
Abstract
DNA methylation, an epigenetic modification, regulates gene transcription and maintains genome stability. DNA methyltransferase (DNMT) inhibitors can activate silenced genes at low doses and cause cytotoxicity at high doses. The ability of DNMT inhibitors to reverse epimutations is the basis of their use in novel strategies for cancer therapy. In this review, we examined the literature on DNA methyltransferase inhibitors. We summarized the mechanisms underlying combination therapy using DNMT inhibitors and clinical trials based on combining hypomethylation agents with other chemotherapeutic drugs. We also discussed the efficacy of such compounds as antitumor agents, the need to optimize treatment schedules and the regimens for maximal biologic effectiveness. Notably, the combination of DNMT inhibitors and chemotherapy and/or immune checkpoint inhibitors may provide helpful insights into the development of efficient therapeutic approaches.
Similar content being viewed by others


Background
Surgery, chemotherapy and radiotherapy are the mainstays of cancer treatment. Radical operation is most often the first treatment for solid tumors. Patients for whom surgery is not an option usually receive chemotherapy and radiotherapy. Chemotherapy has limited applicability in tumor therapy because of the associated complications, including nausea, vomiting, myelosuppression and resistance. With the development of precision medicine, researchers are applying new therapies to target certain molecules within tumor cells to induce cell death. Immunotherapy has gained worldwide attention and is regarded as one of the most radical anticancer treatments to be applied to the clinic. However, immune evasion and immunosuppression complicate the immune response to tumors [1,2,3,4]. It is clear that cancer treatment has various challenges and that it is necessary to continually strive to develop new therapeutic approaches.
Epigenetics refers to inherited altered gene expression that does not involve DNA sequence alteration. Epigenetic alterations include DNA methylation, histone modification and microRNA (miRNA) alteration. Subtle epigenetic regulation controls the activity of genes to affect cancer initiation or progression [5]. Understanding the molecular mechanisms involved in the initiation and maintenance of epigenetic abnormalities in cancer has great potential for clinical translation [6].
DNA methylation is catalyzed by a group of enzymes called DNA methyltransferases (DNMTs) [7]. In mammals, the DNMT family has four members, DNMT1, DNMT3A, DNMT3B and DNMT3L. DNMT1 is required for the maintenance of methylation across the genome. DNMT3A and DNMT3B are referred to as de novo methyltransferases [8]. DNMT3L acts as a stimulator of the catalytic activity of DNMT3A and DNMT3B [9]. De novo DNA methyltransferases DNMT3A and DNMT3B in combination with DNMT3L establish a pattern of methylation that is then faithfully maintained through cell division by the maintenance methyltransferase DNMT1 [10]. DNMT alterations have been frequently observed in various types of tumors, indicating that these alterations accompany the occurrence and development of tumors [11].
DNA methylation occurs by the covalent addition of a methyl group at the 5-carbon of the cytosine ring, resulting in 5-methylcytosine formation in CpG regions, and this process is inhibited by DNMT inhibitors [12]. DNMT inhibitors activate the expression of silenced genes at low doses and are able to kill cancer cells at high doses [13, 14]. The hepatotoxicity caused by DNMT inhibitors limits their application in solid tumor treatment. However, DNMT inhibitors can be used to treat a variety of hematological tumors, including myelodysplastic syndrome (MDS), acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) [15,16,17].
The ability of DNMT inhibitors to kill tumor cells has been acknowledged since Monparlar et al. [18] performed their seminal work, which also found that decitabine is an effective cytostatic inhibitor of tumor cells in vitro. In recent years, some studies have shown that interactions between DNMT inhibitors and chemotherapeutic drugs make combining epigenetic therapy and chemotherapy an attractive approach to circumvent the limitations of chemotherapy alone [19]. Moreover, DNMT inhibitors can reverse epidermal growth factor (EGF) receptor (EGFR) methylation, which may enhance EGFR expression and reverse EGFR tyrosine kinase inhibitor (TKI) resistance [20, 21]. Understanding epigenetics helps us to develop new mechanistic insights into pathways of immune resistance so that immunotherapy may become more widely applied as a therapeutic option in common malignancies [22, 23].
This review describes some of the most recent and promising advances in DNMT inhibitor therapy with an emphasis on the likely implications of the application of DNMT inhibitors combined with other drugs for treating solid tumors.
DNA methylation in cancer epigenomics
In mammals, DNA methylation occurs almost exclusively in CpG regions. While 70%–80% of CpG sites are methylated, the remaining unmethylated CpG sites mostly occur in dense clusters referred to as CpG islands [24,25,26,27,28]. In cancer, aberrant methylation is characterized by the hypermethylation of CpG islands in tumor suppressor genes. There is a wealth of evidence that the hypermethylation of CpG islands in the promoter regions of tumor suppressor genes leads to their inactivation, and this modification is highly implicated in cancer development growth. In contrast, the upregulation of prometastatic genes induced by DNA hypomethylation promotes invasion and metastasis pathways, one of the most morbid aspects of cancer. Therefore, DNA hypermethylation and hypomethylation trigger different cellular mechanisms involved in cancer [29].
To ensure genomic integrity and stability, pericentromeric heterochromatin is highly methylated and satellite sequences and repetitive genomic sequences are silenced [30]. The loss of DNA methylation in these regions may be related to tumor development. Additionally, hypomethylated DNA may also activate latent, genome-incorporated viral sequences. For example, DNA methylation represses the expression of genital human papillomavirus (HPV) and Epstein-Barr virus proteins, which are associated with cervical cancer and nasopharyngeal carcinoma (NPC) progression, respectively [29, 30].
Methylation-associated gene silencing plays a critical role in tumor progression. Hypermethylated genes in regulatory regions are involved in a variety of important cellular pathways [30]. Taken together, these findings indicate that small noncoding RNAs and miRNAs play an important role in tumorigenesis. miRNA hypermethylation and hypomethylation frequently occur in human cancers. Understanding the cross talk between miRNAs and DNA may lead to the discovery of novel therapeutic targets [33, 34].
DNA hypomethylating drugs and their clinical application in solid tumors
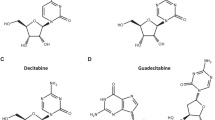
In the early 1960s, two nucleoside DNMT inhibitors were discovered. These were 5-azacytidine (azacitidine, AZA, Vidaza) and its derivative, 5-2′-deoxycytidine (decitabine, DAC, Dacogen). Over the last several decades, the anticancer activity of these agents has been examined [35]. Recently, some new nucleoside DNMT inhibitors and nonnucleoside DNMT inhibitors, including hydralazine, procaine and MG98, have been identified and are currently being investigated as antitumor drugs (Fig. 1).

Chemical structures of nucleoside and nonnucleoside DNA inhibitors
Nucleoside analogs
Azacitidine and decitabine are the most commonly used nucleoside agents in cancer. After cellular uptake, the first limiting step is the ATP-dependent phosphorylation of nucleosides to form monophosphorylated nucleotides [36]. These monophosphorylated nucleotides are incorporated into DNA in the place of cytosine. Then, DNMTs recognize the azacytosine-guanine dinucleotide and catalyze the methylation reaction by forming a covalent bond with the cytosine ring [37]. The covalent complex at C6 cannot be resolved through b-elimination, because of the presence of a nitrogen atom at position 5. Covalently trapped DNMTs are degraded, resulting in the depletion of cellular DNMTs [36, 38]. High-dose DNMT inhibitors facilitate the formation of bulky adducts, leading to replication fork stalling and DNA replication inhibition, which causes cell death [39]. When cells are treated with low DNMT inhibitor doses, the agents are still incorporated into DNA and bind DNMTs, leading to DNMT degradation. Without DNMTs to maintain DNA methylation, CpG sites lose their methylation after cell replication, and the transcription of genes previously silenced by promoter methylation is restored [40, 41]. Decitabine can decrease DNMT1 and DNMT3A expression, reversing abnormal transcription activation, while azacitidine only targets DNMT1 [42]. Another difference between these two drugs is that azacitidine can be incorporated into both DNA and RNA, whereas decitabine can only be incorporated into DNA [18].
Azacitidine, an analog of the cytidine pyrimidine nucleoside, has received approval by the US Food and Drug Administration for the treatment of all subtypes of MDS [43]. Despite marked activity in myeloid malignancy, the use of azacitidine in patients with solid tumors is limited by toxicity, myelosuppression; and low complete and partial response rates (Table 1) [44, 45]. Recently, a two-part phase I study evaluated CC-486 (an oral formulation of azacitidine) in combination with cytotoxic agents or as monotherapy for patients with advanced solid tumors. CC-486 monotherapy resulted in partial responses (three of eight patients) and stable disease (four of eight patients) in patients with nasopharyngeal cancer. Considering the potential benefit of CC-486 as monotherapy in this study, the combination of CC-486 with immune checkpoint inhibitors could be a promising area of clinical investigation [46].
