
Abstract
Background
Cyanobacteria are potential sources for third generation biofuels. Their capacity for biofuel production has been widely improved using metabolically engineered strains. In this study, we employed metabolic engineering design with target genes involved in selected processes including the fatty acid synthesis (a cassette of accD, accA, accC and accB encoding acetyl-CoA carboxylase, ACC), phospholipid hydrolysis (lipA encoding lipase A), alkane synthesis (aar encoding acyl-ACP reductase, AAR), and recycling of free fatty acid (FFA) (aas encoding acyl–acyl carrier protein synthetase, AAS) in the unicellular cyanobacterium Synechocystis sp. PCC 6803.
Results
To enhance lipid production, engineered strains were successfully obtained including an aas-overexpressing strain (OXAas), an aas-overexpressing strain with aar knockout (OXAas/KOAar), and an accDACB-overexpressing strain with lipA knockout (OXAccDACB/KOLipA). All engineered strains grew slightly slower than wild-type (WT), as well as with reduced levels of intracellular pigment levels of chlorophyll a and carotenoids. A higher lipid content was noted in all the engineered strains compared to WT cells, especially in OXAas, with maximal content and production rate of 34.5% w/DCW and 41.4 mg/L/day, respectively, during growth phase at day 4. The OXAccDACB/KOLipA strain, with an impediment of phospholipid hydrolysis to FFA, also showed a similarly high content of total lipid of about 32.5% w/DCW but a lower production rate of 31.5 mg/L/day due to a reduced cell growth. The knockout interruptions generated, upon a downstream flow from intermediate fatty acyl-ACP, an induced unsaturated lipid production as observed in OXAas/KOAar and OXAccDACB/KOLipA strains with 5.4% and 3.1% w/DCW, respectively.
Conclusions
Among the three metabolically engineered Synechocystis strains, the OXAas with enhanced free fatty acid recycling had the highest efficiency to increase lipid production.
Similar content being viewed by others
Background
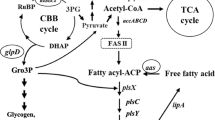
Cyanobacteria have recently been used as the third-generation biofuel resources [1] due to their availability of various valuable precursors such as lipids, alkenes, alkanes, PHB and fatty alcohols for biofuel and biodiesel syntheses [2,3,4]. In addition, they possess a prominent photosynthetic machinery and minimal utilization of basic nutritional requirement with further converting and recycling CO2 into fuels and chemicals [1]. The oil productivity of several microalgae greatly exceeds that of oil crops, which allows them to have economic competitiveness with petro-diesel for transportation fuel [5]. Metabolic engineering technology approach and genome sequence databases of cyanobacteria may be used as potential tools for developing cell production competency of energy containing biomolecules or biofuel products. For the lipid synthetic pathway in cyanobacteria (as shown in Fig. 1), the core metabolite acetyl-CoA is converted to fatty acyl–acyl carrier protein (fatty acyl-ACP) via fatty acid synthesis II (FAS II). The first limiting step of lipid biosynthesis begins with acetyl-CoA carboxylase (ACC) catalyzing a carboxylation reaction of acetyl-CoA to malonyl-CoA. In higher plants, the acetyl-CoA pool, which originates from the Calvin cycle and the breakdown of both carbohydrates and lipids, remained relatively unchanged in the range of 30–50 μM except the fatty acid synthesis whose rates varied significantly [6]. Previously, an engineered ACC overexpressing strain of Escherichia coli showed a sixfold increased fatty acid level [7]. In the cyanobacterial FAS II system, long-chain acyl-ACP or fatty acyl-ACP is mainly used as a key precursor for phospholipid production [8, 9]. The biochemical balance of fatty acyl-ACP is either gained or reduced as described via neighboring pathways (Fig. 1). The key enzyme for free fatty acid recycling to fatty acyl-ACP in Synechocystis is acyl–acyl carrier protein synthetase (AAS) encoded by aas which requires ATP, ACP-SH (acyl carrier protein-SH) and cofactors including Mg2+ and Ca2+ [3, 10]. However, an intermediate flux limitation exists when excess levels of fatty acyl-ACP cause a decreased activity of acetyl-CoA carboxylase (ACC) via a feedback regulation in the fatty acid synthetic processes [11, 12]. An efficient in vivo flow of fatty acyl-ACP intermediate is directed not only to phospholipid production but also, indirectly, to alk(e)ane production [4]. A previous report revealed that overexpression of both aar/ado, encoding acyl-ACP reductase and aldehyde dehydrogenase, in alk(e)ane synthetic pathway resulted in an enhanced alk(e)ane production, especially heptadecane, in Synechococcus sp. NKBG15041c strain [13]. The direct conversion from fatty acyl-ACP to phospholipids in cyanobacteria has been addressed via a set of PlsX/PlsY/PlsC acyltransferase catalytic systems [14]. The phospholipid homeostasis is maintained via both synthesis and degradation. Recently, the key enzyme for phospholipid hydrolysis to FFA in Synechocystis sp. PCC 6803 was identified, a lipase A encoded by sll1969, although its regulatory or inducible mechanism remains unclear [15]. Interestingly, many recent reports revealed the competency of modern metabolic engineering to overcome those intracellular-biochemical limitation, in particular feedback inhibitions. For instance, to decrease the costly fatty acid recovery, a so-called damaging cyanobacterial cell membranes strategy was employed, e.g., an acyl-ACP thioesterase overexpression in order to secrete FFA into culture medium [16, 17].

