
Abstract
Background
Pancreatic cancer is one of the most lethal type of cancers, with an overall five-year survival rate of less than 5%. It is usually diagnosed at an advanced stage with limited therapeutic options. To date, no effective treatment options have demonstrated long-term benefits in advanced pancreatic cancer patients. Compared with other cancers, pancreatic cancer exhibits remarkable resistance to conventional therapy and possesses a highly immunosuppressive tumor microenvironment (TME).
Main body
In this review, we summarized the evidence and unique properties of TME in pancreatic cancer that may contribute to its resistance towards immunotherapies as well as strategies to overcome those barriers. We reviewed the current strategies and future perspectives of combination therapies that (1) promote T cell priming through tumor associated antigen presentation; (2) inhibit tumor immunosuppressive environment; and (3) break-down the desmoplastic barrier which improves tumor infiltrating lymphocytes entry into the TME.
Conclusions
It is imperative for clinicians and scientists to understand tumor immunology, identify novel biomarkers, and optimize the position of immunotherapy in therapeutic sequence, in order to improve pancreatic cancer clinical trial outcomes. Our collaborative efforts in targeting pancreatic TME will be the mainstay of achieving better clinical prognosis among pancreatic cancer patients. Ultimately, pancreatic cancer will be a treatable medical condition instead of a death sentence for a patient.
Similar content being viewed by others


Background
Pancreatic cancer is an aggressive malignancy usually diagnosed at an advanced stage with very limited therapeutic options. According to GLOBOCAN 2018, pancreatic cancer is the seventh leading cause of cancer death in both males and females [1]. The estimated 5-year survival rate for pancreatic cancer is less than 5%, which is the lowest among other cancers [2]. Pancreatic cancer is expected to become the second leading cause of cancer death by 2030 in the United States (US), surpassing breast, prostate and colorectal cancers [3]. One of the backbone chemotherapeutic agents that has been used since the late nineties for pancreatic cancer is gemcitabine [4]. However, clinical data have shown that a large number of patients do not respond to gemcitabine monotherapy, and thus it is believed that the tumor cells have acquired intrinsic or chemoresistance towards gemcitabine treatment [5]. Since then, combinational therapies such as FOLFIRINOX [6] and the combination of gemcitabine with albumin-bound paclitaxel (nab-paclitaxel) [7], have been shown to be an alternative strategy, with only a marginal increase in overall survival (OS) but patients would then suffer with increased toxicity compared to gemcitabine alone.
Recently, the application of immunotherapies to boost effector T cells to kill cancer cells has generated much excitement. Particularly, strategies targeting immune checkpoint molecules through inhibition of programmed death 1 (PD-1) and cytotoxic T lymphocyte antigen-4 (CTLA-4) have demonstrated clinical benefit in several malignancies, such as melanoma [8, 9], Hodgkin’s lymphoma [10], and non-small cell lung cancer (NSCLC) [11]. This has therefore raised hope for pancreatic cancer patients. However, clinical studies have shown that checkpoint inhibition therapy alone is insufficient in treating patients with pancreatic cancer [12, 13]. The tumor microenvironment (TME) of pancreatic cancer is unique and may promote tumor evasion as well as conferring resistance to therapeutic agents including the immune therapies [14]. Based on the literature, compounds, or therapeutic approaches that targeting cytochromes [15] or immune mediators such as legumain [16] and Toll-like receptors [17] may reduce the influence of the tumor microenvironment on tumor progression. Some studies also suggested that nanotechnology or micronized chemotherapy deliveries may enhance the clinical outcomes among cancer patients [18]. However, the evidence for the effectiveness such approaches in targeting pancreatic tumor microenvironment is not clearly defined due to the lack of in-depth studies. Therefore, more thorough clinical research concerning the pancreatic TME is greatly needed.
In this review, we will explore the unique TME of pancreatic cancer that may act to limit the treatment efficacy of immunotherapy. We critically discuss the available treatment strategies for this disease. We will summarize findings on recent and ongoing combination immunotherapies currently being evaluated in clinical trial settings that focused on improving the effectiveness of immunotherapy in pancreatic cancer.