Decitabine is a unique cytosine analog and has recently emerged as a therapy for MDS and CML. Although the promise of these hypomethylating drugs has not been realized for solid tumor cancer therapy, researchers contend that decitabine can achieve optimal biological effects at low doses [47]. In the 2000s, decitabine monotherapy produced unsatisfactory results for patients with solid tumors [48, 49]. Ten patients with refractory solid tumors were included in a phase I study, where decitabine was administered via continuous infusion at 2 mg/m2/day for 168 h. After the treatment cycles, no objective responses were observed, and seven of ten patients exhibited disease progression after one or two cycles. Samlowski et al. [49] examined the expression of select genes after the start of treatment, and their results showed MAGE-1 promoter hypomethylation.
Zebularine is a cytidine analog that lacks the amino group at position 4 of the pyrimidine ring. Zebularine has high stability and low toxicity, and it is stable at acidic and neutral pHs, enabling oral administration [37, 50]. When zebularine traps DNMT on DNA, zebularine becomes an obstacle for the second round of replication. This results in a collapsed replication fork and the formation of replication-dependent double stand breaks (DSBs) [51]. Moreover, zebularine can suppress the interaction of DNMT1 with G9a histone methyltransferases, which may regulate the survival and apoptosis of human cancer cells [52]. Transient zebularine exposure produces differential cell density-dependent responses and correlates with the overexpression of genes related to cancer stem cells and the key epithelial–mesenchymal transition process [53]. Although zebularine is more stable and less toxic than azacitidine and decitabine, clinical trials are required to demonstrate its therapeutic effect in solid tumors.
Guadecitabine (SGI-110) is a second-generation decitabine and deoxyguanosine compound with prolonged half-life and activity in AML and high-risk AML. Guadecitabine addresses the shortcomings of first-generation DNMT inhibitors that are susceptible to deamination by cytidine deaminase (CDA). CDA is found in multiple organs in the body, causing first-generation DNMT inhibitors to have short plasma half-lives. Guadecitabine has improved stability that confers enhanced DNA incorporation into dividing cells and is more resistant to CDA [54]. Based on these factors, it is believed that guadecitabine may be a more appropriate DNMT inhibitor than azacitidine and decitabine [55, 56]. Guadecitabine has been demonstrated to have clinical activity in MDS and AML [57, 58]. However, a substantial difference in cost in combination with a marginal difference in survival benefit might limit its use in the clinical setting [59].
Another cytosine analog, 4′-thio-2′-deoxycytidine (TdCyd), has been used in clinical trials for patients with advanced solid tumors [60]. This compound incorporates into the DNA sequence recognized by the bacterial C5 DNA methyltransferase M. 5-Fluoro-2′-deoxycytidine (FdCyd) has also been assessed in clinical trials for the treatment of advanced solid tumors, AML and multiple sclerosis (MS) [61, 62]. In both in vitro and in vivo models, TdCyd and FdCyd potently deplete DNMT1 in cancer and concomitantly inhibit tumor growth [63].
Nonnucleoside analogs
To overcome the disadvantages of nucleoside analogs, including poor bioavailability, chemical instability under physiological conditions and a lack of selectivity, nonnucleoside analogs have been developed over the last decades [64]. The structures of nonnucleoside analogs are very heterogeneous, but their mechanisms of action are independent of DNA incorporation. Some drugs (including procainamide, an amide, and its ester analog procaine) have been repurposed after they were shown to have demethylating effects. These agents show affinity for CpG-rich regions of DNA, blocking the activity of DNMTs and reactivating some tumor suppressor genes [65].
SGI-1027 was synthesized as a quinoline-based compound and was described for against DNMT1, DNMT3A and DNMT3B [66, 67]. After that, Valente et al. [68] and Rilova et al. [67] described two analogues of SGI-1027, which are MC3343 and MC3353. SGI-1027 and its analogue share DNA-competitive and AdoMet non-competitive behavior on DNMT1 [64]. SGI-1027 may inhibit DNMT activity, induce the degradation of DNMT1 and reactivate tumor suppressor genes [69]. SGI-1027 can also impair cervical cancer cell and hepatocellular carcinoma cell propagation by dramatically increasing apoptotic cell death and cell cycle arrest [69, 70]. As a novel DNMT inhibitor, MC3343 is more potent and selective than SGI-1027 toward other S-adenosylhomocysteine-dependent (SAM-dependent) methyltransferases [71, 72]. Zwergel et al. reported that MC3353 displays a stronger in cell demethylating ability than both azacitidine and decitabine. Besides, this compound proved antiproliferative activity in several cancer cell line types [73].
In addition to SGI-1027, some oligonucleotides are accommodated in the catalytic pocket of DNMTs, where they effectively function as competitive inhibitors. MG98 has shown interesting preclinical evidence that it can inhibit DNMT1 [74], allowing for the re-expression of tumor suppressor genes and tumor growth inhibition [75, 76]. In an open-label phase I study, patients with advanced solid malignancies were treated with escalating MG98 doses administered as a continuous infusion over 7 days repeated every 14 days. After two cycles, suppression of DNMT1 expression was observed in 26 of the 32 patients studied. One patient achieved a partial response, and another achieved prolonged disease stabilization [76].
N-Phthaloyl-L-tryptophan (RG108), a DNMT1 inhibitor [77], targets DNMT1 SAM cofactor binding. RG108 significantly inhibits the proliferation of endometrial cancer cells, blocks the cell cycle in the G2/M phase and induces apoptosis by increasing hMLH1 expression and inhibiting DNMT3B expression [78]. Selective nonnucleoside DNMT1 inhibitors in the DC_05 series of compounds can also play an anticancer role by inducing DNA hypomethylation to restore tumor suppressor gene expression [40]. Interestingly, the histone H3 lysine 9 methyltransferases (G9a/GLP) inhibitor BIX-01294 showed novel ability to inhibit the DNA methyltransferase DNMT3A at low micromolar levels without inhibition of DNMT1 and G9a [80]. Nanaomycin A is the first selective DNMT3B inhibitor that can induce genomic demethylation. Nanaomycin A interacts with DNMT3B amino acid residues that are involved in methylation, preventing DNMT3B from participating in normal DNA methylation [81]. Nanaomycin A treatment reduces global methylation levels in cancer cell lines and reactivates transcription of the RASSF1A tumor suppressor gene [82].
Experimental studies on the effects of combination therapies using DNMT inhibitors
Enhanced radiation sensitivity
Kumar et al. [83] examined γ-radiation-resistant and γ-radiation-sensitive cell lines to determine the relationship between radiation sensitivity and DNA methylation. They demonstrated that treating cells with decitabine and trichostatin A (TSA) before irradiation enhanced DNA strand breakage, G2/M phase arrest, apoptosis and cell death. Moreover, γ-radiation increased the transcriptional activity of the p16INK4a and ATM gene promoters by altering DNA methylation levels. 111Indium-labeled human epidermal growth factor (111In-DTPA-hEGF) is an auger electron-emitting agent that targets EGFR-overexpressing cells. Together with (111) In-DTPA-hEGF, decitabine can sensitize breast cancer to ionizing radiation and induce DNA destruction [84]. Kim et al. [85] investigated the underlying cellular mechanisms of combination treatment using ionizing irradiation and decitabine in human colon cancer cells. After this treatment, colon cancer cell growth was significantly lower than that with decitabine or radiotherapy alone, and increases in the number of G1-phase cells and the apoptosis rate were observed for colon cancer cells. Recently, Ou et al. [86] found that RG108 increased the radiosensitivity of esophageal cancer cells. Esophageal cancer cell apoptosis and G2/M-phase arrest were induced by X-ray irradiation and were significantly enhanced by RG108.
Increased sensitivity to anticancer drugs
In the 1990s, Fost. et al. [87] explored the combined use of decitabine and cisplatin in vitro. They demonstrated the synergistic cytotoxicity of this drug combination against a panel of six human cell lines. Epigenetic priming with decitabine can improve the sensitivity of gastric cancer cells to SN38 (doxorubicin) and cisplatin [88]. Low-nanomolar doses of decitabine and azacitidine induce sustained antitumor responses [89]. In myeloma cell lines, researchers observed a significant phenomenon of cell proliferation inhibition after combination therapy of decitabine with adriamycin [90]. Several studies have investigated the molecular mechanisms through which DNMT inhibitors affect the efficacy of other drugs (Fig. 2).

