
Abstract
Background
Cyanobacteria can utilize solar energy and convert carbon dioxide into biofuel molecules in one single biological system. Synechocystis sp. PCC 6803 is a model cyanobacterium for basic and applied research. Alkanes are the major constituents of gasoline, diesel and jet fuels. A two-step alkane biosynthetic pathway was identified in cyanobacteria recently. It opens a door to achieve photosynthetic production of alka(e)nes with high efficiency by genetically engineering cyanobacteria.
Results
A series of Synechocystis sp. PCC6803 mutant strains have been constructed and confirmed. Overexpression of both acyl-acyl carrier protein reductase and aldehyde-deformylating oxygenase from several cyanobacteria strains led to a doubled alka(e)ne production. Redirecting the carbon flux to acyl- ACP can provide larger precursor pool for further conversion to alka(e)nes. In combination with the overexpression of alkane biosynthetic genes, alka(e)ne production was significantly improved in these engineered strains. Alka(e)ne content in a Synechocystis mutant harboring alkane biosynthetic genes over-expressed in both slr0168 and slr1556 gene loci (LX56) was 1.3% of cell dry weight, which was enhanced by 8.3 times compared with wildtype strain (0.14% of cell dry weight) cultivated in shake flasks. Both LX56 mutant and the wildtype strain were cultivated in column photo-bioreactors, and the alka(e)ne production in LX56 mutant was 26 mg/L (1.1% of cell dry weight), which was enhanced by 8 times compared with wildtype strain (0.13% of cell dry weight).
Conclusions
The extent of alka(e)ne production could correlate positively with the expression level of alkane biosynthetic genes. Redirecting the carbon flux to acyl-ACP and overexpressing alkane biosynthetic genes simultaneously can enhance alka(e)ne production in cyanobacteria effectively.
Similar content being viewed by others

Background
Interest in engineering cyanobacteria for biofuel production has increased recently driven by using photosynthesis to directly convert carbon dioxide into a desirable fuel [1–6]. Additionally, cyanobacteria exhibit higher solar conversion efficiency and growth rate compared to plants and eukaryotic microalgae [7, 8]. Synechocystis sp. PCC6803 was the first cyanobacterium for which the complete genome was sequenced in 1996 [9]. So far 126 genomic sequences of cyanobacteria strains are available [10]. Well established genetic manipulation techniques have been applied for cyanobacteria. The techniques make cyanobacteria highly tractable platforms to build efficient biosynthetic pathways for biofuel production by genetic engineering [11].
Alkanes with C4-C23 carbon chain length possess higher energy density, hydrophobic property and compatibility with existing liquid fuel infrastructure, which are the predominant constituents of gasoline, diesel, and jet fuels [12]. They can be produced by various organisms such as bacteria, yeasts, plants and insects [13]. In the late 1960s, production of alka(e)nes was reported in a diversity of cyanobacteria [14]. In 2010, a two-step alkane biosynthetic pathway in cyanobacteria was identified. Acyl-acyl carrier protein (ACP) can be reduced to aldehyde by an acyl-ACP reductase (AAR, EC 1.2.1.50), and then aldehyde can be oxidized to alkane or alkene by an aldehyde-deformylating oxygenase (ADO) [15].
Fatty acid substrates as acyl chains of membrane lipids are biosynthesized by fatty acid synthase (FAS). Acetyl-CoA is converted to malonyl-CoA by a multi-subunit acetyl-CoA carboxylase consisting of AccA, AccB, AccC and AccD, which is the rate-limiting step of fatty acid biosynthesis [16]. Acyl-ACPs synthesized by FAS can be incorporated to membrane lipids. Free fatty acids (FFAs) generated by lipolytic enzymes during degradation of membrane lipids can also be activated to acyl-ACPs by an acyl-ACP synthetase (AAS, EC 6.2.1.20) [17].
Kaczmarzyk and Fulda (2010) established that the only AAS gene in Synechocystis sp. PCC 6803 is slr1609[17]. The slr1609-knockout mutant was incapable of importing exogenous fatty acids and secreted fatty acids released from membrane lipids into the medium. This suggests a remarkable role for this cyanobacterial AAS in recycling released fatty acids [17]. Our previous study showed that the alka(e)ne production was significantly reduced in slr1609 deletion mutant of Synechocystis sp. PCC6803, which indicates AAS plays an essential role in alka(e)ne production [18].
