
Abstract
Background
Clostridium butyricum is a butyric acid-producing anaerobic bacteriuma, and commonly present as gut microbiota in humans. This species has been used as a probiotic for the prevention of diarrhea in humans. In this study, we report the draft genome of C. butyricum DKU-01, which was isolated from infant feces, to better understand the characteristics of this strain so that it can later be used in the development of probiotic products.
Results
A total of 79 contigs generated by hybrid assembly of sequences obtained from Roche 454 and Illumina Miseq sequencing systems were investigated. The assembled genome of strain DKU-01 consisted of 4,519,722 bp (28.62% G + C content) with a N50 contig length of 108,221 bp and 4,037 predicted CDSs. The extracted 16S rRNA gene from genome sequences of DKU-01 was similar to Clostridium butyricum with 99.63% pairwise similarity. The sequence of strain DKU-01 was compared with previously reported genome sequences of C. butyricum. The value of average nucleotide identity between strains DKU-01 and C. butyricum 60E3 was 98.74%, making it the most similar strain to DKU-01.
Conclusions
We sequenced the DKU-01 strain isolated from infant feces, and compared it with the available genomes of C. butyricum on a public database. Genes related to Fructooligosaccharide utilization were detected in the genome of strain DKU-01 and compared with other genera, such as Bifidobacterium and Streptococcus. We found that strain DKU-01 can metabolize a wide range of carbohydrates in comparative genome result. Further analyses of the comparative genome and fermentation study can provide the information necessary for the development of strain DKU-01 for probiotics.
Similar content being viewed by others


Background
The intestinal tract harbors various microbes that play a key role in the nutrition, health, and physiology of the host. Probiotic bacteria have been defined as live micro-organisms, when administered in sufficient amounts, which give a health benefit on the host organisms. In practice, probiotics can improve the balance of the intestinal microbiota ecosystem of the host, which is beneficial in treating variety of infant conditions including, infant diarrhea, upset stomach, eczema, and many health conditions of pre-term infants. It is also an effective alternative to antibiotics for growth promotion in a diverse number of livestock species [1]. In addition, it is reported that various immune responses are influenced by probiotics and these immunomodulatory effects have been proposed for use in several potential applications, including management of hypersensitivity reactions, prevention of infectious diarrhea, and tumor suppression [2]. A number of scientific progressions have led to the current situation, in which many probiotic strains exist and are in widespread use. For example, Lactobacillus delbrueckii subsp bulgaricus, Lactobacillus acidophilus, Lactobacillus rhamnosus, Bifidobacterium bifidum, Bifidobacterium adolescentis, Bifidobacterium infantis, Streptococcus thermophilus, Enterococcus faecalis, and Enterococcus faecium are currently being used in probiotic preparations either singly or in combination [3].
Clostridium butyricum is a butyric acid-producing gram-positive anaerobe which is found in soil and the intestines of healthy animals and humans, and the strain MIYAIRI 588 of C. butyricum has been used as a probiotic in Asia for treating and preventing non-antimicrobial induced diarrhea, antimicrobial-associated diarrhea, and constipation in humans [4,5]. In addition, C. butyricum produces short chain fatty acids (SCFAs), such as butyric acid, propionic acid, and acetic acid. Several reports have suggested that SCFAs have potential beneficial effects; in particular, butyric acid has a proliferative effect on intestinal mucosal cells [6] and anti-inflammatory effects [7]. Moreover, C. butyricum can ferment sugar and glycerol to several biofuels and precursors of biomaterials, such as H2, butanol, butyric acid, and 1,3-propanediol [8-11]. Due to the exhaustion of fossil fuels, the conversion of renewable biomass to valuable fuels and chemicals by microorganisms has drawn global interest [12]. For this reason, C. butyricum is receiving renewed attention, and it is very important to isolate the stain. Therefore, investigation of the genetic information and characteristics of C. butyricum DKU-01 is desired. The genome sequence analysis has been proven very useful information for understanding of C. butyricum DKU-01, which exhibits unique physiological and metabolic properties, and its potential metabolic engineering applications.
Methods
Sample collection
Fecal samples were collected from breast-feeding infants less than three months old at a hospital and postpartum care center in nearby Cheonan-Asan from 2012 to 2013. None of the infants had received antibiotics or other drugs in the months prior to sampling. Fecal samples (1 g) were collected on sterilized cotton-tipped swabs and these swabs were placed in sterile 15 ml conical tubes containing 5 ml of GYP liquid medium overlaying the top of the medium with liquid paraffin. These tubes were transferred to GasPak anaerobic jars (Becton, Dickinson and Company, NJ, USA) for further study within 30 min of the collection.
