
Abstract
Background
The Anopheles hyrcanus group, which includes 25 species, is widely distributed in the Oriental and Palaearctic regions. Given the difficulty in identifying cryptic or sibling species based on their morphological characteristics, molecular identification is regarded as an important complementary approach to traditional morphological taxonomy. The aim of this study was to reconstruct the phylogeny of the Hyrcanus group using DNA barcoding markers in order to determine the phylogenetic correlations of closely related taxa and to compare these markers in terms of identification efficiency and genetic divergence among species.
Methods
Based on data extracted from the GenBank database and data from the present study, we used 399 rDNA–ITS2 sequences of 19 species and 392 mtDNA–COII sequences of 14 species to reconstruct the molecular phylogeny of the Hyrcanus group across its worldwide range. We also compared the performance of rDNA–ITS2 against that of mtDNA–COII to assess the genetic divergence of closely related species within the Hyrcanus group.
Results
Average interspecific divergence for the rDNA–ITS2 sequence (0.376) was 125-fold higher than the average intraspecies divergence (0.003), and average interspecific divergence for the mtDNA–COII sequence (0.055) was eightfold higher than the average intraspecies divergence (0.007). The barcoding gap ranged from 0.015 to 0.073 for rDNA–ITS2, and from 0.017 to 0.025 for mtDNA–COII. Two sets of closely related species, namely, Anophels lesteri and An. paraliae, and An. sinensis, An. belenrae and An. kleini, were resolved by rDNA–ITS2. In contrast, the relationship of An. sinensis/An. belenrae/An. kleini was poorly defined in the COII tree. The neutrality test and mismatch distribution revealed that An. peditaeniatus, An. hyrcanus, An. sinensis and An. lesteri were likely to undergo hitchhiking or population expansion in accordance with both markers. In addition, the population of an important vivax malaria vector, An. sinensis, has experienced an expansion after a bottleneck in northern and southern Laos.
Conclusions
The topology of the Hyrcanus group rDNA–ITS2 and mtDNA–COII trees conformed to the morphology-based taxonomy for species classification rather than for that for subgroup division. rDNA–ITS2 is considered to be a more reliable diagnostic tool than mtDNA–COII in terms of investigating the phylogenetic correlation between closely related mosquito species in the Hyrcanus group. Moreover, the population expansion of an important vivax malaria vector, An. sinensis, has underlined a potential risk of malaria transmission in northern and southern Laos. This study contributes to the molecular identification of the Anopheles hyrcanus group in vector surveillance.
Graphical abstract

Similar content being viewed by others
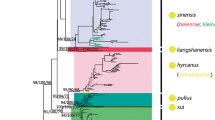

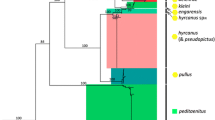
Background
The Anopheles hyrcanus group consists of at least 25 species and is classified into the Myzorhynchus series of Anopheles, with one provisional designated member [1, 2]. The members of this group are extensively distributed within the Oriental and Palaearctic regions, including a number of species capable of transmitting not only malaria [3,4,5,6] and filariasis [7, 8], but also Japanese encephalitis virus [9,10,11]. According to previous studies, Anopheles sinensis and An. lesteri are the major malaria vector present in China [12]; An. hyrcanus acts as a potential malaria vector in the south of France [13, 14]; An. kleini and An. pullus are the primary malaria vectors in South Korea [3]; An. sinensis, An. nigerrimus and An. peditaeniatus are the potential malaria vectors in Thailand [4]; An. hyrcanus group is among the major Anopheles species found across eight provinces in Laos [15]. As suggested in our recent study, An. sinensis is the predominant Anopheles species and suspected to be extensively distributed in the north of Laos (Phoshaly Province) [16]. Since the primary malaria vectors are classified mainly into the Hyrcanus group, it is essential to devise an efficient and precise method to identify the members of this group [17], which is an essential requirement for malaria vector surveillance in practice [18, 19]. However, even trained taxonomists are unlikely—or find it extremely difficult— to accurately distinguish species within the Hyrcanus group based only on morphological properties [6, 7] due to the significant variation in morphology and the adults of some closely related species exhibiting nearly identical adult morphological properties [8, 9].
DNA barcoding refers to an important addition to conventional approaches based on morphology and an effective tool that is used to identify species without the need to consider life stages. A DNA marker that is evolving at the species level can contribute toward accurate phylogenetic reconstruction in the Hyrcanus group and elucidate the ambiguity arising from an improper classification process [20, 21]. The internal transcribed spacer 2 (ITS2) has been commonly employed to address taxonomic problems in the Hyrcanus group due to its low intraspecific and high interspecific variability, as suggested in an abundance of studies [8, 9, 18, 22,23,24]. Using this marker, three newly proposed lineages revealed, including two species, An. belenrae and An. kleini, separated from An. sinensis [25], and one species showing a close relation to An. hyrcanus, with the provisional designation of An. hyrcanus spIR [18]. The mitochondrial cytochrome c oxidase subunit region (e.g. COI and COII) was taken as the standard barcode for identifying species within an extensive range of animal taxa [18, 19] and may be effective in providing barcoding data, in particular for assessing interspecific hybridization. Nevertheless, introgression in animals is considered to frequently involve mitochondrial DNA (mtDNA), as evidenced by recently appearing hybridization events among species [26, 27].
In order to reconstruct the molecular phylogeny of the Hyrcanus group, it is necessary to identify the barcoding gap of ITS2 and COII. Accordingly, the specimens of identical species in different geography sites must be examined [19, 28] for calculating the intraspecific and interspecific variations of COII and ITS2 within the group. The database of COII and ITS2 sequences in GenBank enables reference sequences to be used for identifying Hyrcanus group species based on a comparatively extensive geographic distribution [29]. Therefore, in the present study, GenBank sequences and data from our original study in northern and southern Laos were used to reconstruct a phylogeny for the Hyrcanus group on the basis of ITS2 and COII, with the aim to determine the phylogenetic correlations between taxa with close relations. In addition, we compared rDNA–ITS2 and mtDNA–COII in terms of their efficiency to distinguish different species and to determine the genetic divergence among different species in the Hyrcanus group, thereby contributing to the identification of molecular data on mosquitoes for use in malaria vector surveillance.