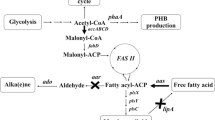
Alka(e)ne biosynthesis was reported in a diversity of cyanobacteria [19]. Heptadecane and heptadecene are the major constituents of alka(e)nes in Synechocystis sp. PCC6803, and the total alka(e)ne content was about 0.1% of the cell dry weight (DW) [5, 20]. Alkane biosynthetic genes from cyanobacteria were heterologously expressed in Escherichia coli and Synechococcus sp. PCC 7002, and alka(e)ne production ranged from 5–40 mg/L in E. coli and reached 5% of DW in Synechococcus sp. PCC 7002 [13, 21]. Redirecting the glyceraldehyde 3-phosphate (3-PGA) originated from Calvin-Benson-Bassham cycle to acyl-ACP and enhancing the expression of alkane biosynthetic genes should improve efficiency and yield of alka(e)ne production in Synechocystis sp. PCC6803 (Figure 1).

Schematic overview of fatty acid, alkane (alkene) and main competing metabolic pathways in Synechocystis sp. PCC6803. Key enzyme genes in those pathways are indicated. 3-PGA, glyceraldehyde 3-phosphate; PYR, pyruvate; PHB, poly-β-hydroxybutyrate; acyl-ACP, acyl- acyl carrier protein; ddh, 2-hydroxyacid dehydrogenase gene; phaA, polyhydroxyalkanoates-specific beta-ketothiolase gene; accBCDA, multi-subunit acetyl-CoA carboxylase gene;lipA, lipolytic enzyme gene; aas, acyl-ACP synthetase gene; aar, acyl-ACP reductase gene; ado, aldehyde-deformylating oxygenase.
In this study, metabolic engineering approaches were employed to construct a series of Synechocystis sp. PCC6803 mutant strains. Alka(e)ne production was enhanced by 8.3 times in one of these modified strains by overexpressing alkane biosynthetic genes in two different loci of the genome.
Results and discussion
Alka(e)ne production can be doubled in Synechocystismutants overexpressing cyanobacteria alkane biosynthetic genes
Synechocystis mutants overexpressing either or both native alkane biosynthetic genes (sll0208 and sll0209) were constructed. The production of alka(e)ne can be doubled in the mutant overexpressing both sll0208 and sll0209 (LX32, about 700 μg/L/OD) compared with parent strain (6803yu, about 300 μg/L/OD), and no significant changes was detected for the mutant strain only expressing either sll0208 (LX31) or sll0209 (LX33) as shown in Figure 2A.

Alka(e)ne production in Synechocystis mutants overexpressing cyanobacteria alkane biosynthetic genes. (A) Alka(e)ne production in Synechocystis mutants overexpressing sll0208 (LX31), sll0209 (LX33) and both genes (LX32) compared with Synechocystis sp. PCC6803 (6803yu). Error bars represent the standard deviation of three replicates. (B) Alka(e)ne production in Synechocystis mutants overexpressing orf1594 and Npun_R1711 (LX39), alkane biosynthetic genes from Synechococcus elongatus PCC7942 (LX34) and Nostoc punctiforme PCC73102 (LX35) compared with wildtype strain (6803yu) and LX32 mutant. Error bars represent the standard deviation of three replicates.
Alkane biosynthetic genes from Synechococcus elongatus PCC7942 (orf1593 and orf1594) and Nostoc punctiforme PCC73102 (npun1710 and npun 1711) were also overexpressed in Synechocystis sp. PCC6803. Alka(e)ne production can also be doubled in these two mutants (LX34 and LX35, Figure 2B). These results indicate that enhanced activity of AAR and ADO resulting from overexpression can convert more acy-ACP available to alka(e)ne.
Pentadecane can be produced in Synechococcus elongatus PCC7942, while no pentadecane was detected in the Synechocystis mutant overexpessing orf1593 and orf1594 (LX34). The in vitro enzyme activity assays performed by Eser et al. (2011) suggested that the Nostoc punctiforme PCC73102 ADO may possess higher activity than the Synechocystis sp. PCC6803 ADO [22]. The highest reported titers of alka(e)nes from this pathway involved E. coli overexpressing orf1594 (aar) and Npun_R1711 (ado) among 16 different combinations of the two enzymes from a variety of cyanobacteria [13]. In the Synechocystis mutant overexpressing orf1594 and Npun_R1711 (LX39), alka(e)ne production was not improved significantly compared with other three mutants (LX32, LX34 and LX35), while about 60 μg/L/OD730 pentadecane can be produced in this mutant (Figure 2B).