The fatty acid biosynthesis and its neighboring pathways in Synechocystis sp. PCC 6803. Key enzyme genes include accABCD, multi-subunit acetyl-CoA carboxylase gene; aar, acyl-ACP reductase gene; aas, acyl-ACP synthetase; ado, aldehyde oxidase; fabD, malonyl coenzyme A-acyl carrier protein transacylase; lipA, lipolytic enzyme genes; plsX, plsY, plsC, putative phosphate acyl-transferases; phaA, polyhydroxyalkanoates specific beta-ketothiolase gene. The thick arrow is represented as the overexpression (OX) of that gene whereas the cross symbol is represented the knockout (KO) of that gene
In this study, we generated three metabolically engineered Synechocystis 6803 strains: OXAas—aas-overexpression, OXAas/KOAar—aas-overexpression with aar gene interruption, and OXAccDACB/KOLipA—accDACB-overexpression with lipA gene interruption (Fig. 1). Our results demonstrate a significant increase of lipid production in all engineered Synechocystis 6803 strains.
Methods
Strains and growth conditions
Synechocystis sp. strain PCC 6803 was grown under normal growth condition of BG11 medium at 28 °C under a continuous light illumination intensity of 40 µmol photons/m2/s. All engineered strains, OXAas, OXAccDACB/KOLipA and OXAas/KOAar (Table 1), were grown in a BG11 medium containing 35 µg/mL of chloramphenicol. Escherichia coli DH5α strain was used as a host propagation and grown at 37 °C on the Luria–Bertani (LB) agar medium containing 30 µg/mL of chloramphenicol. The pre-cultivation was performed initially on BG11 agar plates and transferred to 100 mL-liquid medium until cells reaching mid-log phase of growth before starting the experiment. The initial cell density of Synechocystis cells for a culture experiment was set at the optical density at 730 nm (OD730) of about 0.15. Growth measurement was monitored by a spectrophotometer at OD730. Dry cell weight (DCW) was performed by incubating the harvested cells in 60 °C oven until obtaining a constant dry weight.
Construction of recombinant plasmids
The pEERM plasmid [18] was used as a cloning and expression vector in this study. pEERM mainly contains various crucial regions including the flanking region of upstream PsbA2 sequence, promoter sequence of PsbA2 (PpsbA2), multiple cloning sites of XbaI, PstI and SpeI, chloramphenicol resistance cassette and the flanking region of downstream PsbA2 sequence, respectively. Construction of a recombinant pEERM_aas plasmid (Table 1) firstly started by PCR amplifying the homologous aas gene fragment encoding AAS from Synechocystis sp. PCC 6803 genomic DNA template using a specific pair of primers, Aas_F and Aas_R (Table 2). The amplified aas fragment was then ligated into pEERM vector between the sites of XbaI and SpeI locating downstream of PsbA2 promoter. For a recombinant pEERM_LipA/AccDACB (Table 1), the flanking region replacements in pEERM vector of both upstream and downstream PsbA2 sequences were performed with the flanking regions of both upstream and downstream lipA gene sequences (encoding lipase A) obtained from PCR using two pairs of primers including USlipA_F and USlipA_R and DWlipA_F and DWlipA_R (Table 2), respectively. On the other hand, the inserted accDACB gene fragments encoding ACC were obtained by PCR (primer sequences shown in Table 2). All gene fragments were ligated with end-terminal sequence removing and sequentially cloned into pEERM plasmid between the XbaI and SpeI sites. Moreover, the recombinant pEERM_Aar/Aas plasmid (Table 1) was constructed by replacing both upstream and downstream regions in pEERM vector with both upstream and downstream regions of aar gene obtained by PCR using specific pairs of primers including USaar_F and USaar_R and DWaar_F and DWaar_R (Table 2), respectively.
Natural transformation of recombinant plasmid into Synechocystis cells
Synechocystis wild-type cells, grown in 50 mL-BG11 medium for 2–3 days until reaching an OD730 of about 0.5, were harvested by centrifugation at 6000 rpm (4025×g). Obtained cell pellet was resuspended in 500 µL of new BG11 medium followed by the addition of 10 µg of each recombinant plasmid. The cell suspension was incubated at 28 °C for 6 h by inverting the mixture tube every 2 h before spreading on a 0.45 µm sterile nitrocellulose membrane placed over the normal BG11-agar plate. After 24 h incubation, the membrane was transferred onto BG11-agar containing 35 µg/mL of chloramphenicol. Normally, survived colonies were obtained within 3–4 weeks of incubation. Generated transformants were further examined for their gene location by PCR using selected, specific primers (Table 2).
Determination of intracellular pigment content
Total chlorophyll a (Chl a) and carotenoid (Car) contents were extracted by N,N-dimethylformamide (DMF), and their contents were determined by measuring the absorbance at 461, 625 and 664 nm using a spectrophotometer [19, 20]. The Chl a and Car contents were normalized to a cell number corresponding to 1.0 × 108 of the cells [19,20,21].
Measurement of oxygen evolution
Harvested cells were incubated in the dark for 30 min before measuring their relative O2 evolution rate of cells under saturated white light illumination using Clark-type oxygen electrode (Hansatech instruments, UK) at 25 °C. The O2 evolution rate was represented as µmol/mg Chl a/h [22].
