
Abstract
Background
The Anopheles hyrcanus group includes 25 species, and is widely distributed in the Oriental and Palaearctic regions. Several species within this group are vectors of malaria, lymphatic filariasis and Japanese encephalitis. It is difficult or impossible to identify cryptic species based on their morphological characteristics, with some closely related species of the Hyrcanus Group have similar adult morphological characteristics. Thus, their molecular identification has been an important complementary method to traditional morphological taxonomy.
Methods
We used 461 ribosomal DNA (rDNA) internal transcribed spacer 2 (ITS2) sequences relating to 19 species to reconstruct the molecular phylogeny of the Hyrcanus Group across its range. In addition, we compared the performance of rDNA ITS2 to that of mitochondrial DNA (mtDNA) cytochrome c oxidase subunit 1 gene (cox1) to assess the genetic divergence of Hyrcanus Group sibling species.
Results
Based on Kimura’s 2-parameter (K2P) distance model, the average conspecific ITS2 divergence was 0.003, whereas sequence divergence between species averaged 0.480. Average ITS2 sequence divergences were almost 160 times higher among the Hyrcanus Group members than within each species. Two sets of sibling species, An. lesteri Baisas & Hu, 1936 and An. paraliae Sandosham, 1959; and An. sinensis Wiedemann, 1828, An. belenrae Rueda, 2005, and An. kleini Rueda, 2005, were resolved by ITS2. Each of these species was represented as an independent lineage in the phylogenetic tree. Results suggest that An. pseudopictus Grassi, 1899 and An. hyrcanus (Pallas, 1771) are most likely a single species. We uncovered two new ITS2 lineages that require further study before resolving their true taxonomic status, and designed a diagnostic polymerase chain reaction (PCR) assay to distinguish five morphologically similar species.
Conclusions
Nuclear and mitochondrial genes generally provided consistent results for subgroup division. Compared to cox1, ITS2 is a more reliable tool for studying phylogenetic relationships among closely related mosquito taxa. Based on species-specific differences in ITS2 sequences, the multiplex PCR assay developed here can be used to improve the efficiency of vector identification. Thus, this research will promote the progress of malaria vector surveillance in both epidemic and non-epidemic areas of South and East Asia.
Similar content being viewed by others


Background
The Anopheles hyrcanus group (hereinafter Hyrcanus Group) includes (at least) 25 species and belongs to the Anopheles Myzorhynchus Series, with one provisionally designated member [1, 2]. The group is widely distributed in Oriental and Palaearctic regions. Anopheles sinensis and An. lesteri are the main malaria vectors in China [3], An. hyrcanus is a potential malaria vector in southern France [4, 5], An. kleini and An. pullus Yamada, 1937 are primary vectors of malaria in the Republic of Korea [6], and An. sinensis, An. nigerrimus Giles, 1900, and An. peditaeniatus (Leicester, 1908) are potential vectors of malaria in Thailand [7, 8].
Because many of the primary malaria vectors belong to the Hyrcanus Group, accurate species identification and phylogenetic relationship evaluation in this group are essential for understanding malaria transmission and its relationship with the evolution of Plasmodium spp. However, species within the Hyrcanus Group are difficult or impossible to be accurately distinguished using only morphological characteristics, even for trained taxonomists [9, 10], due to highly variable morphologies and to adults of some species possessing almost identical adult morphological features, as is the case of the most closely related species [11, 12].
DNA barcoding is an important addition to traditional morphology-based methods and a highly useful tool for species recognition, regardless of life stage [13,14,15]. The mitochondrial cytochrome c oxidase subunit 1 gene (cox1) is the standard barcode for species identification in a wide range of animal taxa, and is approximately 658 bp long [16, 17]. However, genomic introgression frequently involves mitochondrial DNA (mtDNA), because of recent hybridization events between species [18, 19]. Recent hybridization transfers mtDNA from one species to another and can lead to mtDNA variation [18, 20, 21]. In our previous study [22], cox1 failed to distinguish between recently diverged taxa, suggesting that mtDNA may not be appropriate for investigating the molecular phylogeny of the Hyrcanus Group.
A DNA marker that evolves at a species-level rate would be able to accurately reconstruct phylogenetic relationships within the Hyrcanus Group, and elucidate the ambiguity that has arisen from improper classification [23, 24]. Internal transcribed spacer 2 (ITS2) has been used for addressing taxonomic issues within the Hyrcanus Group because it has high interspecific, and low intraspecific variability [11, 12, 16, 25,26,27]. Using this marker, three newly proposed lineages have been revealed: two separated from An. sinensis, namely An. belenrae and An. kleini [28], and one closely related to An. hyrcanus, provisionally designated as An. hyrcanus spIR [16]. In addition, based on interspecific comparisons of ITS2, An. yatsushiroensis was synonymized with An. pullus [29, 30], An. kunmingensis and An. liangshanensis Kang, Tan, Cao, Cheng, Yang & Huang, 1984 were found to be synonymous [31, 32], An. paraliae was considered a synonym of An. lesteri [33], An. anthropophagus and An. lesteri were classified as conspecific [34, 35], and An. kleini was presumed a synonym of An. engarensis Kanda & Oguma, 1978 [12].