Methods
Mosquito collection and identification
Adult mosquitoes were collected by overnight trapping with battery-operated CDC light traps (model 1012; John W. Hock Inc., Gainesville, FL, USA) in cattle/pig pens or human residences (rooms) from 20:00 h to 08:00 h in Pathoomphone County (Champasak Province), Pak lay County (Xaignabouli Province) and Yot Ou County (Phongsaly Province), Laos, in 2018 (Additional file 1: Fig. S1). The live adult mosquitoes were killed by freezing. Subsequent isolation and identification procedures were carried out based on gender, species and subgroup, under a dissecting microscope using standard techniques [30, 31]. All mosquitoes were first morphologically sorted in the field using the keys of Das et al. [32]. Each morphologically identified specimen was kept individually in a 1.5-ml microcentrifuge tube filled with 75% ethanol and then stored at 4 ℃ for molecular confirmation of species and further processing.
DNA extraction, ITS2/COII amplification and sequencing
Genomic DNA was isolated from individual mosquitoes using the QIAamp® DNA Mini Kit (QIAGEN, Hilden, Germany) following the manufacturer’s instructions. Approximately 650 bp of the COII gene and a 550-bp PCR product of the ITS2 region was amplified using primer pairs LYS-R (5′-ACTTGCTTTCAGTCATCTAATG-3′) and LEU-F (5′-TCTAATATGGCAGATTAGTGCA-3′) and ITS2-R (5′-TATGCTTAAATTCAGGGGGT-3′) and ITS2-F (5′-TGTGAACTGCAGGACACAT-3′), respectively. ITS2 was amplified in a PCR reaction volume of 25 µl with the following cycling parameters: 94 ℃, 2 min; then 94 ℃/30 s, 50 ℃/30 s, 72 ℃/40 s for 40 cycles; and a final extension at 72 ℃ for 10 min. COII was amplified in a PCR reaction volume of 25 µl with the following cycling parameters: 95 ℃, 5 min; then 95 ℃/1 min, 51 ℃/1 min, 72 ℃/ 2 min for 35 cycles; wiath a final extension 72 ℃ for 10 min. The PCR products were then analyzed by 1.5% agarose gel electrophoresis stained with GoldView (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China), under UV transillumination. The sequencing reaction proceeded in both directions with the assistance of an ABI Big Dye Terminator Kit v.3.1 (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA); analysis was conducted using ABI Prism 3500xL-Avant Genetic Analyzer (Applied Biosystems, Thermo Fisher Scientific) in Shanghai (Sangon Biotech).
Sequence alignment and phylogenetic analysis
The keywords “(species name)” and “ITS2/COII” were used to search for ITS2 or COII sequences of members of the Hyrcanus group deposited in GenBank. ITS2 and COII sequences that were distant from conspecific sequences after initial sequence alignment were eventually excluded from further analyses. In total, 691 ITS2 sequences and 368 COII sequences of the Hyrcanus group were ultimately extracted from GenBank and used in this study (Additional file 2: Tables S1, S2). These sequences were subsequently aligned and identical sequences obtained from the same dataset or species were excluded from further analysis. Ultimately, 267 ITS2 and 260 COII sequences (haplotypes) were further screened in a genetic divergence analysis and phylogenetic analysis (Additional file 2: Tables S3, S4). A total of 132 COII and ITS2 sequences from 89 An. sinensis, 18 An. peditaeniatus, 3 An. nitidus, 8 An. argyropus and 14 An. nigerrimus were generated in this study. Table 1 provides detailed information on the ITSE and COII sequences.
The ITS2 and COII sequence dataset was combined with data on fragments from our original study and records retrieved from GenBank. A multiple sequence alignment was conducted in MEGA-X [33], while the manual adjustment was made using BioEdit V7.0.9 if required [34]. Gaps were excluded from the analysis and characters were unweighted. Both the maximum likelihood (ML) tree and the Neighbor-Joining (NJ) tree were performed with 1000 bootstraps in MEGA X [33]. The NJ method generally reveals shallow intraspecific and deep interspecific divergences [19, 35], for which a bootstrapped NJ tree was constructed using 1000 replicates [36] to provide a graphical representation of the phylogenetic correlations among the Hyrcanus group members. Anopheles lindesayi (GenBank accession no. AJ620898) and An. claviger (GenBank accession nos. AY129232 and DQ229313) were taken as outgroup taxa to the Hyrcanus group, in line with a prior study [37]. The visualization and the editing of the tree were performed using FigTree v1.4.2 [38].
Genetic diversity analysis, demographic history analysis and neutrality test
Intra- and interspecific ITS2/COII divergences were tested using the Kimura’s 2-parameter (K2P) distance model [39] in MEGA X [33]. Genetic divergence was determined using Nei’s distance model [40], in Arlequin v3.5.2.2 [41]. Genetic diversity indices were calculated and neutrality tests (Fu’s Fs [42] and Tajima’s D [43]) were performed using DnaSP v5.10 [44]. The mismatch distribution (with the simulation in Arlequin v.3.5) was achieved to distinguish a multimodal or ragged distribution from a smooth unimodal distribution [45,46,47].
Results
Intra- and interspecific ITS2/COII variation
The mean intra- and interspecific K2P distances of the ITS2 sequence in 19 Hyrcanus group members and those of the COII sequence in 14 Hyrcanus group members were computed and compared using the K2P distance model [39] in this study. Tables 2 and 3 show the intra- and interspecific divergences of ITS2 and COII in the Hyrcanus group. Individual species were represented as few as one and as many as 143 individuals, for a total of 399 ITS2 sequences, and by one to 140 individuals for a total of 392 COII sequences. The distribution of the pairwise K2P genetic distance of ITS2 and COII is illustrated in Fig. 1, which shows a distinct barcoding gap.