Determinations of total lipid and unsaturated lipid contents
During cultivation, fifteen mL-cell cultures of either WT cells or engineered strains were harvested by centrifugation at 6000 rpm (4025×g), at room temperature for 10 min. Ten mL of a CHCL3:MeOH (3:1 ratio) mixture was added and incubated in a 55 °C water bath for 2 h. After that, ten mL of distilled water was added into the reaction tube and mixed. The sample mixtures were further incubated at room temperature for 10 min and separated by centrifugation at 6000 rpm (4025×g), room temperature for 10 min. The aqueous phase was discarded whereas the chloroform phase was collected for lipid determination. All lipids dissolved in chloroform were determined by acid–dichromate oxidation method [23]. One mL of dissolved lipid sample was added into 2 mL of concentrated sulfuric acid (H2SO4, 98%) and mixed vigorously using vortex. After that, 2 mL of 0.167 M potassium dichromate (K2CrO7) solution was added before boiling the mixture for 30 min. After the mixture was cooled down to room temperature, 2 mL of distilled water was added. The total lipid content was determined spectrophotometrically by measuring its absorbance at 600 nm. A commercial standard canola oil was prepared as control. The calculated content of total lipid was represented as % w/DCW.
The unsaturated lipid content was determined by a colorimetric sulfo-phosphovanillin (SPV) reaction method [24]. One mL of dissolved lipid was added into 2 mL of concentrated H2SO4 (98%), mixed and vigorously vortexed. Then, the mixture was boiled for 30 min and cooled down to room temperature. The 2 mL mixture of 17% H3PO4 and 0.2 mg/mL vanillin (1:1) was added into the solution and mixed. The total unsaturated lipid content was then determined by measuring absorbance of the reaction mixture at 540 nm using spectrophotometer. The commercial standard γ-linoleic acid (C18:3) was prepared in the same way as sample. The calculated content of total unsaturated lipid was represented as % w/DCW.
Reverse transcription PCR
Total RNA was extracted from cells using TRIzol® Reagent (Invitrogen) and treated with RNase-free DNaseI (Fermentas) to remove the genomic DNA contamination before converting to cDNA using SuperScript™ III First-Strand Synthesis Kit (Invitrogen). The obtained cDNA was used as a template in PCR of genes involved in lipid biosynthesis including accA, aas, plsX, lipA and aar using corresponding RT-PCR primers listed in Table 3. The PCR products were checked by 1% (w/v) agarose gel electrophoresis. Band intensity quantification was also performed using Syngene® Gel Documentation (Syngene, Frederick, MD).
Nile red staining
To investigate the presence of neutral lipids, the Nile red method [25] was used. One hundred µL of cell culture was stained with 30 µg/mL of Nile red solution containing 0.9% (w/v) NaCl and further incubated in the dark overnight. After that, the stained cells were smeared on the glass slide and visualized under the fluorescent microscope (Olympus DP72, USA).
Analysis of fatty acid composition
For analysis of intracellular fatty acid composition, total lipids were extracted from 500 mL of cell culture with OD730 of about 0.5. The method was modified according to O’Fallon et al. [26] in order to generate fatty acid methyl esters (FAMEs). Mixture of methanol and 1 N KOH (1:3 ratio) was added to cell pellet and incubated in a 55 °C water bath for 1.5 h. Then, concentrated sulfuric acid (98%) was added and immediately mixed by inverting the tube. Equal volume of hexane was then added to the reaction tube and mixed with vortex. The hexane fraction was transferred to gas vials for GC–MS/MS detection. The data are shown as the percentage of fatty acid composition in Synechocystis cells.
Results
After the recombinant plasmids pEERM_aas, pEERM_Aar/Aas and pEERM_LipA/AccDACB (Table 1) were successfully constructed, they were separately transformed into Synechocystis WT cells generating the strains OXAas, OXAas/KOAar and OXAccDACB/KOLipA, respectively. The obtained transformants grown on BG11 agar plate containing 35 µg/mL chloramphenicol were randomly selected and examined for their respective gene locations by PCR using various specific pairs of primers (Table 2). Obtained PCR products when using selected primers of each strain are shown in Fig. 3. The data revealed that the engineered strains OXAas, OXAccDACB/KOLipA and OXAas/KOAar were successfully obtained. In Fig. 3A.a, the pEERM core structure was examined using primers pE_SF and pE_SR generating a DNA fragment of 350 bp (Table 2 and Fig. 2). WT (lane 1) contained no pEERM vector whereas the vector was observed in transformants or OXAas strain (lanes 6, 8, 9 and 10). Interestingly, OXAas possessed a single homologous recombination since a size of 2.5 kb between cmr and aas locus was observed in OXAas (lanes 2–6) except in WT (lane 1) (Fig. 3A.b). Additionally, 1.4 kb and 2.5 kb fragments were observed in OXAas except in WT (lane 1) by PCR using the two pairs of Aas_F6 and Cm_SR primers (Fig. 3A.c), and UUPSF_Aas and Cm_SR (Fig. 3A.d) primers, respectively (Table 2 and Fig. 2). An interruption of the aar gene by inserting aas gene fragment generated the OXAas/KOAar strain (Fig. 2). By PCR amplification using a pair of CAar_F and Aas_SR primers and another pair of UUPSF_Aar and Aas_SR primers (Table 2 and Fig. 2), 600 bp and 1.4 kb fragments were observed in strain OXAas/KOAar (Fig. 3B.a, b). For strain OXAccDACB/KOLipA, the native lipA gene was disrupted by a cassette fragment of accD, accA, accC and accB with homologous recombination using the flanking region of lipA gene (Fig. 2). A correct gene location was demonstrated for strain OXAccDACB/KOLipA (lanes 1–5) after being examined by PCR using a pair of UUPSF_lipA and AccD_SR primers (Fig. 3C).

