
Abstract
Background
Mycoparasitism, a lifestyle where one fungus is parasitic on another fungus, has special relevance when the prey is a plant pathogen, providing a strategy for biological control of pests for plant protection. Probably, the most studied biocontrol agents are species of the genus Hypocrea/Trichoderma.
Results
Here we report an analysis of the genome sequences of the two biocontrol species Trichoderma atroviride (teleomorph Hypocrea atroviridis) and Trichoderma virens (formerly Gliocladium virens, teleomorph Hypocrea virens), and a comparison with Trichoderma reesei (teleomorph Hypocrea jecorina). These three Trichoderma species display a remarkable conservation of gene order (78 to 96%), and a lack of active mobile elements probably due to repeat-induced point mutation. Several gene families are expanded in the two mycoparasitic species relative to T. reesei or other ascomycetes, and are overrepresented in non-syntenic genome regions. A phylogenetic analysis shows that T. reesei and T. virens are derived relative to T. atroviride. The mycoparasitism-specific genes thus arose in a common Trichoderma ancestor but were subsequently lost in T. reesei.
Conclusions
The data offer a better understanding of mycoparasitism, and thus enforce the development of improved biocontrol strains for efficient and environmentally friendly protection of plants.
Similar content being viewed by others


Background
Mycoparasitism is the phenomenon whereby one fungus is parasitic on another fungus, a lifestyle that can be dated to at least 400 million years ago by fossil evidence [1]. This has special relevance when the prey is a plant pathogen, providing a strategy for biological control of pests for plant protection ('biocontrol'). The movement toward environmentally friendly agricultural practices over the past two decades has thus accelerated research in the use of biocontrol fungi [2]. Probably the most studied biocontrol agents are species of the genus Hypocrea/Trichoderma, Trichoderma atroviride (Ta) and Trichoderma virens (Tv) - teleomorphs Hypocrea atroviridis and Hypocrea virens, respectively - being among the best mycoparasitic biocontrol agents used in agriculture [3]. The beneficial effects of Trichoderma spp. on plants comprise traits such as the ability to antagonize soil-borne pathogens by a combination of enzymatic lysis, secretion of antibiotics, and competition for space and substrates [4, 5]. In addition, it is now known that some Trichoderma biocontrol strains also interact intimately with plant roots, colonizing the outer epidermis layers, and acting as opportunistic, avirulent plant symbionts [6].
Science-based improvement of biocontrol agents for agricultural applications requires an understanding of the biological principles of their actions. So far, some of the molecular aspects - such as the regulation and role of cell wall hydrolytic enzymes and antagonistic secondary metabolites - have been studied in Trichoderma [3–5]. More comprehensive analyses (for example, by the use of subtractive hybridization techniques, proteomics or EST approaches) have also been performed with different Trichoderma species, but the interpretation of the data obtained is complicated by the lack of genome sequence information for the species used (reviewed in [7]).
Recently, the genome of another Trichoderma, Trichoderma reesei (Tr, teleomorph H. jecorina), which has a saprotrophic lifestyle and is an industrial producer of plant biomass hydrolyzing enzymes, has been sequenced and analyzed [8]. Here we report the genome sequencing and comparative analysis of two widely used biocontrol species of Trichoderma, that is, Ta and Tv. These two were chosen because they are distantly related to Tr [9] and represent well defined phylogenetic species [10, 11], in contrast to Trichoderma harzianum sensu lato, which is also commonly used in biocontrol but constitutes a complex of several cryptic species [12].
Results
Properties of the T. atroviride and T. virensgenomes
The genomes of Ta IMI 206040 and Tv Gv29-8 were sequenced using a whole genome shotgun approach to approximately eight-fold coverage and further improved using finishing reactions and gap closing. Their genome sizes were 36.1 (Ta) and 38.8 Mbp (Tv), and thus larger than the 34 Mbp determined for the genome of Tr [8]. Gene modeling, using a combination of homology and ab initio methods, yielded 11,865 gene models for Ta and 12,428 gene models for Tv, respectively (Table 1), both greater than the estimate for Tr (9,143). As shown in Figure 1, the vast majority of the genes (7,915) occur in all three Trichoderma species. Yet Tv and Ta contain about 2,756 and 2,510 genes, respectively, that have no true orthologue in any of the other species, whereas Tr has only 577 unique genes. Tv and Ta share 1,273 orthologues that are not present in Tr, which could thus be part of the factors that make Ta and Tv mycoparasites (for analysis, see below).

Distribution of orthologues of T. atroviride, T. virens and T. reesei. The Venn diagram shows the distribution found for the three species of Trichoderma.
With respect to other ascomycetes, Tr, Ta and Tv share 6,306/7,091, 6,515/7,549, and 6,564/7,733 orthologues with N. crassa and Gibberella zeae, respectively. Thus, approximately a third of the genes in the three Trichoderma species are not shared in even the relatively close relative G. zeae and are thus unique to Trichoderma.
Genome synteny
A comparison of the genomic organization of genes in Ta, Tv and Tr showed that most genes are in synteny: only 367 (4%) genes of Tr, but 2,515 (22%) of genes of Tv and 2,690 (21%) genes of Ta are located in non-syntenic regions (identified as a break in synteny by a series of three or more genes (Table 2); a global visual survey can be obtained at the genome websites of the three Trichoderma species (see Materials and methods) by clicking 'Synteny' and 'Dot Plot'). As observed for other fungal genomes [13–15], extensive rearrangements have occurred since the separation of these three fungi but with the prevalence of small inversions [16]. The numbers of the synteny blocks increased with their decreased size, compatible with the random breakage model [14] as in aspergilli [15, 17]. Sequence identity between syntenic orthologs was 70% (Tr versus Ta), 78% (Tr versus Tv), and 74% (Tv versus Ta), values that are similar to those calculated for aspergilli (for example, Aspergillus fumigatus versus Aspergillus niger (69%) and versus Aspergillus nidulans (68%) and comparable to those between fish and man [17, 18].
Transposons
A scan of the genome sequences with the de novo repeat finding program 'Piler' [19] - which can detect repetitive elements that are least 400 bp in length, have more than 92% identity and are present in at least three copies - was unsuccessful at detecting repetitive elements. The lack of repetitive elements detected in this analysis is unusual in filamentous fungi and suggests that, like the Tr genome [8], but unlike most other filamentous fungi, the Ta and Tv genomes lack a significant repetitive DNA component.
Because of the paucity of transposable elements (TEs) in the Trichoderma genomes, we wondered whether simple sequence repeats and minisatellite sequences may also be rare. To this end, we surveyed the genomes of the Trichoderma species using the program Tandem Repeat Finder [20]. We also included the genomes of three additional members of the Sordariomycetes and one of the Eurotiomycetes as reference (Table S1 in Additional file 1). Satellite DNA content varied from as little as 2,371 loci (0.53% of the genome) in A. nidulans to 9,893 (1.46% of the genome) in Neurospora crassa. Satellite DNA content of the Trichoderma genomes ranged from 5,249 (0.94%) in Ta to 7,743 (1.54%) in Tr. Since these values are within the range that we found in the reference species, we conclude that there is no unusual variation in the satellite DNA content of the Trichoderma genomes.
We also scanned the genomes with RepeatMasker and RepeatProteinMask [21] to identify sequences with similarity to known TEs from other organisms. Thereby, sequences with significant similarity to known TEs from other eukaryotes were identified (Table 3). In most cases, the TE families that we detected were fragmented and highly divergent from one another, suggesting that they did not arise from recent transposition events. Based on these results, we conclude that no extant, functional TEs exist in the Trichoderma genomes. The presence of ancient, degenerate TE copies suggests that Trichoderma species are occasionally subject to infection, or invasion by TEs, but that the TEs are rapidly rendered unable to replicate and rapidly accumulate mutations.