Intra- and interspecific divergences in the Hyrcanus group members determined using Kimura’s 2-parameter distance. Genetic divergence is shown on the Y-axis, and the Hyrcanus group members are shown on the X-axis. a Genetic divergence of ITS2. The barcoding gap ranged from 0.015 to 0.073. b Genetic divergence of COII. The barcoding gap ranged from 0.017 to 0.025. arg. An. argyropus, bel. An. belenrae, cra. An. crawfordi, eng. An. engarensis, hyr. An. hyrcanus, hyr. spIR An. hyrcanus spIR, kle. An. kleini, kwe. An. kweiyangensis, les. An. lesteri, lia. An. liangshanensis, nig. An. nigerrimus, nit. An. nitidus, par. An. paraliae, ped. An. peditaeniatus, pse. An. pseudopictus, pul. An. pullus, pur. An. pursati, siner. An. sineroides, sin. An. sinensis
The intraspecific K2P distance of ITS2 reached 0.003 on average. However, no intraspecific variation was found for An. belenrae, An. kleini, An. paraliae or An. pursati. A high level of divergence was detected in two species: An. lesteri (0.015) and An. crawfordi (0.013). A short distance was detected in a number of species pairs, including An. hyrcanus/An. pseudopictus (0.001), An. hyrcanus/An. hyrcanus spIR (0.020), An. hyrcanus spIR/An. pseudopictus (0.020), An. lesteri/An. paraliae (0.042) and An. kleini/An. engarensis (0.069) (Table 2). The taxonomic validity of An. pseudopictus, An. hyrcanus, An. hyrcanus spIR, An. paraliae and An. kleini remains debatable [1, 9, 14, 18, 29], and further studies are required before a definitive conclusion can be drawn. Accordingly, the interspecific K2P distance varied from 0.073 between An. kleini and An. sinensis to 0.704 between An. nigerrimus and An. pseudopictus, with an average of 0.376. Based on these findings, the intragroup species divergence in the ITS2 sequence was approximately 125-fold higher than the average within-species divergence.
The intraspecific K2P distance of COII was 0.007 on average. A high level of divergence was detected in three species: An. lesteri (0.017), An. crawfordi (0.012) and An. hyrcanus (0.011). Nevertheless, lesteri/paraliae and sinensis/belenrae/kleini exhibited a poorly stated relationship, with a significantly low value of pairwise distance between An. lesteri and An. paraliae (0.017), An. kleini and An. sinensis (0.009), An. belenrae and An. sinensis (0.007) and An. kleini and An. belenrae (0.010) (Table 3). Accordingly, the interspecific K2P distance varied from 0.025 between An. sineroides and An. paraliae to 0.081 between An. crawfordi and An. argyropus, with an average of 0.055. Based on these findings, the intragroup species divergence in the COII sequence was approximately eightfold higher than the average within-species divergence.
The ITS2 barcoding gap ranged from 0.015 to 0.073 (Fig. 1a), while the COII barcoding gap ranged from 0.017 to 0.025 (Fig. 1b), suggesting that the ITS2 spacer can serve as a more effective marker than COII for differentiating members of the Hyrcanus group. In Fig. 2, each dot represents a species, with interspecific distance on the Y-axis and intraspecific distance on the X-axis. Notably, there are more ITS2 dots than COII dots close to the top left-hand corner of the graph.

ITS2 and COII sequence divergences in the Hyrcanus group. The minimum interspecific (intergroup) divergence is plotted against the maximum intraspecific divergence. Red dots indicate the ITS2 sequence divergences of 16 species, including An. argyropus, An. belenrae, An. crawfordi, An. hyrcanus, An. hyrcanus spIR, An. kleini, An. lesteri, An. liangshanensis, An. nigerrimus, An. nitidus, An. paraliae, An. peditaeniatus, An. pullus, An. pursati, An. sineroides and An. sinensis. Green dots indicate the COII sequence divergences of 13 species, including An. argyropus, An. belenrae, An. crawfordi, An. hyrcanus, An. kleini, An. lesteri, An. nigerrimus, An. nitidus, An. paraliae, An. peditaeniatus, An. pullus, An. pursati and An. sinensis
Phylogenetic analysis
ITS2 and COII records obtained from GenBank were combined with sequences obtained in the original study and suspicious fragments (those distant from conspecific sequences after initial sequence alignment) were excluded, leaving 399 ITS2 sequences of 19 members within the Hyrcanus group, together with 392 COII sequences of 14 Hyrcanus group members to reconstruct a phylogenetic tree. The topology of the NJ tree and ML tree showed an approximate consistency in terms of main lineage, despite a slight difference in node confidence data between the two (Additional file 3: Fig. S2; Additional file 4: Fig. S3). Although the ITS2- or COII-based phylogenetic tree was consistent with conventional morphology taxonomy in terms of species recognition, its subgroup arrangement failed to comply with that achieved under morphology-based grouping.
The NJ–K2P analysis of the ITS2 sequences resulted in the identification of two major clusters in the Hyrcanus group: the Nigerrimus and Lesteri-Unassigned species subgroups, respectively. The Nigerrimus subgroup includes An. nigerrimus, An. nitidus, An. argyropus and An. pursati, and the Lesteri-Unassigned species subgroup includes An. lesteri, An. paraliae, An. crawford, An. peditaeniatus (the Lesteri-subgroup), An. sinensis, An. engarensis, An. belenrae, An. kleini, An. liangshanensis, An. kweiyangensis, An. sineroides, An. hyrcanus, An. hyrcanus spIR, An. pseudopictus and An. pullus (the Unassigned species subgroup). Each of these species was arranged on a single branch and had the homolog to its closest taxon in the tree, demonstrating their potential role as the candidate species or recent divergence. However, one An. kweiyangensis (GenBank accession no. AF261150.2) was classified into the An. liangshanensis clade, one An. engarensis (GenBank accession no. AB159604.1) was classified into the An. klein clade and one An. hyrcanus spIR was classified into the An. hyrcanus clade (Additional file 3: Fig. S2a). All lineages covering individuals representing the same species were supported by high bootstrap data, with the exceptions of An. pseudopictus and An. hyrcanus, which exhibited barcode congruence with a significantly small interspecific distance (0.001) (Tables 2, 3). Moreover, slight genetic divergence was also observed between An. lesteri and An. paraliae (0.042), between An. kleini and An. engarensis (0.069), between An. liangshanensis and An. kweiyangensis (0.098), between An. hyrcanus and An. hyrcanus spIR (0.020) and between An. hyrcanus spIR and An. pseudopictus (0.020) (Tables 2, 3).