Main text
Characteristics of TME in pancreatic cancer
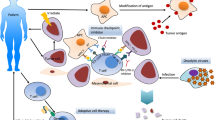
Pancreatic cancer features a highly immunosuppressive microenvironment, characterized by a dense desmoplastic stroma, which impedes blood flow to the area, inhibits drug delivery, and suppresses antitumor immune response [19]. This favors cancer progression by protecting pancreatic tumors from immune surveillance as well as regional and distant metastasis [20]. Additionally, the hypoxic environment, acidic extracellular pH, and high interstitial fluid pressure in the TME also act to enhance tumorigenesis and tumor progression [21]. In order to create an environment that is conducive for tumor growth, tumor supporting cells are upregulated, whereas the immune cells are downregulated in the TME of pancreatic cancer. Cells such as myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory T cells (Tregs), fibroblasts, and mast cells are upregulated in the TME, ultimately protecting tumor cells from being eliminated by the immune system; on the other hand, natural killer (NK) cells and CD8+ T cells that act to destroy tumor cells are downregulated [22]. The interaction between the tumor cells and TME components acts to facilitate the development and progression of tumors, as well as invasion, and metastasis (Fig. 1) [23].

Crosstalk of pancreatic cancer cells with other cells in the tumor microenvironment
Cancer-associated fibroblasts (CAFs)
CAFs are an important component within the tumor stroma. They develop from bone marrow-derived mesenchymal stem cells (MSCs), pancreatic stellate cells (PSCs), and resting fibroblasts in the pancreas through epithelial-mesenchymal transition (EMT). Activation of CAFs is induced by cancer-secreting cytokines such as TGF-β, sonic hedgehog (SHH), TNF-α, IL-6, and IL-10 [24]. CAFs have been shown to be associated with tumor progression by stimulating the production of growth factors, inflammatory cytokines and chemokines, pro-angiogenic factors, and metabolites that stimulate signaling pathways in cancer cells [25]. Additionally, CAFs are also involved in remodeling of the ECM to form a pro-cancerous microenvironment by producing excessive amounts of structural matrix components, such as collagen, fibronectin, and proteoglycans [26, 27]. This in turn promotes the aggressive biology of pancreatic cancer, resulting in tumor proliferation, angiogenesis, metastasis, survival, and resistance to therapy.
Additionally, the migration and proliferation of pancreatic cancer is also supported by CAFs expressing fibroblast activation proteins (FAP). Overexpression of FAP in tumor cells resulted in increased tumorigenicity and enhanced tumor progression [28, 29]; the enzymatic activity of FAP plays an essential role in FAP-driven tumor growth in a murine xenograft model, as evidenced by inhibition of FAP enzymatic activity being associated with tumor attenuation [30]. As expected, higher levels of FAP expression in patients were associated with shorter overall survival compared with patients showing lower FAP expression, suggesting that FAP is important for tumor progression and metastasis in pancreatic cancer [31].Deletion of FAP gene and pharmacologic inhibition of FAP enzymatic activity reduced the tumor burden probably through disturbing stromagenesis, angiogenesis, and promotes nti-tumor immune responses. [32,33,34]. Administration of FAP-CAR T cells into tumor-bearing mice significantly reduced tumor growth and induced apoptosis of tumor cells [31]. The degree of desmoplasia was found to be reduced in highly desmoplastic lung cancer xenografts, with a marked disruption of adenocarcinoma ductal-like structure of the tumor nodules, a decrease in collagen and fibronectin content, and an increase in necrosis in FAP-CAR T cell-treated tumors. This in turn promotes the recruitment of immune cells, consequently augmenting antitumor immunity [31]. Similar results were observed in murine models of pancreatic cancer, where FAP-CAR T cells significantly inhibited the growth of non-immunogenic tumor [31,32,33,34]. Depletion of FAP-expressing stromal cells resulted in a better immunological response and a lower tumor burden [29, 35],These findings may suggest therapeutics that selectively target FAP-expressing cells, but not other cancer-associated stromal cells, may result in a better prognosis.
