
Abstract
Background
Piroplasmosis is an important disease of domestic animals and wildlife and is caused by organisms from the genera Theileria and Babesia. Wildlife such as sika deer play an important role as reservoir hosts for several species of Theileria and Babesia. Using blood samples collected from sika deer, we investigated the epidemiology of Theileria spp. and Babesia spp. in sika deer from Jilin Province in China and identified those species that cause pathogenic infections in sika deer.
Methods
Sixty-eight blood samples of sika deer were collected from three areas of the Jilin Province in Northeast China. Genomic DNA was extracted, and the V4 hypervariable region of the 18S rRNA of the piroplasms was amplified using the nested PCR method. The selected positive samples were sequenced to identify species of Babesia and Theileria.
Results
PCR detection revealed that 24 samples were positive for Theileria and Babesia spp. (35.29 %, 95 % CI = 11.8-46.8). After alignment, a sequenced fragment for Theileria cervi was found to be the most prevalent from the obtained samples (22.06 %, 95 % CI = 11.8-49.6). Six sika deer samples were identified as being infected with a Theileria sp. that was similar to a Theileria sp. found from spotted deer in India. In addition to the results above, for the first time, we identified T. annulata infection from one sample of sika deer and Babesia sp. from two samples, which showed high identity with Babesia motasi found in sheep from China.
Conclusion
The present study offers new data on the pathogens of piroplasmosis in sika deer in northeastern China. For the first time, sika deer was confirmed as a reservoir host for the T. annulata of cattle and the B. motasi of sheep, which was found in China.
Similar content being viewed by others


Background
Piroplasmosis is an important tick-borne disease of domestic and wild ruminants and is caused by different species of Theileira and Babesia all around the world. Theileria spp. are cosmopolitan tick-borne protozoan pathogens that can infect both livestock and wild ruminants [1]. Depending on the pathogenicity, T. annulata and T. parva, transmitted by species of Hyalomma and Rhipicephalus appendiculatus, were thought to be the most pathogenic species, which cause lymphoproliferative disease with high mortality rates [2], whereas other Theileira species are described as benign pathogenic parasites transmitted by Rhipicephalus and Haemaphysalis ticks to cattle and small ruminants. Many studies have reported that cervids could be infected with many species of Theileria. Theileria cervi is a nonpathogenic parasite found in different deer species including white-tail deer, elk, mule deer and pampas deer, which is transmitted by Amblyomma americanum in the USA [3, 4]. Theileria capreoli Rukhlyadev was first morphologically described from roe deer; similar Theileria species, including Theileria sp. ZS TO4, have been described in red deer, roe deer and chamois; and Theileria sp. 3185/02 has been described in red deer and roe deer based on analyses of 18S rRNA gene [5–7]. In sika deer, at least three Theileria spp. that are different from the species identified in cattle, have been reported in Japan and China [3, 8, 9]. In addition, similar species of Theileria were also reported in Chinese water deer from North Korea [1]. In reindeer, two Theileria genotypes have been found that are similar to a Theileria sp. from white-tailed deer but different from T. cervi [10]. Thus, cervids could be the host for many species of Theileria and may play important roles in their epidemiology in the wild.
Many of the tick species including Ixodes ricinus, I. scapularis, Rhipicephalus microplus and Haemaphysalis longicornis were described as the vector that transmitted the pathogens of babesisosis to livestock and wild animals. Babesia divergens, an Ixodes ricinus-transmitted parasite of cattle and humans, has been reported in several deer species including reindeer, roe deer and red deer and is widely distributed in Europe [7, 11–13]. Babesia capreoli is another species that is prevalent in free-living roe deer and sika deer on the European continent [11, 14, 15]. In America, the main pathogen of deer babesiosis is Babesia odocoilei, which has been detected in roe deer, reindeer and elk [16, 17]. Some novel Babesia spp. have been identified in different deer species; these include B. venatorum (formerly identified as Babesia sp. EU1) in roe deer and B. pecorum sp. in red deer [18, 19]. Additionally, the pathogens of bovine babesiosis, B. bigemina and B. bovis have been identified in white-tailed deer [18].
The sika deer, which is a first-grade state protected animal in China, numbers approximately 550,000 head and is mainly concentrated in Northeast China [19]. Theileria cervi and another Theileria sp. were the first pathogens to be reported as causing cervid theileriosis in sika deer in central China [9]. In Northwest China, Theileria uilenbergi and T. capreoli have been described in sika deer [20]. However, there have been no epidemiological studies on cervid piroplasmosis in northeastern China. In the present study, polymerase chain reaction assay was carried out to amplify the V4 hypervariable region of the 18S rDNA of the piroplasms, and the sequenced fragments were used for phylogenetic analysis to identify species of Theileria and Babesia.