Redirecting the carbon flux to acyl-ACP can enhance alka(e)ne production in cyanobacteria effectively
Since acyl-ACP is the immediate substrate for alka(e)ne biosynthesis, redirecting the carbon flux to acyl-ACP may enhance downstream alka(e)ne production in cyanobacteria [20]. AAS plays an essential role in recycling the released fatty acids to acyl-ACP [17]. Our previous work showed that native alka(e)ne production was not enhanced by overexpressing slr1609 alone. Maybe activities of AAR and ADO are too low to convert acyl-ACP to alka(e)ne efficiently [18]. A Synechocystis mutant overexpressing alkane biosynthetic genes and slr1609 (LX38) showed enhanced productivity of alka(e)ne by 130% and 60% when compared to Synechocystis sp. PCC6803 and LX32 mutant, respectively (Figure 3A). Overexpressing AAS, AAR and ADO gene simultaneously may improve acyl-ACP pool and downstream alka(e)ne production. Considering our previous observation of a 90% reduction of alka(e)ne content in slr1609 deletion mutant [18], acyl-ACPs from FFA activation by AAS may be predominant source of alka(e)ne in Synechocystis strains.

Alka(e)ne production can be enhanced effectively by redirecting the carbon flux to acyl-ACP. (A) Alka(e)ne production in Synechocystis mutants overexpressing sll0208 and sll0209 in slr1609 over-producing strain (LX38) and phaA gene deletion mutant (LX40) compared with wildtype strain (6803yu) and LX32 mutant. Error bars represent the standard deviation of three replicates. (B) Alka(e)ne production in Synechocystis mutants overexpressing sll0208 and sll0209 in acetyl-CoA carboxylase genes (LX57) and lipolytic enzyme gene (LX58) over-producing strain compared with wildtype strain (6803yu) and LX32 mutant. Error bars represent the standard deviation of three replicates.
Synechocystis sp. PCC6803 can accumulate poly-β-hydroxybutyrate (PHB) as carbon and energy storage compound [23]. Acetyl-CoA and NADPH are required for PHB synthesis. The β-ketothiolase encoded by phaA (slr1993) condenses two molecules of acetyl-CoA to acetoacetyl-CoA, which is the first step of PHB biosynthesis. Alka(e)ne profiles of the Synechocystis mutant overexpressing sll0208 and sll0209 with slr1993 deletion (LX40) was analyzed, and the alka(e)ne productivity was enhanced by 150% and 70% compared with Synechocystis sp. PCC6803 and LX32 mutant, respectively (Figure 3A). Overexpression of AAR and ADO gene and deletion of PHB biosynthetic gene(s) simultaneously can divert acetyl-CoA and NADPH into production of fatty acid and enhance production of fatty acid-derived alka(e)nes.
Acyl-ACP pool may also be improved by increasing activity of acetyl-CoA carboxylase (ACC), which is the bottleneck of fatty acid biosynthesis. In a Synechocystis mutant overexpressing accBCDA genes from our previous work, a 56% increase of native alka(e)ne production was obtained [5]. With this mutant, the sll0208 and sll0209 were overexpressed to yield LX57 strain. The alka(e)ne production of LX57 mutant was enhanced by 3.6 times when compared to Synechocystis sp. PCC6803 (Figure 3B).
The lipolytic enzymes are capable of hydrolyzing acyl chains from membrane lipids. FFAs released from membrane lipids can be activated to acyl-ACPs by an AAS. Cyanobacteria have plenty of membrane lipids and a dynamic lipid metabolism. Based on sequence identity analysis, sll1969 was annotated as a putative lipolytic enzyme gene (lipA). Liu and Curtiss (2012) showed that less FFAs were released in Synechocystis sll1969-deletion mutant than the wildtype strain [24]. Alka(e)ne productivity of the Synechocystis mutant overexpressing sll0208, sll0209 and sll1969 (LX58) was enhanced by 3 times in comparison with wildtype strain (Figure 3B). Increasing activities of LipA can degrade more membrane lipids into FFAs. FFA can be activated to acyl-ACP by AAS. Since acyl-ACP is the immediate substrate for alka(e)ne biosynthesis, overexpressing sll1969 would lead to an increased alka(e)ne biosynthesis.
Alka(e)ne production can be improved significantly in Synechocystismutants overexpressing multiple copies of alkane biosynthetic genes
To investigate whether alka(e)ne productivity could be further enhanced by overexpressing multiple copies of alkane biosynthetic genes, Synechocystis mutants overexpressing two copies of sll0208 and sll0209 were constructed. Alka(e)ne productivity of a Synechocystis mutant overexpressing two copies of sll0208 and sll0209 in slr0168 site (LX70) was 1.2 mg/L/OD730 (Figure 4A). Alka(e)ne productivity of LX56 strain overexpressing sll0208 and sll0209 in both slr0168 and slr1556 (2-hydroxyacid dehydrogenase gene, ddh) loci was 2.3 mg/L/OD730 (Figure 4A). The final DW of wildtype and LX56 strain in shake flasks was 0.44 and 0.5 g/L, respectively. Alka(e)ne production of wildtype strain in shake flasks was 0.14% of DW (0.64 mg/L). Alka(e)ne production of LX56 strain was enhanced by 8.3 times, up to 1.3% of DW (6.5 mg/L).

