
Abstract
Background
Species of the Anopheles hyrcanus group are widely distributed in Palearctic and Oriental regions and some of them are important malaria vectors. The cryptic species of An. hyrcanus group was almost impossible to identify based only on their morphology. The phylogenetic relationship of An. hyrcanus group was also not clear.
Methods
Five members of An. hyrcanus group were identified by rDNA ITS2 sequencing as An. yatsushiroensis, An. belenrae, An. kleini, An. lesteri and An. sineroides. The mitochondrial genome fragments were sequenced and annotated using the mitochondrial genome of An. sinensis as reference. Based on the four segments and Joint Data sequences of these species, and other four anopheline species downloaded from GenBank, intraspecific as well as interspecific genetic distances were calculated and the phylogenetic trees were reconstructed by the methods of neighbor joining, maximum parsimony, minimum evolution and maximum likelihood.
Findings
Four parts of mitochondrial genomes, which were partial fragments COI + tRNA + COII (F5), ATP6 + COIII(F7 + F8), ND1(F19) and lrRNA (F21), were obtained. All fragments were connected as one sequence (referred as Joint Data), which had a total length of 3393 bp. All fragment sequences were highly conservative within species, with the maximum p distance (0.026) calculated by F19 of An. belenrae. The pairwise interspecific p distance calculated by each fragment showed minor or even no difference among An. sinensis, An. kleini and An. belenrae. However, interspecific p distances calculated by the Joint Data sequence ranged from 0.004 (An. belenrae vs An. kleini) to 0.089 (An. sineroides vs An. minimus), and the p distances of the six members of An. hyrcanus group were all less than 0.029. The phylogenetic tree showed two major clades: all subgenus Anopheles species (including six members of An. hyrcanus group, An. atroparvus and An. quadrimaculatus A) and subgenus Cellia (including An. dirus and An. minimus). The An. hyrcanus group was divided into two clusters as ((An. lesteri, An. sineroides) An. yatsushiroensis) and ((An. belenrae, An. sinensis) An. kleini)).
Conclusions
The An. hyrcanus group in this study could be divided into two clusters, in one of which An. belenrae, An. sinensis and An. kleini were most closely related. More molecular markers would make greater contribution to phylogenetic analysis.
Similar content being viewed by others

Multilingual abstracts
Please see Additional file 1 for translations of the abstract into the five official working languages of the United Nations.
Background
Anopheles hyrcanus group belongs to the subgenus Anopheles, genus Anopheles. It is widely distributed in Palearctic and Oriental regions, including 25 species with valid reported mosquito species [1]. There were 22 species of An. hyrcanus group distributed in China, including three unnamed ones [2]. Some cryptic species of An. hyrcanus group have similar morphological characteristics, making it difficult to identify them based only on their morphology [3]. Moreover, quite a few hybridized individuals were found in field [4, 5] and reestablished phylogenetic trees of An. hyrcanus group showed disparity according to various molecular markers [3, 6, 7]. These facts illustrated the complex phylogenetic relationships within An. hyrcanus group.
Mitochondrial genome strictly followed maternal inheritance in structure as well as in evolution, with abundant information for genetic and phylogenetic population studies. The mitochondrial genome of Anopheles mosquitoes consisted of 13 protein-coding genes, 22 transfer RNA (tRNA) genes, two ribosomal RNA (rRNA) genes and an AT-rich control region [8,9,10]. At present, certain genes of mitochondrial genome were employed to analyze the interspecific or intraspecific differences. For instance, COI sequence was used as DNA barcoding to distinguish mosquito species [11, 12], while genes such as COI [13], COII [14], ND5 [15] and control region [16] were utilized to detect the genetic population structure of Hyrcanus group members.
So far, we have reported the complete mitochondrial genome of An. sinensis in Hyrcanus group [9]. In this study, we sequenced and analyzed mitochondrial genome fragments of An. hyrcanus group members in China in order to reestablish the phylogenetic relationships and determine the taxonomic status of cryptic species. Moreover, we discussed the contribution of mitochondrial genome fragments in phylogenetic study.
Materials and methods
Ethics statement
No permits were required for the described field studies. Adult mosquito collection in chicken farms and livestock pens was agreed by the owners at each location.
Mosquito collection and species identification
With the consent of the owners, mini light traps (MYFS-HJY-1, Houji Shenzhen, China) were set up in chicken farms and livestock pens between 6:30 pm and 7:30 am. Then the captured Anopheles specimens were collected by the mini light traps and manually by entomological aspirators in the evening and killed by freezing before being individually transferred to laboratory in centrifuge tubes for further analysis (Table 1). In the light of the taxonomic key by Lu et al. [17], the samples were morphologically identified as members of An. hyrcanus group.