Pancreatic stellate cells (PSCs)
More than 80% of human pancreatic cancer tissues are associated with a highly desmoplastic stroma, and pancreatic stellate cells (PSCs) are creators of this stroma in pancreatic cancer [23]. In the non-inflamed pancreas, quiescent PSCs have a lower mitotic index [21] and are involve in maintaining tissue homeostasis [36]. In pancreatic ductal adenocarcinoma (PDAC), quiescent PSCs are activated by environmental stress (oxidative stress and hypoxia), cellular factors [IL-1, IL-6, hypoxia inducible factor 1-α (HIF1-α), and TGF-β], as well as molecular signaling pathway such as the PI3K pathway, and are transformed into myofibroblast-like cells [21, 37, 38]. These activated PSCs acquire proliferative capability and are associated with an upregulation of matrix metalloproteinases (MMPs) and extracellular matrix proteins (ECMs) [38], which enhance sustained fibrosis and tumor angiogenesis via the production of vascular endothelial growth factors (VEGFs) [21], creating a physical barrier to therapeutic agents [36]. Additionally, activated PSCs promote tumor progression by secreting IL-6, TGF-β, stromal cell-derived factor-1 (SDF-1), hepatocyte growth factor (HGF), and galectin-1 (Gal-1, 38]. Notably, PSCs play a key role in inducing desmoplastic reactions in the TME of pancreatic cancer. Recent evidence has demonstrated that PSCs can drive desmoplasia via several signaling pathway, such as IL-6/JAK/STAT signaling, paracrine Sonic Hedgehog (SHH) signaling, the vitamin D Receptor (VDR) pathway, and CXCL12/CXCR4 signaling axis. In addition, the secretion of CXCL12 by PSCs results in limiting the migration of CD8+ T cells to juxtatumoral stromal compartments, protecting the tumor cells from the cytotoxicity of CD8+ T cells [39]. Overexpression of Gal-1 in PSCs promotes immunosuppression by inducing apoptosis of CD4+ and CD8+ T cells and increasing the secretion of Th2 cytokines (IL-4 and IL-5), while reducing the secretion of Th1 cytokines (IL-2 and IFN-γ) [40].
CD4+CD25+Foxp3+ regulatory T cells (Tregs)
CD4+CD25+Foxp3+ regulatory T cells (Tregs) also defined as suppressor T cells, play an important role in immunosuppression via expression of CTLA-4 and secretion of IL-10 and TGF-β [41]. In the physiological state, Tregs help to regulate immunological tolerance to self-antigens and prevent autoimmunity, whereas, in tumors, they suppress antitumor immune responses by inhibiting effector T cell functions [41]. The migration of circulating Tregs into the pancreatic cancer TME is controlled by the interactions between tumor chemokines and their ligands/receptors. It was also shown that in both human PDAC and a mouse pancreatic tumor model, the cancer cells produced higher level of ligands for chemokine receptor 5 (CCR5), while Tregs expressed CCR5. When CCR5/CCL5 interaction is diminished or blocked, migration of Tregs to tumor is reduced, and even the size of tumor became smaller [42]. In addition, TGF-β was found to be involved in the recruitment of Tregs in pancreatic cancer. Tregs secrete TGF-β to suppress antitumor cytotoxic activity; on the other hand, pancreatic cancer induces Tregs in the presence of TGF-β [43]. In a murine model of pancreatic cancer, the conversion of CD4+CD25− naïve T cells into Foxp3+ Tregs was shown to be mediated by TGF-β [44]. In PDAC, poorer prognosis of patients is associated with a higher proportions of Tregs in tumor-infiltrating lymphocytes (TILs) [45]. The increased infiltration of Tregs into the TME is also positively correlated with tumor progression [42] and inversely correlated with the presence of CD8+ T cells [45]. In pre-clinical studies, the depletion of Tregs with anti-CTLA-4, anti-CD25 or CCR5 inhibitor r reduced tumor growth and prolonged the animals’ survival by enhancing the activation of tumor specific T cells. [41, 42, 46, 47].
