Stem Cell Res Ther. 2014;5:53.
Jackson WM, Nesti LJ, Tuan RS. Potential therapeutic applications of muscle-derived mesenchymal stem and progenitor cells. Expert Opin Biol Ther. 2010;10:505–17.
Jackson WM, Aragon AB, Bulken-Hoover JD, Nesti LJ, Tuan RS. Putative heterotopic ossification progenitor cells derived from traumatized muscle. J Orthop Res. 2009;27:1645–51.
Jackson WM, Lozito TP, Djouad F, Kuhn NZ, Nesti LJ, Tuan RS. Differentiation and regeneration potential of mesenchymal progenitor cells derived from traumatized muscle tissue. J Cell Mol Med. 2011;15:2377–88.
Jackson WM, Aragon AB, Djouad F, Song Y, Koehler SM, Nesti LJ, et al. Mesenchymal progenitor cells derived from traumatized human muscle. J Tissue Eng Regen Med. 2009;3:129–38.
Jackson WM, Alexander PG, Bulken-Hoover JD, Vogler JA, Ji Y, McKay P, et al. Mesenchymal progenitor cells derived from traumatized muscle enhance neurite growth. J Tissue Eng Regen Med. 2013;7:443–51.
Jackson WM, Aragon AB, Onodera J, Koehler SM, Ji Y, Bulken-Hoover JD, et al. Cytokine expression in muscle following traumatic injury. J Orthop Res. 2011;29:1613–20.
Kluk MW, Ji Y, Shin EH, Amrani O, Onodera J, Jackson WM, et al. Fibroregulation of mesenchymal progenitor cells by BMP-4 after traumatic muscle injury. J Orthop Trauma. 2012;26:693–8.
Smith LR. Influencing the secretion of myogenic factors from mesenchymal stem cells. Stem Cell Res Ther. 2014;5:96.
Sohn J, Lu A, Tang Y, Wang B, Huard J. Activation of non-myogenic mesenchymal stem cells during the disease progression in dystrophic dystrophin/utrophin knockout mice. Hum Mol Genet. 2015;24:3814–29.
Gurgis C, Mokbel N, DiGirolamo DJ. Therapies for musculoskeletal disease: can we treat two birds with one stone? Curr Osteoporos Rep. 2014;12:142–53.
Sassoli C, Nosi D, Tani A, Chellini F, Mazzanti B, Quercioli F, et al. Defining the role of mesenchymal stromal cells on the regulation of matrix metalloproteinases in skeletal muscle cells. Exp Cell Res. 2014;323:297–313.
Verhoekx JSN, Mudera V, Walbeehm ET, Hovius SER. Adipose-derived stem cells inhibit the contractile myofibroblast in Dupuytren’s disease. Plast Reconstr Surg. 2013;132:1139–48.
Brew K, Nagase H. The tissue inhibitors of metalloproteinases (TIMPs): an ancient family with structural and functional diversity. Biochim Biophys Acta Mol Cell Res. 2010;1803:55–71.
Seebach E, Holschbach J, Buchta N, Bitsch RG, Kleinschmidt K, Richter W. Mesenchymal stromal cell implantation for stimulation of long bone healing aggravates Staphylococcus aureus induced osteomyelitis. Acta Biomater. 2015;21:165–77.
Aoyama T, Goto K, Kakinoki R, Ikeguchi R, Ueda M, Kasai Y, et al. An exploratory clinical trial for idiopathic osteonecrosis of femoral head by cultured autologous multipotent mesenchymal stromal cells augmented with vascularized bone grafts. Tissue Eng Part B Rev. 2014;20:233–42.
Aoyama T, Fujita Y, Madoba K, Nankaku M, Yamada M, Tomita M, et al. Rehabilitation program after mesenchymal stromal cell transplantation augmented by vascularized bone grafts for idiopathic osteonecrosis of the femoral head: a preliminary study. Arch Phys Med Rehabil. 2015;96:532–9.
Vogel S, Börger V, Peters C, Förster M, Liebfried P, Metzger K, et al. Necrotic cell-derived high mobility group box 1 attracts antigen-presenting cells but inhibits hepatocyte growth factor-mediated tropism of mesenchymal stem cells for apoptotic cell death. Cell Death Differ. 2015;22:1219–30.
Docheva D, Müller SA, Majewski M, Evans CH. Biologics for tendon repair. Adv Drug Deliv Rev. 2014;84:222–39.
Gaspar D, Holladay C, Pandit A, Zeugolis D. Progress in cell based therapies for tendon repair. Trends Biotechnol. 2015;84:240–56.
Angele P, Kujat R, Koch M, Zellner J. Role of mesenchymal stem cells in meniscal repair. J Exp Orthop. 2014;1:12.
Zellner J, Taeger CD, Schaffer M, Roldan JC, Loibl M, Mueller MB, et al. Are applied growth factors able to mimic the positive effects of mesenchymal stem cells on the regeneration of meniscus in the avascular zone? Biomed Res Int. 2014. doi:10.1155/2014/537686.
Ding Z, Huang H. Mesenchymal stem cells in rabbit meniscus and bone marrow exhibit a similar feature but a heterogeneous multi-differentiation potential: superiority of meniscus as a cell source for meniscus repair. BMC Musculoskelet Disord. 2015;16:65.
Centeno CJ, Pitts J, Al-sayegh H, Freeman MD. Anterior cruciate ligament tears treated with percutaneous injection of autologous bone marrow nucleated cells: a case series. J Pain Res. 2015;8:437–47.
Takayama K, Kawakami Y, Mifune Y, Matsumoto T, Tang Y, Cummins JH, et al. The effect of blocking angiogenesis on anterior cruciate ligament healing following stem cell transplantation. Biomaterials. 2015;60:9–19.