The member species of An. hyrcanus group was further identified by molecular markers with rDNA ITS2 sequences. Single mosquito genomic DNA was extracted using DNAzol (Life Technologies, USA) following the manufacturer’s instructions. DNA pellet was dissolved in 80 μl H2O. The rDNA ITS2 fragment was amplified according to the method by Lin et al. and Ma et al. [18, 19]. An ABI 3730 (Boshang Biotech Co., Ltd. Shanghai, China) was applied to purify and sequence the PCR products. Finally, the sequences were Blast aligned in Genebank on the NCBI website to determine the species [18, 19].
Amplification and sequencing of mitochondrial genome fragments
Some fragments of the mitochondrial genome were amplified referring to the universal primers designed for mitochondrial genome of Diptera [20] (Table 2, Additional file 7: Figure S1). The length range of the amplified product was from 500 bp to 1200 bp and the overlapping length between adjacent sequences was between 50 bp and 485 bp. The range of percentage of GC was from 40 to 60%, while the annealing temperature was either 45 °C to 47 °C. PCR reaction was run in a 25 μl mixture containing 1 μl DNA template, 0.2 μmol/L primers and other PCR reagents (Aidlab Biotechnologies, China). PCR thermal cycling included a 2 min initial denaturation at 94 °C, followed by 30–35 cycles of denaturation at 94 °C for 30 s, annealing at 45 °C/47 °C for 30 s, elongation at 72 °C for 1 min, and a final extension for 8 min at 72 °C. The PCR products were purified and sequenced by an ABI 3730 machine.
Phylogenetic analysis
The sequences were compared using DNAstar 7.1 (https://www.dnastar.com/software/lasergene/) [21] and annotation as well as splicing was completed referring to the mitochondrial genome of An. sinensis (GenBank accession No. KT218684.1) [9]. The intraspecific differences (p distance) were calculated by MEGA 7.0 (https://mega.software.informer.com/7.0/) [22] before further analysis was conducted using consensus sequence as the species-specific identity.
The complete mitochondrial genome of An. sinensis (KT218684.1), An. dirus A (JX219731.1), An. atroparvus (KT382817.1), An. quadrimaculatus (AL04272.1) and An. minimus (KT895423.1) were downloaded from GenBank database. Mitochondrial genome fragment sequences from a total of 10 species were aligned using MEGA 7.0, including An. yatsushiroensis, An. belenrae, An. kleini, An. lesteri and An. sineroides, in addition to the aforementioned five species downloaded from GenBank. The variable bases, parsimony information bases and nucleotide composition were analyzed by MEGA 7.0. In light of the Joint Data (JD) and the separated segments (F5 (COI + tRNA + COII), F7 + F8 (ATP6 + COIII), F19 (ND1) and F21 (lrRNA)), phylogenetic trees were constructed by the methods of neighbor joining (NJ), maximum parsimony (MP) and minimum evolution (ME), respectively. On the other hand, maximum likelihood (ML) tree was reconstructed by PhyML 3.0 (http://phylogeny.lirmm.fr/phylo_cgi/one_task.cgi?task_type=phyml) [23]. The best-fit nucleotide substitution model was obtained by Modeltest 3.7 (http://evomics.org/resources/software/molecular-evolution-software/modeltest/) [24] before ML tree construction. Moreover, bootstrap values for 1000 replicates of all trees were calculated. A congruence length test for JD was performed before analysis [25].
Results and discussion
Sequence characters of mitochondrial genome fragments
Five mitochondrial genome fragments out of the five members in An. hyrcanus group were obtained. Fragment F5 (893 bp in length) comprised segmental COI, full-length tRNA-Leu plus segmental COII (MK690504–MK690508). Due to a partial overlapping found in Fragment F7 and F8, they were connected (denoted as F7 + F8) for further analysis. Its length was 1407 bp and included segmental ATP6 and segmental COIII (MK825734–MK825738). F19, 583 bp in length, was segmental ND1 (MK825739–MK825748). F21 was segmental 16 lrRNA (MK825744–MK825748) with 510 bp in length. As the combined sequence of all fragments, JD had an aggregate length of 3393 bp.