Evidence for the operation of repeat-induced point mutation in Trichoderma
The paucity of transposons in Trichoderma could be due to repeat-induced point mutation (RIP), a gene silencing mechanism. In N. crass a and many other filamentous fungi, RIP preferentially acts on CA dinucleotides, changing them to TA [22]. Thus, in sequences that have been subject to RIP, one should expect to find a decrease in the proportion of CA dinucleotides and its complement dinucleotide TG as well as a corresponding increase in the proportion of TA dinucleotides. The RIP indices TA/AT and (CA + TG)/(AC + GT) developed by Margolin et al. [22] can be used to detect sequences that have been subject to RIP. Sequences that have been subjected to RIP are expected to have a high TA/AT ratio and low (CA + TG)/(AC + GT) ratio, with values >0.89 and <1.03, respectively, being indicative of RIP [22, 23].
To identify evidence for RIP in the TE sequences, we computed RIP indices for four of the most prevalent TE families in each of the three species (Table 4). Since many of the sequences are very short, we computed the sum of the dinucleotide values within each TE family within each species, and used the sums to compute the RIP ratios. In only one of the 12 families did we find that both RIP indices were within the ranges that are typically used as criteria for RIP. Most of the TE sequences that we identified in the Trichoderma genomes are highly degenerate and have likely continued to accumulate mutations after the RIP process has acted on them. We suspect that these mutations have masked the underlying bias in dinucleotide frequencies, making the RIP indices ineffective at identifying the presence of RIP. To overcome this, we also prepared manually curated multiple sequence alignments of the TE families, selecting only sequences that had the highest sequence similarity, and thus should represent the most recent transposon insertion events in the genomes. We were able to prepare curated alignments for all four of the test TE families of Tr and Tv only for the long terminal repeat element Gypsy and the long interpersed nuclear element R1 in Ta (Table S2 in Additional file 1). Among DNA sequences that make up these ten alignments, we detected RIP indices within the parameters that are indicative of RIP in seven alignments. In addition, all seven alignments have high transition/transversion ratios, as is expected in sequences that are subject to RIP.
Finally, screening of the genome sequences of Tr, Ta and Tv identified orthologues of all genes required for RIP in N. crassa (Table 5).
Paralogous gene expansion in T. atroviride and T. virens
We used Marcov cluster algorithm (MCL) analysis [24] and included ten additional ascomycete genomes present in the Joint Genome Institute (JGI) genome database (including Eurotiomycetes, Sordariomycetes and Dothidiomycetes) to identify paralogous gene families that have become expanded either in all three Trichoderma species or only in the two mycoparasitic Trichoderma species. Forty-six such families were identified for all three species, of which 26 were expanded only in Ta and Tv. The largest paralogous expansions in all three Trichoderma species have occurred with genes encoding Zn(2)Cys(6) transcription factors, solute transporters of the major facilitator superfamily, short chain alcohol dehydrogenases, S8 peptidases and proteins bearing ankyrin domains (Table 6). The most expanded protein sets, however, were those that were considerably smaller in Tr (P < 0.05). These included ankyrin proteins with CCHC zinc finger domains, proteins with WD40, heteroincompatibility (HET) and NACHT domains, NAD-dependent epimerases, and sugar transporters.
Genes with possible relevance for mycoparasitism are expanded in Trichoderma
Mycoparasitism depends on a combination of events that include lysis of the prey's cell walls [3, 4, 7]. The necessity to degrade the carbohydrate armor of the prey's hyphae is reflected in an abundance of chitinolytic enzymes (composing most of the CAZy (Carbohydrate-Active enZYmes database) glycoside hydrolase (GH) family GH18 fungal proteins along with more rare endo-β-N-acetylglucosaminidases) and β-1,3-glucanases (families GH17, GH55, GH64, and GH81) in Trichoderma relative to other fungi. Family GH18, containing enzymes involved in chitin degradation, is also strongly expanded in Trichoderma, but particularly in Tv and Ta, which contain the highest number of chitinolytic enzymes of all described fungi (Table 7). Chitin is a substantial component of fungal cell walls and chitinases are therefore an integral part of the mycoparasitic attack [3, 25]. It is conspicuous that not only was the number of chitinolytic enzymes elevated but that many of these chitinases contain carbohydrate binding domains (CBMs). Mycoparasitic Trichoderma species are particularly rich in subgroup B chitinases that contain CBM1 modules, historically described as cellulose binding modules, but binding to chitin has also been demonstrated [26]. Tv and Ta each have a total of five CBM1-containing GH18 enzymes. Subgroup C chitinases possess CBM18 (chitin-binding) and CBM50 modules (also known as LysM modules; described as peptidoglycan- and chitin-binding modules). Interestingly, CBM50 modules in Trichoderma are found not only in chitinases but also frequently as multiple copies in proteins containing a signal peptide, but with no identifiable hydrolase domain. In most cases these genes can be found adjacent to chitinases in the genome.
Together with the expanded presence of chitinases, the number of GH75 chitosanases is also significantly expanded in all three analyzed Trichoderma species. As with plant pathogenic fungi [27, 28], we have also observed an expansion of plant cell wall degrading enzyme gene families. A full account of all the carbohydrate active enzymes is presented in Tables S3 to S8 in Additional file 1. Additional details about the Trichoderma CAZome (the genome-wide inventory of CAZy) are given in Chapter 1 of Additional file 2.
Another class of genes of possible relevance to mycoparasitism are those involved in the formation of secondary metabolites (Chapter 2 of Additional file 2). With respect to these, the three Trichoderma species contained a varying assortment of non-ribosomal peptide synthetases (NRPS) and polyketide synthases (PKS) (Table 8; see also Tables S9 and S10 in Additional file 1). While Tr (10 NRPS, 11 PKS and 2 NRPS/PKS fusion genes [8]) ranked at the lower end when compared to other ascomycetes, Tv exhibited the highest number (50) of PKS, NRPS and PKS-NRPS fusion genes, mainly due to the abundance of NRPS genes (28, twice as much as in other fungi). A phylogenetic analysis showed that this was due to recent duplications of genes encoding cyclodipeptide synthases, cyclosporin/enniatin synthase-like proteins, and NRPS-hybrid proteins (Figure S1 in Additional file 3). Most of the secondary metabolite gene clusters present in Tr were also found in Tv and Ta, but about half of the genes remaining in the latter two are unique for the respective species, and are localized on non-syntenic islands of the genome (see below). Within the NRPS, all three Trichoderma species contained two peptaibol synthases, one for short (10 to 14 amino acids) and one for long (18 to 25 amino acids) peptaibols. The genes encoding long peptaibol synthetase lack introns and produce an mRNA that is 60 to 80 kb long that encodes proteins of approximately 25,000 amino acids, the largest fungal proteins known.
Besides PKS and NRPS, Ta and Tv have further augmented their antibiotic arsenal with genes for cytolytic peptides such as aegerolysins, pore-forming cytolysins typically present in bacteria, fungi and plants, yeast-like killer toxins and cyanovirins (Chapter 2 of Additional file 2). In addition, we found two high molecular weight toxins in Ta and Tv that bear high similarity (E-value 0 for 97% coverage) to the Tc ('toxin complex') toxins of Photorhabdus luminescens, a bacterium that is mutualistic with entomophagous nematodes [29] (Table S11 in Additional file 1). Apart from Trichoderma, they are also present in G. zeae and Podospora anserina. Yet there may be several more secondary metabolite genes to be detected: Trichoderma species contain expanded arrays of cytochrome P450 CYP4/CYP19/CYP26 subfamilies (Table S12 in Additional file 1), and of soluble epoxide hydrolases that could act on the epoxides produced by the latter (Figure S2 in Additional file 3).
The Hypocrea/Trichoderma genomes also contain an abundant arsenal of putatively secreted proteins of 300 amino acids or less that contain at least four cysteine residues (small secreted cysteine-rich proteins (SSCPs); Chapter 3 of Additional file 2). They contained both unique and shared sets of SSCPs, with a higher complexity in Tv and Ta than in Tr (Table S13 in Additional file 1).