According to the NJ-K2P analysis conducted on COII sequences, the group fell into a minimum of three major clusters. The first cluster comprised only An. nigerrimus; the second cluster included An. nitidus, An. pursati and An. argyropus; the third cluster included An. sinensis, An. belenrae, An. kleini, An. lesteri, An. paraliae, An. crawfordi, An. hyrcanus, An. peditaeniatus, An. sineroides and An. pullus. Nearly all those node-linking sequences of individuals pertaining to the identical species showed high bootstrap value; however, the correlation of An. sinensis/An. belenrae/An. kleini remained unclear. Instead, they exhibited extremely small pairwise distance data (Tables 2, 3) which led to the formation of a distinct clade with high node confidence data (Additional file 3: Fig. S2b). It is worth noting that five An. lesteri individuals (GenBank accession nos. EU699070.1, EU699071.1, EU699072.1, EU699073.1 and EU699065.1) were found much closer to An. nitidus individuals (GenBank accession nos. AB777833.1 and AB777824.1) than to some of their conspecifics; these were classified as An. nitidus lineage in the phylogenetic tree. Among these, EU699070.1 and EU699072.1 showed a 100% sequence similarity to AB777833.1;e EU699071.1 was 99.85% similar to AB777833.1; and EU699073.1 and EU699065.1 showed a 99.27% sequence similarity to AB777824.1. Nevertheless, the interspecific distance was 0.051 between An. lesteri and An. nitidus, which is close to the average interspecific distance (0.055). Thus, it is practically possible for these sequences from An. lesteri individuals to be incorrect, these results can presumably be attributed to the misidentification of original specimens.
Demographic history and neutrality test on the basis of ITS2 and COII sequences
A demographic history and neutrality test was further conducted using a total of 823 ITS2 and 500 COII sequences of the Hyrcanus group extracted from GenBank and our original data. Tables 4 and 5 list the sequence numbers according to the respective species, haplotype diversity, haplotype number, polymorphic site and Fu’s Fs and Tajima’s D. The presence of positive neutrality test data indicates either balancing selection or population size reduction. In contrast, as suggested by a negative neutrality test value, the group underwent an expansion after a bottleneck was reached, which could be attributable to purifying the selection process or a selective sweep under the context of genetic hitchhiking.
The significant negative values of the neutrality test were identified within An. lesteri (Tajima’s D = − 1.88497, P < 0.02), An. sinensis (Tajima’s D = − 1.60196, P < 0.05; Fu’s Fs = − 16.474, P < 0.001), An. nigerrimus (Tajima’s D = − 2.105, P < 0.02), An. hyrcanus (Tajima’s D = − 1.78638, P < 0.02; Fu’s Fs = − 4.462, P < 0.02) and An. peditaeniatus (Tajima’s D = − 2.67451, P < 0.001) on the basis of ITS2 (Table 4), as were An. lesteri (Tajima’s D = − 1.68623, P < 0.05; Fu’s Fs = − 24.744, P < 0.001), An. sinensis (Tajima’s D = − 1.97306, P < 0.02; Fu’s Fs = − 55.03, P < 0.001), An. nigerrimus (Tajima’s D = − 2.38098, P < 0.02; Fu’s Fs = − 8.892, P < 0.02), An. hyrcanus (Fu’s Fs = − 9.744, P < 0.02), An. nitidus (Fu’s Fs = − 18.427, P < 0.001), An. peditaeniatus (Tajima’s D = − 1.64465, P < 0.05; Fu’s Fs = − 7.932, P < 0.05) and An. kleini (Fu’s Fs = − 9.839, P < 0.02) on the basis of COII (Table 5).
A smooth and unimodal mismatch distribution was detected in An. lesteri, An. sinensis, An. hyrcanus and An. peditaeniatus using both markers, which conforms to the expected mismatch distributions under the sudden expansion model. In addition, a smooth and unimodal mismatch distribution was also detected not only in the population of An. liangshanensis on the basis of ITS2, but also in the population of An. nigerrimus and An. nitidus on the basis of COII (Additional file 5: Fig. S4).
Furthermore, the An. sinensis samples collected from Laos in this study showed significant negative values in the neutrality test on the basis of both markers: ITS2 (Tajima’s D = − 1.52504, P < 0.05; Fu’s Fs = − 8.158, P < 0.02), COII (Tajima’s D = − 1.82131, P < 0.05; Fu’s Fs = − 17.607, P < 0.001) (data not shown). A smooth and unimodal mismatch distribution of ITS2 and COII was detected in the An. sinensis population from Laos. As mentioned above, this population experienced expansion after reaching a bottleneck (Additional file 6: Fig. S5)
Discussion
Subdivision of the Hyrcanus group
Based on morphological properties, the Hyrcanus group can be classified into three subgroups [48, 49]: the Nigerrimus subgroup, including An. nigerrimus, An. nitidus, An. pursati and An. pseudosinensis; the Lesteri subgroup, including An. lesteri, An. paraliae, An. peditaeniatus, An. crawfordi and An. vietnamensis; and species in an unassigned subgroup. Given the difficulty in identifying cryptic species by morphological characteristics alone, molecular methods have been used as powerful tools to complement traditional morphological taxonomy [10, 11, 17]. However, the barcoding of DNA for all members of the Hyrcanus group has scarcely been performed. Fang et al. were the first to have reconstructed the molecular phylogeny and analyzed the genetic divergence of the Hyrcanus group through two DNA barcoding markers, namely, ITS2 and COI [50, 51]. Based on the GenBank database and their original study data, they used 461 ITS2 sequences of 19 species and 466 COI sequences of 18 species to reconstruct the molecular phylogeny of the Hyrcanus group across its worldwide geographic range [50, 51].