Intraspecific differences
The five fragments’ sequences of mitochondrial genome were aligned among the five members of An. hyrcanus group in this study. The averages of the nucleotide composition as well as the numbers of conserved and variable bases are shown in Additional file 2: Table S1. All fragments’ sequences were highly conservative within species, with the maximum p distance (0.026) calculated by F19 of An. belenrae (Table 3). The reported intraspecific differences of mitochondrial genomes in An. hyrcanus group were as follows: An. lesteri (COII: h = 0.000–0.005; Cyt B: h = 0.000–0.005) [14], An. sinensis (COI: p = 0.0088, Pi = 0.0039–0.0105; COII: p = 0.0047; control region: h = 0.00453–0.01617) [13, 16, 26]. The maximum intraspecific distance of COI in 17 members of Hyrcanus group was 0.008 (range: 0.002–0.017) [7]. The results indicated varied intraspecific differences among the fragments, suggesting that prudence would be required in the analysis of interspecies relationships within An. hyrcanus group. The differences between COI sequences increased in higher taxonomic categories [27], while the COI barcoding gap was usually 2% within species [28]. High divergence of intraspecific distance was probably caused by recent geographic isolation, indicating the presence of cryptic species [27].
Interspecific differences
The four different mitochondrial genome fragments and the JD sequences of the five members of An. hyrcanus group in this study, together with the five anopheline species downloaded from the GenBank were analyzed using MEGA 7.0. The ranges of the variable and parsimony information bases were from 46 (F21) to 636 (JD) and 18 (F21) to 363 (JD), respectively, and the averages of GC content varied from 24.6 to 27.4% (Table 4).
The pairwise interspecific p distance based on the four fragments (F5, F7 + F8, F19, F21) of mitochondrial genome showed that F21 sequence was completely conserved between An. kleini and An. belenrae, suggesting that F21 fragment had no interspecific resolution. Moreover, other fragments showed minor difference among An. sinensis, An. kleini and An. belenrae. Meanwhile, the p distance between subgenus Cellia and Anopheles species was greater than that among the species within the same subgenus (Additional file 3: Table S2, Additional file 4: Table S3, Additional file 5: Table S4 and Additional file 6: Table S5).
The interspecific p distances calculated by the JD sequence ranged from 0.004 (An. belenrae vs An. kleini) to 0.089 (An. sineroides vs An. minimus), and the p distances of the 6 members of An. hyrcanus group were all less than 0.029 (Table 5).
COI gene was the most common mitochondrial genome fragment for mosquito identification and genetic relationship analysis [7, 12, 13, 29,30,31,32,33,34]. In addition, the vast majority of intraspecific distances in 122 mosquito species (15 genera) were at levels from 6 to 15% [12]. On the basis of mtDNA COI sequence, the average intraspecific K2P distance of Hyrcanus group 17 species was 0.008 (range: 0.002–0.017) [7]. However, compared with the intraspecific distance, the interspecific distance among cryptic species was smaller, such as An. sinensis vs. An. belenrae (0.009) [7], An. liangshanensis vs An. kunmingensis (0.002), and An. yatsushiroensis vs An. junlianensis (0.003) [35]. The F21 fragments were even the same in both An. kleini and An. belenrae. Therefore, only partial mitochondrial genome fragment sequences were insufficient to accurately explain the phylogenetic relationship within An. hyrcanus group, especially among cryptic species.
Phylogenetic analysis
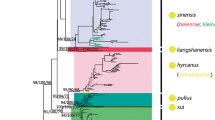
The phylogenetic trees were reconstructed using four methods based on the three independent fragments (except F21). All topological structures of the phylogenetic trees based on F7 + F8 and F19 were consistent and split up into two clades. One clade consisted of An. dirus and An. hyrcanus group six species, while the other included the rest three species (Additional file 8: Figure S2). The topology based on F5 and JD was also consistent. There were two major clades, one of which included all nine species (An. hyrcanus group six species, An. atroparvus and An. quadrimaculatus A) that belonged to subgenus Anopheles, and the other contained An. dirus and An. minimus that belonged to subgenus Cellia (Fig. 1). The 6 species of An. hyrcanus group were divided into two clusters: ((An. lesteri, An. sineroides) An. yatsushiroensis)) and ((An. belenrae, An. sinensis) An. kleini)). The optimal nucleotide substitution models for F5 and JD were GTR + G and GTR + G + I, respectively. The test result of congruence length for JD showed there was not a congruence length data (P = 0.01). The bootstrap values of ML tree were almost above 47% (Fig. 1).