Alka(e)ne production in Synechocystis mutants overexpressing two copies of alkane biosynthetic genes. (A) Alka(e)ne production in the Synechocystis mutant overexpressing sll0208 and sll0209 in both slr0168 and ddh gene sites (LX56) compared with wildtype strain (6803yu) and the Synechocystis mutant overexpressing two copies of sll0208 and sll0209 in slr0168 site (LX70). Error bars represent the standard deviation of three replicates. (B) Growth curves of wildtype strain and the LX56 strain in the bubble column photo-bioreactors. Error bars represent the standard deviation of three replicates. (C) Alka(e)ne production calculated as a percentage of DW of LX56 strain was enhanced by 8 times compared with wildtype strain when cultivated in the bubble column photo-bioreactors. Error bars represent the standard deviation of three replicates.
The transcriptional levels of sll0208 and sll0209 were steadily increased in wildtype, LX32, LX70 and LX56 mutant in semi-quantitative reverse transcription PCR analysis (Figure 5), which indicated alka(e)ne production could correlate positively with the expression of alkane biosynthetic genes to some extent. Transcription of two adjacent copies of sll0208 and sll0209 may interfere with each other, so transcription level of two copies of sll0208 and sll0209 in tandem in LX70 is lower than that of two copies of sll0208 and sll0209 in separate gene locus in LX56. Overexpressing alkane biosynthetic genes in multiple gene loci can significantly improve the efficiency of alka(e)ne production in cyanobacteria. Similar effects can also be found when multiple-site overexpression was applied to cyanobacteria ethanol or ethylene production [6, 25].

Semi-quantitative reverse transcription PCR analysis of the transcriptional levels of sll0208 and sll0209 in wildtype, LX32, LX70 and LX56 mutant. The rnpB gene was used as the external standards. Lane 1, LX56; Lane 2, LX70; Lane 3, LX32; Lane 4: wildtype.
Small-scale photo-bioreactors for cultivation of cyanobacteria are required for precise characterization of wildtype and engineered strains to optimize the culture conditions and alka(e)ne production. The LX56 mutant and wildtype strain exhibited similar growth characteristics when cultivated in the bubble column photo-bioreactors. Both of the cultures in photo-bioreactors reached a much higher density (OD730~13) than cultures in shake flasks (OD730 ~3 to 4) at stationary phase (Figure 4B), and the final DW of wildtype and LX56 strain in photo-bioreactors was 2.2 and 2.3 g/L, respectively. The alka(e)ne production of wildtype strain was 2.8 mg/L, which was 0.13% of DW. The alka(e)ne production of LX56 strain was enhanced by 8 times compared with the wildtype strain, up to 26 mg/L, which was 1.1% of DW (Figure 4C).
In the previous reports, alka(e)ne production ranged from 5 to 40 mg/L in E. coli[13] and reached 5% of DW in Synechococcus sp. PCC 7002 [21] by heterologously expressing cyanobacterial AAR and ADO genes. Integrating different strategies of genetic modifications used in these studies into a Synechocystis mutant strain will further enhance yield of alka(e)nes. Medium chain alkane is less toxic to cyanobacteria than alcohol, acid and other non-native products [26], which leaves huge room for engineering cyanobacteria to improve alka(e)ne production.
Conclusions
Overexpressing cyanobacteria alkane biosynthetic genes and redirecting the carbon flux to acyl-ACP can improve alka(e)ne production in cyanobacteria significantly. The results indicate that metabolic engineering strategies are powerful for engineering cyanobacteria to overproduce alka(e)nes. Low activities of AAR and ADO need to be enhanced by protein engineering for further improvement of alka(e)ne production. It is also important to understand the physiological roles and regulatory mechanism of native alka(e)nes in cyanobacterial cell.
Methods
Chemicals and reagents
Eicosane was purchased from Sigma-Aldrich (USA). Other chemicals were from Merck (Germany) or Ameresco (USA). Oligonucleotides and gene synthesis were carried out by Sangon (Shanghai, China). Taq DNA polymerase and all restriction endonucleases were obtained from Fermentas (Canada) or Takara (Japan). The DNA ladders were from Takara (Japan). The kits used for molecular cloning were from Omega (USA) or Takara (Japan).
