
Abstract
Background
Filamentous fungi are frequently used as production platforms in industrial biotechnology. Most of the strains involved were known as reproducing exclusively asexually thereby preventing the application of conventional strain breeding techniques. In the last decade, evidence was obtained that a number of these imperfect fungi possess a sexual life cycle, too. Trichoderma reesei, an industrial producer of enzymes for food, feed and biorefinery purposes, is heterothallic and takes a special position among industrially utilized species as all industrial strains are derived from the single MAT1-2 isolate QM6a. Consequently, strain improvement by crossing is not feasible within this strain line as this necessitates a MAT1-1 mating partner. Simply switching the mating type in one of the mating partners to MAT1-1, however, is not sufficient to produce a genotype capable of sexual reproduction with QM6a MAT1-2.
Results
We have used a systems biology approach to identify genes restoring sexual reproduction in the QM6a strain line. To this end, T. reesei QM6a was crossed with the MAT1-1 wild-type strain CBS999.97. The descendants were backcrossed 8-times in two lineages with QM6a to obtain mating competent MAT1-1 strains with a minimal set of CBS999.97 specific genes. Comparative genome analysis identified a total of 73 genes of which two—encoding an unknown C2H2/ankyrin protein and a homolog of the WD-protein HAM5—were identified to be essential for fruiting body formation. The introduction of a functional ham5 allele in a mating type switched T. reesei QM6a allowed sexual crossing with the parental strain QM6a.
Conclusion
The finding that Trichoderma reesei is generally capable of undergoing sexual reproduction even under laboratory conditions raised hope for the applicability of classical breeding techniques with this fungus as known for plants and certain yeasts. The discovery that the wild-type isolate QM6a was female sterile, however, precluded any progress along that line. With the discovery of the genetic cause of female sterility and the creation of an engineered fertile strain we now provide the basis to establish sexual crossing in this fungus and herald a new era of strain improvement in T. reesei.
Similar content being viewed by others


Background
Filamentous fungi are key organisms in biotechnology, being at the core of many industrial processes, such as the production of enzymes, vitamins, polysaccharides, polyhydric alcohols, pigments, lipids, organic acids and glycolipids [1, 2]. Strain improvement of these organisms to obtain commercially acceptable production yields has for a long time been mostly relying on classical mutagenesis and only in the last two decades recombinant DNA technologies increasingly gained importance. This is because—in contrast to plants and yeasts, for which strain breeding by crossing has been an essential strategy for strain improvement—the majority of them were isolated in their asexual form and their sexual form and life cycle had been unknown for many years. In the last decade, evidence was obtained that also a number of these fungi possess a sexual cycle [3–5]. The ability to mate industrially relevant fungi under laboratory conditions has been demonstrated, amongst others, for the organic acid producer Aspergillus tubingensis [6] and the penicillin producer Penicillium chrysogenum [7]. Crossing of different industrial strains of a given species would have the significant advantage of being a “natural” strategy for rapidly introducing multiple favorable traits even if their genetic foundation is unknown or to remove mutations or undesired genes like secondary metabolite clusters or antibiotic resistance markers whose presence may interfere with regulatory requirements.
Trichoderma reesei (syn. Hypocrea jecorina) is a major industrial enzyme producer, particularly of cellulases and hemicellulases that are used for applications in the food, feed and biorefinery businesses [8–11]. This fungus too was for a long time considered to be asexual. Seidl et al. [12] first described the ability of T. reesei to perform sexual reproduction, and to be heterothallic. Heterothallic species require two compatible mating partners each of them carrying a different mating type locus—either MAT1-1 or MAT1-2—which are required for successful sexual reproduction [13, 14].
All strains of T. reesei which are nowadays used in industry and most strains used in academic research originate from a single isolate—T. reesei QM6a [15] which carries the mating type MAT1-2. This implies that for crossing industrial lineages of these enzyme producers, a mating type switch from MAT1-2 to MAT1-1 is needed in one of the mating partners. However, a simple mating type switch proved to be insufficient to enable sexual reproduction within the T. reesei QM6a strain line [12]. This finding is in contrast to, e.g., Magnaporthe grisea or Neurospora crassa, where strains in which the MAT locus was exchanged were fertile in crossings with the respective parental strains [16, 17]. Interestingly, the mating type switched T. reesei QM6a strain was able to mate with MAT1-2 wild isolates other than QM6a. From these findings Seidl et al. [12] concluded that T. reesei QM6a is very well able to act as a male partner in crossings but that it cannot produce fruiting bodies and hence is female sterile, probably because of its maintenance in laboratories for over 60 years without selective pressure to sustain mating competence.
In this study, we describe the identification of the cause for the female sterility of T. reesei QM6a. By comparing the genomes of female fertile MAT1-1 QM6a offspring—obtained after repeated backcrossing with QM6a—to that of their parental strain QM6a, we identified genes putatively required for female fertility and being either mutated or lacking in QM6a. Systematic gene knock-out of the candidate genes for female fertility and their reintroduction led to the deciphering of this malfunction on a genetic basis and enables us to rescue female fertility in T. reesei QM6a. This puts us now in the position to establish strain breeding programs by sexual crossing of QM6a descendants.