Genes present in T. atroviride and T. virens but not in T. reesei
As mentioned above, 1,273 orthologous genes were shared between Ta and Tv but absent from Tr. When the encoded proteins were classified according to their PFAM domains, fungal specific Zn(2)Cys(6) transcription factors (PF00172, PF04082) and solute transporters (PF07690, PF00083), all of unknown function, were most abundant (Table S14 in Additional file 1). However, the presence of several PFAM groups of oxidoreductases and monooxygenases, and of enzymes for AMP activation of acids, phosphopathetheine attachment and synthesis of isoquinoline alkaloids was also intriguing. This suggests that Ta and Tv may contain an as yet undiscovered reservoir of secondary metabolites that may contribute to their success as mycoparasites.
We also annotated the 577 genes that are unique in T. reesei: the vast majority of them (465; 80.6%) encoded proteins of unknown function or proteins with no homologues in other fungi. The remaining identified 112 genes exhibited no significant abundance in particular groups, except for four Zn(2)Cys(6) transcription factors, four ankyrins, four HET-domain proteins and three WD40-domain containing proteins.
Evolution of the non-syntenic regions
A search for overrepresentation of PFAM domains and Gene Ontology terms in the non-syntenic regions described above revealed that all retroposon hot spot repeat domains [30] are found in the non-syntenic regions. In most eukaryotes, these regions are located in subtelomeric areas that exhibit a high recombination frequency [31]. In addition, the genes for the protein families in Tv and Ta that were significantly more abundant compared to Tr were enriched in the non-syntenic areas (Table 9). In addition, the number of paralogous genes was significantly increased in the non-syntenic regions. We considered three possible explanations for this: the non-syntenic genes were present in the last common ancestor of all three Trichoderma species but were then selectively and independently lost; the non-syntenic areas arose from the core genome by duplication and divergence during evolution of the genus Trichoderma; and the non-syntenic genes were acquired by horizontal transfer. To distinguish among these hypotheses for their origin, we compared the sequence characteristics of the genes in the non-syntenic regions to those present in the syntenic regions in Trichoderma and genes in other filamentous fungi. We found that the majority (>78%) of the syntenic as well as non-syntenic encoded proteins have their best BLAST hit to other ascomycete fungi, indicating that the non-syntenic regions are also of fungal origin. Also, a high number of proteins encoded in the non-syntenic regions of Ta and Tv have paralogs in the syntenic region. Finally, codon usage tables and codon adaptation index analysis [32] indicate that the non-syntenic genes exhibit a similar codon usage (Figure S3 in Additional file 3). Taken together, the most parsimonious explanation for the presence of the paralogous genes in Ta and Tv is that the non-syntenic genes arose by gene duplication within a Trichoderma ancestor, followed by gene loss in the three lineages, which was much stronger in Tr.
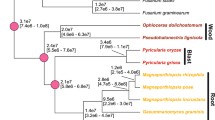
Tr, Ta and Tv each occupy very diverse phylogenetic positions in the genus Trichoderma, as shown by a Bayesian rpb2 tree of 110 Trichoderma taxa (Figure 2). In order to determine which of the three species more likely resembles the ancestral state of Trichoderma, we performed a Bayesian phylogenetic analysis [33] using a concatenated set of 100 proteins that were encoded by orthologous genes in syntenic areas in the three Trichoderma species and also G. zeae and Chaetomium globosum. The result (Figure 2) shows that Ta occurs in a well-supported basal position to Tv and Tr. These data indicate that Ta resembles the more ancient state of Trichoderma and that both Tv and Tr evolved later. The lineage to Tr thus appears to have lost a significant number of genes present in Ta and maintained in Tv. The long genetic distance of Tr further suggests that it was apparently evolving faster then Ta and Tv since the time of divergence.

Mycoparasitism is an ancient life style of Trichoderma. (a) Position of Ta, Tv and Tr within the genus Hypocrea/Trichoderma. The positions of Tr, Tv and Ta are 4, 29 and 97, respectively - shown in bold), and a few hallmark species are given by their names. For the identities of the other species, see the gene accession numbers (Materials and methods). (b) Bayesian phylogram based on the analysis of amino acid sequences of 100 orthologous syntenic proteins (MCMC, 1 million generations, 10,449 characters) in Tr, Tv, Ta, Gibberella zeae and Chaetomium globosum. Circles above nodes indicate 100% posterior probabilities and significant bootstrap coefficients. The numbers in the boxes between (a) and (b) indicate the genome sizes and gene counts and percentage net gain regarding Ta. Photoplates show the mycoparasitic reaction after the contact between Trichoderma species and Rhizoctonia solani. Trichoderma species are always on the left side; dashed lines indicate the position of Trichoderma overgrowth of R. solani.
To test this assumption, we compared the evolutionary rates of the 100 orthologous and syntenic gene families between the three Trichoderma species. The median values of the evolutionary rates (Ks and Ka) of Ta-Tr and Tv-Tr were all significantly higher (1.77 and 1.47, and 1.33 and 1.19, respectively) than those of Ta-Tv (1.13 and 0.96; all P values <0.05 by the two-tailed Wilcoxon rank sum test). This result supports the above suggestion that Tr has been evolving faster than Ta and Tv.
Discussion
Comparison of the genomes of two mycoparasitic and one saprotrophic Trichoderma species revealed remarkable differences: in contrast to the genomes of other multicellular ascomycetes, such as aspergilli [15, 17], those of Trichoderma appear to be have the highest level of synteny of all genomes investigated (96% for Tr and still 78/79% for Tv and Ta, respectively, versus 68 to 75% in aspergilli), and most of the differences between Ta and Tv versus Tr or other ascomycetes occur in the non-syntenic areas. Nevertheless, at a molecular level the three species are as distant from each other as apes from Pices (fishes) or Aves (birds) [17], suggesting a mechanism maintaining this high genomic synteny. Espagne et al. [13] proposed that a discrepancy of genome evolution between P. anserina, N. crassa and the aspergilli and saccharomycotina yeasts is based on the difference between heterothallic and homothallic fungi: in heterothallics the presence of interchromosomal translocation could result in chromosome breakage during meiosis and reduced fertility, whereas homothallism allows translocations to be present in both partners and thus have fewer consequences on fertility. Since Trichoderma is heterothallic [34], this explanation is also applicable to it. However, another mechanism, meiotic silencing of unpaired DNA [35] - which has also been proposed for P. anserina [13], and which eliminates progeny in crosses involving rearranged chromosomes in one of the partners - may not function in Trichoderma because one of the essential genes (SAD2 [36]) is missing.
Our data also suggest that the ancestral state of Hypocrea/Trichoderma was mycoparasitic. This supports an earlier speculation [37] that the ancestors of Trichoderma were mycoparasites on wood-degrading basidiomycetes and acquired saprotrophic characteristics to follow their prey into their substrate. Indirect evidence for this habitat shift in Tr was also presented by Slot and Hibbett [38], who demonstrated that Tr - after switching to a specialization on a nitrogen-poor habitat (decaying wood) - has acquired a nitrate reductase gene (which was apparently lost earlier somewhere in the Sordariomycetes lineage) by horizontal gene transfer from basidiomycetes.
Furthermore, the three Trichoderma species have the lowest number of transposons reported so far. This is unusual for filamentous fungi, as most species contain approximately 10 to 15% repetitive DNA, primarily composed of TEs. A notable exception is Fusarium graminearum [27], which, like the Trichoderma species, contains less than 1% repetitive DNA [8]. The paucity of repetitive DNA may be attributed to RIP, which has been suggested to occur in Tr [8] and for which we have here provided evidence that it also occurs in Ta and Tv. It is likely that this process also contributes to prevent the accumulation of repetitive elements.