Plasmid construction
Plasmids constructed and used in this study were listed in Table 1.
E. coli strain DH5a was used for molecular cloning. Synechocystis sp. PCC6803 and Synechococcus elongatus PCC7942 were generous gifts from Prof. Xudong Xu of Institute of Hydrobiology, Chinese Academy of Sciences. Nostoc punctiforme PCC73102 was a generous gift from Prof. John C. Meeks of UC Davis, USA.
Alkane biosynthetic genes sll0208 along with sll0209 were amplified from the genomic DNA of Synechocystis sp. PCC6803 with the primers 020809F1/020809R1. The primers 0208F1/0208R1 and 0209F1/0209R1 were used to amplify sll0208 and sll0209 gene respectively. Alkane biosynthetic genes orf1593 along with orf1594 were amplified from the genomic DNA of Synechococcus elongatus PCC7942 with the primers 9394F1/9394R1. The primers 1594F1/1594R1 were used to amplify orf1594 gene. Alkane biosynthetic genes npun1710 and npun1711 were amplified from the genomic DNA of Nostoc punctiforme PCC73102 with the primers 1011F1/1011R1. The primers 1711F1/1711R1 were used to amplify npun1711. The sll0208 gene, sll0208 along with sll0209, orf1593 along with orf1594, npun1710 along with npun1711, orf1594 along with npun1711 were subcloned into XbaI/SmaI site of the plasmid pFQ9R [5], resulting in pLX1, pLX3, pLX5, pLX6, pLX9 plasmid, respectively. The sll0209 gene was subcloned into NdeI/XhoI site of the plasmid pXT37b [5] to generate plasmid pLX4.
The ddh gene were amplified from the genomic DNA of Synechocystis sp PCC6803 by PCR using the primers ddh-F/ddh-R and inserted into the TA cloning site of pMD18-T-Simple, to generate the plasmids pXT119. The plasmid pLX3 was used as the template to amplify the 2.5 kb fragment of P rbc promoter , sll0208 and sll0209 and Rubisco terminator (T rbc ) [5] using the primers rbcNF/rbcNR. The 2.5 kb fragment was inserted into the TA cloning site of pGEM-T Easy to generate the plasmids pLX27. The ck2 cassette was excised with SalI and XbaI from pRL446 [27] and inserted into the SalI/SpeI site of pLX27, to generate the plasmid pLX28. The 3.6kb fragment containing ck2, P rbc , T rbc , sll0208 and sll0209 was digested with NdeI and SphI from pLX28 and cloned into BglII site of pXT119 with blunt ends, to generate the plasmid pLX13.
The P rbc promoter and sll0208 along with sll0209 were amplified from the plasmid pLX3 by fusion PCR using the primers PrbcBX-F/PrbcK-R, 0809K-F/0809B-R. The above fragment with XbaI site and SpeI/SalI sites on the 5’ and 3’ ends respectively was inserted into the TA cloning site of pMD19-T-Simple to generate the plasmids pLX14. To utilize the isocaudarner pair XbaI and SpeI, the fragment containing two copies of P rbc , sll0208 and sll0209 gene in tandem was digested with XbaI and SalI, and cloned into XbaI/SalI site of pFQ9R, to generate the plasmid pLX59. Plasmid maps were listed in Additional file 1: Figure S1.
Transformation and construction of Synechocystissp. PCC6803 mutant strains
Strains constructed and used in this study were listed in Table 2.
All of the above plasmids were checked by enzyme digestion and then transformed to Synechocystis cells. The transformations of Synechocystis strains with plasmids were performed as described [28]. The plasmids pLX1, pLX3, pLX4, pLX5, pLX6, pLX9, pLX13 and pLX59 were transformed to Synechocystis sp. PCC6803 to generate the mutant strains LX31, LX32, LX33, LX34, LX35, LX39, LX55 and LX70, respectively. The plasmid pLX3 was transformed to Synechocystis mutant strain XT203 and GQ4 to generate LX40 and LX38 mutant strains, respectively. The plasmid pLX13 was transformed to Synechocystis mutant strain GQ10, Syn-20ACC and LX32 to generate LX58, LX57, and LX56 mutant strains, respectively. For the initial selection of transformants, the DNA/cell mixture was applied to BG11 agar plates. After 18 h the membrane filters were applied to fresh BG11 agar plates containing following antibiotics (10μg mL-1 spectinomycin, 10 μg mL-1 erythromycin or 5 μg mL-1 spectinomycin/kanamycin). Homogeneous mutants were obtained by successive streaking on BG11 plates with appropriate antibiotics. Homologous integration of the expressing cassette and complete segregation were confirmed by PCR using primers listed in Additional file 1: Table S1. Schematic diagrams for homogeneous recombination of different plasmids were listed in Additional file 1: Figure S2. PCR analysis of the genotype of Synechocystis mutant strains were displayed in Additional file 1: Figure S3.