Results
Preparation of two T. reesei MAT1-1 inbred strain lines
In a first attempt to identify genes that are absent or mutated in T. reesei QM6a but functional in sexually reproducing wild-type isolates, and thus are potentially responsible for the observed female sterility of strain QM6a, we sequenced and analyzed the genomes of the T. reesei strains C.P.K. 1282 (MAT1-1), C.P.K. 170 (MAT1-1) and C.P.K. 938 (MAT1-2). The two MAT1-1 strains were chosen as they reproduce sexually with QM6a and C.P.K. 938 mates with the two MAT1-1 strains. Comparative genome analysis of these strains revealed cumulative sequence differences between them and T. reesei QM6a of a magnitude ranging from 21,000 to 141,000 SNVs (single nucleotide variants) in all exon regions preventing the straightforward identification of genes responsible for female sterility. Therefore, we used an inbreeding strategy first to narrow down the number of candidate genes. We crossed QM6a with different available MAT1-1 wild-type strains followed by backcrossing of the MAT1-1 progenies with QM6a to establish inbred strain lines (see Additional file 1: Table S1). Thereby, strain CBS999.97 was identified as the ideal candidate since these crossings were highly reproducible. Fruiting bodies were formed within 5–10 days vs. 10–20 days for the other tested wild-type strains. Furthermore, the sexual fertility of the progenies was highest from crosses with CBS999.97 whereas the progenies of the other crosses lost their sexual fertility to cross with QM6a after one or two generations.
MAT1-1 progenies from the cross of QM6a and CBS999.97 were backcrossed over eight generations in two separate lines with QM6a, resulting in the two MAT1-1 inbred strains RL1/A8-02 and RL2/A8-11 that were phenotypically similar to QM6a. These two strains were still able to mate with different MAT1-2 wild-type strains (see Additional file 2: Figure S1) as well as with strain QM6a.
Genome analysis of T. reesei RL1/A8-02 and RL2/A8-11
Whole genome comparison of T. reesei RL1/A8-02, RL2/A8-11 and QM6a (Tables 1, 2) revealed that strain RL1/A8-02 shows a higher degree of similarity with the parental strain QM6a than strain RL2/A8-11, as both the number of SNVs and DIPs (deletion/insertion polymorphism) is smaller. We identified a total number of 427 genes, in which a SNV or DIP caused a change of the amino acid sequence of the encoded protein in QM6a. The actual number of affected genes is lower than the sum of SNVs and DIPs (497) since most genes carrying a DIP are also affected by SNV(s). To increase the likelihood of tracking genes causing female sterility in strain QM6a, we only focused on those SNVs and DIPs which the two inbred strains had in common and which differentiated these strains from strain QM6a. Out of these, we selected those genes in which the SNVs and DIPs resulted in a change of the amino acid sequence of the corresponding proteins, excluded those genes in which SNVs and DIPs resulted in the occurrence of a premature stop codon in strains RL1/A8-02 and RL2/A8-11, and excluded all genes in which SNVs resulted in a conserved amino acid exchange (e.g., A- > V, I- > L, K- > E, etc.). For the remaining genes a BLASTX search was performed against the NCBI database to exclude all genes for which the amino acid changes in the proteins were located in non-conserved regions. Using this approach, the number of candidates was reduced to 73 genes (Additional file 3: Table S2). The majority of them encoded unknown proteins that were conserved in other Pezizomycotina and enzymes of intermediary and secondary metabolism (Fig. 1). All the other gene groups (encoding carbohydrate active enzymes, solute transporters, transcription factors, proteases and components of signal transduction pathways) comprised only 3 (in one case 5) members.

FunCat categorization of the 73 female sterility candidate genes of T. reesei QM6a. CAZymes: carbohydrate active enzymes, which include glycosyl hydrolases, glycosyl transferases and accessory enzymes. The corresponding FunCat numbers are: 01, metabolism; 01.25.01, CAZymes; 11.02.03.04, transcription factors; 20.01, solute transport; 01.25.03, proteases; 30, signal transduction; 99, protein without described function, i.e., orphan (if the encoded protein has no homolog outside the genus Trichoderma), or unknown (if the encoded protein has orthologs in other species than Trichoderma). “Others” comprise genes that do not fall into any of the listed categories
Most of the sequence differences are clustered on the mating type chromosome
An intriguing feature, which emerged during the functional annotation of the 73 genes, was that their genomic location was strongly biased: the majority of them (52) were located on scaffold 6 (Additional file 3: Table S2; Fig. 2) which harbors the mating type locus and an over proportionally high number (4 and 7) were located on scaffolds 21 and 26, respectively. A recently released computational model of the assembled T. reesei chromosomes [18] suggests that these scaffolds are not located on the same chromosome which corroborates our data that scaffold 6, 21 and 26 do not reside in direct neighborhood (Linke et al. unpublished data).

Plot of scaffold size (full squares) and the selected 73 genes differing in their sequence from T. reesei QM6a on the respective scaffolds (full triangles) vs. scaffold number. The gray circle around the triangle corresponds to scaffold 6 and notes the in relation to scaffold size disproportionately high number of genes differing in their sequence from strain T. reesei QM6a
Identification of genes involved in mating of T. reesei
For the identification of those genes among the 73 candidates (vide supra) that were crucial for mating, we systematically produced gene knock-outs in a tku70 deleted derivative of T. reesei strain RL1/A8-02 and tested whether the resulting deletion strains are still capable of mating and fruiting body formation with QM6a (MAT1-2). Deletion of 2 of the 73 genes—Trire2:67350 and Trire2:59270—consistently resulted in a fruiting-body deficient phenotype. Their reintroduction into the respective knock-out strain restored the mating competence and fruiting body formation. For three genes Trire2:3422, Trire2:47930 and Trire2:81593 we were not able to obtain a deletion strain.