Outline maps representing gene locations in all Synechocystis engineered strains. OXAas strain (upper) was singly recombined with Aas gene locus whereas OXAas/KOAar strain (middle) was generated by interrupting aar gene with aas gene fragment insertion. Finally, OXAccDACB/KOLipA strain (bottom) was constructed by inserting a cassette fragment of accD, accA, accC and accB to disrupt lipA gene

Confirmation of each gene location by PCR analysis using specific pairs of primers (Table 2) in each engineered strain including OXAas (A), OXAas/KOAar (B) and OXAccDACB/KOLipA (C) strains in this study. The location of aas gene fragment in OXAas was checked using a pair of pE_SF and pE_SR primers for pEERM core structure (a). Lane M: GeneRuler™ DNA ladder (Fermentas), lane 1: negative control using WT as template and lanes 2–10: clone numbers 1 to 9, For Cm_SF and Aas_SR (b) primer, lane M: GeneRuler™ DNA ladder (Fermentas), lane 1: negative control using WT as template and lanes 2–6: clone numbers 1 to 5. In (c), the pair of Aas_F6 and Cm_SR (c) primers was used, lane M: GeneRuler™ DNA ladder (Fermentas), lane 1: negative control using WT as template and lanes 2–6: clone numbers 1 to 5. The UUPSF_Aas and Cm_SR (d) primer, lane M: GeneRuler™ DNA ladder (Fermentas), lane 1: negative control using WT as template and lanes 2–6: clone numbers 1 to 5. Confirmation of gene location in OXAas/KOAar (B) using a pair of CAar_F and Aas_SR (a) primer, Lane M: GeneRuler™ DNA ladder (Fermentas), lane 1: negative control using WT as template and lanes 2–9; clone numbers 1 to 8 whereas UUPSF_Aar and Aas_SR (b) primers was used. Lane M: GeneRuler™ DNA ladder (Fermentas), lane 1: negative control using WT as template and lanes 2–11: clone numbers 1 to 10. The gene location in OXAccDACB/KOLipA (C) using pair of UUPSF_lipA and AccD_SR primer, Lane M: GeneRuler™ DNA ladder (Fermentas) and lanes 1–5; clone numbers 1 to 5
Cell growth of WT and all engineered strains is shown in Fig. 4a. All engineered strains grew slightly slower than WT, in particular OXAccDACB/KOLipA. Oxygen evolution rates, representing the photosynthetic efficiency of the cells, were monitored in three growth stages including start, day 4 and day 8 of cultivation (Fig. 4b). WT cells gave a slight decrease of oxygen evolution rate at day 8 of growth whereas the oxygen evolution rates of all engineered strains showed no changes at both day 4 and day 8. The intracellular pigments including chlorophyll a and carotenoid contents during cultivation (Fig. 4c, d respectively) depicted the significant differences of WT and engineered strains, which were apparent during 8–16 days cultivation. Chlorophyll a and carotenoid contents of the engineered strains were significantly lower when compared to WT. Interestingly, the OXAas/KOAar strain showed a constant level of carotenoids throughout the cultivation period.

Growth curve (a), oxygen evolution rate (b), chlorophyll a content (c) and carotenoid content (d) of wild type, OXAas, OXAccDACB/KOLipA and OXAas/KOAar Synechocystis strains grown in BG11 medium. The error bars represent standard deviations of means (mean ± SD, n = 3). Means with the same letter are not significantly different (in b) with the significance level at P < 0.05
Total lipid contents in all strains are shown in Fig. 5a. At the start of cultivation, WT cells accumulated total lipids about 16.8% w/DCW and showed a slight increase at day 8 of cell growth. We noticed that at the start of cultivation the OXAas produced the highest level of total lipids among all strains examined with about 23.5% w/DCW. Cells at day 4 increased the accumulation of total lipids in all engineered strains, especially OXAas and OXAccDACB/KOLipA showing 34.5 and 32.5%w lipids/DCW, respectively. At day 8 of cultivation, the total lipid contents of both OXAas and OXAccDACB/KOLipA decreased to similar level as that of WT. Additionally, although OXAas/KOAar did not induce a sharp increase of total lipid content, an increase of total lipid level was observed along all growth phases when compared to WT. This was substantiated by the highest production rate of lipids observed in OXAas strain at day 4 of cultivation (Table 4). It should be noted that the lipid titer of OXAas was increased at a slower rate compared with the other two engineered strains after 4 days. Total unsaturated lipid contents produced by all strains are shown in Fig. 5b. In our observation, the intracellular amount of total unsaturated lipid in WT was 14-fold lower than total lipids. Results revealed that all engineered strains had significantly increased a growth-dependent unsaturated lipid production. When compared with that of WT, OXAccDACB/KOLipA showed a 2.3-fold higher unsaturated lipid content at day 4 of growth whereas OXAas/KOAar gave the highest level of about unsaturated lipid 5.4% w/DCW (Fig. 5b). Additionally, there was a notable increase of saturated palmitic acid (C16:0) in the engineered strains, especially in OXAas showing higher than 70% (Table 5) when compared to WT [27]. The unsaturated oleic acid (C18:1) was induced in OXAccDACB/KOLipA and OXAas/KOAar.