Molecular regulatory mechanisms of DNMT inhibitors in increasing the sensitivity to drugs. DNMT inhibitor treatment can increase the sensitivity of chemotherapeutic drugs via the methylation status of ARNTL, RASS1, MLH1, hMLH1, WT1 and BCL-2. DNMT inhibitors are able to sensitize tumor-targeting drugs through the induction of various proteins, such as EREG, EGFR and XAF1. They can also enhance immunotherapy by targeting EZH2 and MAGE-3
A comparative study showed that platinum-resistant cell lines exhibited more epigenetic alterations than platinum-sensitive cell lines, and the hypermethylation of promoter regions was significantly increased. The authors identified 14 genes that were hypermethylated in cisplatin-resistant cell lines but not in cisplatin-sensitive parental cell lines. Six of 14 genes (SAT, C8orf4, LAMB3, TUBB, G0S2 and MCAM) were cisplatin inducible in sensitive cell lines but not in resistant cell lines [91]. DNMT inhibitors demethylated the promoter CpG regions of ARNTL. The ARNTL protein suppressed NPC cell proliferation and enhanced cell sensitivity to cisplatin by targeting CDK5. ARNTL overexpression suppressed NPC cell proliferation in vitro and in vivo, and the opposite effect was observed following ARNTL silencing. Gene set enrichment analysis (GSEA) revealed that ARNTL is associated with the cell cycle and that ectopic expression and overexpression of ARNTL could induce G2-M phase arrest [92]. Moreover, in an in vivo melanoma model, DNMT inhibitors augmented the hypermethylation status of the RASSF1 gene promoter, targeted the CTGF and CYR61 genes through the hippocampal pathway and increased the sensitivity of bladder cancer cells to cisplatin and adriamycin [15]. Moreover, MLH1 expression was closely related to the methylation status of the hMLH1 promoter [93]. Ovarian cancer cell lines showed increased hMLH1 promoter methylation after developing drug resistance, and a correlation was observed between hMLH1 methylation and the general survival rate (p < 0.01) [94]. Several studies have shown that decitabine can reverse cisplatin resistance by inhibiting hMLH1 in human non-small cell lung cancer (NSCLC) and esophageal carcinoma [95, 96]. DNMT inhibitors augment the sensitivity of tumor cells to irinotecan drugs (CPT11/SN38) by targeting the BCL-2 oncogene and increasing BCL-2 protein expression [97, 98]. VHL-TGFBI hypomethylation was found to be related to the sensitivity to paclitaxel (PTX) [99].
Methylation of the EGFR promoter inhibits EGFR expression in a variety of tumor cells. Three NSCLC cell lines (H1650, H1299 and PC-9) with different EGFR mutation statuses and levels of EGFR-TKI sensitivity were used in this study. The results showed that the EGFR promoter region was unmethylated in PC-9 cells and that these cells were sensitive to gefitinib (an EGFR-TKI drug). In contrast, the EGFR promoter region was methylated in H1650 and H1299 cells, and the cells were resistant to gefitinib [100]. Treatment with decitabine resulted in the re-expression of EGFR in CAMA1 and MB453 cell lines, which are relatively resistant to gefitinib. However, after cotreatment with decitabine and gefitinib, a significant effect was observed on apoptosis induction. DNMT inhibitors can reverse the hypermethylation status of EGFR promoters in different cancers, which may enhance EGFR expression and reverse EGFR-TKI resistance [20, 21, 101]. Qu et al. [102] confirmed that upregulated EGFR expression through promoter demethylation was associated with the adenoma–carcinoma transition, and this was accompanied by an increase in EGFR phosphorylation, as assessed by reverse-phase protein analysis. Jiyoeu et al. [103] found that the hypomethylation of epidermal regulatory protein (EREG) binding with EGFR-induced gastric cancer cells grew. DNMT3b knockdown significantly increased EREG expression and did not significantly affect EREG promoter methylation. In another study, combined treatment with decitabine and gefitinib increased XIAP-associated factor 1 (XAF1) expression, which plays an important role in apoptosis [104].
Significantly tumor growth inhibition and prolonged survival were observed in the CT26 mouse model after treatment with a combination of PD-1 blockade and decitabine versus treatment with decitabine or PD-1 blockade alone. Decitabine may provide clinical benefits to patients with colorectal cancer and low microsatellite instability or microsatellite stability [105]. In NSCLC, combining the DNA hypomethylating agent azacytidine with anti-PD-1 therapy significantly reduced tumor size compared with that with anti-PD-1 therapy alone. This combination might therefore be a promising approach to overcoming anti-PD-1 resistance [106].
Identification of biomarkers
High levels of methylated CFTR are observed in breast cancer, and CFTR overexpression can inhibit breast cancer cell growth. Increased cell invasion was observed following CFTR knockdown. These results suggest that CFTR might be a diagnostic marker of breast cancer [107]. DACT2 is frequently inactivated by CpG methylation in NPC. DNMT inhibitors inhibit NPC cell proliferation and metastasis through the suppression of β-catenin/Cdc25c signaling. A study suggested that DACT2 promoter methylation was a potential epigenetic biomarker for the detection of NPC and for chemotherapy guidance [96]. Stewart et al. [98] showed that KRAS genomic status predicted decitabine sensitivity in ovarian cancer cells. Pretreatment with decitabine decreased the cytotoxic activity of MEK inhibitors in KRAS-mutant ovarian cancer cells, with reciprocal downregulation of DNMT1 and MEK/ERK phosphorylation. This study implicated KRAS status as a biomarker of drug response in ovarian cancer. BRAFV600e plays an important role in melanoma tumorigenesis. Hou et al. [109] investigated the role of BRAFV600E signaling in altering gene methylation in the genome of melanoma cells and identified genes coupled to BRAFV600E signaling through examination of methylation aberrations. The results indicate that a wide range of genes with broad functions are linked to BRAFV600E signaling through hypermethylation or hypomethylation. Low-dose decitabine therapy promotes antitumor T cell responses by promoting T cell proliferation, and an increased proportion of IFNγ + T cells may act as a prognostic biomarker of the decitabine-based antitumor therapy response [110].
Cancer cell reprogramming
Low-dose decitabine treatment remarkably enhanced the effects of cisplatin and gemcitabine on basal-like bladder cancer in vivo and in vitro. These effects were accompanied by decreases in genome-wide DNA methylation, gene re-expression and changes in key cellular regulatory pathways, including STAT3 signaling [111]. DNA methylation status sequencing at different time points during colitis-associated cancer (CAC) revealed that 811 genes were hypermethylated at different time points during CAC initiation and progression. These hypermethylated genes, including BAD and inositol polyphosphate phosphatase-like 1 (INPPL1) hub genes, are involved in the MARK and EGF/EGFR pathways [112]. Tumor growth and drug response were assessed in PANC-1 cells (pancreatic ductal adenocarcinoma, PDAC) after exposure to a noncytotoxic dose of azacitidine. The authors observed that unique peptides (SST and SSTR2) were expressed in the pancreas and confirmed that azacitidine epigenetically reprogrammed PANC-1 cells to induce anticancer effects [113].
DNMT inhibitors promoted MIG-6 re-expression by inhibiting MIG-6 promoter methylation. The negative feedback of MIG-6 expression increased the number of EGFR receptors [114]. Chou-Talalay analysis showed that, in bladder cancer cells, the combination of decitabine with an entinostat (ENT) histone deacetylase inhibitor could not reverse chemoresistance. However, the combination treatment between decitabine and ENT led to forkhead box class O1 (FoxO1) upregulation, and FoxO1 expression resulted in increased relapse-free survival in patients with bladder cancer. Moreover, this combination further activated proapoptotic Bim and p21, cell cycle regulators [115]. These results show that low FoxO1 expression in tumor specimens may be associated with resistance to cisplatin first-line therapy in patients with bladder cancer.
Eliciting an immune response against cancer
The immune system maintains the function of the body when attacked by external substances through its two roles as a "monitor" and "protector" [116, 117]. Deregulated immune systems cannot effectively kill tumor cells, leading to immune evasion [118]. There is evidence that tumor immune evasion is mediated by nonmutational epigenetic events involving chromatin and that epigenetics and mutations collaborate to determine the state of tumor progression. Therefore, epigenetic therapy has become a “double-edged sword” with potential value in immune therapy (Fig. 3) [119, 120].

DNMT inhibitors in immune-oncology
Although human endogenous retroviral sequences (ERVs) make up approximately 8.5% of our DNA, they have not been extensively studied because their repetitive nature complicates mapping [121]. Several studies have highlighted the importance of DNA methylation in the suppression of ERVs [122]. It is possible that DNMT inhibitors can reactive ERVs. After reactivation, repeat elements produced by ERVs may form nucleic acid molecules of various configurations that are then sensed by the innate immune machinery to trigger an immune response [120].
Decitabine treatment may result in the production of the antigen encoded by MAGE-1 (a cancer testis antigen (CTA) member). MAGE-1 is associated with major histocompatibility complex class I molecules at the cell surface for T-cell recognition [123]. Thus, CTAs are a potential source of new tumor cell surface antigens and are widely used in CAR T cell production [124, 125]. The efficacy of coupling an immune checkpoint blockade approach with a DNMT inhibitor may be increased by taking advantage of a bystander effect by attracting T cells to the tumor and simultaneously enforcing the uniform expression and display of CTAs [120].
Decitabine treatment enhances human IFNγ + T cell activation and proliferation and promotes Th1 polarization and the activity of cytotoxic T cells in vivo and in vitro. DNA hypomethylation directly enhances PD-L1 expression in tumor cells and increases the expression of immune-related genes and T cell infiltration [110]. Overexpression of DNMT1 and EZH2 can result in the consumption of B cells and prevent macrophage production. This may explain why decitabine can increase the antitumor T cell response [126]. In another study, Peng et al. announced that DNMT inhibitors may improve the clinical efficacy of MAGE-A3-specific T cell therapy by increasing target gene expression [127].