To test whether any of these five genes are associated with female sterility of T. reesei QM6a we first constructed a recombinant QM6a MAT1-1 strain by replacing the MAT1-2 locus with the MAT1-1 locus of strain C.P.K. 1282. The integration of the respective genes of the MAT1-1 locus, MAT1-1-1, MAT1-1-2 and MAT1-1-3, into the chromosomal DNA of mitotically stable transformants was confirmed by diagnostic PCR analysis (Additional file 4: Figure S2). Mating assays with other (non QM6a) MAT1-2 wild-type strains confirmed that the strain is able to reproduce sexually with these strains. As expected from earlier results [12], this strain could not mate with QM6a. It was subsequently used as recipient strain for complementation with the five genes (the two genes whose knockout resulted in a fruiting body deficient phenotype and those for which no deletion strains could be obtained). The CBS999.97 alleles of the single genes as well as different combinations of them were transformed into QM6a MAT1-1. Single spore isolated transformants were then tested for their ability to produce fruiting bodies with the MAT1-2 wild-type isolate QM6a. A diagnostic PCR analysis of the fertile transformants identified the CBS999.97 Trire2:67350 allele to be responsible for the recovery of female fertility in a QM6a MAT1-1 strain as this allele was present in all mating positive transformants recorded (Fig. 3).

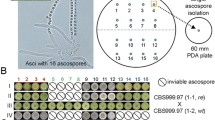
Mating assays of T. reesei QM6a MAT1-2 with a mating type switched T. reesei QM6a MAT1-1 (left) and with a mating type switched T. reesei QM6a MAT1-1 which was additionally complemented with the functional ham5 allele of CBS999.97 (right). Due to the effect of female sterility no fruiting bodies are formed in the contact zone of a QM6a MAT1-2 strain with a mating type switched QM6a. After complementation with a functional ham5 allele the phenotype is altered, sporulation is reduced and fruiting bodies (indicated by black arrows) appear in the contact zone of the two strains
Trire2:67350 is the orthologue of N. crassa HAM-5 and P. anserina IDC1
Trire2:67350 was originally annotated in the T. reesei genome database v2.0 (http://www.genome.jgipsf.org/Trire2/Trire2.home.html) as a hypothetical protein and the corresponding gene harbors five introns. Its sequence is moderately conserved among Ascomycetes (Additional file 5: Table S3). In order to compare the amino acid sequence of the functional allele of Trire2:67350 with that of its orthologs from other fungi, we isolated RNA and sequenced the corresponding cDNA from strains RL1/A8-02 and RL2/A8-11. This showed that the T. reesei gene is 5121 nt long and interrupted by three short introns residing in the 5′-terminal fifth of its sequence (Fig. 4a). In silico translation yields a 1625 amino acids protein with a Mw of 178.133 kDa. Using this sequence its orthologs were identified as ham-5 (Neurospora crassa) and idc1 (Podospora anserina). The T. reesei HAM5 protein, similar to its orthologs in other Pezizomycota, carries a serine-rich WD40 repeat (IPR015943) region between amino acids 40 and 320, suggesting a possible protein–protein interaction site. In addition, and also consistent with its orthologs in other fungi, the protein exhibits a proline-rich region (C-terminal) which is also conserved among different species. Interestingly, the T. reesei HAM5 contains an additional stretch of glutamine residues (35 of 38 residues) close to the proline-rich region (Fig. 4b). This polyglutamine stretch is also present in T. virens, although much shorter than that in T. reesei and half of it replaced by a proline repeat. In T. atroviride it is even shorter and flanked by glutaminic acid repeats at its N-terminal side and proline repeats on its C-terminal side (Additional file 6: Figure S4). However, the polyglutamine stretch is completely absent in other fungal species even in phylogenetically close ones such as Beauveria spp. or Metarhizium spp.

The ham5 locus (a), the in silico translated and cDNA-confirmed HAM5 protein (b), and a comparison of the amino acid sequence of HAM5 (c) of T. reesei QM6a (red) and of T. reesei RL2/A8-02 (black). The vertical lines serve to relate the respective regions in a, b and c
To identify the mutation that renders ham5 in T. reesei QM6a non-functional, we compared its genomic sequence with that of strains RL1/A8-02 and RL2/A8-11. The ham5 gene of T. reesei QM6a differed in a total of 91 nucleotide positions of which 9 occurred in introns (Additional file 7: Figure S3). One of them (G531 → T) occurred at the first position of the second intron, thus changing the invariant G+1 of the donor splicing site [19] to a T. Sequencing of the cDNA formed by strain QM6a confirmed that as a consequence of this mutation, this second intron is not spliced, which results—due to the subsequent in frame occurrence of a TAA stop codon formed by the internal donor splicing site (5′-TTAAGT-3′)—in the formation of a truncated protein of only 151 amino acids in T. reesei QM6a (Fig. 4c).
Discussion
In this study, we have identified the genetic basis for the female sterility of T. reesei QM6a [12] and strains derived from it. The availability of mating competent T. reesei QM6a derived strains now allows, as shown here, crossing of strains from relevant lineages of T. reesei to combine industrially important genetic traits, and thus to include strain crossing in industrial strain improvement programs.