Cultivation of Synechocystisstrains
Normal liquid cultures of all Synechocystis strains in this study were grown at 30°C in 500 mL shake flasks containing 300 mL BG11 medium with aeration by sterile air under constant illumination at a photosynthetic photon flux density of approximately 30 μmol photons m-2 s-1 of white light. When necessary, the following antibiotics were added: kanamycin (20 μg mL-1) and spectinomycin (20 μg mL-1). Cell growth of each culture was monitored by measuring OD730. Synechocystis sp. PCC6803 wildtype and the mutant strains exhibited similar growth rate and final cell density. The cultures cultivated in shake flasks were harvested at OD730 of 3 to 4 after 14 days, when the stationary phase reached. All Synechocystis strains in this study were cultivated in shake flasks first to evaluate yields of alka(e)nes. The alka(e)ne content of LX56 mutant with the highest alka(e)ne yield in this work and the wildtype control cultivated in shake flasks were calculated as a percentage of DW. Conversion between OD730 and DW of LX56 mutant and Synechocystis sp. PCC6803 cultivated in shake flasks was performed by regression analysis (Additional file 1: Figure S4).
The bubble column photo-bioreactor was a 580 mm×30 mm glass column with a silica gel plug. Synechocystis sp. PCC6803 and the LX56 mutant strain were grown in flasks to exponential phase and harvested by centrifugation. The harvested cells were re-suspended in 200 mL fresh BG11 media, and transferred to the column photobioreactors at 30°C under 50 μEm-2 s-1 of white light with air bubbling for 24h, after which the light intensity was adjusted to 100 μEm-2 s-1 and the aeration was switched to 5% (v/v) CO2-enriched air. LX56 mutant was grown in the presence of 10 μg mL-1 kanamycin and 10 μg mL-1 spectinomycin. Cell growth of Synechocystis sp. PCC6803 and LX56 mutant was monitored by measuring OD730.
Extraction and GC-MS analysis ofalka(e)nes
Alka(e)nes were extracted from Synechocystis cells. 200 mL normal culture or 50 mL culture of column photo-bioreactor at stationary phase was harvested by centrifugation. The cells were resuspended in 10 mL of TE buffer (pH8.0) and then lysed by sonication. 50 μL eciosane (1 mg/mL) was added to the cell lysate as the internal standard for alka(e)ne analysis. The lysate was extracted for 1h at room temperature with 10 mL chloroform–methanol (v/v, 2:1) [29]. A two-phase system (top: aqueous, bottom: organic) was generated after shaking for 1 h and centrifugation at 8000 rpm at room temperature for 15 min. The bottom organic phase was transferred to a new glass tube and evaporated to dryness under a stream of nitrogen at 55°C. The residue was dissolved in 1mL of n-hexane. Aliquots of this mixture were analyzed by GC-MS using an Agilent 7890A-5975C system equipped with a HP-INNOWax (30 m×250 μm×0.25 μm). Helium (constant flow 1 mL/min) was used as the carrier gas. The temperature of the injector was 250°C and the following temperature program was applied: 100°C for 1 min, increase of 5°C min-1 to 150°C then increase of 10°C min-1 to 250°C for 15 min. The internal standard was used to determine alka(e)ne yield, which was reported as the mean based on three independent experiments.
Semi-quantitative reverse transcription PCR
RNA was isolated from 50 ml cultures of Synechocystis cells in mid-exponential phase by using TRIzol Reagent (Life Technologies). The first-strand cDNA was synthesized from 1μg of total RNA using a RevertAid First Strand cDNA synthesis Kit (Thermo SCIENTIFIC) according to the manufacturer’s protocol. PCR was performed using primers 0809RTF1/R1 (Additional file 1: Table S1) to amplify 350 bp of internal coding region of sll0208 and sll0209. The RNase P subunit B (rnpB) gene-specific primer pairs rnpB1/2 (Additional file 1: Table S1) were designed to amplify rnpB as external standards. Thirty cycles were used for rnpB cDNA, and 35 cycles were used for sll0208 and sll0209 cDNA.
Abbreviations
- ACP:
-
Acyl carrier protein
- AAR:
-
Acyl-ACP reductase
- ADO:
-
Aldehyde-deformylating oxygenase
- ACS:
-
Acyl-CoA synthetase
- AAS:
-
Acyl-ACP synthetase
- ACC:
-
Acetyl-CoA carboxylase
- FAS:
-
Fatty acid synthase
- FFA:
-
Free fatty acid
- DW:
-
Cell dry weight
- PCR:
-
Polymerase chain reaction
- OD:
-
Optical density.