Isolation and identification of strain
For the isolation of C. butyricum from infant feces, collected fecal samples were incubated at 30°C in an anaerobic chamber (Forma Anaerobic System; Thermo Fisher Scientific Inc., Waltham, MA, USA) using a gas phase of N2/H2/CO2 (80:10:10%, v/v) for 3 days. After enrichment, they were further serially diluted and plated on RCM (Reinforced Clostridial Medium, BD) for anaerobes in an anaerobic chamber at 30°C for 48 h. Morphologically different colonies were selected randomly. The selected colonies were purified by repeated streaking on the agar media and subcultured periodically.
All isolated strains used in this work were tested for catalase. Single colonies were selected and grown in RCM broth in order to establish pure cultures of the gas bubble-producing strains. One hundred and two isolates showing a bubble with soap skin were selected as the candidates and their biochemical profiles were confirmed using API 20A (BioMerieux, Basingstoke, UK) in order to identify the isolates. One strain, DKU-01, which showed the active formation of gas bubbles, was chosen for further studies and its identification was confirmed by 16S rRNA sequencing.
For the identification of 16S rRNA sequences, genomic DNA was extracted from the strain using the phenol extraction method. The 16S rRNA gene was amplified using a polymerase chain reaction (PCR) with the universal primers 27F (5′-AGAGTTTGATCMTGGCTCAG-3′) and 1492R (5′-TACGGTYACCTTGTTGTTACGACTT-3′). The conditions of amplification were: 94°C for 5 min denaturation, followed by 30 cycles at 94°C for 1 min, 50°C for 1 min, and 72°C for 1 min and 50 sec, with a final 4 min extension at 72°C. After amplification, the PCR products were purified using an AccuPrep PCR purification kit (BIONEER, Daejeon, Korea) and sequenced with the ABI prism™ Bigdye™ Terminator Cycle sequencing ready reaction kit in an automated ABI 3730XL capillary DNA sequencer (Applied Biosystems, Foster City, CA, USA). Strain DKU-01 was identified as Clostridium butyricum and was deposited in the strain collection of the Korean Culture Center of Microorganisms (Daejeon, Korea) under accession number KCTC 12367BP.
Genome sequencing for strain DKU-01
The extracted genomic DNA was confirmed by 1% agarose gel electrophoresis and visualized under a Gel Doc system (Bio-Rad, Hercules, CA, USA) and the concentration of DNA was quantified using a PicoGreen dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). To verify contamination of DNA or isolate culture, 16S rRNA gene (about 1.4kb) was amplified and sequenced again. After confirmation of the isolated DNA, a sequencing library for the Illumina machine was prepared using Truseq DNA LT Sample Prep Kit (Illumina, San Diego, CA, USA) and a library for the Roche 454 machine was constructed using the GS FLX Titanium Rapid Library Preparation Kit (454 Life Science, Branford, CT, USA). The genome sequencing of strain DKU-01 was performed by the Illumina Miseq (250 bp paired end sequencing) and Roche 454 (8Kb-insert paired end sequencing) sequencing systems.
Assembly and annotation of genome sequence
The de novo assembly was conducted using the CLC genomic workbench 6.0 (CLC Bio, Denmark) with sequences obtained from the Miseq system and the GS Assembler 2.6 (Roche Diagnostics, Branford, CT, USA) to sequences from the Roche 454 system. The hybrid assembly of generating contigs from both systems was performed using the CodonCode Aligner (CodonCode Co. Dedham, MA, USA). Gene prediction was performed using Glimmer 3 [13], and the annotations were conducted by a homology search against Clusters of Orthologous Groups (COG) and the SEED database [14,15]. The comparative analyses of gene contents were performed using the RAST server [16]. The chromosomal region focusing fructooligosaccharide utilization in contig number 22 of strain DKU-01 was compared with synteny in similar organism using browse genome on the RAST server. The nucleotide similarities of msm operon were obtained by webcat (www.webact.org/WebACT/home) using BLASTN.
Quality assurance
The potential contamination was confirmed by identification of 16S rRNA gene and comparison of the genome sequence with the published genomes of the same genus. 16S rRNA genes were extracted from assembled contigs using the rRNA selector [17] and identified by using the EzTaxon-e database (http://eztaxon-e.ezbiocloud.net) [18]. Genome sequences were compared with available genomes of Clostridium spp. on the NCBI database using the average nucleotide identity (ANI) values [19]. The genomic distance between genome sequences were calculated using ANI values. In a pair of genomes, the query sequence was cut into small fragments (1020bp). Then, high-scoring segment pairs between genomes were determined by the BLAST algorithm [19]. For genome tree, the calculated ANI values were converted into distances between genomes of Clostridium. The distance values were obtained from ANI values complement to 1. This pairwise distance matrix was used to construct a dendrogram by the Unweighted Pair Group Method with Arithmetic Mean clustering method. The digital DNA-DNA hybridization values were calculated using Genome-to-Genome Distance Calculator (GGDC; http://ggdc.dsmz.de).