Strain crossing has only rarely been used with filamentous fungi [cf. 5], probably due to the fact that most of the respective fungi were isolated as asexual individuals or rapidly lost the ability for sexual development upon cultivation in the laboratory. Fungal field populations have a female-sterile/hermaphrodite status and female-sterile mutants may comprise >50 % of the population [20]. Saleh et al. [21] have shown that in Magnaporthe oryzae asexual propagation results in loss of sexual reproduction after only a couple of generations. They interpreted this loss as the result of genetic drift after the introduction of the fungus into a new environment. This is essentially what happens when a fungus is purified from an environmental sample and maintained in a laboratory environment. We, therefore, expect that loss of sexual propagation may also have occurred with most fungi used in biotechnology. In fact, recent success in crossing of industrially applied strains of P. chrysogenum also revealed strains that failed to recombine [7], and thus these may also have acquired mutations in genes essential for sexual reproduction. Therefore, the strategy used in this paper may stimulate respective research also in other industrial fungi.
One gene that was identified in this study as the cause for the observed female sterility in T. reesei QM6a is highly conserved in Ascomycetes and an ortholog of the recently identified P. anserina idc1 and N. crassa ham-5 [22, 23]. The loss of function of this gene has been reported in N. crassa to result in defective conidial anastomosis tube formation and impairment of hyphal fusion during colony initiation and in the mature colony [22, 24]. In addition it caused a lack of formation of pigment and aerial hyphae, female sterility, and the inability to trigger cell degeneration in P. anserina [23]. The above-described cell fusion that occurs between vegetative cells as a prerequisite for colony establishment is also required for mating [25–27], and thus several N. crassa mutants deficient in hyphal fusion and/or signaling are also female sterile [27]. Therefore, our identification of ham5 as a gene defective in T. reesei QM6a and being responsible for its female sterility agrees well with these findings.
While this paper was prepared for submission, HAM5 was shown to physically interact with a conserved MAP kinase cascade that includes NRC-1, MEK-2 and MAK-2, suggesting that it functions as a scaffold/transport hub for the MAP kinase cascade members for oscillation and chemotropic interactions during germling and hyphal fusion in N. crassa [28]. This finding is further supported by Dettmann et al. [29] who just recently identified HAM5 as a cell–cell communication-specific scaffold protein of the Neurospora crassa MAK-2 cascade (homologous to the budding yeast pheromone pathway).
The most striking difference in the protein sequence of T. reesei HAM5 from that of other Ascomycetes is the occurrence of the polyglutamine stretch in T. reesei which is lacking in other fungal genera. Since the nucleotide sequence encoding this polyglutamine repeat is not replaced by an intron in other fungi, it is important to note that the apparent lack of occurrence of the polyglutamine stretch in these fungi is not due to a potentially wrong intron prediction. The function of polyglutamine sequences is still under debate. One hypothesis postulates that they simply form a flexible spacer between protein domains [30]. On the other hand, polyglutamine sequences are generally believed to mediate protein–protein interaction [31], e.g., in transcription factors [32], where they can activate gene transcription [33, 34] and nuclear localization [35, 36]. Schaefer et al. [30] also reported that polyglutamine tracts are often accompanied by proline-rich regions in their C-terminal vicinity, which is the case in T. reesei as well. It is also intriguing to note that the polyglutamine stretch is longest in T. reesei and shortest in T. atroviride. The latter is phylogenetically more ancient than T. reesei [37] which lends to speculate that the polyglutamine stretch in HAM5 arose during the evolution of Trichoderma. Whether this endowed HAM5 with genus-specific functions not present in other fungi, or simply served to adapt its function to Trichoderma physiology will be an intriguing subject for further studies.
Conclusion
The discovery of the sexual life cycle of T. reesei by Seidl et al. [12] was the first step towards the application of classical strain breeding in T. reesei. The identification of the strain’s female sterility, however, precluded the use of classical genetic tools for strain improvement. By identifying the cause for female sterility on a genetic basis we have now provided the basis for the use of strain improvement by sexual crossing in T. reesei. To the best of our knowledge, we here provide the first example where advanced sequencing technologies in combination with a back-crossing strategy and a systematic knockout program allowed to identify relevant mutations in an industrial fungus. We strongly believe that the strategy used in this paper can be applied to resolve many other unclear genotype phenotype correlations and so help to overcome bottlenecks in further improvement of strains.
Methods
Strains and cultivations
The strains used throughout this work are listed in Additional file 1: Table S1. Strains were cultivated on potato dextrose agar (PDA; Difco, Sparks, MD, USA) and maintained on 28 °C in darkness. Crossing of strains was performed by opposing the two mating partners on a PDA plate on the benchtop (temperature variations between 18 and 22 °C under day/night light fluctuations) for 10–14 days until formation of fruiting bodies and ascospore discharge could be detected.
Construction of inbred strains RL1/A8-02 and RL2/A8-11
Sexual crossings of QM6a (MAT1-2) with strain CBS999.97 (MAT1-1) were performed followed by repeated cycles of backcrossing of the MAT1-1 progenies with QM6a (MAT1-2). Fruiting bodies ejected their ascospores to the cover of the Petri dish from where they were collected. The mating type of several single spore isolated offsprings was tested by diagnostic PCR (sequence of oligonucleotides is given in Additional file 8: Table S4). Approximately 5 MAT1-1 carrying individuals were then used for a mating assay to produce another generation of offsprings. By this method, we could obtain fertile strains carrying the MAT1-1 locus with a maximum of QM6a-specific genes. The resultant progenies from the eighth generation were then tested for their ability to mate with T. reesei QM6a and other MAT1-2 strains. One strain of two independent inbred lines each (RL1/A8-02 and RL2/A8-11) was then used for genome sequencing.