To reconstruct the molecular phylogeny of the Hyrcanus Group, the barcoding gap of ITS2 should be identified. Therefore, it is necessary to include specimens of the same species collected from different geographical locations [17, 36], in order to calculate the intra- and interspecific variation of ITS2 within the group. The ITS2 sequence database in GenBank (http://www.ncbi.nlm.nih.gov/genbank) permits the use of reference sequences for Hyrcanus Group species identification across a relatively wide geographical distribution [33]. Thus, in the present study, GenBank sequences and our original data were used to reconstruct a phylogeny for the Hyrcanus Group based on ITS2, to resolve phylogenetic relationships between closely related taxa. In addition, we compared rDNA ITS2 and mtDNA cox1 in terms of genetic divergence and distinction efficiency among Hyrcanus Group species. Using conserved ITS2 fragments from Hyrcanus Group species, we also designed a diagnostic polymerase chain reaction (PCR) assay for distinguishing five morphologically similar species. The present study will help improve the efficiency of mosquito molecular identification in malaria vector surveillance.
Methods
ITS2 sequence retrieval from GenBank
We used the keywords “(species name) & ITS2” to search for ITS2 sequences of Hyrcanus Group members deposited in GenBank. Details of these sequences are provided in Additional file 1: Table S1.
DNA extraction, ITS2 amplification and sequencing
Dry, pin-mounted museum specimens of the Hyrcanus Group collected after the year 2000, and identified using standard taxonomic keys [37, 38], were used for DNA extraction following the protocol specified in a previous paper [22]. One leg was removed from each adult specimen, transferred to a dry 1.5 ml Eppendorf tube, ground to powder, and incubated in lysis buffer overnight at 56 °C. Bind, wash, and elution steps followed the manufacturer’s instructions for the Qiagen DNA Blood and Tissue Kit (Qiagen, Hilden, Germany). Voucher specimens were stored in the Herbarium of the National Institute of Parasitic Diseases, Chinese Center for Disease Control and Prevention (NIPD, China CDC).
Approximately 550 bp PCR products were amplified using the forward primer ITS2a (5′-TGT GAA CTG CAG GAC ACA T-3′) and the reverse primer ITS2b (5′-TAT GCT TAA ATT CAG GGG GT-3′) [39]. The 25 μl reaction mixture contained 12.5 μl of 2× Taq PCR Master Mix with dyes (DBI Bioscience, Shanghai, China), 1 μl of each 10 μM primer, 4 μl of extracted DNA, and 6.5 μl of ddH2O. The thermocycling profile consisted of an initial denaturation at 95 °C for 2 min followed by 35 cycles at 95 °C for 1 min, 55 °C for 1 min, and 72 °C for 2 min, with a final extension at 72 °C for 7 min. Amplicons were subjected to electrophoresis on a 1.2% agarose gel stained with GoldView dye (Solarbio, Beijing, China), and cleaned and sequenced by Sangon (Shanghai, China).
Sequence annotation, alignment, and phylogenetic analysis
Our original ITS2 sequences, and those deposited in GenBank, were annotated using the ITS2 annotation tool (http://its2.bioapps.biozentrum.uni-wuerzburg.de) [40]. A multiple sequence alignment was conducted in ClustalW2 [41] and manually adjusted where necessary. Gaps were excluded from the analysis and characters were unweighted. Anopheles lindesayi Giles, 1900 (AJ620898) and An. claviger (Meigen, 1804) (AY129232 and DQ229313) were used as outgroup taxa to the Hyrcanus Group, following previous studies [42]. The neighbour-joining (NJ) method generally reveals shallow intraspecific and deep interspecific divergences [17, 43]; thus, a bootstrapped NJ tree was constructed using 1000 replicates by ClustalW2 to provide a graphical representation of the phylogenetic relationships among Hyrcanus Group members. This phylogram was visualized using Figtree v1.4.2 [44].
Genetic diversity analysis and neutrality test
Both intraspecific and interspecific ITS2 divergences were examined using Kimura’s 2-parameter (K2P) distance model [45] in MEGA v5.1 [46]. Genetic divergence, according to Nei’s distance model [47], was determined in Arlequin v3.5.2.2 [48]. Genetic diversity indices and neutrality tests (Fu’s Fs [49] and Tajima’s D [50]) were calculated using DnaSP v5.10 [51].
Primer design and conditions of the multiplex PCR assay
We developed a multiplex PCR-based assay to simultaneously identify five Hyrcanus Group species, An. sinensis, An. lesteri, An. peditaeniatus, An. hycanus and An. pullus, most of which are sympatric. Species-specific reverse primers were designed using Primer3web (http://primer3.ut.ee/), based on the distinctive set of ITS2 sequences of each species. Forward primers applied the universal primer ITS2a, which is located on the conserved 5.8S gene. The species-specific reverse primers were as follows: An. peditaeniatus, 5′-ACG GCG TAG GTT ATT GTC TCT-3′; An. hyrcanus, 5′-GGY TTT ACA CCG CAG TTC TT-3′; An. lesteri, 5′-GCC CAT TCC MCT ATC TCG AA-3′; An. pullus, 5′-CGC TCT CTC AAC AAC TGG GT-3′; and An. sinensis, 5′-GAG TGG CCT CAC TCT TGG AG-3′. The multiplex PCR was conducted in 25 μl total volume, containing 12.5 μl of 2× Taq PCR Master Mix (with dyes), 1.5 μl of 10 μM ITS2a, 0.75 μl of each 10 μM species-specific reverse primer, 2 μl of extracted DNA, and 5.25 μl of ddH2O. Conditions for the amplification comprised an initial denaturation at 95 °C for 2 min followed by 35 cycles at 95 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s, with a final extension at 72 °C for 5 min. The amplicons were separated by electrophoresis on 1.2% agarose gels in 0.5× Tris-borate-EDTA (TBE) buffer and stained with GoldView dye (Solarbio, Beijing, China).