Similarly, both rDNA and mtDNA were used in the present study to perform DNA barcoding. So far, there are 691 ITS2 of 19 species and 368 COII of 14 species of the Hyrcanus group deposited in GenBank (Additional file 2: Table S1). The NJ tree obtained in this study supported the monophyly of the Hyrcanus group, but the subgroup arrangement based on the rDNA and mtDNA DNA markers failed to comply with that indicated by morphological characteristics. Based on ITS2 sequences, there were two major subgroups that could be recognized: the first group includes An. nigerrimus, An. nitidus, An. argyropus and An. pursati, and the second group includes An. sinensis, An. engarensis, An. belenrae, An. kleini, An. liangshanensis, An. kweiyangensis, An. sineroides, An. lesteri, An. paraliae, An. crawford, An. peditaeniatus, An. hyrcanus, An. pseudopictus and An. pullus. This result is consistent with the study conducted by Fang based on ITS2 sequences and suggests that the morphology-based Lesteri subgroup is not monophyletic [51]. In addition, as indicated by the tree topologies in prior studies [8, 9, 18], An. crawfordi had the smallest distance to An. peditaeniatus; however, according to the results of the study of Fang [51] and the present study, An. crawfordi was at a distance from An. peditaeniatus, but approached An. lesteri and An. paraliae to form a single clade in the NJ tree based on ITS2. Moreover, An. pseudopictus individuals were embedded in the An. hyrcanus lineage in the ITS2 tree, implying the probability that An. hyrcanus and An. pseudopictus are the same species, consistent with the findings of prior studies [14, 51]. Similarly, An. engarensis and An. kleini were found to be of the identical species according to the study of Hwang [9] and the current study. However, the results allow for some speculation on the relationship between An. kweiyangensis and An. liangshanensis. In the ITS2 tree built in the study of Fang [51] and in the present study, the two species were classified into the same clade, but they were classified into two different clades in the COI tree [50].
Notably, three major clusters were found using the COII sequences in this study: An. nigerrimus was separated from the Nigerrimus group to form the first cluster; An. nitidus, An. pursati and An. argyropus were included in the second cluster; and all remaining species were included in the third cluster. Fang et al. reported that the pairwise distance of An. paraliae and An. lesteri reached 0.019, but that it was impossible to distinguish the two species in the phylogenetic tree based on COI [50]. In this study, however, the two species were easily distinguishable in the NJ tree based on COII, despite the pairwise distance of An. lesteri and An. paraliae reaching as low as 0.017. In their study, Taai et al. considered An. paraliae to be a synonym of An. lesteri, based on the results of crossing experiments performed between these two species using data on species distributions, morphological variants, cytology and comparative DNA sequence analyses [29]. Therefore, An. paraliae and An. lesteri should be treated as two closely related species in this study. In addition, the relationship between An. sinensis, An. belenrae and An. kleini remained inconclusive in the COII tree. Showing significantly low pairwise distance values (Tables 2, 3), these species contributed to the formation of a monoclade with a high achievement of node confidence (Additional file 3: Fig. S2), which suggests the potential occurrence of ancient hybridization in the aforementioned three species with close correlations.
Due to the failure in previous studies to cover an appropriate number of species, it is difficult to detect subgroups in the phylogenetic tree, as a result of which no consistent results were produced in the current and prior studies on the genetic relationships of Hyrcanus group members. The genes submitted to GenBank were not properly presented, with a number of error sequences possibly in the database [52, 53]. In this study, some error sequences related to the authors who submitted these sequences to GenBank were used to conduct the phylogenetic analysis, which led to misidentification (See Results section Phylogenetic analysis). These error sequences must be removed from future phylogenetic analyses.
Phylogenetic reconstructions with the use of COII and ITS2
An effective DNA marker must exhibit low intraspecific distances and large interspecific distances [54]. In this study, we found that the COII barcoding gap varied from 0.017 to 0.025, whereas that of ITS2 ranged from 0.015 to 0.073. The ITS2 interspecific genetic distance was 125-fold higher than that of the average intraspecies difference; in comparison, the average COII divergence among group of species was merely eightfold higher than that within species.
Since mtDNA is characterized by maternal inheritance, any offspring or hybrid would have only the mtDNA of the maternal species. Accordingly, hybridization can lead to shared or highly consistent sequences in the mitochondrial genome. Therefore, the major downside of employing COII for phylogenetics research of the Hyrcanus group is that COII fails to distinguish between those species with a close relationship, i.e. An. sinensis, An. belenrae and An. kleini. As suggested by the divergent mtDNA of An. belenrae and An. kleini, the mitochondrial genomes of incipient sibling species were sympatrically replaced by those of An. sinensis extensively in many regions. In general, recent hybridization can play a role in transferring mtDNA from one species to another to cause mtDNA variation [26, 55, 56]. Thus, results on mtDNA are considered to be more useful when speculating about the possibility of ancient hybridization in mosquito molecular phylogeny. In contrast, rDNA has been suggested to be more reliable than mtDNA in addressing evolutionary problems with the recently diverged taxa or cryptic species of mosquitoes [51] and in building species boundaries when these can not be addressed by using mtDNA. Hence, the ITS2 marker has been widely used for species identification and phylogenetic reconstruction for the Hyrcanus group [8, 9, 18, 22, 23].
Demographic history of An. sinensis population
According to the results of the neutrality tests and mismatch distribution, a number of members of the Hyrcanus group, namely An. peditaeniatus, An. hyrcanus, An. sinensis and An. lesteri, are suspected to have experienced hitchhiking or population expansion based on both markers. These four species show an extensive distribution, and some have the potential to transmit malaria. In addition, the An. sinensis samples collected from Laos in this study showed significant negative values in the neutrality tests, according to both markers. A smooth and unimodal mismatch distribution of ITS2 and COII was observed in the population of An. sinensis from Laos, suggesting that the expansion occurred after a bottleneck was reached. According to several recent studies, An. sinensis acted as an important vector of local malaria outbreak, which highlights a potential risk of malaria transmission in endemic areas [57, 58]. Therefore, the potential transmission of vivax malaria by An. sinensis in the northern and southern regions of Laos deserves more attention.
Conclusions
The topology of the ITS2 and COII trees of the Hyrcanus group conformed to the morphology-based taxonomy for species classification rather than for subgroup division. Compared to COII within the Hyrcanus group, rDNA–ITS2 can be considered to be the more reliable tool for investigating the phylogenetic correlation of closely related mosquito species, such as An. belenrae, An. kleini and An. sinensis. Moreover, the population expansion of an important vivax malaria vector, An. sinensis, has been emphasized as a potential risk factor in malaria transmission in northern and southern Laos. The results of this study support the efficacy of molecular identification of the Anopheles hyrcanus group in terms of vector surveillance.
Availability of data and materials
Data supporting the conclusions of this article are included within the article and its additional files. The datasets generated and/or analysed during the current study are available in the GenBank (http://www.ncbi.nlm.nih.gov/). The raw datasets used and/or analysed during this study are available from the corresponding author upon reasonable request.