Genome sequencing of T. reesei RL1/A8-02 MAT1-1 and RL2/A8-11 MAT1-1
Two libraries, with 320 bp and 8 kbp insert size, were prepared for each strain, respectively. To this end, DNA was fragmented using a Covaris S2 system (Covaris, Inc. Woburn, MA, USA) and fragments were purified using the QIAquick PCR purification kit (Qiagen, Hilden, Germany). Paired-end libraries were prepared using the NEBNext DNA Sample Prep modules (New England Biolabs, Ipswich, MA, USA) following the manufacturer’s instructions. Briefly, fragments were end-repaired using Klenow and T4 DNA polymerases and phosphorylated with T4 polynucleotide kinase. Fragments were then 3′-adenylated using Klenow exo-DNA polymerase, and Illumina adapters were added using DNA ligase. Ligation products of ~400 bp were gel-purified using the Qiagen gel extraction kit (Qiagen, Hilden, Germany). To avoid the introduction of a guanine–cytosine (GC) bias during the gel-purification step in the standard Illumina library preparation protocol, the gel slice was dissolved rather at room temperature than with heating. The size-selected, adapter-modified DNA fragments were PCR amplified using PE PCR primers 1.0 and 2.0 (Illumina, San Diego, CA, USA) using Phusion DNA polymerase (New England Biolabs, Ipswich, MA, USA) and the following protocol: polymerase activation (98 °C for 30 s), followed by 10 cycles (denaturation at 98 °C for 10 s, annealing at 65 °C for 30 s and extension at 72 °C for 50 s) with a final, 5-min extension at 72 °C. Libraries were purified and quantified using the Qubit HS Assay Kit (Invitrogen, Carlsbad, CA, USA).
Cluster amplification was performed using the TruSeq PE Cluster Kit v5 on a cluster station, and all libraries were sequenced on a single Illumina HiSeq 2000 lane using TruSeq SBS 36 Cycle Kits v5 (Illumina, San Diego, CA, USA) using a 2 × 107 bp paired-end protocol. Sequencing image files were processed using Illumina’s Sequencing Control Software (SCS) by Illumina’s Real Time Analysis (RTA) v2.6 and CASAVA v1.7 (Illumina Inc., San Diego, CA, USA) to generate base calls and Phred-like base quality scores and to remove failed reads.
Sequences were quality filtered using CLC Genomic Workbench (version 5.1, CLC bio, Arhus, Denmark) and de novo assembled with Newbler (version 2.60, Roche/454, Brandford, CT, USA) and CLC Genomic Workbench. To identify large insertions in the two backcrossed genomes which are not present in the published T. reesei QM6a genome sequence, the obtained contigs were mapped to the scaffolds of T. reesei QM6a (accessions: GL985056-GL985132; NC_003388) using BLAST [38] and r2cat [39]. The BLAST hits were subsequently processed with a custom R script. Single nucleotide variation (SNVs) and insertions and deletions (DIPs) between the QM6a reference sequence and the aligned sequences of the two backcrossed lines were identified using CLC Genomic Workbench. Candidate genes were then manually tested for silent mutations. These and sequence differences that lead to a conserved amino acid exchange that does not interfere with the function of the putative protein were excluded from further analysis and discarded.
Genes were then classified according to their annotation in the T. reesei genome database v2.0 (Joint Genome Institute, http://www.genome.jgi.doe.gov, [40]), the MIPS Functional Catalogue (http://www.mips.helmholtz-muenchen.de/proj/funcatDB, [41]) and a manually curated completely annotated genome database of T. reesei present proprietary to the authors at TU Vienna.
Data deposition
The sequences reported in this paper have been deposited in the EMBL-EBI short read archive under the accession number ERP009132. The T. reesei HAM5 protein sequence derived from cDNA sequencing is available in GenBank with accession number KM677955.
Fungal strain constructions
Trichoderma reesei was transformed via protoplast transformation [42]. Deletion cassettes consisting of 1.0–1.5 kb fragments of gene-specific flanking regions interrupted by the selection marker were assembled by yeast recombinational cloning as described in detail by Schuster et al. [43]. Individual flanking regions of candidate genes were amplified using oligonucleotides 5F + 5R as well as 3F + 3R from genomic DNA of strain CBS999.97 with Phusion Polymerase (Thermo Fisher Scientific/Fermentas, St. Leon-Rot, Germany). As marker the hph expression cassette (2.7 kb) was amplified by PCR from the plasmid pLHhph [44] using the oligonucleotides hph fw and hph rv. All sequences of the oligonucleotides used are given in Additional file 8: Table S4.
To construct a T. reesei QM6a MAT1-1 strain, the resident MAT1-2 locus was replaced by the MAT1-1 locus from T. reesei C.P.K. 1282. A 5500 bp fragment of this MAT1-1 locus, including additional 3300-bp upstream and 1300-bp downstream sequences was amplified from genomic DNA using the primers MAT1-1_fw and MAT1-1_rv (Additional file 8: Table S4), and ligated into vector pCR blunt (Life Technologies/Invitrogen; Carlsbad, CA, USA). The hph selection cassette [44] was cloned into the AvrII site, situated in the intergenic region between MAT1-1-3 and Trire2:59147 (encoding a DNA lyase). For transformation of T. reesei QM6a, the replacement cassette was PCR amplified using the primer pair 1-2_replace_cassette_fw and 1-2_replace_cassette_rv (Additional file 8: Table S4). Strains were tested for integration of the cassette by PCR and mitotic stability. Positive strains were further tested for their ability to mate with MAT1-2 wild-type isolates.