Evaluation of the multiplex PCR assay
Anopheles liangshanensis, An. jeyporiensis James, 1902, An. minimus Theobald, 1901, An. harrisoni Harbach & Manguin, 2007, An. maculatus Theobald, 1901, An. aconitus Dönitz, 1902, An. splendidus Koidzumi, 1920 and An. dravidicus Christophers, 1924 are common in China. The DNA of several Anopheles species (see Additional file 2: Table S2) previously identified by molecular methods in our lab was used to validate the novel multiplex PCR assay. To determine the sensitivity of the assay, it was further tested using a DNA dilution series for each of the five target species from 10 ng/μl to 1 × 10-5 ng/μl or 1 × 10-6 ng/μl.
Results
Sequence analysis
Four hundred and forty one ITS2 sequences of the Hyrcanus Group were extracted from GenBank. Details of the ITS2 sequences used for the phylogenetic analysis are given in Additional file 1: Table S1. Seven ITS2 sequences were distant from conspecific sequences, and therefore were excluded from further phylogenetic analyses. We could not find identical sequences in GenBank for two of these sequences. One questionable sequence for An. crawfordi Reid, 1953 (AF261949) was distant from conspecific individuals; its closest sequences were from An. peditaeniatus, with a similarity of 83–84%. In our previous study [22], two peculiar cox1 sequences for An. crawfordi (KF830735 and KF830736) clustered with sequences for An. xui Dong, Zhou, Dong & Mao, 2007. Unfortunately, there is no current ITS2 record of An. xui. The sequence AF261949 might have been obtained from an An. xui specimen, but more rigorous morphological and genetic examinations are needed to confirm this. The other sequence for An. hyrcanus was detected as accession number KC769647, and its closest sequences were from An. sineroides Yamada, 1924 (90% similarity). The original specimen was from China; however, no detailed information about the collection site was available. We obtained four ITS2 sequences of An. hyrcanus from Xinjiang province, the only region of China in which it is found [37]; therefore, specimen KC769647 was probably collected in Xinjiang Province as well. It is unlikely that specimens from the same province would have such large genetic distances between them, so it is possible that the specimen KC769647 belongs to a cryptic species, or to another Hyrcanus Group member that has no available ITS2 data.
Five An. sinensis individuals (KJ462254–KJ462258) were much closer to An. crawfordi individuals (99% similarity with AB779132) than to some of their conspecifics, and clustered with the An. crawfordi lineage in the phylogenetic tree. Thus, these sequences might be incorrect, presumably due to the misidentification of original specimens.
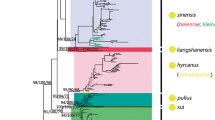
The ITS2 sequences KJ960222–KJ960226, collected from Iran, were originally identified as An. hyrcanus. In the alignment of the Hyrcanus Group members, their nucleotide substitutions were more similar to those of An. hyrcanus spIR sequences than to An. hyrcanus sequences, particularly when considering that all had an 11 bp insertion [16]. In the NJ tree (Fig. 1), they clustered with the An. hyrcanus spIR lineage and were separated from An. hyrcanus, suggesting they might be sequences of An. hyrcanus spIR.

Neighbour-joining phylogenetic tree of the Hyrcanus Group based on ITS2 sequences from GenBank and our original data. Bootstrap values (1000 replicates) of neighbour-joining analyses are shown above/below the main lineages. Lineage designation is indicated on the right. The geometric shapes correspond to different subgroups of the Hyrcanus Group, according to the classification of Harbach [1], Lesteri Subgroup; Nigerrimus Subgroup; unclassified species. Bars represent 0.04 substitutions per site. Anopheles lindesayi and An. claviger were used as the outgroup taxa
Twenty-seven ITS2 sequences belonging to An. sinensis, An. peditaeniatus, An. pullus, An. hyrcanus, An. lesteri, and An. liangshanensis were generated during this study and have been submitted to GenBank; their accession numbers and collection sites are presented in Table 1.
Intra- and interspecific ITS2 variation
Individual species were represented by one to 83 individuals, for a total of 461 ITS2 sequences (Table 2). Almost all species possessed a distinctive set of ITS2 sequences. The intra- and interspecific divergences of ITS2 in the Hyrcanus Group are shown in Table 2, and Fig. 2 shows the distribution of pairwise K2P genetic distances between ITS2 sequences, revealing a distinct barcoding gap. The average intraspecific K2P distance was 0.003. No intraspecific variation was found for An. belenrae, An. kleini, An. paraliae or An. pursati Laveran, 1902. Deep divergences were detected in two species, An. lesteri (0.0142) and An. crawfordi (0.0143). Short distances were observed between some pairs of species: An. hyrcanus and An. pseudopictus (0.001), An. hyrcanus spIR and An. pseudopictus (0.020), An. hyrcanus and An. hyrcanus spIR (0.021), An. lesteri and An. paraliae (0.048), and An. kleini and An. engarensis (0.072). This was confirmed by the Nei’s genetic distance obtained (Additional file 3: Figure S1). The taxonomic validity of An. pseudopictus, An. hyrcanus spIR, An. paraliae, and An. kleini is still controversial [1, 4, 12, 16, 33], and further studies are needed to resolve these uncertainties; we excluded the above data in the calculation of the mean interspecific distances. Therefore, interspecific K2P distances ranged from 0.081 between An. pullus and An. pseudopictus to 0.920 between An. nigerrimus and An. lesteri, with an average of 0.480. Hence, the ITS2 sequence divergences among intragroup species were, on average, almost 160 times higher than the average divergences within species. The ITS2 barcoding gap was 0.014–0.081 (Fig. 2), suggesting that the ITS2 spacer is a good marker for differentiating Hyrcanus Group members.