Myeloid-derived suppressor cells (MDSCs)
MDSCs are immature myeloid cells that suppress the immune response in pancreatic cancer. Granulocytic MDSCs express CD33, CD11b, and IL-4Rα, with low levels of CD15 and high levels of arginase. On the other hand, monocytic MDSCs express the same markers as granulocytic MDSCs, but with lower levels of CD15, and also express CD14 apart from arginase. They also express inducible nitric oxide synthase (iNOS) [48]. In pancreatic cancer, the proliferation and migration of MDSCs from bone marrow into the TME is consistently induced by granulocyte macrophage colony-stimulating factor (GM-CSF) [38]. In tumors, MDSCs suppress the antitumor activity of both CD8+ and CD4+ T cells, and expand immunosuppressive Tregs. Moreover, MDSCs can block innate immunity by converting M1 macrophages which promote tumor regression into M2 phenotypes which facilitate tumor progression [49] as well as suppressing NK cells antitumor cytotoxicity [50]. This repolarization is the result of cross-talk between MDSCs and macrophages, in which the production of IL-10 by MDSCs is increased, while increasing the production of IL-12 by macrophages [49]. MDSCs can suppress T cell activity via multiple mechanisms; these mechanisms include the depletion of arginine (Arg) [51], the secretion of reactive oxygen species (ROS) [52] and downregulation of L-selectin [53]. In tumors, MDSCs synthesize high levels of arginase-1 (Arg-1) to deplete Arg in the TME [51]. Depletion of Arg results in decreased expression of the CD3ζ chain, leading to the reduction of IL-2 and IFN-γ, and, consequently, inhibition of the proliferation of T cells and induction of T cell tolerance [23]. The secretion of cytokines such as IL-10 and TGF-β [38] and increased activation of STAT 3 and NADPH [54] can induce MDSCs to release ROS, resulting in oxidative stress in T cells [38] as well as the suppression of CD8+ T cell responses [52]. The production of free radical peroxynitrite (PNT) was shown to stimulate MDSCs to mediate tumor cells resistance to CTLs, through T cell tolerance and nitration of T cell receptors (TCRs) on the T cell surface. This causes TCRs to lose the ability to recognize and bind to specific peptide/MHC complexes and perform subsequent antitumor activity. Another mechanism employed by MDSCs to inhibit antitumor immunity is by impairing the homing of T cells to lymph nodes via the downregulation of L-selectin. Adaptive T cell-mediated antitumor immunity requires the activation of antigen-naïve T cells; L-selectin is important in facilitating the extravasation of leucocytes to lymph nodes where they become activated prior to being directed to inflammatory sites such as the TME [53, 54]. By downregulating L-selectin expression in CD8+ and CD4+ T cells, MDSCs impair the T cell trafficking pattern, thereby inhibiting the activation of T cells [54].
Tumor associated macrophages (TAMs)
Macrophages in tumors are usually defined as TAMs and often express the M2 phenotype. In general, M1 macrophages facilitate tumor regression and Th1 responses by secreting tumor necrosis factor-α (TNF-α) and IL-12; on the other hand, M2 macrophages display an immune suppressive phenotype and release IL-10 which promotes a Th2 response [55]. In human PDAC, macrophages are prominent compared with the healthy pancreas. Macrophages were found to infiltrate in low grade, pre-invasive pancreatic tumor lesions and persist in invasive pancreatic cancer in a mouse model [56]. The percentages of MDSCs and TAMs are elevated significantly with the progression of pancreatic cancer; conversely, the percentages of CD8+ and CD4+ T cells are significantly reduced [23]. As a result, macrophages play a critical role in facilitating tumor progression, angiogenesis, stromal remodeling, and metastasis in pancreatic cancer [57]. TAMs can facilitate tumor metastasis by secreting matrix proteins and proteases such as serine proteases, matrix metalloproteinases (MMPs), and cathepsins which act to modify the extracellular matrix (ECM) composition [58]. The overexpression of MMP9 induced via the interaction of macrophage inflammatory protein-3 alpha (MIP-3α) with its receptor, increases the expression of CCR6 on pancreatic cancer cells, consequently increasing the invasion of pancreatic cancer cells [59]. Macrophages also drive the development of an immunosuppressive environment by secreting angiogenic factors such as thymidine phosphorylase (TP), vascular endothelial growth factor (VEGF), MMPs, cyclooxygenase-2 (Cox-2), CXCL12, and CCL2, as well as immunosuppressive factors such as IL-10, TGF-α, Arg-1, CCL17 and CCL22 [58]. TAMs also promote apoptosis of T cells by expressing programmed death-ligand 1 (PD-L1) on their cell surface [58]. As a result, TAMs may contribute to the pancreatic tumor immune evasion, resulting in the survival of these tumors, despite aggressive chemotherapy .