Strain QM6a (MAT1-1) was complemented with the five putatively functional alleles of genes Trire2:67350, Trire59270, Trire2:3422, Trire2:47930 and Trire2:81593 of strain CBS999.97 (MAT1-1). The genes were PCR amplified (sequences of the oligonucleotides are given in Additional file 8: Table S4) and ligated/cloned by In-Fusion recombination (Clontech, Mountainview, CA, USA) in a derivative of pPki-Gen [45] where the pki1 promoter was removed by an EcoRI and XbaI digest (Thermo Fisher Scientific/Fermentas, St. Leon-Rot, Germany). Transformants were selected and purified on PDA plates containing 100 µg/ml geneticindisulfate G418 (Carl Roth GmbH + Co. KG, Karlsruhe, Germany). A scheme of the vector construct to complement strain QM6a MAT1-1 with the 5 final candidate genes is given in Additional file 9: Figure S5. The vector construction for the complementation with the functional allele of Trire2:67350 (pFSG_ID67350) serves as an example since all plasmids were constructed in the same manner. The integration of the five respective genes was verified by PCR using each a gene specific primer and a second primer binding within the selection marker to exclusively screen for the integration of the CBS999.97 specific allele. Sequences of the oligonucleotides used for PCR analysis are given in Additional file 8: Table S4.
RNA extraction and cDNA synthesis of Trire2:67350
To obtain cDNAs of ham5 for determining the respective protein sequence of T. reesei QM6a, RL1/A8-02 and RL2/A8-11, strains were cultivated on PDA plates covered with cellophane. Total RNA was isolated according to Gruber et al. [46]. Briefly, mycelium was ground in liquid nitrogen and transferred to reaction tubes containing a mixture of 650 µl lysis buffer (0.6 M NaCl, 10 mM EDTA, 100 mM Tris pH 8 and 4 % SDS) and 650 µl phenol (pH 8). Samples were vortexed well and placed on ice for 15 min. Thereafter, samples were centrifuged at >13,000 rpm at 4 °C for 20 min. The supernatant was transferred to a fresh reaction tube containing 8 M LiCl, vortexed well and put on 4 °C overnight. The samples were centrifuged again at >13,000 rpm at 4 °C for 20 min and the pellet was dissolved in 750 µl isopropanol. For precipitation the samples were placed at −20 °C for 2 h, then centrifuged at >13,000 rpm at 4 °C for 20 min. The RNA pellet was finally dissolved in deionized water.
DNase treated (DNase I, RNase free; Thermo Fisher Scientific/Fermentas, St. Leon-Rot, Germany) RNA (5 µg) was reverse transcribed with the RevertAid™ First Strand cDNA Kit (Thermo Fisher Scientific/Fermentas, St. Leon-Rot, Germany) according to the manufacturer’s protocol with a combination of the provided oligo-dT and random hexamer primers.
Abbreviations
- DIP:
-
deletion/insertion polymorphism
- MAT1-1 :
-
mating type 1-1
- MAT1-2 :
-
mating type 1-2
- SNV:
-
single nucleotide variant
- T. reesei :
-
Trichoderma reesei
References
Adrio JL, Demain AL (2003) Fungal biotechnology. Int Microbiol 6:191–199
Rokem JS (2010) Industrial Mycology. In: Doelle HW, Rokem S, Berovic M (eds) Biotechnology, vol 6. Encyclopedia of Life Support Systems Publishers, Developed under the Auspices of the UNESCO. Eolss Publishers, Paris, pp 75–97
Dyer PS, O’Gorman CM (2011) A fungal sexual revolution: Aspergillus and Penicillium show the way. Curr Opin Microbiol 14(6):649–654
Dyer PS, O’Gorman CM (2012) Sexual development and cryptic sexuality in fungi: insights from Aspergillus species. FEMS Microbiol Rev 36(1):165–192
Kück U, Böhm J (2013) Mating type genes and cryptic sexuality as tools for genetically manipulating industrial molds. Appl Microbiol Biotechnol 97(22):9609–9620
Horn BW, Olarte RA, Peterson SW, Carbone I (2013) Sexual reproduction in Aspergillus tubingensis from section Nigri. Mycologia 105(5):1153–1163
Böhm J, Hoff B, O’Gorman CM, Wolfers S, Klix V, Binger D, Zadra I, Kürnsteiner H, Pöggeler S, Dyer PS, Kück U (2013) Sexual reproduction and mating-type-mediated strain development in the penicillin-producing fungus Penicillium chrysogenum. Proc Natl Acad Sci USA 110(4):1476–1481
Ferreira NL, Margeot A, Blanquet S, Berrin JG (2014) Use of cellulases from Trichoderma reesei in the twenty-first century part I: Current industrial uses and future applications in the production of second ethanol generation. In: Gupta VK, Schmoll M, Herrera-Estrella A, Upadhyay RS, Druzhinina I, Tuohy M (eds) Biotechnology and biology of Trichoderma. Elsevier, The Netherlands, pp 245–261
Kubicek CP (2013) Systems biological approaches towards understanding cellulase production by Trichoderma reesei. J Biotechnol 163(2):133–142
Kunamneni A, Plou FJ, Alcalde M, Ballesteros A (2014) Trichoderma enzymes for food industries. In: Gupta VK, Schmoll M, Herrera-Estrella A, Upadhyay RS, Druzhinina I, Tuohy M (eds) Biotechnology and biology of Trichoderma. Elsevier, The Neterlands, pp 339–344