Abbreviations
- COII:
-
Cytochrome c oxidase subunit 2
- K2P:
-
Kimura’s 2-parameter
- ITS2:
-
Internal transcribed spacer 2
- mtDNA:
-
Mitrochondrial DNA
- ML:
-
Maximum likelihood
- NJ:
-
Neighbor-joining
References
Harbach RE. The phylogeny and classification of Anopheles. In: Manguin S, editor. Anopheles mosquitoes-new insights into malaria vectors. Rijeka: InTech; 2013. p. 3–55.
Fang Y, Shi WQ, Zhang Y. Research progress in classification of Anopheles hyrcanus group (Diptera: Culicidae). Chin J Parasitol Paras Dis. 2016;34(6):565–70 (in Chinese).
Lee WJ, Klein TA, Kim HC, Choi YM, Yoon SH, Chang KS, et al. Anopheles kleini, Anopheles pullus, and Anopheles sinensis: potential vectors of Plasmodium vivax in the Republic of Korea. J Med Entomol. 2007;44(6):1086–90. https://doi.org/10.1603/0022-2585(2007)44[1086:akapaa]2.0.co;2.
Gingrich JB, Weatherhead A, Sattabongkot J, Pilakasiri C, Wirtz RA. Hyperendemic malaria in a Thai village: dependence of year-round transmission on focal and seasonally circumscribed mosquito (Diptera: Culicidae) habitats. J Med Entomol. 1990;27(6):1016–26. https://doi.org/10.1093/jmedent/27.6.1016.
Alam MS, Khan MG, Chaudhury N, Deloer S, Nazib F, Bangali AM, et al. Prevalence of anopheline species and their Plasmodium infection status in epidemic-prone border areas of Bangladesh. Malar J. 2010;9:15. https://doi.org/10.1186/1475-2875-9-15.
Paredes-Esquivel C, Harbach RE, Townson H. Molecular taxonomy of members of the Anopheles hyrcanus group from Thailand and Indonesia. Med Vet Entomol. 2011;25(3):348–52. https://doi.org/10.1111/j.1365-2915.2010.00937.x.
Jeong KY, Un S, Lee J, Lee IY, Yong TS, Ree HI. Population dynamics of five Anopheles species of the Hyrcanus group in northern Gyeonggi-do, Korea. Korean J Parasitol. 2010;48(4):351–3. https://doi.org/10.3347/kjp.2010.48.4.351.
Yajun M, Xu J. The Hyrcanus group of Anopheles (Anopheles) in China (Diptera: Culicidae): species discrimination and phylogenetic relationships inferred by ribosomal DNA internal transcribed spacer 2 sequences. J Med Entomol. 2005;42(4):610–9.
Hwang UW. Revisited ITS2 phylogeny of Anopheles (Anopheles) Hyrcanus group mosquitoes: reexamination of unidentified and misidentified ITS2 sequences. Parasitol Res. 2007;101(4):885–94. https://doi.org/10.1007/s00436-007-0553-4.
Hebert PD, Cywinska A, Ball SL, deWaard JR. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;270(1512):313–21. https://doi.org/10.1098/rspb.2002.2218.
Hebert PD, Ratnasingham S, deWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci. 2003;270(Suppl 1):S96–9. https://doi.org/10.1098/rsbl.2003.0025.
Zhou ZJ. The malaria situation in the People’s Republic of China. Bull World Health Organ. 1981;59(6):931–6.
Ponçon N, Toty C, Kengne P, Alten B, Fontenille D. Molecular evidence for similarity between Anopheles hyrcanus (Diptera: Culicidae) and Anopheles pseudopictus (Diptera: Culicidae), sympatric potential vectors of malaria in France. J Med Entomol. 2008;45(3):576–80. https://doi.org/10.1603/0022-2585(2008)45[576:mefsba]2.0.co;2.
Ponçon N, Toty C, L’Ambert G, Le Goff G, Brengues C, Schaffner F, et al. Biology and dynamics of potential malaria vectors in Southern France. Malar J. 2007;6:18. https://doi.org/10.1186/1475-2875-6-18.
Vythilingam I, Keokenchan K, Phommakot S, Nambanya S, Inthakone S. Preliminary studies of Anopheles mosquitos in eight provinces in Lao PDR. Southeast Asian J Trop Med Public Health. 2001;32(1):83–7.
Zhang C, Sorchampa S, Zhou H, Jiang J, Yang R, Zhang Y. Survey of asymptomatic malaria and mosquito vectors in Muang Khua District of Phongsaly Province, China-Laos Border. Int J Infect Dis. 2020;96:141–7. https://doi.org/10.1016/j.ijid.2020.03.066.
Schmidt S, Schmid-Egger C, Morinière J, Haszprunar G, Hebert PD. DNA barcoding largely supports 250 years of classical taxonomy: identifications for Central European bees (Hymenoptera, Apoidea partim). Mol Ecol Resour. 2015;15(4):985–1000. https://doi.org/10.1111/1755-0998.12363.
Djadid ND, Jazayeri H, Gholizadeh S, Rad ShP, Zakeri S. First record of a new member of Anopheles Hyrcanus Group from Iran: molecular identification, diagnosis, phylogeny, status of kdr resistance and Plasmodium infection. J Med Entomol. 2009;46(5):1084–93. https://doi.org/10.1603/033.046.0515.
Hebert PD, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA Barcodes. PLoS Biol. 2004;2(10):e312. https://doi.org/10.1371/journal.pbio.0020312.
Cunningham EP, Meghen CM. Biological identification systems: genetic markers. Rev Sci Tech. 2001;20(2):491–9. https://doi.org/10.20506/rst.20.2.1284.
Mohanty A, Swain S, Kar SK, Hazra RK. Analysis of the phylogenetic relationship of Anopheles species, subgenus Cellia (Diptera: Culicidae) and using it to define the relationship of morphologically similar species. Infect Genet Evol. 2009;9(6):1204–24. https://doi.org/10.1016/j.meegid.2009.06.021.
Gao Q, Beebe NW, Cooper RD. Molecular identification of the malaria vectors Anopheles anthropophagus and Anopheles sinensis (Diptera: Culicidae) in central China using polymerase chain reaction and appraisal of their position within the Hyrcanus Group. J Med Entomol. 2004;41(1):5–11. https://doi.org/10.1603/0022-2585-41.1.5.