Clinical findings on DNMT inhibitor combination therapy in solid tumors
Combinations with platinum-based chemotherapy
The majority of combination DNMT inhibitor therapies assessed to date have involved the combination of decitabine and platinum drugs. We collected decitabine-based clinical trials from the National Center for Biotechnology Information (NCBI) database in April 2021 (Table 2).
In 2000, Schwartsmann et al. designed a clinical trial using a fixed dose of 33 mg/m2 cisplatin and four escalating doses of decitabine (45, 67, 90 and 120 mg/m2). However, only a short-lasting partial response was observed in a single patient with cervical cancer, and two minor responses were documented in patients with NSCLC and cervical cancer [128]. Pohlmann et al. also reported the administration of a decitabine-based combination in 2003. Patients with advanced cervical cancer received decitabine (50 mg/m2/day) during a 3-h continuous infusion on day 1, which was followed by the administration of cisplatin (33 g/m2/day) on day 4 of a 21-day cycle. Evaluation after 2 cycles revealed a satisfactory response rate, with eight patients (38.1%) achieving a partial response and five patients (23.8%) achieving stable disease [129].
Patients with ovarian cancer are often administered a platinum compound and a taxane. Several phase I or phase II clinical trials used a low dose of decitabine combined with carboplatin to treat platinum-resistant ovarian cancer or relapsed ovarian cancer (Table 2) [130,131,132,133,134,135]. Among those regimens, a clinical trial administered decitabine (7 mg/m2/day) on days 1–5 followed by reduced taxane and carboplatin. This approach achieved an effective clinical response, with nine patients (22.5%) achieving complete or partial response and nineteen patients (47.5%) achieving stable disease. Notably, MLH1, RASSF1A, HOXA10 and HOXA11 demethylation in tumors was positively correlated with progression-free survival (p < 0.05). Low-dose decitabine altered gene DNA methylation and cancer pathways, restored carboplatin sensitivity in patients with heavily pretreated ovarian cancer and resulted in a high objective response rate and prolonged progression-free survival [132].
Combinations with other chemotherapeutic drugs
A phase I clinical trial recruited pediatric patients with solid tumors. These patients were treated with decitabine (5 mg/m2/day) during a 1-h continuous infusion on days 1–7 and then with doxorubicin (45 mg/m2/day) and cyclophosphamide (1 g/m2/day) on day 7. In total, 60% (12/20) and 87.5% (14/16) of patients displayed significant MAGE-1 and HbF demethylation, respectively, in peripheral blood mononuclear cells [136]. In another phase I study, Stathis et al. studied different doses of decitabine and vorinostat (six sequential and three concurrent doses). The maximum tolerated dose on the sequential schedule was 10 mg/m2/day decitabine on days 1–5 and 200 mg vorinostat three times a day on days 6–12. The results showed that 11 of the 38 patients with solid tumors and non-Hodgkin's lymphoma had a stable response after four treatment cycles [137].
Combinations with molecular targeted therapy
Garrido-Laguna et al. conducted a phase I study to evaluate decitabine in combination with panitumumab (an antibody against EGFR) in wild-type KRAS metastatic colorectal cancer (mCRC) patients. Two of 20 patients (10%) had a partial response, but both had previously received cetuximab and another treatment. Ten patients had stable disease (three of them had stable disease longer than 16 weeks). Decreased MAGE promoter methylation was not observed in peripheral blood mononuclear cells [138].
The BRAF gene regulates the methylation of a wide number of genes and affects multiple cellular functions [109]. A phase Ib study used 3 + 3 dose escalation combining different doses and schedules of subcutaneous decitabine administration with the standard oral dose of vemurafenib (960 mg) twice daily. Fourteen V600EBRAF-positive patients with metastatic melanoma were placed into four groups, and each group received a different regimen. Three patients achieved a complete response, three had a partial response, and five had stable disease. Preclinical assessment demonstrated that this combination treatment delayed the development of acquired resistance and improved the duration of treatment sensitivity [139].
Combinations with immunotherapy
In NSCLC, immunotherapy produced an astounding result. An objective response (a complete or partial response) was observed in 5 of 49 patients with NSCLC. These patients passed the 24-week point without progression with subsequent immune checkpoint therapy, and three of the five developed high-grade partial responses (according to the Response Evaluation Criteria in Solid Tumors (RECIST)) that remained durable over 2.5 years [140, 141]. Eighty-six anti-PD-1 treatment-naïve patients were randomly assigned (1:2) to camrelizumab (200 mg) monotherapy or decitabine (10 mg/d, days 1–5) plus camrelizumab (200 mg, day 8) combination therapy administered every 3 weeks. At the time of analysis, the response duration rates of camrelizumab monotherapy and decitabine plus camrelizumab combined therapy at 6 months were 76% and 100%, respectively. The complete response rate was 32% (6 of 19 patients) with camrelizumab monotherapy versus 71% (30 of 42 patients) for those administered decitabine plus camrelizumab (p = 0.003). Researchers concluded that decitabine plus camrelizumab may reverse the resistance to PD-1 inhibitors in patients with relapsed/refractory classical Hodgkin lymphoma (cHL) [142].
Two different clinical trials combined decitabine and cytokine-induced killer (CIK) cells. The first study divided 52 recurrent ovarian cancer patients with platinum resistance into two groups. Patients in the paclitaxel and carboplatin (DTC) group were treated with decitabine and a reduced dose of paclitaxel and carboplatin. Patients in the DTC + CIK cell group were treated with the same regimens and received CIK cell therapy. Notably, DTC + CIK cell treatment in platinum-resistant/refractory patients led to an overall response rate of 87.50%, a progression-free survival tome of 8 months and an overall survival time of 19 months. DTC treatment in platinum-resistant/refractory patients led to an overall response rate of 22.5%, a progression-free survival time of 4 months and an overall survival time of 12 months. These data indicate that decitabine might show a remarkable clinical response when combined with adoptive immunotherapy in patients with platinum-resistant/refractory ovarian cancer [143]. Another clinical trial enrolled 45 patients with drug-resistant relapsed/refractory esophageal, gastric or colorectal cancers. Patients received decitabine on days 1–5 and were then divided into two groups. Some patients were treated with previous chemotherapy (the DC cohort), while others received CIK cell therapy after previous chemotherapy (DC + CIK cell cohort). In the DC cohort, patients had an overall response rate of 20%, a disease control rate of 70%, a progression-free survival time of 4 months and an overall survival time of 12 months. However, in the DC + CIK cell cohort, the patients had an overall response rate of 28%, a disease control rate of 92%, a progression-free survival time of 6 months and an overall survival time of 11 months [144]. The toxicity and overall response rate observed did not significantly differ between cancer types and treatment cohorts.
Conclusion
The mechanism by which DNMT inhibitors function in combination with antitumor drugs has not yet been fully elucidated. However, the studies explored in this review show that, in most cases, combination treatment with DNMT inhibitors and antitumor drugs has higher efficacy than treatment using antitumor drugs alone. However, there are many hurdles to overcome before the routine clinical application of this therapeutic approach. The sample size for clinical trials is small, with most studies involving fewer than 50 patients. Moreover, there are very few studies that use randomized, blind, controlled designs.
Although combination treatments using DNMT inhibitors and antitumor drugs may provide helpful insights into the development of efficient therapeutic approaches for cancer treatment, further investigation is needed. Such studies should include randomized controlled trials with large sample sizes.
Availability of data and materials
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Abbreviations
- DNMT:
-
DNA methylation transferase
- miRNA:
-
MicroRNA
- EGFR:
-
Epidermal growth factor receptor
- NPC:
-
Nasopharyngeal carcinoma
- MDS:
-
Myelodysplastic syndrome
- CML:
-
Chronic myeloid leukemia
- AML:
-
Acute myeloid leukemia
- TKI:
-
Tyrosine kinase inhibitor
- AdoMet:
-
S-adoenosyl-L-methionine
- SAM:
-
S-adenosylhomosteine
- TSA:
-
Trichostatin A
- NSCLC:
-
Non-small cell lung cancer
- RR:
-
Response rate
- PFS:
-
Progression-free survival
- CTAs:
-
Cancer testis antigen
- INPPL1:
-
Inositol polyphosphate phosphatase-like 1
- CR:
-
Complete response
- PR:
-
Partial response
- SD:
-
Stable disease
- PD:
-
Progressive disease
References
Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discov. 2019;18(3):175–96. https://doi.org/10.1038/s41573-018-0006-z.
Fesnak AD, June CH, Levine BL. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat Rev Cancer. 2016;16(9):566–81. https://doi.org/10.1038/nrc.2016.97.