Potential factors that limit the efficacy of immunotherapy
Pancreatic cancers have an intrinsically low mutational burden, and thereby exhibit low levels of neoantigen-expression. As the mutational load and neoantigen burden are positively correlated with the efficacy of immunotherapy [60, 61], cancers with higher mutational loads, which generate more neoantigens, could elicit enhanced T cell recognition. In contrast, cancers with a low mutational load, for instance pancreatic cancer, only occasionally produce neoantigens, as their average mutation rate accounts only for one mutation per megabase (Mb), compared to 11 mutations per Mb for melanomas [62]. Tumor immunogenicity is the key initial step in launching effective antitumor responses to immune checkpoint blockade. As a result of the lack of sufficient neoantigen targets, the lower level of TILs in the TME creates a non-immunogenic or ‘cold’ microenvironment, thereby limiting effective T cell responses and impeding the efficacy of immunotherapy [63].
Other mechanisms of resistance to immune checkpoint blockade in pancreatic cancer include aberrant expression of immune checkpoints such as PD-L1 on the tumor cell surface, downregulation of antigen presenting MHC molecules, reduced Fas receptor signaling and therefore a reduction in counterattack by T cells via the expression of Fas ligands [64, 65]. In addition, the establishment of a highly desmoplastic TME by stromal cells creates a therapeutic barrier in treating pancreatic cancer [66]. For instance, it is especially difficult to deliver drugs to pancreatic tumors compared with other solid tumors due to their hypovascular and poorly perfused nature [67]. The presence of stromal components in pancreatic cancer increases the interstitial fluid pressure, consequently inhibits the drug from penetrating the interstitial tissue [66, 67]. Furthermore, the formation and function of the blood vasculature can be inhibited by fibroblasts and the fibrotic stroma in pancreatic cancer [67], thereby diminishing drug delivery via the blood and reducing the effectiveness of chemotherapy.
Strategies for Cancer immunotherapy
In recent years, cancer immunotherapy is gaining much attention in view of its promising efficacy. One cancer immunotherapy comprises antibodies that target immune checkpoints. Ipilimumab, the first anti-CTLA-4 antibody was approved by the US Food and Drug Administration (FDA) in 2011 for melanoma [60]. Compared with placebo, ipilimumab significantly improved overall survival, this being, respectively, 9.1 and 11.2 months in patients with previously untreated metastatic melanoma [68]. PD-1 inhibitors such as nivolumab and pembrolizumab have been approved for melanoma treatment [8, 9] and are still being tested in pancreatic cancer clinical trials [12, 69]. Overall survival of melanoma patients who received nivolumab was considerably longer than progression-free survival, with an acceptable long-term safety profile [9]. Similarly, pembrolizumab also showed promising results in decreasing tumor size in melanoma patients [70, 71]. Another three anti-PD-L1 antibodies, atezolizumab, durvalumab and avelumab, have also been approved by the FDA [72]. The safety profile and clinical activity of atezolizumab were studied in renal cell carcinoma (RCC) where the drug showed promising antitumor activity in patients with metastatic disease. About 46% of patients with clear cell RCC had tumor shrinkage with an overall survival of 23.9 months [73]. Durvalumab was approved by the FDA in 2018 to treat patients with unresectable, stage III NSCLC. Patients receiving durvalumab demonstrated significant improvement in progression-free survival (16.8 months) compared with patients who received placebo, 16.8 months (5.6 months) [74]. Avelumab received accelerated approval by the FDA for the treatment of metastatic Merkel cell carcinoma (MCC), a rare type of skin cancer in 2017 [75]. The binding of avelumab to PD-L1 can inhibit the interaction of PD-L1 with PD-1, consequently restoring the immune response as well as antitumor activity [75]. The overall response rate (ORR) was 33%, while the estimated one-year overall and progression-free survival were 52 and 30%, respectively [75].
Durable clinical responses and prolonged survival rate have been shown in patients with melanoma and highly immunogenic cancers using monoclonal antibodies (mAb) targeting CTLA-4 or PD-1 [68]. However, based on the early clinical trials, checkpoint inhibitors, such as anti-CTLA-4, anti-PD-1 or anti-PD-L1, are ineffective when used as monotherapy in the treatment of pancreatic cancer [76]. This inefficacy is likely due to the low immunogenicity and non-inflamed phenotype (low levels of TILs) of pancreatic cancer as mentioned previously [61]. No objective responses were observed in advanced and metastatic pancreatic cancer patients treated with ipilimumab, indicating that ipilimumab alone is not an effective therapy for advanced pancreatic cancer [77]. Similarly, in a phase I study with anti-PD-L1 mAb alone, a 0% overall response rate (ORR) was observed in advanced pancreatic cancer patients [12]. Therefore, immunotherapy is not always effective and requires further development along with new combination strategies in order to enhance its efficacy. These combination therapies can be classified based on their strategic targets: firstly, to promote T cell priming by enhancing TAA (tumor associated antigen) presentation; secondly, to target the immunosuppressive environment thus relieving immunosuppression; and thirdly, to bring more TILs into the TME by breaking down the desmoplastic barrier [60].