Intra- and interspecific ITS2 divergences in 19 Hyrcanus Group members determined using Kimura’s 2-parameter distance. The barcoding gap ranged 0.014–0.081. Y-axis, genetic divergence; x-axis, Hyrcanus Group members
Genetic diversity indices and neutrality tests
Additional file 4: Figure S2 displays the haplotype frequencies in Hyrcanus Group members. Sequence numbers for each species, polymorphic sites, haplotype numbers, haplotype diversity, and the Fu’s Fs and Tajima’s D results are given in Table 3. The presence of high haplotype diversity, combined with a positive neutrality test value, evidences balancing selection or a decrease in population size (e.g. An. crawfordi). Conversely, a relatively low haplotype diversity with a negative neutrality test value indicates that the population has experienced an expansion after a bottleneck, probably caused by purifying selection or a selective sweep due to genetic hitchhiking (e.g. An. peditaeniatus).
Phylogenetic analysis
The Hyrcanus Group is monophyletic (Fig. 1). All lineages, including individuals representing the same species, were supported by high bootstrap values, except for An. pseudopictus, An. pseudopictus and An. hyrcanus, which exhibited barcode congruence with an extremely low interspecific distance (0.001). Low genetic divergences were also observed between An. hyrcanus and An. hyrcanus spIR (0.021), An. lesteri and An. paraliae (0.048), and An. kleini and An. engarensis (0.072). Each of these species was placed in an independent branch and was homologous to its closest taxon in the tree, suggesting that they might be candidate species or have diverged recently.
Although the ITS2-based phylogenetic tree was concordant with traditional morphological taxonomy in terms of species recognition, its subgroup arrangement did not match that obtained with morphology-based grouping. Following the NJ-K2P analysis, two main clusters were identified within the Hyrcanus Group. One consisted of the Nigerrimus Subgroup plus An. argyropus (Swellengrebel, 1914), and the other contained the Lesteri Subgroup and the unassigned species An. hyrcanus, An. hyrcanus spIR, An. pseudopictus, An. sinensis, An. belenrae, An. kleini, An. engarensis, An. liangshanensis, An. kweiyangensis Yao & Wu, 1944 and An. pullus.
Multiplex PCR assay
Based on species-specific differences, six primers were combined in a multiplex PCR mixture for the simultaneous amplification of five Hyrcanus Group members from China (Fig. 3). The lengths of the amplified species-specific PCR products were approximately 96 bp for An. peditaeniatus, 189 bp for An. hyrcanus, 364 bp for An. lesteri, 428 bp for An. pullus and 514 bp for An. sinensis. To validate the assay, 92 specimens were tested, including 24 An. peditaeniatus, 10 An. hyrcanus, 3 An. lesteri, 11 An. pullus and 44 An. sinensis. All specimens that were confirmed by molecular identification yielded a specific band for each species, as expected. The specificity of the multiplex PCR was evaluated for its cross-reactivity with closely related organisms. No amplified product was observed with DNA of other species, even on repeated tests (Additional file 5: Figure S3). The sensitivity tests revealed that the minimum detection limit of mosquito DNA by the multiplex PCR system was 10−4 ng/μl (Additional file 6: Figure S4).

Polymerase chain reaction (PCR) products of a multiplex PCR assay. Lane M: PCR marker; Lanes 1 and 2: An. peditaeniatus; Lanes 3 and 4: An. hyrcanus; Lanes 5 and 6: An. lesteri; Lanes 7 and 8: An. pullus; Lanes 9 and 10: An. sinensis; Lanes 11 and 12: negative controls
Discussion
Subdivision of the Hyrcanus group
Morphological features separate the Hyrcanus Group into three subgroups [52, 53]. Anopheles nigerrimus, An. nitidus Harrison, Scanlon & Reid, 1973, An. pursati and An. pseudosinensis Baisas, 1935 were placed in the Nigerrimus Subgroup, and An. lesteri, An. paraliae, An. peditaeniatus, An. crawfordi and An. vietnamensis Nguyen, Tran & Nguyen, 1993 were assigned to the Lesteri Subgroup. The remaining species were unassigned. Unfortunately, DNA barcoding has not been conducted for every Hyrcanus Group member. Until now, no ITS2 sequence data were known for the following seven species: An. pseudosinensis, An. vietnamensis, An. chodukini Martini, 1929, An. hailarensis Xu & Luo, 1998, An. heiheensis Ma, 1981, An. pursati and An. nimpe Nguyen, Tran & Harbach, 2000. The present study included all the ITS2 sequences available for Hyrcanus Group members deposited in GenBank, including 461 individuals from 19 species. The NJ tree obtained supports the monophyly of the Hyrcanus Group; however, the subgroup arrangement did not match that based on morphological characteristics. We could recognize two main subgroups, one consisting of An. argyropus, An. nigerrimus, An. pursati and An. nitidus, and the other containing An. peditaeniatus, An. pullus, An. hyrcanus spIR, An. hyrcanus, An. pseudopictus, An. kleini, An. engarensis, An. belenrae, An. sinensis, An. sineroides, An. kweiyangensis, An. liangshanensis, An. crawfordi, An. lesteri and An. paraliae. This arrangement suggested that the morphology-based Lesteri Subgroup is not monophyletic. Anopheles lesteri, An. paraliae and An. crawfordi formed a single clade, but An. peditaeniatus was far from these species in the NJ tree. In addition, the ITS2 sequences of An. hyrcanus and An. pseudopictus were almost identical, with an extremely low genetic distance (0.001). As in the results based on the cox1 gene [22], the interspecific was only 0.008, which is far below the maximum intraspecific distance, i.e. 0.017. Anopheles pseudopictus individuals were embedded in the An. hyrcanus lineage, in both ITS2 and cox1 trees [22], in agreement with previous findings comparing morphological characters and molecular markers between the two species and morphologically-intermediate forms [4]. Thus, An. hyrcanus and An. pseudopictus are most likely the same species, although further studies including crossing experiments are needed to confirm this hypothesis.