Total lipid content (a) and unsaturated lipid content (b) in wild type, OXAas, OXAccDACB/KOLipA and OXAas/KOAar Synechocystis strains grown in BG11 medium. The error bars represent standard deviations of means (mean ± SD, n = 3). Means with the same letter are not significantly different with the significance level at P < 0.05
Results of gene expressions related to fatty acid biosynthesis and neighboring pathways (Fig. 1) under log growth phase of all strains, including phaA, accA, aas, plsX, aar and lipA, are shown in Fig. 6. The aas gene overexpression was confirmed with about a fivefold increase in both OXAas and OXAas/KOAar compared to that in WT. In addition, a slight increase (about 1.2-fold) of accA transcript level was observed in OXAccDACB/KOLipA. Surprisingly, our results showed a distinct increase of pha gene expression, related to bioplastic PHB synthesis, in all engineered strains (Fig. 6). To check whether the engineered strains contained higher PHB than WT, we stained OXAas, which showed the highest phaA transcript level, with Nile red and clearly observed significantly more PHB granules compared to those in WT cells (Fig. 7). On the other hand, the aas overexpression induced the accA transcript level, related to a gene of the multi-subunit acetyl-CoA carboxylase, in OXAas and OXAas/KOAar (Fig. 6). For the plsX, related to phospholipid synthesis, the relative transcript levels increased in the engineered strains, especially in OXAccDACB/KOLipA and OXAas/KOAar. Moreover, both OXAas and OXAccDACB/KOLipA showed higher relative transcripts levels of aar, related to alkane synthesis. Finally, an increased transcript level of lipA encoding the phospholipid hydrolyzing lipase was found in the strains OXAas and OXAas/KOAar.

The transcript levels (a) of pha A, accA, aas, plsX, aar, lipA and 16S rRNA genes of WT, OXAas, OXAccDACB/KOLipA and OXAas/KOAar Synechocystis strains. The intensity ratios (b) of phaA/16S rRNA, accA/16S rRNA, aas/16S rRNA, plsX/16S rRNA, aar/16S rRNA and lipA/16S rRNA of all studied strains at log phase of cell growth analyzed by GelQuant.NET program

The Nile red staining of neutral lipids in Synechocystis sp. PCC 6803 wild type (a) and OXAas (b) strain cells in BG11 medium at day 4 of cultivation. The stained cells were visualized under light and fluorescent microscopes with a magnification of ×100. It is noted that the focus setting in panel B with Nile Red staining was directed to PHB granules whereas the focus setting in panel A with Nile Red staining was directed to the whole cells
Discussion
In this study, we constructed three engineered strains of the unicellular cyanobacterium Synechocystos PCC 6803: OXAas/KOAar and OXAccDACB/KOLipA segregated by double homologous recombination whereas the OXAas was generated via single recombination (Figs. 2 and 3). The single integrative crossover or single recombination rarely occurs in Synechocystis PCC 6803 but may be more stable than a double recombination [28]. The genetic stability of the three engineered strains was likely to occur since the analysis of transcript levels in these strains was relatively unchanged during a period of over one year. We demonstrated that the metabolic engineering of all modified strains did not severely affect the cell growth except the intracellular pigment contents (Chl a and Car), in particular in strain OXAas/KOAar after 8–10 days of growth (Fig. 4c, d). However, the oxygen evolution rate, partly representing photosynthetic capacity and efficiency, of all strains studied was not significantly disturbed. On the basis of our empirical experiment and other reports, the normal range of oxygen evolution rate of Synechocystis PCC 6803 photoautotrophically cultivated was about 60–200 µmol/mg Chl a/h depending on strain and light intensity during cultivation [29], as well as if any stressful condition was applied [30]. In our study, the overexpression of aas with a simultaneous aar knockout showed the most significant reduction in OD730 as well as in intracellular pigments content. This reduction may partially correlate with the expression vector chosen and gene impact on cell metabolism. Coincidently, a previous study reported that a knockout of aar in Synechocystis caused not only a fourfold decline in growth when compared to Synechocystis WT cells but also a decreased oxygen evolution rate [31]. Due to the fact that the formation of alkane may partly modulate photosynthetic cyclic electron flow in cyanobacterial membranes, the disrupted aar gene may then cause a lowered growth and photosynthetic efficiency [32].