The phylogenetic maximum likelihood tree based on the Joint Data of mitochondrial genome fragments. The codes are the same as those in Table 5, and numbers on the clades denote bootstrap values
In An. hyrcanus group, An. sinensis had a very close genetic relationship with An. belenrae and An. kleini. Their adults shared such similar morphology that there were not enough taxonomic characteristics to distinguish them. Anopheles belenrae and An. kleini were reported in 2005 [36], and natural hybridization between An. sinensis and An. kleini were found later [4, 5], indicating possible gene introgression in sympatric population and incomplete reproductive isolation between these two species as well as ongoing speciation. It was known that rDNA ITS2 sequences were the ideal molecular marker to distinguish cryptic species. However, the length of rDNA ITS2 in different mosquito species varied greatly, which led to alignment difficultly. Therefore, it was impossible to reconstruct different mosquito species simultaneously. Partial sequences of mitochondrial genome fragments were not able to provide adequate resolution for cryptic species. However, if mitochondrial genome sequences could be sufficiently informative, such as the employment of joint data connection, the phylogenetic tree of cryptic and distant genetic species could be reconstructed simultaneously.
Conclusions
Anopheles hyrcanus group is widely distributed in Palearctic and Oriental regions and some of them are important local malaria vectors. The cryptic species of An. hyrcanus group was almost impossible to identify based only on their morphology. In this study, the phylogenetic tree was established by creating mitochondrial genome fragments sequences (JD). It had two major clades, one of which included all Subgenus Anopheles species (An. hyrcanus group six species, An. atroparvus and An. quadrimaculatus A) and the other was comprised of An. dirus and An. minimus that belonged to subgenus Cellia. Anopheles hyrcanus group was divided into two clusters as ((An. lesteri, An. sineroides) An. yatsushiroensis) and ((An. belenrae, An. sinensis) An. kleini)). More molecular markers would make greater contribution to phylogenetic analysis.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding authors on reasonable request, and sequences are available in GenBank.
Abbreviations
- ATP6 :
-
ATPase6
- COI :
-
Cytochrom oxidase subunit I
- COII :
-
Cytochrom oxidase subunit II
- COIII :
-
Cytochrom oxidase subunit III
- Cyt B :
-
Cytochrom B
- ITS2 :
-
Second internal transcribed spacer
- JD:
-
Joint Data
- lrRNA :
-
Large Rrna
- ME:
-
Minimum evolution
- ML:
-
Maximum likelihood
- MP:
-
Maximum parsimony
- ND1 :
-
NADH dehydrogenase 1
- ND5 :
-
NADH dehydrogenase 5
- NJ:
-
Neighbor joining
- rDNA :
-
Ribosomal DNA
- rRNA :
-
Ribosomal RNA
- tRNA :
-
Transfer RNA
References
Harbach RE. The classification of genus Anopheles (Diptera: Culicidae): a working hypothesis of phylogenetic relationships. Bull Entomol Res. 2004;94(6):537–53.
Ma Y, Xu J. Progress of taxonomic study on the Anopheline mosquitoes in China. Chin J Vector Biol Control. 2015;26(5):433–8.
Ma Y, Xu J. The Hyrcanus group of Anopheles (Anopheles) in China (Diptera: Culicidae): species discrimination and phylogenetic relationships inferred by ribosomal DNA internal transcribed spacer 2 sequences. J Med Entomol. 2005;42(4):610–9.
Choochote W, Min GS, Intapan PM, Tantrawatpan C, Saeung A, Lulitanond V. Evidence to support natural hybridization between Anopheles sinensis and Anopheles kleini (Diptera: Culicidae): possibly a significant mechanism for gene introgression in sympatric populations. Parasit Vectors. 2014;7:36.
Joshi D, Choochote W, Min GS. Short report: natural hybrid between Anopheles kleini and Anopheles sinensis. Am J Trop Med Hyg. 2009;81(6):1020–2.
Khrabrova NV, Andreeva YV, Sibataev AK, Alekseeva SS, Esenbekova PA. Mosquitoes of Anopheles hyrcanus (Diptera, Culicidae) group: species diagnostic and phylogenetic relationships. Am J Trop Med Hyg. 2015;93(3):619–22.
Fang Y, Shi WQ, Zhang Y. Molecular phylogeny of Anopheles hyrcanus group (Diptera: Culicidae) based on mtDNA COI. Infect Dis Poverty. 2017;6(1):61.