Hogan MV, Walker GN, Cui LR, Fu FH, Huard J. The role of stem cells and tissue engineering in orthopaedic sports medicine: current evidence and future directions. Arthrosc J Arthrosc Relat Surg. 2015;31:1017–21.
Carvalho ADM, Badial PR, Alvarez LEC, Yamada ALM, Borges AS, Deffune E, et al. Equine tendonitis therapy using mesenchymal stem cells and platelet concentrates: a randomized controlled trial. Stem Cell Res Ther. 2013;4:85.
Steplewski A, Fertala J, Beredjiklian P, Wang ML, Fertala A. Matrix-specific anchors: a new concept for targeted delivery and retention of therapeutic cells. Tissue Eng Part A. 2015;21:1207–16.
De Lisio M, Jensen T, Sukiennik RA, Huntsman HD, Boppart M. Substrate and strain alter the muscle-derived mesenchymal stem cell secretome to promote myogenesis. Stem Cell Res Ther. 2014;5:74.
Meleshko A, Prakharenia I, Kletski S, Isaikina Y. Chimerism of allogeneic mesenchymal cells in bone marrow, liver, and spleen after mesenchymal stem cells infusion. Pediatr Transplant. 2013;17:189–94.
Lozito TP, Taboas JM, Kuo CK, Tuan RS. Mesenchymal stem cell modification of endothelial matrix regulates their vascular differentiation. J Cell Biochem. 2009;107:706–13.
Lozito TP, Kuo CK, Taboas JM, Tuan RS. Human mesenchymal stem cells express vascular cell phenotypes upon interaction with endothelial cell matrix. J Cell Biochem. 2009;107:714–22.
Titorencu I, Pruna V, Jinga VV, Simionescu M. Osteoblast ontogeny and implications for bone pathology: an overview. Cell Tissue Res. 2014;355:23–33.
Chen WCW, Péault B, Huard J. Regenerative translation of human blood-vessel-derived MSC precursors. Stem Cells Int. 2015;2015:11.
Pisciotta A, Riccio M, Carnevale G, Lu A, De Biasi S, Gibellini L, et al. Stem cells isolated from human dental pulp and amniotic fluid improve skeletal muscle histopathology in mdx/SCID mice. Stem Cell Res Ther. 2015;6:156.
Mukherjee P, Mani S. Methodologies to decipher the cell secretome. Biochim Biophys Acta Proteins Proteomics. 2013;1834:2226–32.
Kupcova Skalnikova H. Proteomic techniques for characterisation of mesenchymal stem cell secretome. Biochimie. 2013;95:2196–211.
Caccia D, Dugo M, Callari M, Bongarzone I. Bioinformatics tools for secretome analysis. Biochim Biophys Acta Proteins Proteomics. 2013;1834:2442–53.
Brown KJ, Seol H, Pillai DK, Sankoorikal BJ, Formolo CA, Mac J, et al. The human secretome atlas initiative: implications in health and disease conditions. Biochim Biophys Acta Proteins Proteomics. 2013;1834:2454–61.
Roberts M, Wall IB, Bingham I, Icely D, Reeve B, Bure K, et al. The global intellectual property landscape of induced pluripotent stem cell technologies. Nat Biotechnol. 2014;32:742–8.
Sanz-Nogués C, O’Brien T. MSCs isolated from patients with ischemic vascular disease have normal angiogenic potential. Mol Ther. 2014;22:1888–9.
Lin W, Li M, Li Y, Sun X, Li X, Yang F, et al. Bone marrow stromal cells promote neurite outgrowth of spinal motor neurons by means of neurotrophic factors in vitro. Neurol Sci. 2014;35:449–57.
Lin R-Z, Moreno-Luna R, Li D, Jaminet SC, Greene AK, Melero-Martin JM. Human endothelial colony-forming cells serve as trophic mediators for mesenchymal stem cell engraftment via paracrine signaling. Proc Natl Acad Sci U S A. 2014;111:10137–42.
Seebach E, Freischmidt H, Holschbach J, Fellenberg J, Richter W. Mesenchymal stroma cells trigger early attraction of M1 macrophages and endothelial cells into fibrin hydrogels, stimulating long bone healing without long-term engraftment. Acta Biomater. 2014;10:4730–41.
Tsai TL, Wang B, Squire MW, Guo LW, Li WJ. Endothelial cells direct human mesenchymal stem cells for osteo- and chondro-lineage differentiation through endothelin-1 and AKT signaling. Stem Cell Res Ther. 2015;6:88.
Acknowledgements
This work is supported in part by the Commonwealth of Pennsylvania Department of Health (SAP 4100050913), NIH (5U18 TR000532), and US Department of Defense (W81XWH-08-2-0032, W81XWH-14-2-0003). A portion of the predoctoral training of HRH was supported by a Training Grant funded by the National Institute of Biomedical Imaging and Bioengineering, NIH (T32EB0010216). HRH acknowledges the faculty, staff, and students of CCME, where HRH served as a Research Fellow during a portion of the time required to draft and edit this document.
Authors’ contributions
HRH created the figure and tables, and researched, drafted, and arranged the document. RST suggested the content, edited the document, and was the invited, corresponding author. Both authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Hofer, H.R., Tuan, R.S. Secreted trophic factors of mesenchymal stem cells support neurovascular and musculoskeletal therapies. Stem Cell Res Ther 7, 131 (2016). https://doi.org/10.1186/s13287-016-0394-0
Published:
DOI: https://doi.org/10.1186/s13287-016-0394-0