Enhancing T cell priming
Insufficient T cell priming is a root cause of ‘cold’ tumors and immune checkpoint unresponsiveness [78]. Antigen presenting cells (APCs), particularly dendritic cells (DCs) are essential for T cell priming which generates effective antitumor T cell responses. Of note, higher levels of circulating DCs have been associated with better survival rate in pancreatic cancer patients [79]. Therefore, the TME would first need to be primed with effector T cells before immune checkpoint inhibitors could play their roles. Combining techniques that inhibit immunosuppressive signaling in TME while activating tumor-specific T cells against tumor cells seems to represent the most promising approach for immunotherapy in the treatment of pancreatic cancer.
-
(1)
Combination of immune checkpoint therapy with chemotherapy
Chemotherapy has been recognized as one important treatment strategy in human malignancy. However, the use of chemotherapy along with other clinically use agents may achieve better clinical outcomes. [80] The recruitment and activation of DCs [81] as well as the induction of the release of tumor specific antigens [82] may have a critical role in achieving this synergism. DCs are critical for T cell priming and the activation of a specific CD8+ T cell immune response. Tumor antigens must be presented by APCs such as DCs to naïve CD8+ T cells via cross-presentation. However, tumor-infiltrating DCs may be functionally impaired or may display defective migration into tumor-draining lymph nodes [81]. This failure can be reversed by using a chemotherapeutic agent to induce the recruitment of DCs to the tumor sites, thereby further enhancing the cross-presenting potential of tumor infiltrating DCs, which is crucial for the subsequent tumor antigen-specific cellular priming [83]. Unfortunately, both acute and cumulative toxicities to normal tissues caused by the delivery of cytotoxic agents have limited the dose and duration of treatment [84]. Therefore, the combination of chemotherapy and immunotherapy could potentially enhance the effectiveness of cancer treatment through different mechanisms of actions.
A recent phase I study (85) evaluated the safety profile of the combination of gemcitabine with an anti-CTLA-4 mAb (tremelimumab; CP-675,206) in metastatic pancreatic cancer patients. Tremelimumab is a fully humanized mAb that antagonizes the binding of CTLA-4 to B7–1 as well as B7–2, blocking the co-inhibition signal, thus leading to T cell activation. This combination therapy resulted in tolerable side effects, with a median overall survival of 5.3, 8.0, and 7.5 months for patients who received 6, 10 and 15 mg/kg of tremelimumab, respectively [85]. Among 28 patients, seven patients showed stable disease for more than 10 weeks; two patients who received 15 mg/kg tremelimumab managed to achieve a partial response at 8 weeks. Guo and coworkers also highlighted preliminary results from an ongoing phase Ib study of ipilimumab and gemcitabine on unresectable pancreatic cancer patients; this similarly showed a partial response and stable disease. Immunohistochemistry analysis further showed that the positive expression of PD-L1 was correlated with a worse overall survival [60].
In a murine model of pancreatic cancer, treatment with anti-PD-L1 or anti-PD-1 mAbs enhanced the infiltration of CD8+ T cells and significantly increased the expression of IFN-γ, granzyme B and perforin in implanted tumors. The blockade of PD-L1 promoted infiltration of CD8+ T cells into the tumor site and induced local immune activation. Furthermore, the combination of gemcitabine with anti-PD-L1 mAb exhibited significant synergistic effect, eliciting a complete response without overt toxicity in treated mice [86]. A clinical study (NCT01313416) on the combination of gemcitabine and pidilizumab (CT-011), a humanized mAb against PD-1 has been closed to enrolment. CT-011 is designed to specifically bind to PD-1, thus inhibiting PD-1 activity and attenuating apoptotic processes of effector or memory T lymphocytes, ultimately resulting in the activation of a CTL antitumor immune response [69]. CT-011 has been studied in murine models of other cancers, including leukemia, melanoma, lung cancer and colorectal carcinoma. CT-011 treatment resulted in reduced tumor growth and prolonged survival in tumor bearing nude mice. CT-011 also provided protection against tumor re-challenge.