Initial findings
General features
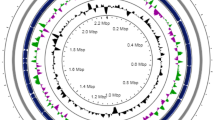
A total of 79 contigs were generated by a hybrid assembly of reads from the Illumina Miseq (12,127,925 reads of 250bp paired end; ˃554-fold coverage) and Roche 454 (319,439 reads of 8kb-insert paired end; ˃13-fold coverage) systems. The genome size of strain DKU-01 was 4,519,722 bases with 28.62% G + C content and 4,037 predicted CDSs, and 59 tRNA and 17 rRNA operons. The length of the largest contig was 334,920 bp, and the N50 contig was 108,221 bp. The distributions of COG and SEED subsystems of strain DKU-01 are depicted in Figure 1. Among the COG category, R (general function prediction only; 399 ORFs) and G (carbohydrate transport and metabolism; 313 ORFs) were abundant, followed by the E category (amino acid transport and metabolism; 288 ORFs), S category (function unknown; 268 ORFs) and K category (transcription; 251 ORFs). In the subsystem distribution, the subsystems of carbohydrates (454 ORFs) and amino acids and derivatives (385 ORFs) were well-represented. The subsystems of di- and oligosaccharides (127 ORFs) and central carbohydrate metabolism (91 ORFs) were prominently detected within the carbohydrate subsystem. Beta-glucoside metabolism (40 ORFs), maltose and maltodextrin utilization (38 ORFs), lactose and galactose uptake and utilization (22 ORFs), and fructooligosaccharides and raffinose utilization (15 ORFs) were abundant categories within the subsystem of di- and oligosaccharides. Pentose phosphate pathway (20 ORFs), glycolysis and gluconeogenesis (16 ORFs), and the entner-Doudoroff pathway (13 ORFs) were prominently detected within the central carbohydrate metabolism subsystem.

Statistics of annotated genes for Clostridium butyricum DKU-01 based on COG (A) and SEED (B) databases.
Comparative analysis of DKU-01 genome with genomes of neighbor strains
Extracted 16S rRNA gene of strain DKU-01 had the highest pairwise similarity with Clostridium butyricum DSM 10702T (99.63%). The genome tree of strain DKU-01 was generated by the ANI values of C. butyricum genomes, which are available on a public database (Figure 2). The results of 16S rRNA gene identification and genome tree indicated that the genome sequences of strain DKU-01 was free of contaminations. Six genome sequences of C. butyricum were compared with the genome sequences of DKU-01, and two strains of C. butyricum, 60E3 and DSM 10702T, were clustered with strain DKU-01. The ANI value of strain DKU-01 to 60E3 was 98.74% and to DSM 10702T was 98.73%. The estimated DNA-DNA hybridization values of strain DHU-01 to 60E3 (90.9 ± 2%) and to DSM 10702T (90.3 ± 2.08%) were obtained by the GGDC. The genomic features and subsystem distributions were compared among three strains (Additional file 1: Table S1). The lowest number of contigs was obtained from strain 60E3 (10), while the highest number of contigs was obtained from strain DSM 10702T (207). The abundances of gene contents in the subsystems were similar in all three genomes.

Genome tree of strain DKU-01 with Clostridium butyricum strains obtained from NCBI database.