Methods
Sample collection
During June and July 2015, 68 blood samples were randomly collected from domesticated sika deer from four ranches (DF n = 13, TH n = 15, SY n = 9, and ZJ n = 31) in Jilin Province, in northeastern China. Blood samples from the jugular veins were collected into tubes containing EDTA and stored at 4 °C until DNA extraction.
DNA extraction and primer design
DNA was extracted from 300 μl blood using the Genomic DNA purification Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The DNA concentration was determined with a NanoDrop 2000 spectrophotometer (Nanodrop Technologies, Wilmington, DE, USA). DNA was stored at -70 °C until further analysis.
PCR amplification of the V4 region of 18S rRNA gene
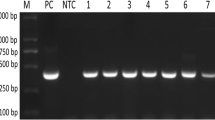
The V4 hypervariable region of the 18S rRNA gene, as the target sequence, was amplified by using nested PCR. Universal primers that can amplify a wide variety of Babesia and Theileria spp. were used [21–23]. The first PCR reaction was conducted using primers RLB-F2 (5’-GACACAGGGAGGTAGTGACAAG-3’) and RLB-R2 (5’-CTAAGAATTTCACCTCTGACAGT-3’). The reactions were performed in a final volume of 25 μl containing 12.5 μl Premix Taq DNA polymerase (TakaRa, China), 1.0 μM of each primer and 1 μl of DNA template. The PCR reaction system comprised one step of initial denaturation at 95 °C for 3 min, followed by 35 cycles of denaturation (95 °C for 1 min), primer annealing (52 °C for 50 s) and extension (72 °C for 1.5 min). The final extension was performed with one step at 72 °C for 5 min. The second PCR was carried out with the primer RLB-FINT (5’-GACAAGAAATAACAATACRGGGC-3’) used as the forward primer together with RLB-R2. The reaction mixture was as in the first PCR reaction, except the template was replaced by 1 μl of the first PCR product. The reaction cycling in the second PCR was optimized for one step of initial denaturation at 95 °C for 3 min, followed by 35 cycles of denaturation (95 °C for 30 s), primer annealing (50 °C for 30 s) and extension (72 °C for 30 s), and the final extension was performed at 72 °C for 5 min. PCR products were electrophoresed on a 1.5 % agarose gel containing 10 μl of Goldview (SolarBio, China) in Tris–acetate-EDTA (TAE) buffer at 120 V for 40 min and visualized under UV light.
The positive PCR products were excised from the gel and purified using an Axygen Gel Purification Kit. The DNA fragment was cloned into the pGEM-T Easy vectors (Promega, 2800 Woods Hollow Road Madison, WI 53711-5399, USA). The Escherichia coli JM 109 (TaKaRa, China) was transformed and plasmid DNA from the selected clones was identified using PCR with primers as indicated (program and reaction mixtures were the same as those used in the PCR amplification described above) to verify the presence of correct inserts in selected clones before proceeding with the sequencing process in the Big Dye Terminator Mix of TaKaRa Company (China).
PCR amplification of a long fragment of the 18S rRNA gene
Samples shown to be positive in the nested PCR were selected for amplification of the long fragment of the 18S rRNA gene, with the aim of confirming the species of Theileria and Babesia. Two published primers, NBab-1f (5’-AAGCCATGCATGTCTAAGTAGAAGCTTTT-3’) and 18SRev-BT (GAATAATTCACCGGATCACTCG), were used to amplify an approximately 1,600-bp fragment [24, 25]. The reaction mixture was performed in a final volume of 50 μl, with 1.0 μM of each primer, 2 μl of DNA template, 5 μl of 10× High Fidelity buffer, 2 μl of 50 mM MgSO4, 4 μl dNTP mixture (2.5 mM of each), and 1 unit of Platinum Taq DNA Polymerase High Fidelity (Invitrogen, USA). The cycling conditions included initial denaturation at 95 °C for 3 min, followed by 35 cycles of 94 °C for 30 s, 58 °C for 30 s and 72 °C for 2 min, with one step of final extension at 72 °C for 10 min. The steps for cloning and sequencing were the same as previously described.