The gene inventory detected in the three Trichoderma species reveals new insights into the physiology of this fungal genus: the strong expansion of genes for solute transport, oxidoreduction, and ankyrins (a family of adaptor proteins that mediate the anchoring of ion channels or transporters in the plasma membrane [39]) could render Trichoderma more compatible in its habitat (for example, to successfully compete with the other saprotrophs for limiting substrates). In addition, the expansion of WD40 domains acting as hubs in cellular networks [40] could aid in more versatile metabolism or response to stimuli. These features correlate well with a saprotrophic lifestyle that makes use of plant biomass that has been pre-degraded by earlier colonizers. The expansion of HET proteins (proteins involved in vegetative incompatibility specificity) on the other hand suggests that Trichoderma species may frequently encounter related yet genetically distinct individuals. In fact, the presence of several different Trichoderma species can be detected in a single soil sample [41]. Unfortunately, vegetative incompatibility has not yet been investigated in any Trichoderma species, and based on the current data, should be a topic of future research.
Finally, the abundance of SSCPs in Trichoderma may be involved in rhizosphere competence: the genome of the ectomycorrhizal basidiomycete Laccaria bicolor also encodes a large set of SSCPs, which accumulate in the hyphae that colonize the host root [42].
Gene expansions in Tv and Ta that do not occur in Tr may comprise genes specific for mycoparasitism. As a prominent example, proteases have expanded in Ta and Tv, supporting the hypothesis that the degradation of proteins is a major trait of mycoparasites [43]. Likewise, the increase in chitinolytic enzymes and some ß-glucanase-containing GH families is remarkable and illustrates the importance of destruction of the prey's cell wall in this process. With respect to the chitinases, the expansion of those bearing CBM50 modules was particularly remarkable: proteins containing these modules were recently classified into several different groups by de Jonge and Thomma [44]. Proteins that consist solely of CBM50 modules are type-A LysM proteins, and there is evidence for the role of these as virulence factors in plant pathogenic fungi. The high numbers of LysM proteins that are found in Trichoderma, however, indicate other/additional roles for these proteins in fungal biology that are not understood yet. Also, the expansion of the GH75 chitosanases was intriguing: chitosan is a partially deacetylated derivative of chitin and, depending on the fungal species and the growth conditions, in mature fungal cell walls chitin is partially deacetylated. It has also been reported that fungi deacetylate chitin as a defense mechanism [45, 46]. Chitosan degradation may therefore be a relevant aspect of mycoparasitism and fungal cell wall degradation that has also not been regarded yet. Overall, the carbohydrate-active enzyme machinery present in Trichoderma is compatible with saprophytic behavior but, interestingly, the set of enzymes involved in the degradation of 'softer' plant cell wall components, such as pectin, is reduced. A possible plant symbiotic relationship [3] might rely on a mycoparasitic capacity along with a reduced specificity for pectin, minimizing the plant defense reaction.
Although the genes encoding proteins for the synthesis of typical fungal secondary metabolites (PKS, NRPS, PKS-NRPS) are also abundant, they are not significantly more expanded than in some other fungi. An exception is Tv and its 28 NRPS genes. However, our genome analysis revealed also a high number of oxidoreductases, cytochrome P450 oxidases, and other enzymes that could be part of as yet unknown pathways for the synthesis of further secondary metabolites. In support of this, several of these genes were found to be clustered in the genome (data not shown), and were more abundant in the two mycoparasitic species Ta and Tv. Together with the expanded set of oxidoreductases, monooxygenases, and enzymes for AMP activation of acids, phosphopathetheine attachment, and synthesis of isoquinoline alkaloids in Ta and Tv, these genes may define new secondary metabolite biosynthetic routes.
Conclusions
Our comparative genome analysis of the three Trichoderma species now opens new opportunities for the development of improved and research-driven strategies to select and improve Trichoderma species as biocontrol agents. The availability of the genome sequences published in this study, as well as of several pathogenic fungi and their potential host plants (for example, [47]) provides a challenging opportunity to develop a deeper understanding of the underlying processes by which Trichoderma interacts with plant pathogens in the presence of living plants within their ecosystem.
Materials and methods
Genome sequencing and assembly
The genomes of T. virens and T. atroviride each were assembled from shotgun reads using the JGI (USA Department of Energy) assembler Jazz (see Table S15 in Additional file 1 for summary of assembly statistics). Each genome was annotated using the JGI Annotation pipeline, which combines several gene prediction, annotation and analysis tools. Genes were predicted using Fgenesh [48], Fgenesh+ [49], and Genewise programs [50]. ESTs from each species (Chapter 4 of Additional file 2) were clustered and either assembled and converted into putative full-length genes directly mapped to genomic sequence or used to extend predicted gene models into full-length genes by adding 5' and/or 3' untranslated regions to the models. From multiple gene models predicted at each locus, a single representative model was chosen based on homology and EST support and used for further analysis. Gene model characteristics and support are summarized in Tables S16 and S17 in Additional file 1.
All predicted gene models were functionally annotated by homology to annotated genes from a NCBI non-redundant set and classified according to Gene Ontology [51], eukaryotic orthologous groups (KOGs) [52], and Kyoto Encyclopedia of Genes and Genomes (KEGG) metabolic pathways [53]. See Tables S18 and S19 in Additional file 1 for a summary of the functional annotation. Automatically predicted genes and functions were further refined by user community-wide manual curation efforts using web-based tools at [54, 55]. The latest version gene set containing manually curated genes is called GeneCatalog.
Assembly and annotation data for Tv and Ta are available through JGI Genome Portals homepage at [54, 55]. The genome assemblies, predicted gene models, and annotations were deposited at GenBank under project accessions [GenBank: ABDF00000000 and ABDG00000000], respectively. GenBank public release of the data described in this paper should coincide with the manuscript publication date.
Genome similarity analysis and genomic synteny
Orthologous genes, as originally defined, imply a reflection of the history of species. In recent years, many studies have examined the concordance between orthologous gene trees and species trees in bacteria. With the purpose of identifying all the orthologous gene pairs for the three Trichoderma species, a best bidirectional blast hit approach as described elsewhere [56, 57] was performed, using the predicted translated gene models for each of the three species as pairwise comparison sets. The areas of relationship known as syntenic regions or syntenic blocks are anchored with orthologs (calculated as mutual best hits or bi-directional best hits) between the two genomes in question, and are built by controlling for the minimum number of genes, minimum density, and maximum gap (genes not from the same genome area) compared with randomized data as described in [56]. While this technique may cause artificial breaks, it highlights regions that are dynamic and picking up a large number of insertions or duplications.
Orthologous and paralogous gene models were identified by first using BLAST to find all pairwise matches between the resulting proteins from the gene models. The pairwise matches from BLAST were then clustered into groups of paralogs using MCL [58]. In parallel we applied orthoMCL [59] to the same pairwise matches to identify the proteins that were orthologous in all of the three genomes. By subtracting all the proteins that were identified as orthologs from the groups of paralogs and unique genes, we were left with only the protein products of gene models that have expanded since the most recent common ancestor (MRCA) of the three Trichoderma genomes. We then calculated the P-value under the null hypothesis that the number of non-orthologous genes that are non-syntenic is less than the number of non-orthologous genes that are syntenic.
Identification of transposable elements
We scanned the Trichoderma genomes with the de novo repeat finding program Piler [19]. Next, we searched for sequences with similarity to known repetitive elements from other eukaryotes with the program RepeatMasker [21] using all eukaryotic repetitive elements in the RepBase (version 13.09) database. After masking repetitive sequences that matched the DNA sequence of known repetitive elements, we scanned the masked genome sequences with RepeatProteinMask (a component of the RepeatMasker application). This search located additional degenerate repetitive sequences with similarity to proteins encoded by TEs in the RepBase database.
CAZome identification and analysis
All protein models for Ta and Tv were compared against the set of libraries of modules derived from CAZy [60, 61]. The identified proteins were subjected to manual analysis for correction of the protein models, for full modular annotation and for functional inference against a library of experimentally characterized enzymes. Comparative analysis was made by the enumeration of all modules identified in the three Trichoderma species and 14 other published fungal genomes.