-
(2)
Combination of immune checkpoint therapy with cancer vaccines
Cancer vaccines are designed to augment antigen presentation and activate effector T cells. When vaccines containing target tumor antigens are given, host APCs would present these antigens to effector T cells which are then primed to kill tumor cells expressing these specific antigens, ultimately stimulating the development of antitumor immunity. One of the most extensively studied cancer vaccines is GVAX. It is made up of allogenic irradiated pancreatic cancer cells that have been genetically engineered to produce GM-CSF, a cytokine that further stimulates antigen presentation, T cell priming, and promotes cytolytic activity against tumor cells [60, 87]. In a phase II adjuvant study, GVAX induced the expansion of pancreatic cancer-specific CD8+ T cells and consequently improved the overall survival of patients. Patients who remained disease free after combination therapy (chemotherapy, radiotherapy and/or immunotherapy) generated lymphocytes that could respond to a greater variety of tumor associated antigens, suggesting that immunotherapy can be used either as an adjuvant treatment or in combination with other conventional therapies [88]. Immunohistochemical analysis (IHC) revealed the formation of intratumoral tertiary lymphoid aggregates in 33 out of 39 GVAX vaccinated pancreatic cancer patients; these aggregates were not observed in tumors of non--vaccinated patients [89]. The aggregates developed in response to antigen exposure and are composed of APCs and B cells, as well as naïve and activated T cells. These aggregates also indicated that vaccine-based immunotherapy plays a role in inducing an adaptive immune response in the TME in which GVAX could alter the pancreatic cancer TME, thus facilitating the infiltration of functional immune effector cells, and cconverting pancreatic cancer from non-immunogenic into immunogenic neoplasms [89].
Conceivably, the combination of an immune checkpoint inhibitor with vaccine therapy may synergistically induce an antitumor immune response. A preclinical study in melanoma indicated that a combination of GVAX with immune checkpoint blockade effectively eradicated tumors in mice which suffered from B16-BL6, an induced, highly non-immunogenic melanoma, that is resistant to immune checkpoint blockade therapy alone [90]. In a randomized phase Ib study, patients receiving the combination of GVAX and anti-CTLA-4 mAb (ipilimumab) had improved overall survival (median 5.7 months) compared to patients receiving ipilimumab alone (3.6 months) Patients with prolonged survival showed a higher number of tumor-infiltrating CD8+ T cells in the TME, indicating an improved antitumor immune response. Compared to ipilimumab alone, the percentage of patients surviving after 1 year was higher (27%) in the combination therapy arm versus 1% [91].These data suggested that T cells first need to be primed for their activation by T cell modulating agents such as ipilimumab.
A similar treatment was tested in preclinical murine models of pancreatic cancer using GVAX plus anti-PD-1 therapy. Combination therapy was found to significantly improve overall survival compared to PD-1 monotherapy. The secretion of interferon (IFN)-γ and the circulation of CD8+ T cells were increased in the TME of mice that received combination therapy with GVAX and PD-1 antibody blockade, compared with PD-1 monotherapy or GVAX therapy alone, indicating that the combination therapy could induce a synergistic effect antitumor immunity [92]. Furthermore, the addition of GVAX and low dose of cyclophosphamide to PD-1 blockade could downregulate the expression of CTLA-4 on T cells [92]. It is also important to highlight that an earlier study showed that PD-L1 was weakly expressed in both human and murine PDACs; the administration of GVAX significantly increased the expression of PD-L1 [92]. GVAX could increase the production of IFN- by infiltrating effector T cells, which may induce the upregulation of immunosuppressive mechanisms such as the overexpression of PD-L1 [89]. The upregulated expression of PD-L1 in tumor cells is associated with increased infiltration of immune cells and the formation of lymphoid aggregates, as well as an enhancement of the response rate of anti-PD-1 and anti-PD-L1 [89]. Lutz et al. also demonstrated an elevated expression of PD-L1 by monocytes and macrophages in the lymphoid aggregates that formed after GVAX therapy. In contrast, pancreatic tumors from non-vaccinated patients were rarely associated with PD-L1 expressing cells, indicating that PD-L1 expression is induced by vaccine treatment. Vaccine-primed patients would be better candidates than non-vaccinated patients for immune checkpoint therapy [89]. Therefore, the ineffectiveness of PD-L1 or PD-1 blockade in pancreatic cancer and the inability of PDAC to respond to a single checkpoint inhibitor therapy could be due to the lack of PD-1/PD-L1 expression and decreased infiltration of immune effector T cells to the tumor site. Thus, vaccine-based immunotherapy may overcome the resistance of pancreatic cancer towards immune checkpoint inhibitors by facilitating infiltration of tumor specific effector cells into the tumor site and upregulating PD-L1 expression, while immune checkpoint inhibitors may enhance the efficacy of vaccine-induced antitumor immune response by targeting PD-L1 signals on tumor cells [93]. To prove this concept, multiple clinical trials of PD-1/PD-L1 blockade in combination with GVAX vaccine therapy are ongoing (NCT02243371; NCT02648282; NCT02451982). For example, a randomized phase I/II clinical trial (NCT02451982) is ongoing to evaluate the efficacy of GVAX with or without anti-PD-1 mAb (nivolumab) as neoadjuvant or adjuvant treatment in resectable pancreatic cancer patients at Johns Hopkins University.