Chu H, Li C, Guo X, Zhang H, Luo P, Wu Z, Wang G, Zhao T. The phylogenetic relationships of known mosquito (Diptera: Culicidae) mitogenomes. Mitochondrial DNA A DNA Mapp Seq Anal. 2018;29(1):31–5.
Chen K, Wang Y, Li XY, Peng H, Ma YJ. Sequencing and analysis of the complete mitochondrial genome in Anopheles sinensis (Diptera: Culicidae). Infect Dis Poverty. 2017;6(1):149.
Demari-Silva B, Foster PG, de Oliveira TM, Bergo ES, Sanabani SS, Pessoa R, Sallum MA. Mitochondrial genomes and comparative analyses of Culex camposi, Culex coronator, Culex usquatus and Culex usquatissimus (Diptera:Culicidae), members of the coronator group. BMC Genomics. 2015;16:831.
Gao B, Fang Y, Zhang J, Wu R, Xu B, Xie L. A DNA barcoding based study to identify Main mosquito species in Taiwan and its difference from those in mainland China. Comb Chem High Throughput Screen. 2017;20(2):147–52.
Wang G, Li C, Guo X, Xing D, Dong Y, Wang Z, Zhang Y, Liu M, Zheng Z, Zhang H, et al. Identifying the main mosquito species in China based on DNA barcoding. PLoS One. 2012;7(10):e47051.
Feng X, Huang L, Lin L, Yang M, Ma Y. Genetic diversity and population structure of the primary malaria vector Anopheles sinensis (Diptera: Culicidae) in China inferred by cox1 gene. Parasit Vectors. 2017;10(1):75.
Yang M, Ma Y, Wu J. Mitochondrial genetic differentiation across populations of the malaria vector Anopheles lesteri from China (Diptera: Culicidae). Malar J. 2011;10:216.
Makhawi AM, Liu XB, Yang SR, Liu QY. Genetic variations of ND5 gene of mtDNA in populations of Anopheles sinensis (Diptera: Culicidae) malaria vector in China. Parasit Vectors. 2013;6:290.
Jung J, Jung Y, Min GS, Kim W. Analysis of the population genetic structure of the malaria vector Anopheles sinensis in South Korea based on mitochondrial sequences. Am J Trop Med Hyg. 2007;77(2):310–5.
Lu B. Fauna Sinica, Insecta Vol. 9: Diptera, Culicidae II. Beijing: Science Press; 1997.
Lin L., Yang M., Nam C., Wu T., Ma Y., Y. M. Molecular taxonomy of Anopheles hyrcanus group in some regions of China. Chin J Vector Biol Control 2013; 24(2):92–97.
Ma Y, Ma YJ, Meng XM, Zhang SB, Liang G. Molecular identification of Anopheles hyrcanus group from Liaoning Province, China. Acta Parasitol Med Entomol Sinica. 2009;16(4):224–7.
Zhang N, Zhang Y, Yu G, Chen B. Structure characteristics of the mitochondrial genomes of Diptera and design and application of universal primers for their sequencing. Acta Entomol Sin. 2013;56(4):398–407.
Burland TG. DNASTAR's Lasergene sequence analysis software. Methods Mol Biol. 2000;132:71–91.
Gu X, Zhang J. A simple method for estimating the parameter of substitution rate variation among sites. Mol Biol Evol. 1997;14(11):1106–13.
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21.
Posada D, Crandall KA. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14(9):817–8.
Leigh JW, Susko E, Baumgartner M, Roger AJ. Testing congruence in phylogenomic analysis. Syst Biol. 2008;57(1):104–15.
Chang X, Zhong D, Lo E, Fang Q, Bonizzoni M, Wang X, Lee MC, Zhou G, Zhu G, Qin Q, et al. Landscape genetic structure and evolutionary genetics of insecticide resistance gene mutations in Anopheles sinensis. Parasit Vectors. 2016;9:228.
Hebert PD, Ratnasingham S, deWaard JR. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species. Proc Biol Sci. 2003;270(Suppl 1):S96–9.
Cywinska A, Hunter FF, Hebert PD. Identifying Canadian mosquito species through DNA barcodes. Med Vet Entomol. 2006;20(4):413–24.
Chan A, Chiang LP, Hapuarachchi HC, Tan CH, Pang SC, Lee R, Lee KS, Ng LC, Lam-Phua SG. DNA barcoding: complementing morphological identification of mosquito species in Singapore. Parasit Vectors. 2014;7:569.