We focused on fructooligosaccharide (FOS) utilization within the carbohydrate subsystem, because fructooligosaccharide has been used as a prebiotic in food products and infant formulas [20]. FOS can be a derivative of simple fructose polymers of fructose moieties attached to a sucrose molecule, and it can be degraded by a variety of lactic acid bacteria in the gut [21,22]. Lactobacillus acidophilus and Bifidobacterium spp. have been studied for the utilization of FOS [22,23], and the utilization of FOS by Clostridium spp. has also been reported [24]. Ten ORFs in the genome of strain DKU-01 were annotated to genes related to FOS utilization (Table 1). The chromosomal region including MsmE, MsmF, and MsmG in contig number 22 of strain DKU-01 was compared with synteny in C. botulinum E3, C. beijerincki NCIMB 8052, Bifidobacterium longum NCC 2705, B. animalis subsp. lactis AD011, B. adolescentis ATCC 15703, Streptococcus pneumoniae TIGR 4, and S. mutans UA159 (Figure 3). Functional analysis of the FOS gene cluster indicated that the ingestion of an oligosaccharide was interceded by an ATP-dependent binding cassette (ABC)-type transport system. Genes encoding the ABC transport system (MsmEFGK) as well as a putative intracellular fructosidase (bfrA) were found to be located in a multiple-sugar metabolism (msm) operon. Although the gene contents surrounding MsmE, MsmF, and MsmG were different between genera, the Msm cluster revealed a high degree of synteny. The divergence of msm operon sequences were analyzed by artemis comparison at nucleotide level. This highest nucleotide similarity value was investigated in MsmF gene from DKU-01 to C. botulinum E3 (85%). The similarity value of msm operon nucleotides from DKU-01 to C. botulinum E3 was 81.6%, and to C. beijerincki NCIMB 8052 was 68%. The nucleotide similarity values of other strains in Figure 3 ranged from 45.18% to 65%. The general structure of the identified gene clusters encoding upregulated genes involved in carbohydrate ingestion and catabolism indicates that typically, a three component system consisting of a regulator, transporter, and glycoside hydrolase(s) can be sufficient for utilization of potential prebiotics, irrespective of the type of transporter identified (phosphoenolpyruvate-dependent sugar phosphotransferase systems (PTS), galactoside pentose hexuronide (GPH) permease, and ABC-type transporters). Most notably, PTS permeases had higher selectivity towards disaccharides e.g. sucrose, lactose, and maltose, whereas ABC and GPH permeases showed to be induced by the longer oligosaccharides e.g. stachyose and β-galactooligosaccharides. Furthermore, similar upregulation patterns of gene expression by widely different prebiotics was interesting, remarkably the FOS-ABC transporter that was also induced by the mixed linkage polydextrose. This suggests that transporters either possess more than one specificity or less stringent molecular recognition of substrates, indicating that a wide range of carbohydrates can be metabolized by C. butyricum DKU-01, and likely similar commensal and probiotic bacteria.

Comparison of the msm cluster in contig number 22 of DKU-01 genome with selected bacteria (based on RAST annotation server). The red arrow (number 1) indicates MsmE; yellow-green arrow (number 2) MsmF; brown arrow (number 3) MsmG; blue arrow (number 4) Oligo-1,6-glucosidase; yellow arrow (number 5) Alpha-galactosidase; blue-green (number 6) Catabolite control protein A; pink arrow (number 7) possible NagC/XylR-type transcriptional regulator; green arrow (number 8) tRNA-specific adenosine-34 deaminase; dark brown arrow (number 9) probable NhaP-type Na(+)/H(+) exchanger; sky-blue arrow (number 10) Sucrose phosphorylase; dark-gray arrow (number 11) MSM operon regulatory protein; pistachio arrow (number 12) Threonine dehydratase; lime-green arrow (number 13) Aldehyde dehydrogenase B; purple arrow (number 14) NAD-dependent protein deacetylase of SIR2 family; primrose arrow (number 15) possible phospholipase. The same number and color in the figure are sets of genes with similar sequences. Genes whose relative position is conserved in at least four other species are functionally coupled and shared gray background boxes.
Future directions
Further analyses of the genome comparisons and FOS fermentation will provide greater information to improve the probiotic effects of strain DKU-01. This information can help to develop improved probiotics for public health.
Availability of supporting data
The obtained genome sequence of Clostridium butyricum DKU-01 was deposited in the DDBJ/EMBL/GenBank under the accession APKZ00000000. The version described in this paper is the first version, APKZ01000000.
References
Khoruts A, Dicksved J, Jansson JK, Sadowsky MJ. Changes in the composition of the human fecal microbiome after bacteriotherapy for recurrent Clostridium difficile-associated diarrhea. J Clin Gastroenterol. 2010;44:354–60.
Kirjavainen PV, El-Nezami HS, Salminen SJ, Ahokas JT, Wright PF. The effect of orally administered viable probiotic and dairy Lactobacilli on mouse lymphocyte proliferation. FEMS Immunol Med Microbiol. 1999;26:131–5.
Fooks LJ, Fuller R, Gibson GR. Prebiotics, probiotics and human gut microbiology. Int Dairy J. 1999;9:53–61.
Seki H, Shiohara M, Matsumura T, Miyagawa N, Tanaka M, Komiyama A, et al. Prevention of antibiotic-associated diarrhea in children by Clostridium butyricum MIYAIRI. Pediatr Int. 2003;45:86–90.
Okamoto T, Sasaki M, Tsujikawa T, Fujiyama Y, Bamba T, Kusunoki M. Preventive efficacy of butyrate enemas and oral administration of Clostridium butyricum M588 in dextran sodium sulfate-induced colitis in rats. J Gastroenterol. 2000;35:341–6.