Sequence analysis
The obtained sequences were aligned using the MegAlign component of the DNAStar software program (Version 4.0 DNAStar, Madison, USA). After alignment with the related Theileria and Babesia spp. 18S rDNA sequences retrieved from the GenBank, parts of the cloning vector region were removed manually. The resulting sequences were then submitted to the GenBank database.
A phylogenetic tree was generated based on the cloned sequences and the related Theileria/Babesia 18S rDNA sequences in GenBank by using the neighbor-joining algorithm in the MEGA 6.0 software [26]. The evolutionary distances were computed using the Kimura two-parameter method [27].
Statistical analysis
The 95 % confidence intervals (95 % CIs) for the overall prevalence values of Theileria/Babesia were calculated using IBM SPSS Statistics version 19.0.
Ethical approval
The present works were approved by the Animal Ethics Committee of Lanzhou Veterinary Research Institute, CAAS (No. LVRIAEC2013-010). The procedures for acquiring the field samples were approved by the Animal Ethics Procedures and Guidelines of China.
Results
The DNA of the Babesia and Theileria spp. samples was detected with nested PCR targeting the V4 region of the 18S rRNA gene. Of the 68 sika deer blood samples, 24 samples were positive for piroplasms (35.29 %, 95 % CI = 11.8-46.8). The PCR products of all 24 positive samples were sequenced and deposited in the GenBank database (accession numbers KT683524-KT683536,KT959214-KT959221, KT970057 and KT970058). After alignment with the related sequences, T. cervi was the most prevalent parasite detected from the sika deer. For the sika deer, 15 out of 68 samples (22.06 %, 95 % CI = 11.8-49.6) were positive for T. cervi. The second most prevalent parasite was a Theileria spp. which had 100 % identity with India spotted deer isolate Theileria. sp (JX112750); 6 samples were positive for this Theileria. For the first time, Theileria annulata was detected in one sika deer (ZJ07) and showed 100 % identity with T. annulata (EU083801). In addition to the Theileria spp. a Babesia sp. was detected in samples DF02 and DF07 and showed 100 % identity with Babesia sp. China-BQ1 (AY260181) from sheep.
The aim of this study was to confirm the parasites identified based on the V4 region of the 18S rRNA gene. A fragment of approximately 1,600 base pairs of the 18S rDNA was amplified and sequenced from selected positive samples, especially the samples positive for T. annulata, Babesia sp. and Theileria sp. The obtained sequences were deposited in GenBank (accession numbers KT959223-KT959232). Clustal W from DNAstar software analysis indicated that two types of Theileria were found. One type contains 5 clones that have at least 99.3 % identity with T. cervi from China and with Theileria sp. (CNY3C, yamaguchi and lwate) from Japan. The other type is similar with Theileria sp. isolated from India spotted deer, having 99.8 % identity with it. Analysis of the long 18S rDNA sequence showed that the sequence of ZJ07 shared 99.4 % identity with T. annulata (EU083801). The sequence of DF02 has 99.3 % identity with Babesia sp. China-BQ1 (AY260181) from sheep. Attempts to amplify the long fragment of the 18S rRNA gene from DF07 failed.
The obtained V4 sequences and long 18S rDNA sequences were subjected to phylogenetic analysis along with the related sequences from GenBank with the MEGA 6.0 software. The two phylogenetic trees yielded were identical. As shown in Figs. 1 and 2, most of the samples fell into the T. cervi clade. However, some samples were different from T. cervi and fell into the Theileria sp. from the India spotted deer group. The sequence from sample ZJ07 was grouped with T. annulata, and DF02 was in the clade of the B. sp from sheep.

Phylogenetic tree of Theileria and Babesia spp. based on the V4 region of 18S rRNA gene sequences. The parasite identified in the present study is marked in bold

Phylogenetic tree of Theileria and Babesia spp. constructed based on the 18S rRNA gene sequences. The parasite identified in the present study is marked in bold
Discussion
In the present study, four different sika deer pastures which are close to some small villages containing free-grazing cattle and sheep, were selected for collection of blood samples. During sample collection, some sika deer were found losing weight and suffering anemia, in addition with the detection of T. annulata and T. sergenti from cattle from the same area [28]. It is possible for sika deer infected with piroplasms. Among wildlife, species of the Cervidae are not only the hosts for piroplasms that affect different species of deer, but could also be the hosts for many bovine piroplasms. The pathogens of bovine babesiosis, including B. bigemina and B. bovis, have been reported in infected white-tailed deer, pampas deer, brown brocket deer and marsh deer from the United States and Brazil [4, 18, 29]. Another bovine Babesia spp. found from deer is B. divergens, which is also a main pathogen of human babesiosis in Europe [30, 31]; many deer species, including reindeer, roe deer and red deer, have been described as infected with this parasite [11, 32, 33]. To date, no report exists of the agent of bovine theileriosis being found in cervids. Only a single Theileria spp. identified from sika deer has shown 97 % identity with T. sergenti, but the species was distinct from the clade of cattle Theileria spp. in the phylogenetic tree based on an analysis of 18S rRNA genes [3].