Ashfaq M, Hebert PD, Mirza JH, Khan AM, Zafar Y, Mirza MS. Analyzing mosquito (Diptera: culicidae) diversity in Pakistan by DNA barcoding. PLoS One. 2014;9(5):e97268.
Murugan K, Vadivalagan C, Karthika P, Panneerselvam C, Paulpandi M, Subramaniam J, Wei H, Aziz AT, Alsalhi MS, Devanesan S, et al. DNA barcoding and molecular evolution of mosquito vectors of medical and veterinary importance. Parasitol Res. 2016;115(1):107–21.
Paramasivan R, Dhananjeyan KJ, Pandian RS. A preliminary report on DNA barcoding and phylogenetic relationships of certain public health important mosquito species recorded in rural areas of South India. J Vector Borne Dis. 2013;50(2):144–6.
Rozo-Lopez P, Mengual X. Mosquito species (Diptera, Culicidae) in three ecosystems from the Colombian Andes: identification through DNA barcoding and adult morphology. Zookeys. 2015;513:39–64.
Versteirt V, Nagy ZT, Roelants P, Denis L, Breman FC, Damiens D, Dekoninck W, Backeljau T, Coosemans M, Van Bortel W. Identification of Belgian mosquito species (Diptera: Culicidae) by DNA barcoding. Mol Ecol Resour. 2015;15(2):449–57.
Ma Y, Ma YJ, Wang Y. Phylogenetic relationship of Anopheles species, subgenus Anopheles in China: based on mtDNA and rDNA sequences. Chin Tropic Med. 2015;15(11):1181–288.
Leopoldo MR. Two new species of Anopheles (Anopheles) Hyrcanus group (Diptera: Culicidae) from the republic of South Korea. Zootaxa. 2005;941:1–26.
Acknowledgements
We are grateful to Mao-Qing Gong and Peng Cheng (Shandong), Ting-Ting Zhang, Tong-Shan Cao, Jun Chen (Liaoning), Jing-Xia Cheng, Pei-Fang Dai, Jun-Ying Zhao, Xiao-Dong Tian (Shanxi), who provided field assistance in the study.
Funding
This work was supported by the National Natural Sciences Foundation of China (No. 81371848) and the Infective Diseases Prevention and Cure Project of China (No. 2017ZX10303404–002).
Author information
Authors and Affiliations
Contributions
All authors read and approved the final version of the manuscript. YM and HP designed the study and conducted morphological identification. HC, JG, XL, YM and HP collected mosquito specimens in the field. HC and JG conducted species molecular identification. HZ, SL and MG conducted mitochondrial genome fragment sequencing. HZ, MG and YM carried out data analysis. SL, TF, HP and YM wrote the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This study was carried out in strict accordance with the National Natural Science Foundation of China ethical guidelines for biomedical research involving living animals and human subjects.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Supplementary information
Additional file 1.
Multilingual abstracts in the five official working languages of the United Nations.
Additional file 2: Table S1.
The mean of nucleotide composition and the numbers of conserved and variable bases of mitochondrial genome fragments in Anopheles hyrcanus group 5 members.
Additional file 3: Table S2.
The pairwise p distance between Subgenus Cellia and Anopheles species in this study calculated by F5 sequences.)
Additional file 4: Table S3.
The pairwise p distance between Subgenus Cellia and Anopheles species in this study calculated by F7 + F8 sequences.
Additional file 5: Table S4.
The pairwise p distance between Subgenus Cellia and Anopheles species in this study calculated by F19 sequences. (DOCX 15 kb)
Additional file 6: Table S5.
The pairwise p distance between Subgenus Cellia and Anopheles species in this study calculated by F21 sequences.
Additional file 7: Figure S1.
Primers for amplifying mitochondrial genome fragments of Anopheles.
Additional file 8: Figure S2.
The phylogenetic ML tree reconstructed based on F7 + F8 fragment of mitochondrial genome. YAT: An. yatsushiroensis; BEL: An. belenrae; KLE: An. kleini; LES: An. lesteri; SINE: An. sineroides; SIN: An. sinensis; DIR: An. dirus A; ATR: An. atroparvus; QUA: An. quadrimaculatus; MIN: An. minimus. The numbers on the clades denote the bootstrap confidence values.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
Cite this article
Zhu, HM., Luo, SH., Gao, M. et al. Phylogeny of certain members of Hyrcanus group (Diptera: Culicidae) in China based on mitochondrial genome fragments. Infect Dis Poverty 8, 91 (2019). https://doi.org/10.1186/s40249-019-0601-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s40249-019-0601-1