References
Atsumi S, Higashide W, Liao JC: Direct photosynthetic recycling of carbon dioxide to isobutyraldehyde. Nat Biotechnol. 2009, 27: 1177-1180. 10.1038/nbt.1586.
Dexter J, Fu PC: Metabolic engineering of cyanobacteria for ethanol production. Energ Environ Sci. 2009, 2: 857-864. 10.1039/b811937f.
Lindberg P, Park S, Melis A: Engineering a platform for photosynthetic isoprene production in cyanobacteria, using Synechocystis as the model organism. Metab Eng. 2010, 12: 70-79. 10.1016/j.ymben.2009.10.001.
Liu XY, Sheng J, Curtiss R: Fatty acid production in genetically modified cyanobacteria. P Natl Acad Sci USA. 2011, 108: 6899-6904. 10.1073/pnas.1103014108.
Tan XM, Yao L, Gao QQ, Wang WH, Qi FX, Lu XF: Photosynthesis driven conversion of carbon dioxide to fatty alcohols and hydrocarbons in cyanobacteria. Metab Eng. 2011, 13: 169-176. 10.1016/j.ymben.2011.01.001.
Ungerer J, Tao L, Davis M, Ghirardi M, Maness PC, Yu JP: Sustained photosynthetic conversion of CO2 to ethylene in recombinant cyanobacterium Synechocystis 6803. Energ Environ Sci. 2012, 5: 8998-9006. 10.1039/c2ee22555g.
Lu XF: A perspective: Photosynthetic production of fatty acid-based biofuels in genetically engineered cyanobacteria. Biotechnol Adv. 2010, 28: 742-746. 10.1016/j.biotechadv.2010.05.021.
Ducat DC, Way JC, Silver PA: Engineering cyanobacteria to generate high-value products. Trends Biotechnol. 2011, 29: 95-103. 10.1016/j.tibtech.2010.12.003.
Kaneko T, Sato S, Kotani H, Tanaka A, Asamizu E, Nakamura Y, Miyajima N, Hirosawa M, Sugiura M, Sasamoto S: Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 1996, 3: 109-136. 10.1093/dnares/3.3.109.
Shih PM, Wu DY, Latifi A, Axen SD, Fewer DP, Talla E, Calteau A, Cai F, de Marsac NT, Rippka R: Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc Natl Acad Sci U S A. 2013, 110: 1053-1058. 10.1073/pnas.1217107110.
Huang HH, Camsund D, Lindblad P, Heidorn T: Design and characterization of molecular tools for a Synthetic Biology approach towards developing cyanobacterial biotechnology. Nucleic Acids Res. 2010, 38: 2577-2593. 10.1093/nar/gkq164.
Peralta-Yahya PP, Zhang FZ, del Cardayre SB, Keasling JD: Microbial engineering for the production of advanced biofuels. Nature. 2012, 488: 320-328. 10.1038/nature11478.
Schirmer A, Rude MA, Li XZ, Popova E, del Cardayre SB: Microbial Biosynthesis of Alkanes. Science. 2010, 329: 559-562. 10.1126/science.1187936.
Han J, McCarthy ED, Hoeven WV, Calvin M, Bradley WH: Organic geochemical studies, ii. A preliminary report on the distribution of aliphatic hydrocarbons in algae, in bacteria, and in a recent lake sediment. Proc Natl Acad Sci U S A. 1968, 59: 29-33. 10.1073/pnas.59.1.29.
Li N, Chang WC, Warui DM, Booker SJ, Krebs C, Bollinger JM: Evidence for Only Oxygenative Cleavage of Aldehydes to Alk(a/e)nes and Formate by Cyanobacterial Aldehyde Decarbonylases. Biochemistry-US. 2012, 51: 7908-7916. 10.1021/bi300912n.
Davis MS, Solbiati J, Cronan JE: Overproduction of acetyl-CoA carboxylase activity increases the rate of fatty acid biosynthesis in Escherichia coli. J Biol Chem. 2000, 275: 28593-28598.
Kaczmarzyk D, Fulda M: Fatty acid activation in cyanobacteria mediated by acyl-acyl carrier protein synthetase enables fatty acid recycling. Plant Physiol. 2010, 152: 1598-1610. 10.1104/pp.109.148007.