Antonarakis ES, Piulats JM, Gross-Goupil M, et al. Pembrolizumab for treatment-refractory metastatic castration-resistant prostate cancer: multicohort, open-label Phase II KEYNOTE-199 study. J Clin Oncol. 2020;38(5):395–405. https://doi.org/10.1200/JCO.19.01638.
Le DT, Kim TW, Van Cutsem E, et al. Phase II open-label study of pembrolizumab in treatment-refractory, microsatellite instability-high/mismatch repair-deficient metastatic colorectal cancer: KEYNOTE-164. J Clin Oncol. 2020;38(1):11–9. https://doi.org/10.1200/JCO.19.02107.
Chan TA, Glockner S, Yi JM, et al. Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis. PLoS Med. 2008;5(5): e114. https://doi.org/10.1371/journal.pmed.0050114.
Tsai H-C, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Cell Res. 2011;21(3):502–17. https://doi.org/10.1038/cr.2011.24.
Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74(1):481–514. https://doi.org/10.1146/annurev.biochem.74.010904.153721.
Zhang W, Xu J. DNA methyltransferases and their roles in tumorigenesis. Biomark Res. 2017;5:1. https://doi.org/10.1186/s40364-017-0081-z.
Jeltsch A. Molecular enzymology of mammalian DNA methyltransferases. Curr Top Microbiol Immunol. 2006;301:203–25. https://doi.org/10.1007/3-540-31390-7_7.
Schübeler D. Function and information content of DNA methylation. Nature. 2015;517(7534):321–6. https://doi.org/10.1038/nature14192.
Zhang J, Yang C, Wu C, Cui W, Wang L. DNA methyltransferases in cancer: biology, paradox, aberrations, and targeted therapy. Cancers. 2020;12(8):2123. https://doi.org/10.3390/cancers12082123.
Kim M, Costello J. DNA methylation: an epigenetic mark of cellular memory. Exp Mol Med. 2017;49(4): e322. https://doi.org/10.1038/emm.2017.10.
Sato T, Issa J-PJ, Kropf P. DNA hypomethylating drugs in cancer therapy. Cold Spring Harb Perspect Med. 2017;7(5):6948. https://doi.org/10.1101/cshperspect.a026948.
Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against cancer. Annu Rev Med. 2016;67(1):73–89. https://doi.org/10.1146/annurev-med-111314-035900.
Khandelwal M, Anand V, Appunni S, et al. Decitabine augments cytotoxicity of cisplatin and doxorubicin to bladder cancer cells by activating hippo pathway through RASSF1A. Mol Cell Biochem. 2018;446(1–2):105–14. https://doi.org/10.1007/s11010-018-3278-z.
Pinto A, Maio M, Attadia V, Zappacosta S, Cimino R. Modulation of HLA-DR antigens expression in human myeloid leukaemia cells by cytarabine and 5-aza-2′-deoxycytidine. Lancet Lond Engl. 1984;2(8407):867–8. https://doi.org/10.1016/s0140-6736(84)90900-0.
Mizuno S, Chijiwa T, Okamura T, et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97(5):1172–9. https://doi.org/10.1182/blood.v97.5.1172.
Momparler RL. Molecular, cellular and animal pharmacology of 5-aza-2′-deoxycytidine. Pharmacol Ther. 1985;30(3):287–99. https://doi.org/10.1016/0163-7258(85)90053-1.
Nie J, Liu L, Li X, Han W. Decitabine, a new star in epigenetic therapy: the clinical application and biological mechanism in solid tumors. Cancer Lett. 2014;354(1):12–20. https://doi.org/10.1016/j.canlet.2014.08.010.
Montero AJ, Díaz-Montero CM, Mao L, et al. Epigenetic inactivation of EGFR by CpG island hypermethylation in cancer. Cancer Biol Ther. 2006;5(11):1494–501. https://doi.org/10.4161/cbt.5.11.3299.
Micevic G, Theodosakis N, Bosenberg M. Aberrant DNA methylation in melanoma: biomarker and therapeutic opportunities. Clin Epigenet. 2017;9:34. https://doi.org/10.1186/s13148-017-0332-8.
Olino K, Park T, Ahuja N. Exposing hidden targets: combining epigenetic and immunotherapy to overcome cancer resistance. Semin Cancer Biol. 2020;65:114–22. https://doi.org/10.1016/j.semcancer.2020.01.001.
Topper MJ, Vaz M, Marrone KA, Brahmer JR, Baylin SB. The emerging role of epigenetic therapeutics in immuno-oncology. Nat Rev Clin Oncol. 2020;17(2):75–90. https://doi.org/10.1038/s41571-019-0266-5.
Ehrlich M, Gama-Sosa MA, Huang LH, et al. Amount and distribution of 5-methylcytosine in human DNA from different types of tissues of cells. Nucleic Acids Res. 1982;10(8):2709–21.
Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16(1):6–21. https://doi.org/10.1101/gad.947102.
Lister R, Pelizzola M, Dowen RH, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462(7271):315–22. https://doi.org/10.1038/nature08514.
Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA. 2000;97(10):5237–42.
Xin Y, O’Donnell AH, Ge Y, et al. Role of CpG context and content in evolutionary signatures of brain DNA methylation. Epigenetics. 2011;6(11):1308–18. https://doi.org/10.4161/epi.6.11.17876.
Cheishvili D, Boureau L, Szyf M. DNA demethylation and invasive cancer: implications for therapeutics. Br J Pharmacol. 2015;172(11):2705–15. https://doi.org/10.1111/bph.12885.
Kulis M, Esteller M. DNA methylation and cancer. Adv Genet. 2010;70:27–56. https://doi.org/10.1016/B978-0-12-380866-0.60002-2.
Takacs M, Banati F, Koroknai A, et al. Epigenetic regulation of latent Epstein-Barr virus promoters. Biochim Biophys Acta. 2010;1799(3–4):228–35. https://doi.org/10.1016/j.bbagrm.2009.10.005.
Badal V, Chuang LSH, Tan EH-H, et al. CpG methylation of human papillomavirus type 16 DNA in cervical cancer cell lines and in clinical specimens: genomic hypomethylation correlates with carcinogenic progression. J Virol. 2003;77(11):6227–34. https://doi.org/10.1128/jvi.77.11.6227-6234.2003.
Lu J, Getz G, Miska EA, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435(7043):834–8. https://doi.org/10.1038/nature03702.
Wang S, Wu W, Claret FX. Mutual regulation of microRNAs and DNA methylation in human cancers. Epigenetics. 2017;12(3):187–97. https://doi.org/10.1080/15592294.2016.1273308.
Linnekamp JF, Butter R, Spijker R, Medema JP, van Laarhoven HWM. Clinical and biological effects of demethylating agents on solid tumours—a systematic review. Cancer Treat Rev. 2017;54:10–23. https://doi.org/10.1016/j.ctrv.2017.01.004.
Stresemann C, Lyko F. Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int J Cancer. 2008;123(1):8–13. https://doi.org/10.1002/ijc.23607.
Nunes SP, Henrique R, Jerónimo C, Paramio JM. DNA methylation as a therapeutic target for bladder cancer. Cells. 2020;9(8):1850. https://doi.org/10.3390/cells9081850.
Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc Natl Acad Sci USA. 1984;81(22):6993–7. https://doi.org/10.1073/pnas.81.22.6993.
Qin T, Youssef EM, Jelinek J, et al. Effect of cytarabine and decitabine in combination in human leukemic cell lines. Clin Cancer Res. 2007;13(14):4225–32. https://doi.org/10.1158/1078-0432.CCR-06-2762.
Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20(1):85–93. https://doi.org/10.1016/0092-8674(80)90237-8.
Momparler RL. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine). Semin Hematol. 2005;42(3 Suppl 2):S9-16.
Ferlay J, Soerjomataram I, Dikshit R, et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359-386. https://doi.org/10.1002/ijc.29210.
Kaminskas E, Farrell AT, Wang Y-C, Sridhara R, Pazdur R. FDA drug approval summary: azacitidine (5-azacytidine, Vidaza) for injectable suspension. Oncologist. 2005;10(3):176–82. https://doi.org/10.1634/theoncologist.10-3-176.
Weiss AJ, Metter GE, Nealon TF, et al. Phase II study of 5-azacytidine in solid tumors. Cancer Treat Rep. 1977;61(1):55–8.
Weiss AJ, Stambaugh JE, Mastrangelo MJ, Laucius JF, Bellet RE. Phase I study of 5-azacytidine (NSC-102816). Cancer Chemother Rep. 1972;56(3):413–9.
Von Hoff DD, Rasco DW, Heath EI, et al. Phase I study of CC-486 alone and in combination with carboplatin or nab-paclitaxel in patients with relapsed or refractory solid tumors. Clin Cancer Res. 2018;24(17):4072–80. https://doi.org/10.1158/1078-0432.CCR-17-3716.
Li X, Mei Q, Nie J, Fu X, Han W. Decitabine: a promising epi-immunotherapeutic agent in solid tumors. Expert Rev Clin Immunol. 2015;11(3):363–75. https://doi.org/10.1586/1744666X.2015.1002397.