We also demonstrated that the day 4-growth phase of all strains was suitable for highest production of lipid metabolites. Our results indicate that the highest levels of lipids were observed in engineered strain OXAas with about 34.5% w/DCW which is twofold higher than WT cells during log growth phase (Fig. 5). Sheng and co-workers previously reported that the intracellular lipid contents in cyanobacterium Synechocystis PCC 6803 were limited to a range between 10 and 15% w/DCW, significantly lower than that observed in the present study, with the majority being diacylglycerol components [33]. We observed an enhanced FFAs incorporation into fatty acyl-ACP, the initial precursor for lipid synthesis, resulting in significantly higher lipid level than WT (Fig. 1). In addition, an overexpression of the multi-subunit acetyl Co-A carboxylase gene (accDACB) in combination with a lipA knockout (strain OXAccDACB/KOLipA) resulted in a lipid content of about 32.5% w/DCW. The ACC encoded by a multi-subunit of accA, accB, accC and accD played a role as the rate-limiting step for the fatty acid biosynthesis [34, 35]. Coincidently, accABCD overexpressing Escherichia coli showed a sixfold increase of the fatty acid biosynthesis rate [8]. In our study, we designed not only an accDACB overexpression strain but also a strain with accDACB overexpression in combination with a lipA knockout (Fig. 1). This was done in order to prevent membrane lipids degradation to FFAs and potentially gain more lipids, as it has been shown that deleting sll1969 (or lipA) encoding a putative lipolytic enzyme significantly decrease membrane lipid degradations [36]. On the other hand, the OXAas/KOAar strain with disrupted alkane production showed no increase of lipid production. Our results suggest that the lipid production in our engineered strains is partially associated with cell growth, in particular at day 4. Among engineered strains OXAas/KOAar showing a slightly lowered growth, a significant reduction of pigment contents and O2 evolution rate, a lower total lipid, had the highest total unsaturated lipids (Fig. 5b). In addition, the homeostasis of lipid balance might adjust the excess synthesized lipid down to normal level either via feedback inhibition of acetyl Co-A carboxylase by the fatty acyl-ACP or via lipid degradation. Additionally, the desaturation activity of the membrane lipids in Synechocystis has been located to the cytoplasmic and thylakoid membranes [37]. The increase of unsaturated lipid levels in OXAccDACB/KOLipA and OXAas/KOAar was noted which may be ascribed to the FA desaturation activity as supported by the decrease of palmitic acid (C16:0) as well as the increase of oleic acid (C18:1) composition when compared to that of OXAas (Fig. 5b and Table 5). On the other hand, due to low molar C/N ratio of about 1/47 in BG11 medium, additional C in the form of acetate was shown to stimulate lipid production [27]. In this regard, the improvement of lipid synthesis in cyanobacteria is very challenging due to the small pool size of acetyl-CoA and the TCA fluxes [38]. Further improvements may redirect the upstream flux towards acetyl-CoA [39] or engineer the CO2-fixing machinery [40, 41].
We also examined relative gene expression detected by RT-PCR of genes related to the fatty acid biosynthesis and neighboring pathways (Figs. 1 and 6). One of the metabolic balance responses for lipid synthesis depends on feedback inhibition, herein fatty acyl-ACP which thereby inhibited back to ACC enzyme [11, 12]. Our results indicate that the aas-overexpressing strains (OXAas and OXAas/KOAar) showed a significantly induced accA transcript level when compared to WT. In addition, the OXAas strain showed an up-regulation of the aar transcript levels compared to WT. Interestingly, all OX strains contained significantly increased levels of phaA transcript related to bioplastic PHB synthesis. Furthermore, Nile-Red staining of strain OXAas showed an increase in PHB granules compared to WT (Fig. 7). Thus, our observations may suggest that the overexpression of acc and aas influenced the acetyl Co-A synthesis enhancing both fatty acid synthesis and PHB production. Interestingly, in Ralstonia eutropha H16, a re-consumption of fatty acids is stimulated through the beta-oxidation pathway which iteratively removes two carbons from both fatty acid to yield acetyl-CoA, and from 3-hydroxyl-acyl-CoA, an intermediate in beta-oxidation, which enters the PHB synthetic pathway [42]. We also noted that increased levels of lipA transcripts were observed in the two strains OXAas and OXAas/KOAar which needs more FFA substrate from phospholipid degradation. Our results are in agreement with a previous finding that lipA encoding lipase A catalyzes phospholipids hydrolysis [3] with a tight correlation with AAS which recycles the free fatty acids into fatty acyl-ACP. On the other hand, we propose that the increased levels of plsX transcript observed in strains OXAccDACB/KOLipA and KOAas/KOAar, compared to WT, and strain OXAas are due to an influence of the lipA and aar knockouts, respectively. In Streptococcus mutans, the deletion of PlsX gene encoding an acyl-ACP:phosphate transcylase, evidently lost the central function of unsaturated fatty acid movement into membrane and the acid-adaptive response [43]. As expected, the transcript levels of aar in strains OXAas and OXAccDACB/KOLipA were induced when compared to WT possibly caused by the aas overexpression resulting in an enhanced flux ability of the substrate fatty acyl-ACP.
Conclusions
Our results of metabolic engineering of various genes involved in the fatty acid synthesis, phospholipid hydrolysis, alkane synthesis, and recycling of free fatty acid (FFA) in cyanobacterium Synechocystis sp. PCC 6803 indicated an increase in acetyl Co-A flux towards both routes of lipid and PHB syntheses as evident by their increased contents. Among the three engineered strains, OXAas with enhanced recycling of FFA had the highest lipid content and lipid production rate after 4 days cultivation.