Songsawatkiat S, Baimai V, Thongsahuan S, Otsuka Y, Taai K, Hempolchom C, et al. Evidence to support a conspecific nature of allopatric cytological races of Anopheles nitidus (Diptera: Culicidae) in Thailand. J Insect Sci. 2014;14:287. https://doi.org/10.1093/jisesa/ieu149.
Li C, Lee J, Groebner J, Kim H, Klein T, Oguinn M, et al. A newly recognized species in the Anopheles Hyrcanus Group and molecular identification of related species from the Republic of South Korea (Diptera: Culicidae). Zootaxa. 2005;939:1–8.
Rueda L. Two new species of Anopheles (Anopheles) Hyrcanus Group (Diptera: Culicidae) from the Republic of South Korea. Zootaxa. 2005;941(941):1–26. https://doi.org/10.5281/zenodo.171166.
Mallet J. Hybridization as an invasion of the genome. Trends Ecol Evol. 2005;20(5):229–37. https://doi.org/10.1016/j.tree.2005.02.010.
Brennan IG, Bauer AM, Jackman TR. Mitochondrial introgression via ancient hybridization, and systematics of the Australian endemic pygopodid gecko genus Delma. Mol Phylogenet Evol. 2016;94(Pt B):577–90. https://doi.org/10.1016/j.ympev.2015.10.005.
Bergsten J, Bilton DT, Fujisawa T, Elliott M, Monaghan MT, Balke M, et al. The effect of geographical scale of sampling on DNA barcoding. Syst Biol. 2012;61(5):851–69. https://doi.org/10.1093/sysbio/sys037.
Taai K, Baimai V, Saeung A, Thongsahuan S, Min GS, Otsuka Y, et al. Genetic compatibility between Anopheles lesteri from Korea and Anopheles paraliae from Thailand. Mem Inst Oswaldo Cruz. 2013;108(3):312–20. https://doi.org/10.1590/S0074-02762013000300009.
Perandin F, Manca N, Calderaro A, Piccolo G, Galati L, Ricci L, et al. Development of a real-time PCR assay for detection of Plasmodium falciparum, Plasmodium vivax, and Plasmodium ovale for routine clinical diagnosis. J Clin Microbiol. 2004;42(3):1214–9. https://doi.org/10.1128/jcm.42.3.1214-1219.2004.
Stresman GH, Kamanga A, Moono P, Hamapumbu H, Mharakurwa S, Kobayashi T, et al. A method of active case detection to target reservoirs of asymptomatic malaria and gametocyte carriers in a rural area in Southern Province, Zambia. Malar J. 2010;9:265. https://doi.org/10.1186/1475-2875-9-265.
Das BP, Rajagopal R, Akiyama J. Pictorial key to the species of Indian Anophline mosquitoes. J Pure Appl Zool. 1990;2(3):131–62.
Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547–9. https://doi.org/10.1093/molbev/msy096.
Hall T. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser. 1999;41:95–8. http://www.mbio.ncsu.edu/BioEdit/bioedit.html.
Nei M, Kumar S, Takahashi K. The optimization principle in phylogenetic analysis tends to give incorrect topologies when the number of nucleotides or amino acids used is small. Proc Natl Acad Sci USA. 1998;95(21):12390–7. https://doi.org/10.1073/pnas.95.21.12390.
Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc Natl Acad Sci USA. 2004;101(30):11030–5. https://doi.org/10.1073/pnas.0404206101.
Wang G, Li C, Guo X, Xing D, Dong Y, Wang Z, et al. Identifying the main mosquito species in China based on DNA barcoding. PLoS ONE. 2012;7(10): e47051. https://doi.org/10.1371/journal.pone.0047051.
Rambaut A. FigTree. 2012. https://github.com/rambaut/figtree/releases. Accessed 30 Jan 2021.
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111–20. https://doi.org/10.1007/BF01731581.
Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol. 1983;19(2):153–70. https://doi.org/10.1007/BF02300753.
Excofer L, Laval G, Schneider S. Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evol Bioinform Online. 2005;1:47–50.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123(3):585–95.
Fu YX. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147(2):915–25.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25(11):1451–2. https://doi.org/10.1093/bioinformatics/btp187.
Slatkin M, Hudson RR. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991;129(2):555–62.
Rogers AR, Harpending H. Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol. 1992;9(3):552–69. https://doi.org/10.1093/oxfordjournals.molbev.a040727.
Rogers AR. Genetic evidence for a Pleistocene population explosion. Evolution. 1995;49(4):608–15. https://doi.org/10.1111/j.1558-5646.1995.tb02297.x.
Harrison BA. A new interpretation of affinities within the Anopheles hyrcanus Complex of southeast Asia. Mosq Syst. 1972;4(3):73–83.
Harbach RE. The classification of genus Anopheles (Diptera: Culicidae): a working hypothesis of phylogenetic relationships. Bull Entomol Res. 2004;94(6):537–53. https://doi.org/10.1079/ber2004321.
Fang Y, Shi WQ, Zhang Y. Molecular phylogeny of Anopheles hyrcanus group (Diptera: Culicidae) based on mtDNA COI. Infect Dis Poverty. 2017;6(1):61. https://doi.org/10.1186/s40249-017-0273-7.
Fang Y, Shi WQ, Zhang Y. Molecular phylogeny of Anopheles hyrcanus group members based on ITS2 rDNA. Parasit Vectors. 2017;10(1):417. https://doi.org/10.1186/s13071-017-2351-x.
Harris DJ. Can you bank on GenBank? Trends Ecol Evol. 2003;18(7):317–9. https://doi.org/10.1016/s0169-5347(03)00150-2.
Bidartondo MI. Preserving accuracy in GenBank. Science. 2008;319(5870):1616. https://doi.org/10.1126/science.319.5870.1616a.
Hemmerter S, Slapeta J, Beebe NW. Resolving genetic diversity in Australasian Culex mosquitoes: incongruence between the mitochondrial cytochrome c oxidase I and nuclear acetylcholine esterase 2. Mol Phylogenet Evol. 2009;50(2):317–25. https://doi.org/10.1016/j.ympev.2008.11.016.