Aparicio A, Eads CA, Leong LA, et al. Phase I trial of continuous infusion 5-aza-2′-deoxycytidine. Cancer Chemother Pharmacol. 2003;51(3):231–9. https://doi.org/10.1007/s00280-002-0563-y.
Samlowski WE, Leachman SA, Wade M, et al. Evaluation of a 7-day continuous intravenous infusion of decitabine: inhibition of promoter-specific and global genomic DNA methylation. J Clin Oncol. 2005;23(17):3897–905. https://doi.org/10.1200/JCO.2005.06.118.
Yoo CB, Cheng JC, Jones PA. Zebularine: a new drug for epigenetic therapy. Biochem Soc Trans. 2004;32(Pt 6):910–2. https://doi.org/10.1042/BST0320910.
Orta ML, Pastor N, Burgos-Morón E, et al. Zebularine induces replication-dependent double-strand breaks which are preferentially repaired by homologous recombination. DNA Repair. 2017;57:116–24. https://doi.org/10.1016/j.dnarep.2017.07.002.
Ye K, Wang S, Wang J, Han H, Ma B, Yang Y. Zebularine enhances apoptosis of human osteosarcoma cells by suppressing methylation of ARHI. Cancer Sci. 2016;107(12):1851–7. https://doi.org/10.1111/cas.13088.
Raggi C, Factor VM, Seo D, et al. Epigenetic reprogramming modulates malignant properties of human liver cancer. Hepatol Baltim Md. 2014;59(6):2251–62. https://doi.org/10.1002/hep.27026.
Wong KK, Hassan R, Yaacob NS. Hypomethylating agents and immunotherapy: therapeutic synergism in acute myeloid leukemia and myelodysplastic syndromes. Front Oncol. 2021. https://doi.org/10.3389/fonc.2021.624742.
Daher-Reyes GS, Merchan BM, Yee KWL. Guadecitabine (SGI-110): an investigational drug for the treatment of myelodysplastic syndrome and acute myeloid leukemia. Expert Opin Investig Drugs. 2019;28(10):835–49. https://doi.org/10.1080/13543784.2019.1667331.
Agrawal K, Das V, Vyas P, Hajdúch M. Nucleosidic DNA demethylating epigenetic drugs—a comprehensive review from discovery to clinic. Pharmacol Ther. 2018;188:45–79. https://doi.org/10.1016/j.pharmthera.2018.02.006.
Garcia-Manero G, Roboz G, Walsh K, et al. Guadecitabine (SGI-110) in patients with intermediate or high-risk myelodysplastic syndromes: phase 2 results from a multicentre, open-label, randomised, phase 1/2 trial. Lancet Haematol. 2019;6(6):e317–27. https://doi.org/10.1016/S2352-3026(19)30029-8.
Chung W, Kelly AD, Kropf P, et al. Genomic and epigenomic predictors of response to guadecitabine in relapsed/refractory acute myelogenous leukemia. Clin Epigenet. 2019;11(1):106. https://doi.org/10.1186/s13148-019-0704-3.
Saygin C, Carraway HE. Emerging therapies for acute myeloid leukemia. J Hematol Oncol. 2017;10(1):93. https://doi.org/10.1186/s13045-017-0463-6.
National Cancer Institute (NCI). Phase I trial of 4′-Thio-2′-Deoxycytidine (TdCyd) in patients with advanced solid tumors. clinicaltrials.gov; 2021. https://clinicaltrials.gov/ct2/show/NCT02423057. Accessed 22 Apr 2021.
City of Hope Medical Center. Phase I trial of 5-Fluoro-2′-Deoxycytidine with tetrahydrouridine. clinicaltrials.gov; 2015. https://clinicaltrials.gov/ct2/show/NCT00359606. Accessed 22 Apr 2021.
City of Hope Medical Center. A phase I study of 5-Fluoro-2′-Deoxycytidine with Tetrahydrouridine (FdCyd + THU) in myeloid leukemia and MDS. clinicaltrials.gov; 2015. https://clinicaltrials.gov/ct2/show/NCT01041443. Accessed 22 Apr 2021.
Thottassery JV, Sambandam V, Allan PW, et al. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4′-thio-2′-deoxycytidine and 5-aza-4′-thio-2′-deoxycytidine. Cancer Chemother Pharmacol. 2014;74(2):291–302. https://doi.org/10.1007/s00280-014-2503-z.
Gros C, Fleury L, Nahoum V, et al. New insights on the mechanism of quinoline-based DNA Methyltransferase inhibitors. J Biol Chem. 2015;290(10):6293–302. https://doi.org/10.1074/jbc.M114.594671.
Castillo-Aguilera O, Depreux P, Halby L, Arimondo PB, Goossens L. DNA methylation targeting: the DNMT/HMT crosstalk challenge. Biomolecules. 2017;7(1):3. https://doi.org/10.3390/biom7010003.
Datta J, Ghoshal K, Denny WA, et al. A new class of quinoline-based DNA hypomethylating agents reactivates tumor suppressor genes by blocking DNA methyltransferase 1 activity and inducing its degradation. Cancer Res. 2009;69(10):4277–85. https://doi.org/10.1158/0008-5472.CAN-08-3669.
Rilova E, Erdmann A, Gros C, et al. Design, synthesis and biological evaluation of 4-amino-N- (4-aminophenyl)benzamide analogues of quinoline-based SGI-1027 as inhibitors of DNA methylation. ChemMedChem. 2014;9(3):590–601. https://doi.org/10.1002/cmdc.201300420.
Valente S, Trisciuoglio D, De Luca T, et al. 1,3,4-Oxadiazole-containing histone deacetylase inhibitors: anticancer activities in cancer cells. J Med Chem. 2014;57(14):6259–65. https://doi.org/10.1021/jm500303u.
Sun N, Zhang J, Zhang C, Zhao B, Jiao A. DNMTs inhibitor SGI-1027 induces apoptosis in Huh7 human hepatocellular carcinoma cells. Oncol Lett. 2018;16(5):5799–806. https://doi.org/10.3892/ol.2018.9390.
She S, Zhao Y, Kang B, et al. Combined inhibition of JAK1/2 and DNMT1 by newly identified small-molecule compounds synergistically suppresses the survival and proliferation of cervical cancer cells. Cell Death Dis. 2020;11(9):724. https://doi.org/10.1038/s41419-020-02934-8.
Valente S, Liu Y, Schnekenburger M, et al. Selective non-nucleoside inhibitors of human DNA methyltransferases active in cancer including in cancer stem cells. J Med Chem. 2014;57(3):701–13. https://doi.org/10.1021/jm4012627.
Zwergel C, Fioravanti R, Stazi G, et al. Novel quinoline compounds active in cancer cells through coupled DNA methyltransferase inhibition and degradation. Cancers. 2020;12(2):447. https://doi.org/10.3390/cancers12020447.
Zwergel C, Schnekenburger M, Sarno F, et al. Identification of a novel quinoline-based DNA demethylating compound highly potent in cancer cells. Clin Epigenet. 2019;11(1):68. https://doi.org/10.1186/s13148-019-0663-8.
Winquist E, Knox J, Ayoub J-P, et al. Phase II trial of DNA methyltransferase 1 inhibition with the antisense oligonucleotide MG98 in patients with metastatic renal carcinoma: a National Cancer Institute of Canada Clinical Trials Group investigational new drug study. Investig New Drugs. 2006;24(2):159–67. https://doi.org/10.1007/s10637-006-5938-1.
Amato RJ, Stephenson J, Hotte S, et al. MG98, a second-generation DNMT1 inhibitor, in the treatment of advanced renal cell carcinoma. Cancer Investig. 2012;30(5):415–21. https://doi.org/10.3109/07357907.2012.675381.
Plummer R, Vidal L, Griffin M, et al. Phase I study of MG98, an oligonucleotide antisense inhibitor of human DNA methyltransferase 1, given as a 7-day infusion in patients with advanced solid tumors. Clin Cancer Res. 2009;15(9):3177–83. https://doi.org/10.1158/1078-0432.CCR-08-2859.
Asgatay S, Champion C, Marloie G, et al. Synthesis and evaluation of analogues of N-phthaloyl-l-tryptophan (RG108) as inhibitors of DNA methyltransferase 1. J Med Chem. 2014;57(2):421–34. https://doi.org/10.1021/jm401419p.
Yang L, Hou J, Cui X-H, Suo L-N, Lv Y-W. RG108 induces the apoptosis of endometrial cancer Ishikawa cell lines by inhibiting the expression of DNMT3B and demethylation of HMLH1. Eur Rev Med Pharmacol Sci. 2017;21(22):5056–64. https://doi.org/10.26355/eurrev_201711_13818.
Chen S, Wang Y, Zhou W, et al. Identifying novel selective non-nucleoside DNA methyltransferase 1 inhibitors through docking-based virtual screening. J Med Chem. 2014;57(21):9028–41. https://doi.org/10.1021/jm501134e.