Abbreviations
- AAR:
-
acyl-ACP reductase
- AAS:
-
acyl–acyl carrier protein synthetase
- ACC:
-
acetyl-CoA carboxylase
- ACP:
-
acyl carrier protein
- Car:
-
carotenoids
- Chl a :
-
chlorophyll a
- CO2 :
-
carbon dioxide
- DCW:
-
dry cell weight
- DMF:
-
N,N-dimethylformamide
- FAS:
-
fatty acid synthase
- FFA:
-
free fatty acid
- h:
-
hour
- m:
-
meter
- μg:
-
microgram
- mL:
-
milliliter
- min:
-
minute
- nm:
-
nanometer
- OD:
-
optical density
- PCR:
-
polymerase chain reaction
- PHB:
-
polyhydroxybutyrate
- rpm:
-
revolutions per minute
- s:
-
seconds
- SPV:
-
sulfo-phosphovanillin
- WT:
-
wild type
References
Quintana N, Van Der Kooy F, Van De Rhee MD, Voshol GP, Verpoorte R. Renewable energy from cyanobacteria: energy production optimization by metabolic pathway engineering. Appl Microbiol Biotechnol. 2011;91:471–90.
Gao Q, Wang W, Zhao H, Lu X. Effects of fatty acid activation on photosynthetic production of fatty acid-based biofuels in Synechocystis sp. PCC 6803. Biotechnol Biofuels. 2012;5:17.
Kaczmarzyk D, Fulda M. Fatty acid activation in cyanobacteria mediated by acyl–acyl carrier protein synthetase enables fatty acid recycling. Plant Physiol. 2010;152:1598–610.
Wang W, Liu X, Lu X. Engineering cyanobacteria to improve photosynthetic production of alka(e)nes. Biotechnol Biofuels. 2013;6:69.
Chisti Y. Biodiesel from microalgae. Biotechnol Adv. 2007;25:294–306.
Post-Beittenmiller D, Roughan G, Ohlrogge JB. Regulation of plant fatty acid biosynthesis analysis of acyl-coenzyme A and acyl–acyl carrier protein substrate pools in spinach and pea chloroplasts. Plant Physiol. 1992;100:923–30.
Kobayashi MA, Watada H, Kawamori R, Maeda S. Overexpression of acetyl-coenzyme A carboxylase beta increases proinflammatory cytokines in cultured human renal proximal tubular epithelial cells. Clin Exp Nephrol. 2010;14:315–24.
Davis MS, Solbiati J, Cronan JE Jr. Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis in Escherichia coli. J Biol Chem. 2000;275:28593–8.
Heath RJ, Jackowski S, Rock CO. Fatty acid and phospholipid metabolism in prokaryotes. New Compr Biochem. 2002;36:55–92.
Ray TK, Cronan JE Jr. Activation of long chain fatty acids with acyl carrier protein: demonstration of a new enzyme, acyl–acyl carrier protein synthetase, in Escherichia coli. Proc Natl Acad Sci USA. 1976;73:4374–8.
Andre C, Haslam RP, Shanklin J. Feedback regulation of plastidic acetyl-CoA carboxylase by 18:1-acyl carrier protein in Brassica napus. Proc Natl Acad Sci USA. 2012;109:10107–12.
Davis MS, Cronan JE Jr. Inhibition of Escherichia coli acetyl coenzyme A carboxylase by acyl–acyl carrier protein. J Bacteriol. 2001;183:1499–503.
Yoshino T, Liang Y, Arai D, Maeda Y, Honda T, Muto M, et al. Alkane production by the marine cyanobacterium Synechococcus sp. NKBG15041c possessing the α-olefin biosynthesis pathway. Appl Microbiol Biotechnol. 2015;99:1521–9.
Zhang YM, Rock CO. Thematic review series: glycerolipids. Acyltransferases in bacterial glycerophospholipid synthesis. J Lipid Res. 2008;49:1867–74.
Gao Q, Tan X, Lu X. Characterization of a key gene in membrane lipid cycle in Synechocystis sp. PCC 6803. Sheng Wu Gong Cheng Xue Bao. 2012;28:1473–81.
Liu X, Curtiss R III. Nickel-inducible lysis system in Synechocystis sp. PCC6803. Proc Natl Acad Sci USA. 2009;106:21550–4.
Liu X, Sheng J, Curtiss R III. Fatty acid production in genetically modified cyanobacteria. Proc Natl Acad Sci USA. 2011;108:6899–904.
Englund E, Andersen-Ranberg J, Miao R, Hamberger B, Lindberg P. Metabolic engineering of Synechocystis sp. PCC 6803 for production of the plant diterpenoid manoyl oxide. ACS Synth Biol. 2015;4:1270–8.
Chamovitz D, Sandmann G, Hirschberg J. Molecular and biochemical characterization of herbicide-resistant mutants of cyanobacteria reveals that phytoene desaturation is a rate-limiting step in carotenoid biosynthesis. J Biol Chem. 1993;268:17348–53.
Moran R. Formulae for determination of chlorophyllous pigments extracted with N,N-dimethylformamide. Plant Physiol. 1982;69:1376–81.
Jantaro S, Mäenpää P, Mulo P, Incharoensakdi A. Content and biosynthesis of polyamines in salt and osmotically stressed cells of Synechocystis sp. PCC 6803. FEMS Microbiol Lett. 2003;228:129–35.
Baebprasert W, Jantaro S, Khetkorn W, Lindblad P, Incharoensakdi A. Increased H2 production in the cyanobacterium Synechocystis sp. strain PCC 6803 by redirecting the electron supply via genetic engineering of the nitrate assimilation pathway. Metab Eng. 2011;13:610–6.
Fales MF. Evaluation of a spectrophotometric method for determination of total fecal lipid. Clin Chem. 1971;17:1103–8.
Cheng YS, Zheng Y, Vandergheynst JS. Rapid quantitative analysis of lipids using a colorimetric method in a microplate format. Lipids. 2011;46:95–103.