Sixty-eight DNA samples of sika deer were first evaluated with a nested PCR method for the detection of piroplasms [21–23]. From the PCR results, 24 samples were found to be positive for Theileria or Babesia. Because an aim of the present study was to clarify the genus of the parasites found, the V4 sequence of all positive samples was sequenced. Sequence analysis allowed the identification of two types of Theileria, which were the same T. cervi and Theileria sp. found in Indian spotted deer. Because T. cervi can infect different deer species and has been found previously in sika deer in China [3, 9, 34], the finding of T. cervi in sika deer in the present study was not surprising. Of more interest was the identification of T. annulata in sika deer based on the analysis of 18S rDNA sequences. In the phylogenetic tree constructed based on the 18S rDNA sequences, the T. annulata isolated from the sika deer fell into the clade including the T. annulata isolates from China and other countries. The percent identity of T. annulata isolated from sika deer with the T. annulata Yining (EU083801) isolate was 99.4 %. To the best of our knowledge, T. annulata had only been isolated previously from infected sheep and bovidae. No other information on the organisms that cause bovine theileriosis has been identified from other ruminants. This is the first identification of T. annulata in sika deer and extends the host range for this parasite.
As previous studies have described, the pathogens of cervine babesiosis include B. capreoli, B. venatorum (formerly as Babesia sp. EU1), B. divergens and B. pecorum [35]. In the present study, none of the Babesia spp. described above were found, but B. motasi infection was identified in two sika deer. B. motasi is one of the most important pathogens of ovine babesiosis in China. At least 6 isolates of B. motasi have been collected by our institute [36]. The Babesia parasite found in the present study showed 100 % identity on the V4 region and 99.3 % identity with the long fragment of the 18S rDNA with Babesia sp. China-BQ1 (Lintan), which indicated the parasite is B. motasi. Previous studies have reported that B. motasi can infect wild Caprinae, such as the Alpine ibex and chamois but no other animals [13]. Our finding is the first to describe sika deer as the host for B. motasi.
From Jilin province, many tick species have been reported including Hyalomma scupense, Hy. detritum Schulze which are the vectors for T. annulata, and Hae. longicornis for the transmission of B. motasi [37]; the vector for T. cervi in China is still unclear. In Japan, Hae. longicornis, Hae. flava, Hae. yeni, Hae. megaspinosa, I. ovatus and Amblyomma testudinarium were detected on sika deer, but the piroplasms were not determined from the ticks [3, 38].
As described before, livestock could introduce disease to wildlife, which can act as a maintenance host and, therefore, as the source of infection for livestock [39]. Therefore, in future studies, confirmation of the ability of the T. annulata and B. motasi that were isolated from sika deer to infect their original hosts is needed, thus helping with the control of the disease in both livestock and wildlife.
References
Han JI, Jang HJ, Na KJ. Molecular detection of Theileria sp. in wild Chinese water deer (Hydropotes inermis argyropus). J Wildl Dis. 2009;45:1213–6.
Onuma M, Kakuda T, Sugimoto C. Theileria parasite infection in East Asia and control of the disease. Comp Immunol Microbiol Infect Dis. 1998;21:165–77.
Inokuma H, Tsuji M, Kim S-J, Fujimoto T, Nagata M, Hosoi E, et al. Phylogenetic analysis of Theileria sp. from sika deer, Cervus nippon, in Japan. Vet Parasitol. 2004;120:339–45.
Silveira JAG, Rabelo EML, Lacerda ACR, Borges PAL, Tomás WM, Pellegrin AO, et al. Molecular detection and identification of hemoparasites in pampas deer (Ozotoceros bezoarticus Linnaeus, 1758) from the Pantanal Brazil. Ticks Tick-Borne Dis. 2013;4:341–5.