Phylogenetic and evolutionary analyses
One-hundred genes were randomly selected from Ta, Tv, Tr and C. globosum based on their property to fulfill two requirements: they were in synteny in all four genomes, and they were true orthologues (no other gene encoding a protein with amino acid similarity >50% present). After alignment, the concatenated 10,449 amino acids were subjected to Bayesian analysis [33] using 1 million generations. The respective cDNA sequences (31,347 nucleotides) were also concatenated, and Ks/Ka ratios determined using DNASp5 [62]. The same file was also used to determine the codon adaptation index [32]. In addition, 80 non-syntenic genes were also selected randomly for this purpose.
The species phylogram of Trichoderma/Hypocrea was constructed by Bayesian analysis of partial exon nucleotide sequences (824 total characters from which 332 were parsimony-informative) of the rpb2 gene (encoding RNA polymerase B II) from 110 ex-type strains, thereby spanning the biodiversity of the whole genus. The tree was obtained after 5 million MCMC generations sampled for every 100 trees, using burnin = 1200 and applying the general time reversible model of nucleotide substitution. The NCBI ENTREZ accession numbers are: 1 [HQ260620]; 3 [DQ08724]; 4 [HM182969]; 5 [HM182984]; 6 [HM182965]; 7 [AF545565]; 8 [AF545517]; 16 [FJ442769]; 17 [AY391900]; 18 [FJ179608]; 19 [FJ442715]; 20 [FJ442771]; 21 [AY391945]; 22 [EU498358]; 23 [DQ834463]; 24 [FJ442725]; 25 [AF545508]; 26 [AY391919]; 27 [AF545557]; 28 [AF545542]; 29 [FJ442738]; 30 [AF545550]; 31 [AY391909]; 32 [AF545516]; 33 [AF545518]; 34 [AF545512]; 35 [AF545510]; 36 [AF545514]; 37 [AY391921]; 38 [AF545513]; 39 [AY391954]; 40 [AY391944]; 41 [AF545534]; 42 [AY391899]; 43 [AY391907]; 44 [AF545511]; 45 [AY391929]; 46 [AF545540]; 47 [AY391958]; 48 [AY391924]; 49 [AF545515]; 50 [AY391957]; 51 [AF545551]; 52 [AF545522]; 53 [FJ442714]; 54 [AF545509]; 55 [AY391959]; 56 [DQ087239]; 57 [AF545553]; 58 [AF545545]; 59 [DQ835518]; 60 [DQ835521]; 61 [DQ835462]; 62 [DQ835465]; 63 [DQ835522]; 64 [AF545560]; 65 [DQ835517]; 66 [DQ345348]; 67 [AF545520]; 68 [DQ835455]; 69 [AF545562]; 70 [AF545563]; 71 [DQ835453]; 72 [FJ179617]; 73 [DQ859031]; 74 [EU341809]; 75 [FJ179614]; 76 [DQ087238]; 77 [AF545564]; 78 [FJ179601]; 79 [FJ179606]; 80 [FJ179612]; 81 [FJ179616]; 82 [EU264004]; 83 [FJ150783]; 84 [FJ150767]; 85 [FJ150786]; 86 [EU883559]; 87 [FJ150785]; 88 [EU248602]; 89 [EU241505]; 90 [FJ442762]; 91 [FJ442741]; 92 [FJ442783]; 93 [EU341805]; 94 [FJ442723]; 95 [FJ442772]; 96 [EU2415023]; 97 [EU341801]; 98 [EU248600]; 99 [EU341808]; 100 [EU3418033]; 101 [EU2485942]; 102 [AF545519]; 103 [EU248603]; 104 [EU248607]; 105 [EU341806]; 106 [DQ086150]; 107 [DQ834460]; 108 [EU711362]; 109 [EU883557]; 110 [FJ150790].
Abbreviations
- CAZy:
-
Carbohydrate-Active enZYmes
- CBM:
-
carbohydrate binding module
- EST:
-
expressed sequence tag
- GH:
-
glycosyl hydrolase
- HET:
-
heteroincompatibility
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- KOG:
-
clusters of eukaryotic orthologous groups
- NRPS:
-
non-ribosomal peptide synthase
- PKS:
-
polyketide synthase
- RIP:
-
repeat-induced point mutation
- SSCP:
-
small secreted cysteine-rich protein
- Ta :
-
Trichoderma atroviride
- TE:
-
transposable element
- Tr:
-
Trichoderma reesei
- Tv:
-
Trichoderma virens.
References
Taylor TN, Hass H, Kerp H, Krings M, Hanlin RT: Perithecial ascomycetes from the 400 million year old Rhynie chert: an example of ancestral polymorphism. Mycologia. 2005, 97: 269–285. 10.3852/mycologia.97.1.269.
Vincent C, Goettel MS, Lazarovits G: Biological Control: A Global Perspective: Case Studies from Around the World. 2007, Wallingford, UK: CAB International
Harman GE, Howell CR, Viterbo A, Chet I, Lorito M: Trichoderma species-opportunistic, avirulent plant symbionts. Nat Rev Microbiol. 2004, 2: 43–56. 10.1038/nrmicro797.
Howell CR: Mechanisms employed by Trichoderma species in the biological control of plant diseases: the history and evolution of current concepts. Plant Disease. 2003, 87: 4–10. 10.1094/PDIS.2003.87.1.4.
Harman GE: Overview of mechanisms and uses of Trichoderma spp. Phytopathology. 2006, 96: 190–194. 10.1094/PHYTO-96-0190.
Shoresh M, Harman GE, Mastouri F: Induced systemic resistance and plant responses to fungal biocontrol agents. Annu Rev Phytopathol. 2010, 48: 21–43. 10.1146/annurev-phyto-073009-114450.
Lorito M, Woo SL, Harman GE, Monte E: Translational research on Trichoderma: from 'omics to the field. Annu Rev Phytopathol. 2010, 48: 395–417. 10.1146/annurev-phyto-073009-114314.
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D, Danchin EG, Grigoriev IV, Harris P, Jackson M, Kubicek CP, Han CS, Ho I, Larrondo LF, de Leon AL, Magnuson JK, Merino S, Misra M, Nelson B, Putnam N, Robbertse B, Salamov AA, Schmoll M, Terry A, Thayer N, Westerholm-Parvinen A, et al: Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotechnol. 2008, 26: 553–560. 10.1038/nbt1403.
Druzhinina IS, Kopchinskiy A, Kubicek CP: The first one hundred Trichoderma species characterized by molecular data. Mycoscience. 2006, 47: 55–64. 10.1007/s10267-006-0279-7.
Chaverri P, Samuels GJ, Stewart EL: Hypocrea virens sp. nov., the teleomorph of Trichoderma virens. Mycologia. 2001, 93: 1113–1124. 10.2307/3761672.
Dodd SL, Lieckfeldt E, Samuels GJ: Hypocrea atroviridis sp. nov., the teleomorph of Trichoderma atroviride. Mycologia. 2003, 95: 27–40. 10.2307/3761959.
Druzhinina IS, Kubicek CP, Komoń-Zelazowska M, Mulaw TB, Bissett J: The Trichoderma harzianum demon: complex speciation history resulting in coexistence of hypothetical biological species, recent agamospecies and numerous relict lineages. BMC Evol Biol. 2010, 10: 94-10.1186/1471-2148-10-94.
Espagne E, Lespinet O, Malagnac F, Da Silva C, Jaillon O, Porcel BM, Couloux A, Aury JM, Ségurens B, Poulain J, Anthouard V, Grossetete S, Khalili H, Coppin E, Déquard-Chablat M, Picard M, Contamine V, Arnaise S, Bourdais A, Berteaux-Lecellier V, Gautheret D, de Vries RP, Battaglia E, Coutinho PM, Danchin EG, Henrissat B, Khoury RE, Sainsard-Chanet A, Boivin A, Pinan-Lucarré B, et al: The genome sequence of the model ascomycete fungus Podospora anserina. Genome Biol. 2008, 9: R77-10.1186/gb-2008-9-5-r77.