Sakata T, Adachi M, Hashida M, Sato N, Kojima T. Effect of n-butyric acid on epithelial cell proliferation of pig colonic mucosa in short-term culture. Dtsch Tierarztl Wochenschr. 1995;102:163–4.
Mishiro T, Kusunoki R, Otani A, Ansary MM, Tongu M, Harashima N, et al. Butyric acid attenuates intestinal inflammation in murine DSS-induced colitis model via milk fat globule-EGF factor 8. Lab Invest. 2013;93:834–43.
Cai G, Jin B, Monis P, Saint C. A genetic and metabolic approach to redirection of biochemical pathways of Clostridium butyricum for enhancing hydrogen production. Biotechnol Bioeng. 2013;110:338–42.
Cheng CL, Che PY, Chen BY, Lee WJ, Chien LJ, Chang JS. High yield bio-butanol production by solvent-producing bacterial microflora. Bioresour Technol. 2012;113:58–64.
Zigova J, Sturdik E, Vandak D, Schlosser S. Butyric acid production by Clostridium butyricum with integrated extraction and pretraction. Process Biochemistry. 1999;34:835–43.
Saint-Amans S, Perlot P, Goma G, Soucaille P. High production of 1,3-propanediol from glycerol by Clostridium butyricum VPI 3266 in a simply controlled fed-batch system. Biotechnol Lett. 1994;16:831–6.
Ragauskas AJ, Williams CK, Davison BH, Britovsek G, Cairney J, Eckert CA, et al. The path forward for biofuels and biomaterials. Science. 2006;311:484–9.
Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–9.
Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–7.
Disz T, Akhter S, Cuevas D, Olson R, Overbeek R, Vonstein V, et al. Accessing the SEED Genome Databases via Web Services API: Tools for Programmers. BMC Bioinformatics. 2010;11:319.
Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014;42:D206–214.
Lee JH, Yi H, Chun J. rRNASelector: a computer program for selecting ribosomal RNA encoding sequences from metagenomic and metatranscriptomic shotgun libraries. J Microbiol. 2011;49:689–91.
Kim OS, Cho YJ, Lee K, Yoon SH, Kim M, Na H, et al. Introducing EzTaxon-e: a prokaryotic 16S rRNA gene sequence database with phylotypes that represent uncultured species. Int J Syst Evol Microbiol. 2012;62:716–21.
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57:81–91.
Euler AR, Mitchell DK, Kline R, Pickering LK. Prebiotic effect of fructooligosaccharide supplemented formula at two concentrations compared with unsupplemented formula and human milk. J Pediat Gastroenterol Nut. 2005;40:157–64.
Kaplan H, Hutkins RW. Fermentation of fructooligosaccharides by lactic acid bacteria and bifidobacteria. Appl Environ Microbiol. 2000;66:2682–4.
Rossi M, Corradini C, Amaretti A, Nicolini M, Pompei A, Zanoni S, et al. Fermentation of fructooligosaccharides and inulin by Bifidobacteria: a comparative study of pure and fecal cultures. Appl Environ Microbiol. 2005;71:6150–8.
Barrangou R, Altermann E, Hutkins R, Cano R, Klaenhammer TR. Functional and comparative genomic analyses of an operon involved in fructooligosaccharide utilization by Lactobacillus acidophilus. Proc Natl Acad Sci U S A. 2003;100:8957–62.
Rada V, Nevoral J, Trojanova I, Tomankova E, Smehilova M, Killer J. Growth of infant faecal Bifidobacteria and Clostridia on prebiotic oligosaccharides in in vitro conditions. Anaerobe. 2008;14:205–8.
Acknowledgements
This research was financially supported by the Ministry of Education (MOE) and National Research Foundation of Korea(NRF) through the Human Resource Training Project for Regional Innovation (No. 2012-12-A-05-067-12010100).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
CH Oh and SJ Mo designed the study. SJ Yun, JJ Lee and SH Yoon performed experiments. BS Kim analyzed the sequencing data. All authors read and approved the final manuscript.
SangJoon Mo and Bong-Soo Kim contributed equally to this work.
Additional file
Additional file 1: Table S1.
Comparison of genomic features and subsystem of strain DKU-01 with strain 6E3 and DSM 10702T.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Mo, S., Kim, BS., Yun, SJ. et al. Genome sequencing of Clostridium butyricum DKU-01, isolated from infant feces. Gut Pathog 7, 8 (2015). https://doi.org/10.1186/s13099-015-0055-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13099-015-0055-3