Fuehrer H-P, Biro N, Harl J, Worliczek HL, Beiglböck C, Farkas R, et al. Molecular detection of Theileria sp. ZS TO4 in red deer (Cervus elaphus) and questing Haemaphysalis concinna ticks in Eastern Austria. Vet Parasitol. 2013;197:653–7.
Sawczuk M, Maciejewska A, Skotarczak B. Identification and molecular characterization of Theileria sp. infecting red deer (Cervus elaphus) in northwestern Poland. Eur J Wildl Res. 2007;54:225–30.
García-Sanmartín J, Aurtenetxe O, Barral M, Marco I, Lavin S, García-Pérez AL, et al. Molecular detection and characterization of piroplasms infecting cervids and chamois in Northern Spain. Parasitology. 2007;134(Pt 3):391–8.
Ikawa K, Aoki M, Ichikawa M, Itagaki T. Occurrence of two distinct Theileria lineages in sika deer (Cervus nippon) of Iwate Prefecture, Japan. J Vet Med Sci Jpn Soc Vet Sci. 2011;73:1371–3.
He L, Khan MK, Zhang WJ, Zhang QL, Zhou YQ, Hu M, et al. Detection and identification of Theileria infection in sika deer (Cervus nippon) in China. J Parasitol. 2012;98:598–603.
Garner BC, Holman P, Berent LM. Theileriosis in a reindeer (Rangifer tarandus tarandus) associated with a potentially novel Theileria sp. Vet Clin Pathol Am Soc Vet Clin Pathol. 2012;41:497–501.
Duh D, Petrovec M, Bidovec A, Avsic-Zupanc T. Cervids as Babesiae hosts, Slovenia. Emerg Infect Dis. 2005;11:1121–3.
Lempereur L, Wirtgen M, Nahayo A, Caron Y, Shiels B, Saegerman C, et al. Wild cervids are host for tick vectors of Babesia species with zoonotic capability in Belgium. Vector Borne Zoonotic Dis Larchmt N. 2012;12:275–80.
Michel AO, Mathis A, Ryser-Degiorgis MP. Babesia spp. in European wild ruminant species: parasite diversity and risk factors for infection. Vet Res. 2014;45:65.
Gray JS, Murphy TM, Waldrup KA, Wagner GG, Blewett DA, Harrington R. Comparative studies of Babesia spp. from white-tailed and sika deer. J Wildl Dis. 1991;27:86–91.
Bastian S, Jouglin M, Brisseau N, Malandrin L, Klegou G, L’Hostis M, et al. Antibody prevalence and molecular identification of Babesia spp. in roe deer in France. J Wildl Dis. 2012;48:416–24.
Holman PJ, Swift PK, Frey RE, Bennett J, Cruz D, Wagner GG. Genotypically unique Babesia spp. isolated from reindeer (Rangifer tarandus tarandus) in the United States. Parasitol Res. 2002;88:405–11.
Holman PJ, Bendele KG, Schoelkopf L, Jones-Witthuhn RL, Jones SO. Ribosomal RNA analysis of Babesia odocoilei isolates from farmed reindeer (Rangifer tarandus tarandus) and elk (Cervus elaphus canadensis) in Wisconsin. Parasitol Res. 2003;91:378–83.
Holman PJ, Carroll JE, Pugh R, Davis DS. Molecular detection of Babesia bovis and Babesia bigemina in white-tailed deer (Odocoileus virginianus) from Tom Green County in central Texas. Vet Parasitol. 2011;177:298–304.
Meng QF, Li Y, Zhou Y, Bai YD, Wang WL, Wang WL, et al. Seroprevalence of Neospora caninum infection in farmed sika deer (Cervus nippon) in China. Vet Parasitol. 2015;211:289–92.
Li Y, Chen Z, Liu Z, Liu J, Yang J, Li Q, et al. Molecular identification of Theileria parasites of northwestern Chinese Cervidae. Parasit Vectors. 2014;7:225.
Gubbels JM, de Vos AP, van der Weide M, Viseras J, Schouls LM, de Vries E, et al. Simultaneous detection of bovine Theileria and Babesia species by reverse line blot hybridization. J Clin Microbiol. 1999;37:1782–9.
Centeno-Lima S, Do Rosário V, Parreira R, Maia AJ, Freudenthal AM, Nijhof AM, et al. A fatal case of human babesiosis in Portugal: molecular and phylogenetic analysis. Trop Med Int Health TM IH. 2003;8:760–4.