Fischer G, Rocha EP, Brunet F, Vergassola M, Dujon B: Highly variable rates of genome rearrangements between hemiascomycetous yeast lineages. PLoS Genet. 2006, 2: e32-10.1371/journal.pgen.0020032.
Galagan JE, Calvo SE, Cuomo C, Ma LJ, Wortman JR, Batzoglou S, Lee SI, Baştürkmen M, Spevak CC, Clutterbuck J, Kapitonov V, Jurka J, Scazzocchio C, Farman M, Butler J, Purcell S, Harris S, Braus GH, Draht O, Busch S, D'Enfert C, Bouchier C, Goldman GH, Bell-Pedersen D, Griffiths-Jones S, Doonan JH, Yu J, Vienken K, Pain A, Freitag M, et al: Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature. 2005, 438: 1105–1115. 10.1038/nature04341.
Seoighe C, Federspiel N, Jones T, Hansen N, Bivolarovic V, Surzycki R, Tamse R, Komp C, Huizar L, Davis RW, Scherer S, Tait E, Shaw DJ, Harris D, Murphy L, Oliver K, Taylor K, Rajandream MA, Barrell BG, Wolfe KH: Prevalence of small inversions in yeast gene order evolution. Proc Natl Acad Sci USA. 2000, 97: 14433–14437. 10.1073/pnas.240462997.
Fedorova ND, Khaldi N, Joardar VS, Maiti R, Amedeo P, Anderson MJ, Crabtree J, Silva JC, Badger JH, Albarraq A, Angiuoli S, Bussey H, Bowyer P, Cotty PJ, Dyer PS, Egan A, Galens K, Fraser-Liggett CM, Haas BJ, Inman JM, Kent R, Lemieux S, Malavazi I, Orvis J, Roemer T, Ronning CM, Sundaram JP, Sutton G, Turner G, Venter JC, et al: Genomic islands in the pathogenic filamentous fungus Aspergillus fumigatus. PLoS Genet. 2008, 4: e1000046-10.1371/journal.pgen.1000046.
Nadeau J, Taylor B: Lengths of chromosomal segments conserved since divergence of man and mouse. Proc Natl Acad Sci USA. 1984, 81: 814–818. 10.1073/pnas.81.3.814.
Edgar RC, Myers EW: PILER: identification and classification of genomic repeats. Bioinformatics. 2005, 21 (Suppl 1): i152–i158. 10.1093/bioinformatics/bti1003.
Benson G: Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 1999, 27: 573–580. 10.1093/nar/27.2.573.
RepeatMasker Open-3.0. [https://doi.org/www.repeatmasker.org]
Margolin BS, Garrett-Engele PW, Stevens JN, Fritz DY, Garrett-Engele C, Metzenberg RL, Selker EU: A methylated Neurospora 5S rRNA pseudogene contains a transposable element inactivated by repeat-induced point mutation. Genetics. 1998, 149: 1787–1797.
Selker EU, Tountas NA, Cross SH, Margolin BS, Murphy JG, Bird AP, Freitag M: The methylated component of the Neurospora crassa genome. Nature. 2003, 422: 893–897. 10.1038/nature01564.
Enright AJ, van Dongen S, Ouzounis CA: An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30: 1575–1584. 10.1093/nar/30.7.1575.
Seidl V: Chitinases of filamentous fungi: a large group of diverse proteins with multiple physiological functions. Fungal Biol Rev. 2008, 22: 36–42. 10.1016/j.fbr.2008.03.002.
Limon MC, Chacón MR, Mejías R, Delgado-Jarana J, Rincón AM, Codón AC, Benítez T: Increased antifungal and chitinase specific activities of Trichoderma harzianum CECT 2413 by addition of a cellulose binding domain. Appl Microbiol Biotechnol. 2004, 64: 675–682. 10.1007/s00253-003-1538-6.
Cuomo CA, Güldener U, Xu JR, Trail F, Turgeon BG, Di Pietro A, Walton JD, Ma LJ, Baker SE, Rep M, Adam G, Antoniw J, Baldwin T, Calvo S, Chang YL, Decaprio D, Gale LR, Gnerre S, Goswami RS, Hammond-Kosack K, Harris LJ, Hilburn K, Kennell JC, Kroken S, Magnuson JK, Mannhaupt G, Mauceli E, Mewes HW, Mitterbauer R, Muehlbauer G, et al: The Fusarium graminearum genome reveals a link between localized polymorphism and pathogen specialization. Science. 2007, 317: 1400–1402. 10.1126/science.1143708.
Dean RA, Talbot NJ, Ebbole DJ, Farman ML, Mitchell TK, Orbach MJ, Thon M, Kulkarni R, Xu JR, Pan H, Read ND, Lee YH, Carbone I, Brown D, Oh YY, Donofrio N, Jeong JS, Soanes DM, Djonovic S, Kolomiets E, Rehmeyer C, Li W, Harding M, Kim S, Lebrun MH, Bohnert H, Coughlan S, Butler J, Calvo S, Ma LJ, et al: The genome sequence of the rice blast fungus Magnaporthe grisea. Nature. 2005, 434: 980–986. 10.1038/nature03449.
Münch A, Stingl L, Jung K, Heermann R: Photorhabdus luminescens genes induced upon insect infection. BMC Genomics. 2008, 9: 229-10.1186/1471-2164-9-229.
Wellinger RJ, Sen H: The DNA structures at the ends of eukaryotic chromosomes. Eur J Cancer. 1997, 33: 735–749. 10.1016/S0959-8049(97)00067-1.
Freitas-Junior LH, Bottius E, Pirrit LA, Deitsch KW, Scheidig C, Guinet F, Nehrbass U, Wellems TE, Scherf A: Frequent ectopic recombination of virulence factor genes in telomeric chromosome clusters of P. falciparum. Nature. 2000, 407: 1018–1022. 10.1038/35039531.
Sharp PM, Li WH: The codon adaptation index - a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 1987, 15: 1281–1295. 10.1093/nar/15.3.1281.
Yang Z, Rannala B: Bayesian phylogenetic inference using DNA sequences: a Markov Chain Monte Carlo Method. Mol Biol Evol. 1997, 14: 717–724.
Seidl V, Seibel C, Kubicek CP, Schmoll M: Sexual development in the industrial workhorse Trichoderma reesei. Proc Natl Acad Sci USA. 2009, 106: 13909–13914. 10.1073/pnas.0904936106.
Shiu PK, Metzenberg RL: Meiotic silencing by unpaired DNA: properties, regulation and suppression. Genetics. 2002, 161: 1483–1495.
Borkovich KA, Alex LA, Yarden O, Freitag M, Turner GE, Read ND, Seiler S, Bell-Pedersen D, Paietta J, Plesofsky N, Plamann M, Goodrich-Tanrikulu M, Schulte U, Mannhaupt G, Nargang FE, Radford A, Selitrennikoff C, Galagan JE, Dunlap JC, Loros JJ, Catcheside D, Inoue H, Aramayo R, Polymenis M, Selker EU, Sachs MS, Marzluf GA, Paulsen I, Davis R, Ebbole DJ, et al: Lessons from the genome sequence of Neurospora crassa: tracing the path from genomic blueprint to multicellular organism. Microbiol Mol Biol Rev. 2004, 68: 1–108. 10.1128/MMBR.68.1.1-108.2004.
Rossmann AY, Samuels GJ, Rogerson CT, Lowen R: Genera of Bionectriaceae, Hypocreaceae and Nectriaceae (Hypocreales, Ascomycetes). Stud Mycol. 1999, 42: 1–83.
Slot JC, Hibbett DS: Horizontal transfer of a nitrate assimilation gene cluster and ecological transitions in fungi: a phylogenetic study. PLoS One. 2007, 2: e1097-10.1371/journal.pone.0001097.
Bennett V, Baines AJ: Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001, 81: 1353–1392.