Results of the present and previous studies on genetic relationships among Hyrcanus Group members do not entirely coincide. In previous studies [11, 12, 16, 25, 26], both the number of species and the number of sequences used were far lower than those used in the present study. In addition, some species have been confirmed as synonymous within the Hyrcanus Group [1, 2]. Because previous studies did not include an appropriate number of species, particularly of the Nigerrimus Subgroup, it has been difficult to detect subgroups in the phylogenetic tree. Hwang [12] classified the Hyrcanus Group into four subgroups, based on ITS2 data; however, in his maximum likelihood tree topology, the four subgroups were not on the same phylogenetic level. It should be noted that the above study [12], and two related studies [11, 16], used the same An. crawfordi ITS2 sequence (AF261949) to reconstruct the molecular phylogeny of the Hyrcanus Group. However, in our analysis, this sequence belongs to an originally misidentified specimen. In these three studies [11, 12, 16], the tree topologies suggested that An. crawfordi is closest to An. peditaeniatus, in contrast to the results of the present study and those of another recent study [26], in which An. crawfordi is distant from An. peditaeniatus but close to An. lesteri.
Deep ITS2 intraspecific divergence in An. lesteri and An. crawfordi
As shown in Table 2, deep intraspecific differences were detected in An. lesteri and An. crawfordi (0.0142 and 0.0143, respectively), while all other species analysed presented intraspecific divergences below 0.005. Deep ITS2 divergences within species might be due to hidden diversity. Hwang [12] stated that An. lesteri could be divided into at least three types: A (the dominant type), B (AJ620899 and AJ620902) and C (AJ620900 and AJ620901). He also suggested that the Philippines type (AY375469) of An. lesteri proposed by Rueda et al. [54] should belong to type A. However, in the current study nucleotide substitution and insertions of AY375469 were not as frequent close to type A as to types B and C sequences, although this accession still represented a separate branch from An. lesteri type A in the tree. Therefore, the Philippines type is probably an independent An. lesteri type. For An. crawfordi, sequences could generally be differentiated into Cambodia and Thailand types, with support values of 98 and 79%, respectively. This genetic divergence probably resulted from geographical isolation [55].
Phylogenetic reconstructions using cox1 and ITS2
Based on the results of the present study and our previous study using cox1 [22], we compared the effectiveness of the nuclear ITS2 and mitochondrial cox1 as markers for Hyrcanus Group members. We found that the cox1 barcoding gap was 0.016–0.026, whereas that of ITS2 was 0.014–0.081. The ITS2 sequence divergences were, on average, almost 160 times higher among groups of species than within a species, whereas the average cox1 divergence between congroup species was only eight times higher than that within species. An effective DNA marker should have a small intraspecific distance and a large interspecific distance [56]. In Fig. 4, each dot represents a species, with intraspecific distance on the x-axis and interspecific distance on the y-axis. It is obvious that there are more ITS2 than cox1 dots close to the top left-hand corner of the graph. The main disadvantage of using cox1 in phylogenetic studies of the Hyrcanus Group is that it cannot differentiate between the closely related: An. lesteri and An. paraliae; and An. sinensis, An. belenrae and An. kleini [22]. In the ITS2 tree, each of the above species belonged to an independent lineage, which form monoclades with their sister species. In addition, two other pairs of sibling species, An. hyrcanus and An. hyrcanus spIR, and An. kleini and An. engarensis, were also differentiated by ITS2. Currently, there are no cox1 records of An. hyrcanus spIR or An. engarensis in GenBank. Thus, rDNA ITS2 seems more reliable than mtDNA cox1 for resolving evolutionary issues of the Hyrcanus Group, including recently diverged taxa, such as cryptic species of mosquitoes. Although cox1 may be useful in barcoding information, and particularly for inferring the possibility of ancient hybridization, in mosquito molecular phylogeny, nuclear ITS2 can establish species boundaries in cases that cannot be resolved by mitochondrial cox1. In fact, ITS2 sequences have been most frequently used in species identification and phylogenetic reconstruction of the Hyrcanus Group [11, 12, 16, 25, 26]. In the animal kingdom, the evolutionary rate of the mitochondrial genome is about 5–10 times faster than that of the nuclear genome, making cox1 potentially more useful than ITS2 for correctly identifying recently diverged species [57, 58]. However, the opposite seems to occur within the Hyrcanus Group, which might be explained by the male-biased dispersal [59]. In the field, females usually mate only once, and store sufficient sperms to fertilize all eggs they produce in their lifetime, whereas males mate repeatedly. Consequently, male mosquitoes occupy a dominant position during population expansions. Because mtDNA is typically maternally inherited, any hybrid or offspring would only have the maternal species’ mtDNA. Therefore, hybridization can result in shared or very similar sequences in the mitochondrial genome. The divergent mtDNA of An. belenrae, An. kleini and An. paraliae suggests that the mitochondrial genomes of incipient sibling species have been sympatrically replaced by those of An. sinensis and An. lesteri over wide areas. In the Hyrcanus Group, nuclear markers are less introgressed, and hence more diagnostic, than mtDNA markers.