Puranen T, Alapuranen M, Vehmaanperä J (2014) Trichoderma enzymes for textile industries. In: Gupta VK, Schmoll M, Herrera-Estrella A, Upadhyay RS, Druzhinina I, Tuohy M (eds) Biotechnology and biology of Trichoderma. Elsevier, The Netherlands, pp 351–362
Seidl V, Seibel C, Kubicek CP, Schmoll M (2009) Sexual development in the industrial workhorse Trichoderma reesei. Proc Natl Acad Sci USA 106(33):13909–13914
Debuchy R, Berteaux-Lecellier V, Silar P (2010) Mating systems and sexual morphogenesis in Ascomycetes. In: Borkovich KA, Ebbole DJ (eds) Cellular and Molecular Biology of Filamentous Fungi. ASM Press, Washington DC, pp 501–535
Turgeon BG, Yoder OC (2000) Proposed nomenclature of mating type genes of filamentous Ascomycetes. Fungal Genet Biol 31(1):1–5
Reese ET (1976) History of the cellulase program at the U.S. army Natick Development Center. Biotechnol Bioeng Symp 6:9–20
Kang S, Chumley FG, Valent B (1994) Isolation of the mating-type genes of the phytopathogenic fungus Magnaporthe grisea using genomic subtraction. s 138(2):289–296
Chang S, Staben C (1994) Directed replacement of mt A by mt a-1 effects a mating type switch in Neurospora crassa. Genetics 138(1):75–81
Marie-Nelly H, Marbouty M, Cournac A, Flot JF, Liti G, Parodi DP, Syan S, Guillén N, Margeot A, Zimmer C, Koszul R (2014) High-quality genome (re)assembly using chromosomal contact data. Nat Commun 5:5695
Kupfer DM, Drabenstot SD, Buchanan KL, Lai H, Zhu H, Dyer DW, Roe BA, Murphy JW (2004) Introns and splicing elements of five diverse fungi. Eukaryot Cell 3(5):1088–1100
Leslie JF, Klein KK (1996) Female fertility and mating type effects on effective population size and evolution in filamentous fungi. Genetics 144(2):557–567
Saleh D, Milazzo J, Adreit H, Tharreau D, Fournier E (2012) Asexual reproduction induces a rapid and permanent loss of sexual reproduction capacity in the rice fungal pathogen Magnaporthe oryzae: results of in vitro experimental evolution assays. BMC Evol Biol 29:12–42
Aldabbous MS, Roca MG, Stout A, Huang IC, Read ND, Free SJ (2010) The ham-5, rcm-1 and rco-1 genes regulate hyphal fusion in Neurospora crassa. Microbiology 156(9):2621–2629
Jamet-Vierny C, Debuchy R, Prigent M, Silar P (2007) IDC1, a Pezizomycotina-specific gene that belongs to the PaMpk1 MAP kinase transduction cascade of the filamentous fungus Podospora anserina. Fungal Genet Biol 44:1219–1230
Fu C, Iyer P, Herkal A, Abdullah J, Stout A, Free SJ (2011) Identification and characterization of genes required for cell-to-cell fusion in Neurospora crassa. Eukaryot Cell 10(8):1100–1109
Fleissner A, Simonin AR, Glass NL (2008) Cell fusion in the filamentous fungus, Neurospora crassa. In: Chen EH, Totowa NJ (eds) Methods in molecular biology. Humana Press, New York, pp 21–38
Glass NL, Jacobson DJ, Shiu PKT (2000) The genetics of hyphal fusion and vegetative incompatibility in filamentous ascomycete fungi. Annu Rev Genet 34:165–186
Read ND, Fleissner A, Roca GM, Glass NL (2010) Hyphal fusion. In: Borkovich KA, Ebbole DJ (eds) Cellular and molecular biology of filamentous fungi. American Society for Microbiology, Washington, DC, pp 260–273
Jonkers W, Leeder AC, Ansong C, Wang Y, Yang F, Starr TL, Camp DG 2nd, Smith RD, Glass NL (2014) HAM-5 functions as a MAP kinase scaffold during cell fusion in Neurospora crassa. PLoS Genet 10:e1004783
Dettmann A, Heilig Y, Valerius O, Ludwig S, Seiler S (2014) Fungal communication requires the MAK-2 pathway elements STE-20 and RAS-2, the NRC-1 adapter STE-50 and the MAP kinase scaffold HAM-5. PLoS Genet 10(11):e1004762
Schaefer MH, Wanker EE, Andrade-Navarro MA (2012) Evolution and function of AG/polyglutamine repeats in protein–protein interaction networks. Nucleic Acids Res 40(10):4273–4287
Hands S, Sinadinos C, Wyttenbach A (2008) Polyglutamine gene function and dysfunction in the ageing brain. Biochim Biophys Acta 1779:507–521
Wetzel R (2012) Physical chemistry of polyglutamine: intriguing tales of a monotonous sequence. J Mol Biol 421(4–5):466–490
Mitchell PJ, Tjian R (1989) Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 245:371–378
Chen C, Dickman MB (2002) Colletotrichum trifolii TB3 kinase, a COT1 homolog, is light inducible and becomes localized in the nucleus during hyphal elongation. Eukaryot Cell 1(4):626–633
Alba MM, Guigo R (2004) Comparative analysis of amino acid repeats in rodents and humans. Genome Res 14:549–554
Harrison PM (2006) Exhaustive assignment of compositional bias reveals universally prevalent biased regions: analysis of functional associations in human and Drosophila. BMC Bioinformatics 7:441
Kubicek CP, Herrera-Estrella A, Seidl-Seiboth V, Martinez DA, Druzhinina IS, Thon M et al (2011) Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol 12(4):R40
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402
Husemann P, Stoye J (2010) r2cat: synteny plots and comparative assembly. Bioinformatics 26(4):570–571
Grigoriev IV, Nordberg H, Shabalov I, Aerts A, Cantor M, Goodstein D et al (2012) The genome portal of the Department of Energy Joint Genome Institute. Nucleic Acid Res 40(D1):D26–D32