Choochote W, Min GS, Intapan PM, Tantrawatpan C, Saeung A, Lulitanond V. Evidence to support natural hybridization between Anopheles sinensis and Anopheles kleini (Diptera: Culicidae): possibly a significant mechanism for gene introgression in sympatric populations. Parasit Vectors. 2014;7:36. https://doi.org/10.1186/1756-3305-7-36.
Toews DP, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol. 2012;21(16):3907–30. https://doi.org/10.1111/j.1365-294X.2012.05664.x.
Pang SC, Andolina C, Malleret B, Christensen PR, Lam-Phua SG, Razak MABA, et al. Singapore’s Anopheles sinensis Form A is susceptible to Plasmodium vivax isolates from the western Thailand-Myanmar border. Malar J. 2017;16(1):465. https://doi.org/10.1186/s12936-017-2114-3.
Zhu G, Xia H, Zhou H, Li J, Lu F, Liu Y, et al. Susceptibility of Anopheles sinensis to Plasmodium vivax in malarial outbreak areas of central China. Parasit Vectors. 2013;6:176. https://doi.org/10.1186/1756-3305-6-176.
Acknowledgements
We greatly thank the National Natural Science Foundation of China for study design, analysis and interpretation of the data, as well as writing of the manuscript and the decision to submit the manuscript for publication. We also thank the Yunnan Provincial Collaborative Innovation Center for Public Health and Disease Prevention and Control (grant number 2014YNPHXT03) and the China-ASEAN Public Health Cooperation Fund and Joint Control of Malaria and Dengue in Lancang-Mekong River Area for collecting samples and data and performing the experiments. We thank Dr. Phoutnalong Vilay at the National Malaria Centre of Lao People's Democratic Republic who participated in the sampling and data collection. We are also grateful to Dr. Phanviengxay Khammavong, Hao Phommasa and Chunfu Li for participating in the fieldwork.
Funding
This study was supported by the National Natural Science Foundation of China (Grant Number 31601002, 81160357, 30960327, 30660160), Yunnan Provincial Collaborative Innovation Center for Public Health and Disease Prevention and Control (Grant Number 2014YNPHXT03) and the China-ASEAN Public Health Cooperation Fund and Joint Control of Malaria and Dengue in Lancang-Mekong River Area.
Author information
Authors and Affiliations
Contributions
Data and statistical analysis, and manuscript preparation: YLZ and CLZ. Data collection: YLZ, CLZ, RY, CHL, LBW and HNZ. Mosquito collection and identification: CLZ, RY, CHL, XFG and CHL. Experimental work: YLZ, YD and CLZ. Procurement of funding: YLZ and HNZ. Study conception/design and critical review of the manuscript: YLZ, CLZ and HNZ. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1: Figure S1.
Map of the three sampling sites in Laos: Pathoomphone County (Champasak Province, Laos-Cambodia border), Pak lay County (Xaignabouli Province, Laos-Thailand border) and Yot Ou County (Phongsaly Province, Laos–China border). The shapefile map of Lao PDR was downloaded and prepared by using Pixelmap Generator-Beta on line (amCharts, Vilnius, Lithuania) (https://pixelmap.amcharts.com/), which is copyright free.
Additional file 2: Table S1.
Full list of the 691 ITS2 sequences of the Anopheles hyrcanus group deposited in GenBank, with GenBank accession numbers, species, geographical location and corresponding authors. Table S2. Full list of 368 COII sequences of the Anopheles hyrcanus group deposited in GenBank, with GenBank accession numbers, species, geographical location and corresponding authors. Table S3. Full list of 267 ITS2 sequences (haplotypes) of the Anopheles hyrcanus group used for genetic divergence analysis and phylogenetic analysis. Table S4. Full list of 260 COII sequences (haplotypes) of the Anopheles hyrcanus group used for genetic divergence analysis and phylogenetic analysis.
Additional file 3: Figure S2.
Neighbor-joining phylogenetic tree of the Hyrcanus group based on ITS2 (a) and COII sequences (b) from GenBank and our original data. Bootstrap values (1000 replicates) of neighbor-joining analyses are shown above/below the main lineages. Lineage designation is indicated on the right. Bars represent 20.00 substitutions per site based on ITS2 and 5.0 substitutions per site based on COII. Anopheles lindesayi and An. claviger were used as the outgroup taxa.
Additional file 4: Figure S3.
Maximum Likelihood phylogenetic tree of the Hyrcanus group based on ITS2 (a) and COII sequences (b) from GenBank and our original data. Bootstrap values (1000 replicates) of maximum-likelihood analyses are shown above/below the main lineages. Lineage designation is indicated on the right. Bars represent 20.00 substitutions per site based on ITS2 and 7.0 substitutions per site based on COII. Anopheles lindesayi and An. claviger were used as the outgroup taxa.
Additional file 5: Figure S4.
The mismatch distribution graphs in the Hyrcanus group based on ITS2 (a) and COII (b). The X- and Y-axis show the number of pairwise differences and the frequency of the pairwise comparisons, respectively. The observed frequencies are represented by a dotted line. The frequency expected under the hypothesis of constant population model is depicted by a solid line.arg. An. argyropus, bel. An. belenrae, cra. An. crawfordi, eng. An. engarensis, hyr. An. hyrcanus, hyr. spIR An. hyrcanus spIR, kle. An. kleini, kwe. An. kweiyangensis, les. An. lesteri, lia. An. liangshanensis, nig. An. nigerrimus, nit. An. nitidus, par. An. paraliae, ped. An. peditaeniatus, pse. An. pseudopictus, pul. An. pullus, pur. An. pursati, siner. An. sineroides, sin. An. sinensis
Additional file 6: Figure S5.
The mismatch distribution graphs in Anopheles sinensis based on ITS2 (a) and COII (b). The X- and Y-axis show the number of pairwise differences and the frequency of the pairwise comparisons, respectively. The observed frequencies are represented by a dotted line. The frequency expected under the hypothesis of constant population model is depicted by a solid line.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, C., Yang, R., Wu, L. et al. Molecular phylogeny of the Anopheles hyrcanus group (Diptera: Culicidae) based on rDNA–ITS2 and mtDNA–COII. Parasites Vectors 14, 454 (2021). https://doi.org/10.1186/s13071-021-04971-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-021-04971-4