Stirnimann CU, Petsalaki E, Russell RB, Müller CW: WD40 proteins propel cellular networks. Trends Biochem Sci. 2010, 35: 565–574. 10.1016/j.tibs.2010.04.003.
Migheli Q, Balmas V, Komoñ-Zelazowska M, Scherm B, Fiori S, Kopchinskiy AG, Kubicek CP, Druzhinina IS: Soils of a Mediterranean hot spot of biodiversity and endemism (Sardinia, Tyrrhenian Islands) are inhabited by pan-European, invasive species of Hypocrea/Trichoderma. Environ Microbiol. 2009, 11: 35–46. 10.1111/j.1462-2920.2008.01736.x.
Martin F, Aerts A, Ahrén D, Brun A, Danchin EG, Duchaussoy F, Gibon J, Kohler A, Lindquist E, Pereda V: The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature. 2008, 452: 88–92. 10.1038/nature06556.
Seidl V, Song L, Lindquist E, Gruber S, Koptchinskiy A, Zeilinger S, Schmoll M, Martínez P, Sun J, Grigoriev I, Herrera-Estrella A, Baker SE, Kubicek CP: Transcriptomic response of the mycoparasitic fungus Trichoderma atroviride to the presence of a fungal prey. BMC Genomics. 2009, 10: 567-10.1186/1471-2164-10-567.
de Jonge R, Thomma BP: Fungal LysM effectors: extinguishers of host immunity?. Trends Microbiol. 2009, 17: 151–154. 10.1016/j.tim.2009.01.002.
Baker LG, Specht CA, Donlin MJ, Lodge JK: Chitosan, the deacetylated form of chitin, is necessary for cell wall integrity in Cryptococcus neoformans. Eukaryotic Cell. 2007, 6: 855–862. 10.1128/EC.00399-06.
El Gueddari NE, Rauchhaus U, Moerschbacher BM, Deising HB: Developmentally regulated conversion of surface-exposed chitin to chitosan in cell walls of plant pathogenic fungi. New Phytol. 2002, 156: 103–111. 10.1046/j.1469-8137.2002.00487.x.
diArk: a resource for eukaryotic genome research. [https://doi.org/www.diark.org/diark/search]
Salamov AA, Solovyev VV: Ab initio gene finding in Drosophila genomic DNA. Genome Res. 2000, 10: 516–522. 10.1101/gr.10.4.516.
Birney E, Durbin R: Using GeneWise in the Drosophila annotation experiment. Genome Res. 2000, 10: 547–548. 10.1101/gr.10.4.547.
Zdobnov EM, Apweiler R: InterProScan - an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 2001, 17: 847–848. 10.1093/bioinformatics/17.9.847.
Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G: Gene Ontology: tool for the unification of biology The Gene Ontology Consortium. Nat Genet. 2000, 25: 25–29. 10.1038/75556.
Koonin EV, Fedorova ND, Jackson JD, Jacobs AR, Krylov DM, Makarova KS, Mazumder R, Mekhedov SL, Nikolskaya AN, Rao BS, Rogozin IB, Smirnov S, Sorokin AV, Sverdlov AV, Vasudevan S, Wolf YI, Yin JJ, Natale DA: A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5: R7-10.1186/gb-2004-5-2-r7.
Kanehisa M, Goto S, Kawashima S, Okuno Y, Hattori M: The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32: D277–D280. 10.1093/nar/gkh063.
Trichoderma virens Gv29-8 v2.0. [https://doi.org/www.jgi.doe.gov/Tvirens]
Trichoderma atroviride v2.0[. [https://doi.org/www.jgi.doe.gov/Tatroviride]
Castillo-Ramirez S, Gonzalez V: Factors affecting the concordance between orthologous gene trees and species tree in bacteria. BMC Evol Biol. 2008, 8: 300-10.1186/1471-2148-8-300.
Moreno-Hagelsieb C, Janga SC: Operons and the effect of genome redundancy in deciphering functional relationships using phylogenetic profiles. Proteins. 2008, 70: 344–352.
MCL - a cluster algorithm for graphs. [https://doi.org/micans.org/mcl/]
Li L, Stoeckert CJ, Roos DS: OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 2003, 13: 2178–2189. 10.1101/gr.1224503.
CAZY - Carbohydrate-Active EnZYmes database. [https://doi.org/www.cazy.org]
Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B: The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 2009, 37: D233–D238. 10.1093/nar/gkn663.
Librado P, Rozas J: DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics. 2009, 25: 1451–1452. 10.1093/bioinformatics/btp187.
JGI genome portal. [https://doi.org/genome.jgi-psf.org]
Latgé JP: The cell wall: a carbohydrate armour for the fungal cell. Mol Microbiol. 2007, 66: 279–290. 10.1111/j.1365-2958.2007.05872.x.
de Groot P, Brandt BW, Horiuchi H, Ram AF, de Koster CG, Klis FM: Comprehensive genomic analysis of cell wall genes in Aspergillus nidulans. Fungal Genet Biol. 2009, 46 (Suppl 1): S72–S81.
Lieckfeldt E, Kullnig CM, Samuels GJ, Kubicek CP: Sexually competent, sucrose- and nitrate-assimilating strains of Hypocrea jecorina (Trichoderma reesei, Hypocreales) from South American soils. Mycologia. 2000, 92: 374–384. 10.2307/3761493.
Vargas WA, Mandawe JC, Kenerley CM: Plant-derived sucrose is a key element in the symbiotic association between Trichoderma virens and maize plants. Plant Physiol. 2009, 151: 792–797. 10.1104/pp.109.141291.
Martens-Uzunova ES, Schaap PJ: Assessment of the pectin degrading enzyme network of Aspergillus niger by functional genomics. Fungal Genet Biol. 2009, 46 (Suppl 1): S170–S179.
Berne S, Lah L, Sepcić K: Aegerolysins: structure, function, and putative biological role. Protein Sci. 2009, 18: 694–706.
Goodrich-Blair H, Clarke DJ: Mutualism and pathogenesis in Xenorhabdus and Photorhabdus: two roads to the same destination. Mol Microbiol. 2007, 64: 260–268. 10.1111/j.1365-2958.2007.05671.x.
Hares MC, Hinchliffe SJ, Strong PC, Eleftherianos I, Dowling AJ, French-Constant RH, Waterfield N: The Yersinia pseudotuberculosis and Yersinia pestis toxin complex is active against cultured mammalian cells. Microbiology. 2008, 154: 3503–3517. 10.1099/mic.0.2008/018440-0.
Karasova D, Havlickova H, Sisak F, Rychlik I: Deletion of sodCI and spvBC in Salmonella enterica serovar Enteritidis reduced its virulence to the natural virulence of serovars Agona, Hadar and Infantis for mice but not for chickens early after infection. Vet Microbiol. 2009, 139: 304–309. 10.1016/j.vetmic.2009.06.023.
McNulty C, Thompson J, Barrett B, Lord L, Andersen C, Roberts IS: The cell surface expression of group 2 capsular polysaccharides in Escherichia coli: the role of KpsD, RhsA and a multi-protein complex at the pole of the cell. Mol Microbiol. 2006, 59: 907–922. 10.1111/j.1365-2958.2005.05010.x.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ: Basic local alignment search tool. J Mol Biol. 1990, 215: 403–410.
SignalP 3.0 Server. [https://doi.org/www.cbs.dtu.dk/services/SignalP/]
Brotman Y, Briff E, Viterbo A, Chet I: Role of swollenin, an expansin-like protein from Trichoderma, in plant root colonization. Plant Physiol. 2008, 147: 779–789. 10.1104/pp.108.116293.
Max Planck Institute of Developmental Biology: Bioinformatics toolkit. [https://doi.org/toolkit.tuebingen.mpg.de/blastclust]
Talbot NJ: Growing into the air. Curr Biol. 1997, 7: R78–R81. 10.1016/S0960-9822(06)00041-8.