Gao QQ, Wang WH, Zhao H, Lu XF: Effects of fatty acid activation on photosynthetic production of fatty acid-based biofuels in Synechocystis sp PCC6803. Biotechnology for Biofuels. 2012, 5:
Ladygina N, Dedyukhina EG, Vainshtein MB: A review on microbial synthesis of hydrocarbons. Process Biochem. 2006, 41: 1001-1014. 10.1016/j.procbio.2005.12.007.
Hu P, Borglin S, Kamennaya NA, Chen L, Park H, Mahoney L, Kijac A, Shan G, Chavarría KL, Zhang C: Metabolic phenotyping of the cyanobacterium Synechocystis 6803 engineered for production of alkanes and free fatty acids. Applied Energy. 2013, 102: 850-859.
Reppas NB, Ridley CP, Reppas N, Ridley C, Rodley CP: Producing hydrocarbons comprises culturing engineered cyanobacterium in culture medium and exposing engineered cyanobacterium to light and carbon dioxide. 2010, US: JOULE UNLIMITED INC, 7794969-B1
Eser BE, Das D, Han J, Jones PR, Marsh ENG: Oxygen-Independent Alkane Formation by Non-Heme Iron-Dependent Cyanobacterial Aldehyde Decarbonylase: Investigation of Kinetics and Requirement for an External Electron Donor. Biochemistry-US. 2011, 50: 10743-10750. 10.1021/bi2012417.
Hein S, Tran H, Steinbuchel A: Synechocystis sp. PCC6803 possesses a two-component polyhydroxyalkanoic acid synthase similar to that of anoxygenic purple sulfur bacteria. Arch Microbiol. 1998, 170: 162-170. 10.1007/s002030050629.
Liu X, Curtiss R: Thermorecovery of cyanobacterial fatty acids at elevated temperatures. J Biotechnol. 2012, 161: 445-449. 10.1016/j.jbiotec.2012.08.013.
Gao ZX, Zhao H, Li ZM, Tan XM, Lu XF: Photosynthetic production of ethanol from carbon dioxide in genetically engineered cyanobacteria. Energ Environ Sci. 2012, 5: 9857-9865. 10.1039/c2ee22675h.
Kamarainen J, Knoop H, Stanford NJ, Guerrero F, Akhtar MK, Aro EM, Steuer R, Jones PR: Physiological tolerance and stoichiometric potential of cyanobacteria for hydrocarbon fuel production. J Biotechnol. 2012, 162: 67-74. 10.1016/j.jbiotec.2012.07.193.
Yin C, Li W, Du Y, Kong R, Xu X: Identification of a gene, ccr-1 (sll1242), required for chill-light tolerance and growth at 15°C in Synechocystis sp. PCC 6803. Microbiology. 2007, 153: 1261-1267. 10.1099/mic.0.2006/005074-0.
Williams JGK: Construction of specific mutations in photosystem II photosynthetic reaction center by genetic engineering methods in Synechocystis-6803. Methods Enzymol. 1988, 167: 766-778.
Bligh EG, Dyer WJ: A rapid method of total lipid extraction and purification. Can J Biochem Physiol. 1959, 37: 911-917. 10.1139/o59-099.
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (973: 2011CBA00907), Knowledge Innovation Program of the Chinese Academy of Sciences (KSCX2-EW-G-1-4), the Joint Research Laboratory for Sustainable Aviation Biofuels (QIBEBT-Boeing), and the “100-Talent Program of the Chinese Academy of Sciences” foundation (Grant O91001110A).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XL (Xuefeng Lu) conceived of the study. XL (Xuefeng Lu), WW and XL (Xufeng Liu) designed the experiments. WW and XL (Xufeng Liu) carried out experiments including the construction and cultivation of Synechocystis sp. PCC683 mutant strains, extraction and analysis of alka(e)nes, and GC-MS analysis. XL (Xuefeng Lu), WW and XL (Xufeng Liu) wrote the manuscript. All authors read and approved the final manuscript.
Weihua Wang, Xufeng Liu contributed equally to this work.
Electronic supplementary material
13068_2012_305_MOESM1_ESM.doc
Additional file 1: Table S1: Primers used in this study. Figure S1. Plasmid maps. Figure S2. Schematic diagrams for homogeneous recombination of different plasmids. Figure S3. PCR analysis of the genotype of Synechocystis mutant strains. Figure S4. Linear regression of cell dry weight (DW) versus OD730 for Synechocystis sp. PCC6803(6803yu)and LX56 strain cultivated in shake flasks. (DOC 1020 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, W., Liu, X. & Lu, X. Engineering cyanobacteria to improve photosynthetic production of alka(e)nes. Biotechnol Biofuels 6, 69 (2013). https://doi.org/10.1186/1754-6834-6-69
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1754-6834-6-69