Rotili D, Tarantino D, Marrocco B, et al. Properly substituted analogues of BIX-01294 lose inhibition of G9a histone methyltransferase and gain selective anti-DNA methyltransferase 3A activity. PLoS ONE. 2014;9(5): e96941. https://doi.org/10.1371/journal.pone.0096941.
Caulfield T, Medina-Franco JL. Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanaomycin A. J Struct Biol. 2011;176(2):185–91. https://doi.org/10.1016/j.jsb.2011.07.015.
Kuck D, Caulfield T, Lyko F, Medina-Franco JL. Nanaomycin A selectively inhibits DNMT3B and reactivates silenced tumor suppressor genes in human cancer cells. Mol Cancer Ther. 2010;9(11):3015–23. https://doi.org/10.1158/1535-7163.MCT-10-0609.
Kumar A, Rai PS, Upadhya R, et al. γ-radiation induces cellular sensitivity and aberrant methylation in human tumor cell lines. Int J Radiat Biol. 2011;87(11):1086–96. https://doi.org/10.3109/09553002.2011.605417.
Terry SYA, Vallis KA. Relationship between chromatin structure and sensitivity to molecularly targeted auger electron radiation therapy. Int J Radiat Oncol Biol Phys. 2012;83(4):1298–305. https://doi.org/10.1016/j.ijrobp.2011.09.051.
Kim J-G, Bae J-H, Kim J-A, Heo K, Yang K, Yi JM. Combination effect of epigenetic regulation and ionizing radiation in colorectal cancer cells. PLoS ONE. 2014;9(8): e105405. https://doi.org/10.1371/journal.pone.0105405.
Ou Y, Zhang Q, Tang Y, et al. DNA methylation enzyme inhibitor RG108 suppresses the radioresistance of esophageal cancer. Oncol Rep. 2018;39(3):993–1002. https://doi.org/10.3892/or.2018.6210.
Frost P, Abbruzzese JL, Hunt B, Lee D, Ellis M. Synergistic cytotoxicity using 2′-deoxy-5-azacytidine and cisplatin or 4-hydroperoxycyclophosphamide with human tumor cells. Cancer Res. 1990;50(15):4572–7.
Moro H, Hattori N, Nakamura Y, et al. Epigenetic priming sensitizes gastric cancer cells to irinotecan and cisplatin by restoring multiple pathways. Gastric Cancer. 2019;23:105–15. https://doi.org/10.1007/s10120-019-01010-1.
Tsai H-C, Li H, Van Neste L, et al. Transient low doses of DNA-demethylating agents exert durable antitumor effects on hematological and epithelial tumor cells. Cancer Cell. 2012;21(3):430–46. https://doi.org/10.1016/j.ccr.2011.12.029.
Roolf C, Richter A, Konkolefski C, et al. Decitabine demonstrates antileukemic activity in B cell precursor acute lymphoblastic leukemia with MLL rearrangements. J Hematol Oncol. 2018;11(1):62. https://doi.org/10.1186/s13045-018-0607-3.
Chang X, Monitto CL, Demokan S, et al. Identification of hypermethylated genes associated with cisplatin resistance in human cancers. Cancer Res. 2010;70(7):2870–9. https://doi.org/10.1158/0008-5472.CAN-09-3427.
Peng H, Zhang J, Zhang P-P, et al. ARNTL hypermethylation promotes tumorigenesis and inhibits cisplatin sensitivity by activating CDK5 transcription in nasopharyngeal carcinoma. J Exp Clin Cancer Res CR. 2019;38(1):11. https://doi.org/10.1186/s13046-018-0997-7.
Plumb JA, Strathdee G, Sludden J, Kaye SB, Brown R. Reversal of drug resistance in human tumor xenografts by 2′-deoxy-5-azacytidine-induced demethylation of the hMLH1 gene promoter. Cancer Res. 2000;60(21):6039–44.
Gifford G, Paul J, Vasey PA, Kaye SB, Brown R. The acquisition of hMLH1 methylation in plasma DNA after chemotherapy predicts poor survival for ovarian cancer patients. Clin Cancer Res. 2004;10(13):4420–6. https://doi.org/10.1158/1078-0432.CCR-03-0732.
Cao Y, Chen Y, Huang Y, Liu Z, Li G. In vitro study of human mutL homolog 1 hypermethylation in inducing drug resistance of esophageal carcinoma. Ir J Med Sci. 2017;186(2):257–63. https://doi.org/10.1007/s11845-016-1401-2.
Wu F, Lu M, Qu L, Li D-Q, Hu C-H. DNA methylation of hMLH1 correlates with the clinical response to cisplatin after a surgical resection in Non-small cell lung cancer. Int J Clin Exp Pathol. 2015;8(5):5457–63.
Hakata S, Terashima J, Shimoyama Y, et al. Differential sensitization of two human colon cancer cell lines to the antitumor effects of irinotecan combined with 5-aza-2′-deoxycytidine. Oncol Lett. 2018;15(4):4641–8. https://doi.org/10.3892/ol.2018.7883.
Stewart ML, Tamayo P, Wilson AJ, et al. KRAS genomic status predicts the sensitivity of ovarian cancer cells to decitabine. Cancer Res. 2015;75(14):2897–906. https://doi.org/10.1158/0008-5472.CAN-14-2860.
Shang D, Xian S, Han T, Li X, Liu Y. VHL-TGFBI signaling is involved in the synergy between 5-aza-2′-deoxycytidine and paclitaxel against human renal cell carcinoma. J BUON Off J Balk Union Oncol. 2017;22(2):500–7.
Li X-Y, Wu J-Z, Cao H-X, et al. Blockade of DNA methylation enhances the therapeutic effect of gefitinib in non-small cell lung cancer cells. Oncol Rep. 2013;29(5):1975–82. https://doi.org/10.3892/or.2013.2298.
Hou T, Ma J, Hu C, et al. Decitabine reverses gefitinib resistance in PC9 lung adenocarcinoma cells by demethylation of RASSF1A and GADD45β promoter. Int J Clin Exp Pathol. 2019;12(11):4002–10.
Qu X, Sandmann T, Frierson H, et al. Integrated genomic analysis of colorectal cancer progression reveals activation of EGFR through demethylation of the EREG promoter. Oncogene. 2016;35(50):6403–15. https://doi.org/10.1038/onc.2016.170.
Yun J, Song S-H, Park J, et al. Gene silencing of EREG mediated by DNA methylation and histone modification in human gastric cancers. Lab Investig J Tech Methods Pathol. 2012;92(7):1033–44. https://doi.org/10.1038/labinvest.2012.61.
Lou Y, Zou Z, Chen P, et al. Combination of gefitinib and DNA methylation inhibitor decitabine exerts synergistic anti-cancer activity in colon cancer cells. PLoS ONE. 2014;9(5): e97719. https://doi.org/10.1371/journal.pone.0097719.
Yu G, Wu Y, Wang W, et al. Correction to: Low-dose decitabine enhances the effect of PD-1 blockade in colorectal cancer with microsatellite stability by re-modulating the tumor microenvironment. Cell Mol Immunol. 2019;17:111–2. https://doi.org/10.1038/s41423-019-0340-z.
Zhang Y, Xiang C, Wang Y, Duan Y, Liu C, Zhang Y. PD-L1 promoter methylation mediates the resistance response to anti-PD-1 therapy in NSCLC patients with EGFR-TKI resistance. Oncotarget. 2017;8(60):101535–44. https://doi.org/10.18632/oncotarget.21328.
Liu K, Dong F, Gao H, et al. Promoter hypermethylation of the CFTR gene as a novel diagnostic and prognostic marker of breast cancer. Cell Biol Int. 2020;44(2):603–9. https://doi.org/10.1002/cbin.11260.
Zhang Y, Fan J, Fan Y, et al. The new 6q27 tumor suppressor DACT2, frequently silenced by CpG methylation, sensitizes nasopharyngeal cancer cells to paclitaxel and 5-FU toxicity via β-catenin/Cdc25c signaling and G2/M arrest. Clin Epigenet. 2018;10(1):26. https://doi.org/10.1186/s13148-018-0459-2.
Hou P, Liu D, Dong J, Xing M. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle Georget Tex. 2012;11(2):286–95. https://doi.org/10.4161/cc.11.2.18707.
Li X, Zhang Y, Chen M, et al. Increased IFNγ+ T cells are responsible for the clinical responses of low-dose DNA-demethylating agent decitabine antitumor therapy. Clin Cancer Res. 2017;23(20):6031–43. https://doi.org/10.1158/1078-0432.CCR-17-1201.
Wu M, Sheng L, Cheng M, et al. Low doses of decitabine improve the chemotherapy efficacy against basal-like bladder cancer by targeting cancer stem cells. Oncogene. 2019;38(27):5425–39. https://doi.org/10.1038/s41388-019-0799-1.
Li J, Su X, Dai L, et al. Temporal DNA methylation pattern and targeted therapy in colitis-associated cancer. Carcinogenesis. 2020;41(2):235–44. https://doi.org/10.1093/carcin/bgz199.