ITS2 and cox1 sequence divergences in the Hyrcanus Group. The minimum interspecific (intergroup) divergence is plotted against the maximum intraspecific divergence. ITS2 has more dots above the diagonal line in the top left-hand corner than cox1, indicating low intraspecific and high interspecific divergence
Conclusions
This study used ITS2 sequences of Hyrcanus Group members distributed worldwide, providing a systematic basis for future studies on malaria transmission and its relationship with the evolution of Plasmodium spp. We found that the topology of the Hyrcanus Group ITS2 tree was generally consistent with the morphology-based taxonomy in terms of species classification, but not in terms of subgroup divisions. Nuclear and mitochondrial gene data were generally consistent in subgroup classification; however, cox1 failed to elucidate the phylogenetic status of the incipient sibling species An. belenrae, An. kleini and An. paraliae, suggesting that cox1 might be unable to resolve the molecular phylogeny of the Hyrcanus Group, unlike ITS2. ITS2 is a reliable tool for the study of phylogenetic relationships between closely related mosquito taxa, and cox1 may be a useful supplement to barcoding information, particularly for inferring interspecific hybridization. Both cox1 and ITS2 results suggested that An. pseudopictus and An. hyrcanus might be the same species. Two new ITS2 lineages, namely accessions AF261949 and KC769647, were uncovered, and require further sampling and detailed morphological, genetic, and ecological studies before resolving their true taxonomic status. In addition, unambiguous ITS2 sequence differences among Hyrcanus Group members facilitated the design of species-specific primers for identifying five species within this group that are the most frequently implicated in disease transmission in China. Thus, this method can identify members of the Hyrcanus Group in a simple, fast, and reliable manner during malaria-vector surveillance procedures.
Abbreviations
- China CDC:
-
Chinese Center for Disease Control and Prevention
- cox1:
-
Cytochrome c oxidase subunit 1 gene
- ITS2:
-
Internal transcribed spacer 2
- K2P:
-
Kimura’s 2-parameter
- NIPD:
-
National Institute of Parasitic Diseases
References
Harbach RE. The phylogeny and classification of Anopheles. In: Manguin S, editor. Anopheles mosquitoes - new insights into malaria vectors. Croatia: InTech; 2013. p. 3–55.
Fang Y, Shi W, Zhang Y. [Research progress in classification of Anopheles hyrcanus group (Diptera: Culicidae)]. Chin J Parasitol Paras Dis. 2016;34:565–70. (In Chinese).
Zhou Z. The malaria situation in the People's Republic of China. Bull World Health Organ. 1981;59:931–6.
Poncon N, Toty C, Kengne P, Alten B, Fontenille D. Molecular evidence for similarity between Anopheles hyrcanus (Diptera: Culicidae) and Anopheles pseudopictus (Diptera: Culicidae), sympatric potential vectors of malaria in France. J Med Entomol. 2008;45:576–80.
Poncon N, Toty C, L'Ambert G, Le Goff G, Brengues C, Schaffner F, et al. Biology and dynamics of potential malaria vectors in southern France. Malar J. 2007;6:18.
Lee W, Klein TA, Kim H, Choi Y, Yoon S, Chang K, et al. Anopheles kleini, Anopheles pullus, and Anopheles sinensis: potential vectors of Plasmodium vivax in the Republic of Korea. J Med Entomol. 2007;44:1086–90.
Gingrich JB, Weatherhead A, Sattabongkot J, Pilakasiri C, Wirtz RA. Hyperendemic malaria in a Thai village: dependence of year-round transmission on focal and seasonally circumscribed mosquito (Diptera: Culicidae) habitats. J Med Entomol. 1990;27:1016–26.
Alam MS, Khan MG, Chaudhury N, Deloer S, Nazib F, Bangali AM, et al. Prevalence of anopheline species and their Plasmodium infection status in epidemic-prone border areas of Bangladesh. Malar J. 2010;9:15.
Paredes-Esquivel C, Harbach RE, Townson H. Molecular taxonomy of members of the Anopheles hyrcanus group from Thailand and Indonesia. Med Vet Entomol. 2011;25:348–52.
Jeong KY, Un S, Lee J, Lee IY, Yong TS, Ree HI. Population dynamics of five Anopheles species of the Hyrcanus Group in northern Gyeonggi-do, Korea. Korean J Parasitol. 2010;48:351–3.
Ma Y, Xu J. The Hyrcanus group of Anopheles (Anopheles) in China (Diptera: Culicidae): species discrimination and phylogenetic relationships inferred by ribosomal DNA internal transcribed spacer 2 sequences. J Med Entomol. 2005;42:610–9.