Zanet S, Trisciuoglio A, Bottero E, de Mera IGF, Gortazar C, Carpignano MG, et al. Piroplasmosis in wildlife: Babesia and Theileria affecting free-ranging ungulates and carnivores in the Italian Alps. Parasit Vectors. 2014;7:70.
Oosthuizen MC, Zweygarth E, Collins NE, Troskie M, Penzhorn BL. Identification of a novel Babesia sp. from a sable antelope (Hippotragus niger Harris, 1838). J Clin Microbiol. 2008;46:2247–51.
Matjila PT, Leisewitz AL, Oosthuizen MC, Jongejan F, Penzhorn BL. Detection of a Theileria species in dogs in South Africa. Vet Parasitol. 2008;157:34–40.
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol Biol Evol. 2013;30:2725–9.
Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16:111–20.
Liu JL, Li YQ, Liu AH, Guan GQ, Xie JR, Yin H, et al. Development of a multiplex PCR assay for detection and discrimination of Theileria annulata and Theileria sergenti in cattle. Parasitol Res. 2015;114:21–2751.
da Silveira JAG, Rabelo EML, Ribeiro MFB. Detection of Theileria and Babesia in brown brocket deer (Mazama gouazoubira) and marsh deer (Blastocerus dichotomus) in the State of Minas Gerais, Brazil. Vet Parasitol. 2011;177:61–6.
Kjemtrup AM, Conrad PA. Human babesiosis: an emerging tick-borne disease. Int J Parasitol. 2000;30:1323–37.
Zintl A, Mulcahy G, Skerrett HE, Taylor SM, Gray JS. Babesia divergens, a bovine blood parasite of veterinary and zoonotic importance. Clin Microbiol Rev. 2003;16:622–36.
Langton C, Gray JS, Waters PF, Holman PJ. Naturally acquired babesiosis in a reindeer (Rangifer tarandus tarandus) herd in Great Britain. Parasitol Res. 2003;89:194–8.
Welc-Falęciak R, Werszko J, Cydzik K, Bajer A, Michalik J, Behnke JM. Co-infection and genetic diversity of tick-borne pathogens in roe deer from Poland. Vector Borne Zoonotic DisLarchmt N. 2013;13:277–88.
Yabsley MJ, Quick TC, Little SE. Theileriosis in a white-tailed deer (Odocoileus virginianus) fawn. J Wildl Dis. 2005;41:806–9.
Jouglin M, Fernández-de-Mera IG, de la Cotte N, Ruiz-Fons F, Gortázar C, Moreau E, et al. Isolation and characterization of Babesia pecorum sp. nov. from farmed red deer (Cervus elaphus). Vet Res. 2014;45:78.
Guan G, Liu J, Liu A, Li Y, Niu Q, Gao J, et al. A member of the HSP90 family from ovine Babesia in China: molecular characterization, phylogenetic analysis and antigenicity. Parasitology. 2015;142:1387–97.
Chen Z, Yang XJ, Bu FJ, Yang XH, Yang XL, Liu JZ. Tikcs (Acari: Ixodoidea: Argasidae, Ixodidae) of China. Exp Appl Acarol. 2010;51:393–404.
Inokumma H, Fujimoto T, Hosoi E, Tanaka S, Fujisaki K, Onishi T. Tick infestation of sika deer (Cervus nippon) in western part of Yamaguchi Prefecture, Japan. J Vet Med Sci. 2003;64:615–7.
Daniels PW, Halpin K, Hyatt A, Middleton D. Infection and disease in reservoir and spillover hosts: determinants of pathogen emergence. Curr Top Microbiol Immunol. 2007;315:113–31.
Acknowledgements
This study was financially supported by the NSFC (№31402189, №31372432), ASTIP, FRIP (2014ZL010), CAAS; NBCIS CARS-38; National Key Basic Research Program (973 Program) of China (2015CB150300); Supporting Program (2013BAD12B03, 2013BAD12B05; Jiangsu Co-innovation Center programme for Prevention and Control of Important Animal Infectious Diseases and Zoonoses,State Key Laboratory of Veterinary Etiological Biology Project.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contribution
JL, JY, BW, and AL did the sample collection and the molecular genetic studies. JL, GG, JL and HY wrote and corrected the manuscript. All authors read and approved the final manuscript.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, J., Yang, J., Guan, G. et al. Molecular detection and identification of piroplasms in sika deer (Cervus nippon) from Jilin Province, China. Parasites Vectors 9, 156 (2016). https://doi.org/10.1186/s13071-016-1435-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13071-016-1435-3