Chen W, Zhang C, Song L, Sommerfeld M, Hu Q. A high throughput Nile red method for quantitative measurement of neutral lipids in microalgae. J Microbiol Methods. 2009;77:41–7.
O’Fallon JV, Busboom JR, Nelson ML, Gaskins CT. A direct method for fatty acid methyl ester synthesis: application to wet meat tissues, oils, and feedstuffs. J Anim Sci. 2007;85:1511–21.
Towijit U, Songruk N, Lindblad P, Incharoensakdi A, Jantaro S. Co-overexpression of native phospholipid-biosynthetic genes plsX and plsC enhances lipid production in Synechocystis sp. PCC 6803. Sci Rep. 2018;8:13510.
Labarre J, Chauvat F, Thuriaux P. Insertional mutagenesis by random cloning of antibiotic resistance genes into the genome of the cyanobacterium Synechocystis strain PCC 6803. J Bacteriol. 1989;171:3449–57.
Page LE, Liberton M, Pakrasi HB. Reduction of photoautotrophic productivity in the cyanobacterium Synechocystis sp. strain PCC 6803 by phycobilisome antenna truncation. Appl Environ Microbiol. 2012;78:6349–51.
Jantaro S, Incharoensakdi A, Jansén T, Mulo P, Mäenpää P. Effects of long-term ionic and osmotic stress conditions on photosynthesis in the cyanobacterium Synechocystis sp. PCC 6803. Funct Plant Biol. 2005;32:807–15.
Lea-Smith DJ, Ortiz-Suarez ML, Lenn T, Nurnberg DJ, Baers LL, Davey MP, et al. Hydrocarbons are essential for optimal cell size, division, and growth of cyanobacteria. Plant Physiol. 2016;172:1928–40.
Berla BM, Saha R, Maranas CD, Pakrasi HB. Cyanobacterial alkanes modulate photosynthetic cyclic electron flow to assist growth under cold stress. Sci Rep. 2015;5:14894.
Sheng J, Vannela R, Rittmann BE. Evaluation of methods to extract and quantify lipids from Synechocystis PCC 6803. Bioresour Technol. 2011;102:1697–703.
Kim KH. Regulation of mammalian acetyl-coenzyme A carboxylase. Annu Rev Nutr. 1997;17:77–99.
Munday MR, Hemingway CJ. The regulation of acetyl-CoA carboxylase—a potential target for the action of hypolipidemic agents. Adv Enzyme Regul. 1999;39:205–34.
Liu X, Curtiss R 3rd. Thermorecovery of cyanobacterial fatty acids at elevated temperatures. J Biotechnol. 2012;161:445–9.
Mustardy L, Los DA, Gombos Z, Murata N. Immunocytochemical localization of acyl-lipid desaturases in cyanobacterial cells: evidence that both thylakoid membranes and cytoplasmic membranes are sites of lipid desaturation. Proc Natl Acad Sci USA. 1996;93:10524–7.
Wan N, DeLorenzo DM, He L, You L, Immethun CM, Wang G, Baidoo EEK, Hollinshead W, Keasling JD, Moon TS, Tang YJ. Cyanobacterial carbon metabolism: fluxome plasticity and oxygen dependence. Biotechnol Bioeng. 2017;114:1593–602.
Anfelt J, Kaczmarzyk D, Shabestary K, Renberg B, Rockberg J, Nielsen J, Uhlén M, Hudson EP. Genetic and nutrient modulation of acetyl-CoA levels in Synechocystis for n-butanol production. Microb Cell Fact. 2015;14:167.
Liang F, Lindblad P. Effects of overexpressing photosynthetic carbon flux control enzymes in the cyanobacterium Synechocystis PCC 6803. Metab Eng. 2016;38:56–64.
Liang F, Lindberg P, Lindblad P. Engineering photoautotrophic carbon fixation for enhanced growth and productivity. Sustain Energy Fuels. 2018. https://doi.org/10.1039/c8se00281a.
Chen JS, Colón B, Duse B, Ziesack M, Way JC, Torella JP. Production of fatty acids in Ralstonia eutropha H16 by engineering ß-oxidation and carbon storage. Peer J. 2015;3:e1468.
Cross B, Garcia A, Faustoferri R, Quivey RG Jr. PlsX deletion impacts fatty acid synthesis and acid adaptation in Streptococcus mutans. Microbiology. 2016;162:662–71.
Authors’ contributions
KE was responsible for study conception, main experimenter, data collection, analysis and draft manuscript writing; RM was responsible for study conception and methodological experiment teaching; PL for study conception, strategic pathway design and manuscript revision; AI for study conception and design and manuscript revision; SJ for study conception, critical revision and manuscript writing, and final approval of the manuscript. All authors read and approved the final manuscript.
Acknowledgements
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Funding
This research was funded by the Ratchadapisek Sompoch Endowment Fund (2016), Chulalongkorn University (CU-59-018-FW) to S.J. Also, the Development and Promotion of Science and Technology Talents Project (DPST)’s scholarship for postgraduate tuition and expenses to K.E.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Eungrasamee, K., Miao, R., Incharoensakdi, A. et al. Improved lipid production via fatty acid biosynthesis and free fatty acid recycling in engineered Synechocystis sp. PCC 6803. Biotechnol Biofuels 12, 8 (2019). https://doi.org/10.1186/s13068-018-1349-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-018-1349-8