Hwang UW. Revisited ITS2 phylogeny of Anopheles (Anopheles) Hyrcanus group mosquitoes: reexamination of unidentified and misidentified ITS2 sequences. Parasitol Res. 2007;101:885–94.
Hebert PD, Ratnasingham S, de Waard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci. 2003;270 Suppl 1:96–9.
Hebert PD, Cywinska A, Ball SL, de Waard JR. Biological identifications through DNA barcodes. Proc Biol Sci. 2003;270:313–21.
Schmidt S, Schmid-Egger C, Moriniere J, Haszprunar G, Hebert PD. DNA barcoding largely supports 250 years of classical taxonomy: identifications for central European bees (Hymenoptera, Apoidea partim). Mol Ecol Resour. 2015;15:985–1000.
Djadid ND, Jazayeri H, Gholizadeh S, Rad Sh P, Zakeri S. First record of a new member of Anopheles Hyrcanus Group from Iran: molecular identification, diagnosis, phylogeny, status of kdr resistance and Plasmodium infection. J Med Entomol. 2009;46:1084–93.
Hebert PD, Stoeckle MY, Zemlak TS, Francis CM. Identification of birds through DNA barcodes. PLoS Biol. 2004;2:e312.
Mallet J. Hybridization as an invasion of the genome. Trends Ecol Evol. 2005;20:229–37.
Brennan IG, Bauer AM, Jackman TR. Mitochondrial introgression via ancient hybridization, and systematics of the Australian endemic pygopodid gecko genus Delma. Mol Phylogenet Evol. 2016;94:577–90.
Choochote W, Min GS, Intapan PM, Tantrawatpan C, Saeung A, Lulitanond V. Evidence to support natural hybridization between Anopheles sinensis and Anopheles kleini (Diptera: Culicidae): possibly a significant mechanism for gene introgression in sympatric populations. Parasit Vectors. 2014;7:36.
Toews DP, Brelsford A. The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol. 2012;21:3907–30.
Fang Y, Shi W, Zhang Y. Molecular phylogeny of Anopheles hyrcanus group (Diptera: Culicidae) based on mtDNA COI. Infect Dis Poverty. 2017;6:61.
Cunningham EP, Meghen CM. Biological identification systems: genetic markers. Rev Sci Tech. 2001;20:491–9.
Mohanty A, Swain S, Kar SK, Hazra RK. Analysis of the phylogenetic relationship of Anopheles species, subgenus Cellia (Diptera: Culicidae) and using it to define the relationship of morphologically similar species. Infect Genet Evol. 2009;9:1204–24.
Gao Q, Beebe NW, Cooper RD. Molecular identification of the malaria vectors Anopheles anthropophagus and Anopheles sinensis (Diptera: Culicidae) in central China using polymerase chain reaction and appraisal of their position within the Hyrcanus Group. J Med Entomol. 2004;41:5–11.
Songsawatkiat S, Baimai V, Thongsahuan S, Otsuka Y, Taai K, Hempolchom C, et al. Evidence to support a conspecific nature of allopatric cytological races of Anopheles nitidus (Diptera: Culicidae) in Thailand. J Insect Sci. 2014;14:287.
Li C, Lee J, Groebner J, Kim H, Klein T, Oguinn M, et al. A newly recognized species in the Anopheles Hyrcanus Group and molecular identification of related species from the Republic of South Korea (Diptera: Culicidae). Zootaxa. 2005;939:1–8.
Rueda L. Two new species of Anopheles (Anopheles) Hyrcanus Group (Diptera: Culicidae) from the Republic of South Korea. Zootaxa. 2005;941:1–26.
Shin E, Hong HK. A new synonym of Anopheles (Anopheles) pullus Yamada, 1937: A. (A.) yatsushiroensis Miyazaki, 1951. Korean J Appl Entomol. 2001;31:1–5.
Hwang UW, Yong TS, Ree H. Molecular evidence for synonymy of Anopheles yatsushiroensis and An. pullus. J Am Mosq Control Assoc. 2004;20:99–104.
Ma YJ, Qu FY, Lei XT, Dong XS. Comparison of rDNA-ITS2 sequences and morphological characters of Anopheles kunmingensis and Anopheles langshanensis in China, with discussion on taxonomic status. Chin J Parasitol Paras Dis. 2000;18:65–8. (In Chinese).
Qu FY, Zhu HM. On a new checklist of the anopheline mosquitoes in China with rectification for some specific names. Chin J Parasitol Paras Dis. 2008;26:210–6. (In Chinese).
Taai K, Baimai V, Saeung A, Thongsahuan S, Min GS, Otsuka Y, et al. Genetic compatibility between Anopheles lesteri from Korea and Anopheles paraliae from Thailand. Mem Inst Oswaldo Cruz. 2013;108:312–20.
Wilkerson RC, Li C, Rueda LM, Kim H-C, Klein TA, Song G-H, et al. Molecular confirmation of Anopheles (Anopheles) lesteri from the republic of South Korea and its genetic identity with An. (Ano.) anthropophagus from China (Diptera: Culicidae). Zootaxa. 2003;378:1–14.
Hwang UW, Tang LH, Kobayashi M, Yong TS, Ree HI. Molecular evidence supports that Anopheles anthropophagus from China and Anopheles lesteri from Japan are the same species. J Am Mosq Control Assoc. 2006;22:324–6.