Ruepp A, Zollner A, Maier D, Albermann K, Hani J, Mokrejs M, Tetko I, Güldener U, Mannhaupt G, Münsterkötter M, Mewes HW (2004) The FunCat, a functional annotation scheme for systematic classification of proteins from whole genomes. Nucleic Acids Res 32(18):5539–5545
Gruber F, Visser J, Kubicek CP, de Graaff LH (1990) The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrG-negative mutant strain. Curr Genet 18(1):71–76
Schuster A, Kenneth SB, Collett JR, Baker SE, Seiboth B, Kubicek CP, Schmoll M (2012) A versatile toolkit for high throughput functional genomics with Trichoderma reesei. Biotechnol Biofuels 5:1
Hartl L, Kubicek CP, Seiboth B (2007) Induction of the gal pathway and cellulase genes involves no transcriptional inducer function of the galactokinase in Hypocrea jecorina. J Biol Chem 282(25):18654–18659
Seiboth B, Karimi RA, Phatale PA, Linke R, Hartl L, Sauer DG, Smith KM, Baker SE, Freitag M, Kubicek CP (2012) The putative protein methyltransferase LAE1 controls cellulase gene expression in Trichoderma reesei. Mol Microbiol 84(6):1150–1164
Gruber S, Vaaje-Kolstad G, Matarese F, López-Mondéjar R, Kubicek CP, Seidl-Seiboth V (2011) Analysis of subgroup C of fungal chitinases containing chitin-binding and LysM modules in the mycoparasite Trichoderma atroviride. Glycobiology 21(1):122–133
Authors’ contributions
RL performed experiments, analyzed and interpreted experimental data and drafted the manuscript. GGT carried out the genome analysis and reviewed the manuscript. TH conceived the study, helped with the gene knockout program and reviewed the manuscript. JE and MS helped with the gene knockout program. PL conceived the study and reviewed the manuscript. BS conceived the study, interpreted experimental data and reviewed the manuscript. CPK conceived the study, interpreted experimental data and drafted the manuscript. All authors read and approved the final manuscript.
Acknowledgments
This work has been supported by the Federal Ministry of Science, Research and Economy (BMWFW), the Federal Ministry of Traffic, Innovation and Technology (bmvit), the Styrian Business Promotion Agency SFG, the Standortagentur Tirol and ZIT—Technology Agency of the City of Vienna through the COMET-Funding Program managed by the Austrian Research Promotion Agency FFG (ACIB-Grant No. 824186) and the EU-FP7 COST Action SeqAhead, EC Grant BM1006 (GGT).
Compliance with ethical guidelines
Competing interests The author(s) declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Table S1. List of strains used in and constructed within this study.
Additional file 2:
Figure S1. Pedigree of the T. reesei inbred strains RL1/A8-02 and RL2/A8-11 subjected to genome sequencing. “X” denotes crossing.
Additional file 3:
Table S2. Genes bearing sequence differences in T. reesei QM6a when compared to T. reesei RL1/A8-02 and RL2/A8-11 and selected for the gene knockout program to identify the cause of female sterility in T. reesei QM6a.
Additional file 4:
Figure S2. PCR confirmation of the exchange of the MAT1-2 locus in strain T. reesei QM6a by the MAT1-1 idiomorph amplified from strain C.P.K. 1282. The longer amplicon corresponds to the MAT1-1-2 gene and the shorter one to the MAT1-2-1 gene. To ascertain these results the strain QM6a (MAT1-1) was further tested in mating assays using strain CBS999.97 (MAT1-2) as mating partner.
Additional file 5:
Table S3. Fifty best BLAST hits (NCBI) for Trire2:67350 using the protein sequence of RL1/A8-02 derived from cDNA sequencing.
Additional file 6:
Figure S4. Alignment of the polyglutamine tracts from T. reesei QM6a with that of the orthologs of T. virens (Trive2:50286) and T. atroviride (Triat2:221851).
Additional file 7:
Figure S3. Nucleotide sequence of the ham5 locus from T. reesei QM6a and T. reesei RL1/A8-02 including the mutation being critical for female fertility. The sequence differences are marked in gray, introns are underlined. The critical mutation (G- > T) in the second intron of strain QM6a is indicated by a red box and the resulting stop codon is labeled in yellow.
Additional file 8:
Table S4. Sequences of the primers used for mating-type determination, the vector construction for the direct MAT locus replacement in T. reesei QM6a MAT1-2, the construction of the deletion cassettes for Trire2:59270 and Trire2:67350 and the plasmid construction for gene complementation in T. reesei QM6a MAT1-1.
Additional file 9:
Figure S5. Schematic drawing of the construction of the plasmid for the complementation of T. reesei QM6a with the wild-type allele of Trire2:67350 (ham5).
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Linke, R., Thallinger, G.G., Haarmann, T. et al. Restoration of female fertility in Trichoderma reesei QM6a provides the basis for inbreeding in this industrial cellulase producing fungus. Biotechnol Biofuels 8, 155 (2015). https://doi.org/10.1186/s13068-015-0311-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13068-015-0311-2