Wösten HA, van Wetter MA, Lugones LG, van der Mei HC, Busscher HJ, Wessels JG: How a fungus escapes the water to grow into the air. Curr Biol. 1999, 9: 85–88. 10.1016/S0960-9822(99)80019-0.
Kubicek CP, Baker S, Gamauf C, Kenerley CM, Druzhinina IS: Purifying selection and birth-and-death evolution in the class II hydrophobin gene families of the ascomycete Trichoderma/Hypocrea. BMC Evol Biol. 2008, 8: 4-10.1186/1471-2148-8-4.
Viterbo A, Chet I: TasHyd1, a new hydrophobin gene from the biocontrol agent Trichoderma asperellum, is involved in plant root colonization. Mol Plant Pathol. 2006, 7: 249–258. 10.1111/j.1364-3703.2006.00335.x.
Djonovic S, Pozo MJ, Dangott LJ, Howell CR, Kenerley CM: Sm1, a proteinaceous elicitor secreted by the biocontrol fungus Trichoderma virens induces plant defense responses and systemic resistance. Mol Plant Microbe Interact. 2006, 19: 838–853. 10.1094/MPMI-19-0838.
Djonovic S, Vargas WA, Kolomiets MV, Horndeski M, Wiest A, Kenerley CM: A proteinaceous elicitor Sm1 from the beneficial fungus Trichoderma virens is required for induced systemic resistance in maize. Plant Physiol. 2007, 145: 875–889. 10.1104/pp.107.103689.
Rep M: Small proteins of plant-pathogenic fungi secreted during host colonization. FEMS Microbiol Lett. 2005, 253: 19–27. 10.1016/j.femsle.2005.09.014.
Mukherjee PK, Hadar R, Pardovitz-Kedmi E, Trushina N, Horwitz BA: MRSP1, encoding a novel Trichoderma secreted protein, is negatively regulated by MAPK. Biochem Biophys Res Commun. 2006, 350: 716–722. 10.1016/j.bbrc.2006.09.120.
Armaleo D, Gross SR: Structural studies on Neurospora RNA polymerases and associated proteins. J Biol Chem. 1985, 260: 16174–16180.
Vogels 50x salts. [https://doi.org/www.fgsc.net/methods/vogels.html]
Jones JDG, Dunsmuir P, Bedbrook J: High-level expression of introduced chimaeric genes in regenerated transformed plants. EMBO J. 1985, 4: 2411–2418.
Berrocal-Tito G, Sametz-Baron L, Eichenberg K, Horwitz BA, Herrera-Estrella A: Rapid blue light regulation of a Trichoderma harzianum photolyase gene. J Biol Chem. 1999, 274: 14288–14294. 10.1074/jbc.274.20.14288.
Detter JC, Jett JM, Lucas SM, Dalin E, Arellano AR, Wang M, Nelson JR, Chapman J, Lou Y, Rokhsar D, Hawkins TL, Richardson PM: Isothermal strand displacement amplification applications for high-throughput genomics. Genomics. 2002, 80: 691–698. 10.1006/geno.2002.7020.
Huang X, Madan A: CAP3: A DNA Sequence Assembling Program. Genome Res. 1999, 9: 868–877. 10.1101/gr.9.9.868.
Papadopoulos JS, Agarwala R: COBALT: constraint-based alignment tool for multiple protein sequences. Bioinformatics. 2007, 23: 1073–1079. 10.1093/bioinformatics/btm076.
Acknowledgements
Genome sequencing and analysis was conducted by the US Department of Energy Joint Genome Institute and supported by the Office of Science of the US Department of Energy under contract number DE-AC02-05CH11231. MGC-B, EYG-R, MH-O, and EEU-R are indebted to Conacyt for doctoral fellowships. SLC and FC was supported by the Infrastructures en Biologie Santé et Agronomie (IBISA). EM and RH work was supported by the grants Junta de Castilla y León GR67, MICINN AGL2008-0512/AGR and AGL2009-13431-C02. The work of ISD, VS-S, LA, BS, BM, SZ, MS, and CPK was supported by the Austrian Science Foundation (grants FWF P17895-B06, P20559, T390, P18109-B12, P-19421, V139B20 and P-19340). The work of PMC and BH was supported by project number AANR-07-BIOE-006 from the French national program PNRB. MF was the recipient of a postdoctoral contract Ramón y Cajal from the Spanish Ministry of Science and Innovation (MCINN: RYC-2004-003005). SZ acknowledges support from the Vienna Science and Technology Fund (WWTF LS09-036).
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
CPK, IVG, BH, EM, SEB, CMK, and AHE contributed equally to this work as senior authors. AA, JC, MM, AS, and IVG performed global annotation and analysis, MZ and HS did the assembly, OC and CH finished the assembly, and EL and SL performed the genome and EST sequencing. SEB, AH-E, CMK and CPK designed the study, and coordinated and supervised the analysis; CPK drafted and submitted the paper. All other authors contributed research (annotations and/or analyses). All authors read and approved the final manuscript.
Electronic supplementary material
13059_2010_9593_MOESM1_ESM.XLSX
Additional file 1: Comparative properties and gene inventory of T. reesei, T. virens and T. atroviride. This file contains additional information on genomic properties and selected gene families from the three Trichoderma species comprising 19 tables. Table S1 summarizes the satellite sequences identified in the Trichoderma genomes and four other fungal genomes. Table S2 summarizes manually curated sequence alignments of transposable element families from the Trichoderma genomes. Table S3 lists the total number of CAZy families in Trichoderma and other fungi. Table S4 lists the glycoside hydrolase (GH) families in Trichoderma and other fungi. Table S5 lists the glycosyltransferase (GT) families in Trichoderma and other fungi. Table S6 lists the polysaccharide lyase (PL) families in Trichoderma and other fungi. Table S7 lists the carbohydrate esterase (CE) families in Trichoderma and other fungi. Table S8 lists the carbohydrate-binding module (CBM) families in Trichoderma and other fungi. Table S9 lists the NRPS, PKS and NRPS-PKS proteins in T. atroviride. Table S10 lists NRPS, PKS and NRPS-PKS proteins in T. virens. Table S11 lists the putative insecticidal toxins in Trichoderma. Table S12 lists the cytochrome P450 CYP4/CYP19/CYP26 class E proteins in Trichoderma. Table S13 lists the small-cysteine rich secreted protein from Trichoderma spp. Table S14 lists the most abundant PFAM domains in those genes that are unique to T. atroviride and T. virens and not present in T. reesei. Table S15 surveys the assembly statistics. Table S16 provides gene model support. Table S17 summarizes gene model statistics. Table S18 provides numbers of genes with functional annotation according to KOG, Gene Ontology, and KEGG classifications. Table S19 lists the largest KOG families responsible for metabolism. (XLSX 57 KB)
13059_2010_9593_MOESM2_ESM.DOCX
Additional file 2: Additional information on selected gene groups of Trichoderma , methods used for genome sequencing, and legends for the figures in Additional file 3. Chapter 1: Carbohydrate-Active enzymes (CAZymes). Chapter 2: Aegerolysins and other toxins. Chapter 3: Small secreted cysteine rich proteins (SSCPs). Chapter 4: EST sequencing and analysis. Chapter 5: Legends to figures. (DOCX 38 KB)
13059_2010_9593_MOESM3_ESM.DOC
Additional file 3: Figures that illustrate selected aspects of the main text. Figure S1 provides a phylogeny of Trichoderma NPRSs. Figure S2 compares the numbers of epoxide hydrolase genes in Trichoderma with that in other fungi. Figure S3 compares the codon usage in genes from syntenic and nonsyntenic regions of the genomes of Trichoderma reesei, T. atroviride and T. virens. (DOC 568 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article
Cite this article
Kubicek, C.P., Herrera-Estrella, A., Seidl-Seiboth, V. et al. Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma. Genome Biol 12, R40 (2011). https://doi.org/10.1186/gb-2011-12-4-r40
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/gb-2011-12-4-r40