Gailhouste L, Liew LC, Hatada I, Nakagama H, Ochiya T. Epigenetic reprogramming using 5-azacytidine promotes an anti-cancer response in pancreatic adenocarcinoma cells. Cell Death Dis. 2018;9(5):468. https://doi.org/10.1038/s41419-018-0487-z.
Zhang Y-W, Staal B, Dykema KJ, Furge KA, Vande Woude GF. Cancer-type regulation of MIG-6 expression by inhibitors of methylation and histone deacetylation. PLoS ONE. 2012;7(6): e38955. https://doi.org/10.1371/journal.pone.0038955.
Wang C, Hamacher A, Petzsch P, et al. Combination of decitabine and entinostat synergistically inhibits urothelial bladder cancer cells via activation of FoxO1. Cancers. 2020;12(2):337. https://doi.org/10.3390/cancers12020337.
Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39(1):1–10. https://doi.org/10.1016/j.immuni.2013.07.012.
Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: towards combination strategies with curative potential. Cell. 2015;161(2):205–14. https://doi.org/10.1016/j.cell.2015.03.030.
Manara MC, Valente S, Cristalli C, et al. A quinoline-based DNA methyltransferase inhibitor as a possible adjuvant in osteosarcoma therapy. Mol Cancer Ther. 2018;17(9):1881–92. https://doi.org/10.1158/1535-7163.MCT-17-0818.
Tomasi TB, Magner WJ, Khan ANH. Epigenetic regulation of immune escape genes in cancer. Cancer Immunol Immunother. 2006;55(10):1159–84. https://doi.org/10.1007/s00262-006-0164-4.
Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer. 2019;19(3):151–61. https://doi.org/10.1038/s41568-019-0109-9.
Lander ES, Linton LM, Birren B, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. https://doi.org/10.1038/35057062.
Tang WWC, Dietmann S, Irie N, et al. A unique gene regulatory network resets the human germline epigenome for development. Cell. 2015;161(6):1453–67. https://doi.org/10.1016/j.cell.2015.04.053.
Odunsi K, Matsuzaki J, Karbach J, et al. Efficacy of vaccination with recombinant vaccinia and fowlpox vectors expressing NY-ESO-1 antigen in ovarian cancer and melanoma patients. Proc Natl Acad Sci USA. 2012;109(15):5797–802. https://doi.org/10.1073/pnas.1117208109.
De Carvalho DD, Binato R, Pereira WO, et al. BCR-ABL-mediated upregulation of PRAME is responsible for knocking down TRAIL in CML patients. Oncogene. 2011;30(2):223–33. https://doi.org/10.1038/onc.2010.409.
Rapoport AP, Stadtmauer EA, Binder-Scholl GK, et al. NY-ESO-1-specific TCR-engineered T cells mediate sustained antigen-specific antitumor effects in myeloma. Nat Med. 2015;21(8):914–21. https://doi.org/10.1038/nm.3910.
Wei Y, Lao X-M, Xiao X, et al. Plasma cell polarization to the Immunoglobulin G phenotype in hepatocellular carcinomas involves epigenetic alterations and promotes hepatoma progression in mice. Gastroenterology. 2019;156(6):1890-1904.e16. https://doi.org/10.1053/j.gastro.2019.01.250.
Shi X, Chen X, Fang B, et al. Decitabine enhances tumor recognition by T cells through upregulating the MAGE-A3 expression in esophageal carcinoma. Biomed Pharmacother Biomedecine Pharmacother. 2019;112: 108632. https://doi.org/10.1016/j.biopha.2019.108632.
Schwartsmann G, Schunemann H, Gorini CN, et al. A phase I trial of cisplatin plus decitabine, a new DNA-hypomethylating agent, in patients with advanced solid tumors and a follow-up early phase II evaluation in patients with inoperable non-small cell lung cancer. Investig New Drugs. 2000;18(1):83–91. https://doi.org/10.1023/a:1006388031954.
Pohlmann P, DiLeone LP, Cancella AI, et al. Phase II trial of cisplatin plus decitabine, a new DNA hypomethylating agent, in patients with advanced squamous cell carcinoma of the cervix. Am J Clin Oncol. 2002;25(5):496–501. https://doi.org/10.1097/00000421-200210000-00015.
Appleton K, Mackay HJ, Judson I, et al. Phase I and pharmacodynamic trial of the DNA methyltransferase inhibitor decitabine and carboplatin in solid tumors. J Clin Oncol. 2007;25(29):4603–9. https://doi.org/10.1200/JCO.2007.10.8688.
Fang F, Balch C, Schilder J, et al. A phase 1 and pharmacodynamic study of decitabine in combination with carboplatin in patients with recurrent, platinum-resistant, epithelial ovarian cancer. Cancer. 2010;116(17):4043–53. https://doi.org/10.1002/cncr.25204.
Matei D, Fang F, Shen C, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Res. 2012;72(9):2197–205. https://doi.org/10.1158/0008-5472.CAN-11-3909.
Zhang Y, Mei Q, Liu Y, et al. Epigenetic resensitization to platinum in ovarian cancer. Oncoimmunology. 2017;6(9): e1323619. https://doi.org/10.1080/2162402X.2017.1323619.
Fan H, Lu X, Wang X, et al. Low-dose decitabine-based chemoimmunotherapy for patients with refractory advanced solid tumors: a phase I/II report. J Immunol Res. 2014;2014: 371087. https://doi.org/10.1155/2014/371087.
Fu X, Zhang Y, Wang X, et al. Low dose decitabine combined with taxol and platinum chemotherapy to treat refractory/recurrent ovarian cancer: an open-label, single-arm, phase I/II study. Curr Protein Pept Sci. 2015;16(4):329–36. https://doi.org/10.2174/138920371604150429155740.
George RE, Lahti JM, Adamson PC, et al. Phase I study of decitabine with doxorubicin and cyclophosphamide in children with neuroblastoma and other solid tumors: a Children’s Oncology Group study. Pediatr Blood Cancer. 2010;55(4):629–38. https://doi.org/10.1002/pbc.22607.
Stathis A, Hotte SJ, Chen EX, et al. Phase I study of decitabine in combination with vorinostat in patients with advanced solid tumors and non-Hodgkin’s lymphomas. Clin Cancer Res. 2011;17(6):1582–90. https://doi.org/10.1158/1078-0432.CCR-10-1893.
Garrido-Laguna I, McGregor KA, Wade M, et al. A phase I/II study of decitabine in combination with panitumumab in patients with wild-type (wt) KRAS metastatic colorectal cancer. Investig New Drugs. 2013;31(5):1257–64. https://doi.org/10.1007/s10637-013-9947-6.
Zakharia Y, Monga V, Swami U, et al. Targeting epigenetics for treatment of BRAF mutated metastatic melanoma with decitabine in combination with vemurafenib: a phase lb study. Oncotarget. 2017;8(51):89182–93. https://doi.org/10.18632/oncotarget.21269.
Chiappinelli KB, Zahnow CA, Ahuja N, Baylin SB. Combining epigenetic and immune therapy to combat cancer. Cancer Res. 2016;76(7):1683–9. https://doi.org/10.1158/0008-5472.CAN-15-2125.
Brahmer JR, Tykodi SS, Chow LQM, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65. https://doi.org/10.1056/NEJMoa1200694.
Nie J, Wang C, Liu Y, et al. Addition of low-dose decitabine to anti-PD-1 antibody camrelizumab in relapsed/refractory classical Hodgkin lymphoma. J Clin Oncol. 2019;37(17):1479–89. https://doi.org/10.1200/JCO.18.02151.
Zhang Y, Mei Q, Liu Y, et al. The safety, efficacy, and treatment outcomes of a combination of low-dose decitabine treatment in patients with recurrent ovarian cancer. Oncoimmunology. 2017;6(9): e1323619. https://doi.org/10.1080/2162402X.2017.1323619.
Chen M, Nie J, Liu Y, et al. Phase Ib/II study of safety and efficacy of low-dose decitabine-primed chemoimmunotherapy in patients with drug-resistant relapsed/refractory alimentary tract cancer. Int J Cancer. 2018;143(6):1530–40. https://doi.org/10.1002/ijc.31531.
Acknowledgements
Not applicable.
Funding
This work was supported by Free exploration program of Central South University (201212200021); National Natural Science Foundation of China (81902351); Natural Science Foundation of Hunan province (2018JJ2586); Beijing Xisike clinical oncology research foundation (Y-HS2017-043); Hunan Health Commission project (B2017016).
Author information
Authors and Affiliations
Contributions
CH provides conceptualization. XL involved in data curation, writing-original draft preparation. YZ took part in writing–reviewing. JL participated in writing–reviewing. FW involved in writing–reviewing and editing. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This review is no need to provide a statement on ethics approval and consent.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests in this section.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Hu, C., Liu, X., Zeng, Y. et al. DNA methyltransferase inhibitors combination therapy for the treatment of solid tumor: mechanism and clinical application. Clin Epigenet 13, 166 (2021). https://doi.org/10.1186/s13148-021-01154-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13148-021-01154-x