Bergsten J, Bilton DT, Fujisawa T, Elliott M, Monaghan MT, Balke M, et al. The effect of geographical scale of sampling on DNA barcoding. Syst Biol. 2012;61:851–69.
Lu BL, Xu JJ, Yu Y, Zhang BH, Dong XS. Fauna Sinica, Insecta, vol.9. Diptera: Culicidae II. 1st ed. Beijing: Science Press; 1997.
Rattanarithikul R, Harrison BA, Harbach RE, Panthusiri P, Coleman RE. Illustrated keys to the mosquitoes of Thailand. IV. Anopheles. Se Asian J Trop Med. 2006;37:1–128.
Beebe NW, Saul A. Discrimination of all members of the Anopheles punctulatus Complex by polymerase chain reaction-restriction fragment length polymorphism analysis. Am J Trop Med Hyg. 1995;53:478–81.
Koetschan C, Forster F, Keller A, Schleicher T, Ruderisch B, Schwarz R, et al. The ITS2 database III - sequences and structures for phylogeny. Nucleic Acids Res. 2010;38:D275–9.
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–8.
Wang G, Li C, Guo X, Xing D, Dong Y, Wang Z, et al. Identifying the main mosquito species in China based on DNA barcoding. PLoS One. 2012;7:e47051.
Nei M, Kumar S, Takahashi K. The optimization principle in phylogenetic analysis tends to give incorrect topologies when the number of nucleotides or amino acids used is small. Proc Natl Acad Sci USA. 1998;95:12390–7.
Morariu VI, Srinivasan BV, Raykar VC, Duraiswami R, Davis LS. Automatic online tuning for fast Gaussian summation. Adv Neural Inf Process Syst. 2008;21:1113–20.
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28:2731–9.
Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. II. Gene frequency data. J Mol Evol. 1983;19:153–70.
Excoffier L, Laval G, Schneider S. Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinformatics Online. 2005;1:47–50.
Fu Y. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997;147:915–25.
Tajima F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989;123:585–95.
Librado P, Rozas J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009;25:1451–2.
Harrison BA. A new interpretation of affinities within the Anopheles hyrcanus Complex of southeast Asia. Mosq Syst. 1972;4:73–83.
Harbach RE. The classification of genus Anopheles (Diptera: Culicidae): a working hypothesis of phylogenetic relationships. Bull Entomol Res. 2004;94:537–53.
Rueda L, Wilkerson R, Li C. Anopheles (Anopheles) lesteri Biases and Hu (Diptera: Culicidae): neotype designation and description. Proc Entomol Soc Wash. 2005;107:604–22.
Paduan K, Araujojunior J, Ribolla P. Genetic variability in geographical populations of Aedes aegypti (Diptera, Culicidae) in Brazil elucidated by molecular markers. Genet Mol Biol. 2006;29:391–5.
Hemmerter S, Slapeta J, Beebe N. Resolving genetic diversity in Australasian Culex mosquitoes: incongruence between the mitochondrial cytochrome c oxidase I and nuclear acetylcholine esterase 2. Mol Phylogenet Evol. 2009;50:317–25.
Castro JA, Picornell A, Ramon M. Mitochondrial DNA: a tool for populational genetics studies. Int Microbiol. 1998;1:327–32.
Moore W. Inferring phylogenies from mtDNA variation: mitochondrial-gene trees versus nuclear-gene trees. Evolution. 1995;49:718–26.
Petit RJ, Excoffier L. Gene flow and species delimitation. Trends Ecol Evol. 2009;24:386–93.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2016YFC1202000, 2016YFC1202002, 2016YFC1202003).
Availability of data and materials
All data generated or analyzed during this study are included in this published article and its additional files. The newly generated sequences were submitted in the GenBank database under the accession numbers KU312198–KU312221, KU682193–KU682195, MF498513–MF498558 and MF535198–MF535239.
Author information
Authors and Affiliations
Contributions
YF, WS and YZ designed the study; YF carried out laboratory experiments, performed phylogenetic analyses, and wrote the manuscript; WS and YZ revised the manuscript and gave approval of the version to be published. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
No specific permits were required for this study. The study did not involve endangered or protected species. Therefore, the local ethics committee deemed that approval was unnecessary.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1: Table S1.
List of the Anopheles hyrcanus group ITS2 sequences deposited in GenBank and obtained from this study, with GenBank accession numbers, geographical location, and corresponding authors. (XLSX 50 kb)
Additional file 2: Table S2.
List of the specimens used in the multiplex PCR validation. (PDF 303 kb)
Additional file 3: Figure S1.
Intra- and interspecific ITS2 divergences among the 19 Hyrcanus Group members as determined by Nei’s distance. (TIFF 779 kb)
Additional file 4: Figure S2.
ITS2 haplotype frequencies in the 19 Hyrcanus Group members. (TIFF 145 kb)
Additional file 5: Figure S3.
Partial results of the specificity test of the Multiplex PCR. (PDF 413 kb)
Additional file 6: Figure S4.
Partial results of the sensitivity test of the Multiplex PCR for the five Hyrcanus Group members. (PDF 475 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Fang, Y., Shi, WQ. & Zhang, Y. Molecular phylogeny of Anopheles hyrcanus group members based on ITS2 rDNA. Parasites Vectors 10, 417 (2017). https://doi.org/10.1186/s13071-017-2351-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-017-2351-x