However, there are some limitations and several challenges have been associated with cancer immunotherapy targeting neoantigens. Antigens used in cancer vaccines should preferably be molecules that are different from normal cells, to ensure that antitumor immune response generated by vaccination only targeted on antigen bearing-tumor cells but not normal cells. Most tumor antigens are derived from mutated or modified self-proteins, leading to a risk of immune tolerance. This creates challenges in designing an appropriate cancer vaccine with reduced immune tolerance while eliciting antitumor immunity [94]. Another major concern is the heterogeneity of tumors. Neoantigens may be expressed in some, but not all, tumor cells in an individual patient, resulting in certain tumor cells escaping from immune surveillance [95]. The effectiveness of an allogeneic vaccine is highly correlated to the number of common tumor-associated antigens expressed by both the cancer and the allogeneic cell line. Therefore, the lack of strong immunogenicity of tumor neoantigens may greatly decrease the efficiency of vaccines [96]. In addition, since tumors frequently express antigens that are not specific to the tumor itself, treatment could ultimately lead to an increased risk of autoimmune-related adverse events, host immune suppression, and T cell exhaustion. Cancer vaccines are designed to target tumor neoantigens; tumor cells can evade destruction via developing antigen loss variants and this could increase the risk of autoimmunity. Therefore, to be recognized as an ideal cancer vaccine candidate, it should elicit strong immune response against the target cells, with antigen expression being restricted within the tumor itself, with minimal expression on normal tissues [97]. The highly immunosuppressive microenvironment of pancreatic cancer also contributes significantly to the unresponsiveness [98]. Furthermore, the use of allogenic therapies may promote tumor escape and drive further mutation. It has been hypothesized that the antigenic characteristics of allogeneic tumor cell vaccines developed from established cell lines might not be entirely the same as those of the tumor [99].
Accumulating evidence suggests that tumor neoantigens are one of the important targets for an antitumor immune response. Indeed, a higher neoantigen load and increased level of TILs are associated with improved survival in patients with colorectal [100] and endometrial cancer [101]. Therefore, the development of neoantigen cancer vaccines is highly dependent on the correct prediction and identification of neoantigens. Neoantigen prediction involves a series of computational steps, starting with the identification of mutations at the DNA level by comparing the whole exome sequences with those matched normal cells, followed by identifying targeted neoepitopes with the help of tumor RNA expression profiling, and finally determining the binding affinity of predicted epitopes to the MHC molecules with the use of software programs such as NetMHC or SYFPEITHI [98, 102]. However, there is the possibility of generating false positives (non-existent epitopes) or false negatives (missed epitopes). Computation of mutant allele coverage at the base level also adds difficulty in choosing the right transcript isoform to translate [102].
Reverse immunology has been postulated to be an efficient, high throughput approach for the discovery of tumor antigens. This approach involves the selection of peptides with strong binding to MHC molecules, such as proteins encoded by mutated oncogenes or genes that are highly expressed by tumors [103, 104]. This is predicted by in silico analysis using affinity prediction algorithms such as BIMAS and SYFPEITHY [103]. The most efficient binders are selected and bound onto APCs, and then used to activate CD8+ T cells that specifically recognize peptide-bound target cells [104]. High-throughput serological analysis of recombinant cDNA expression libraries (SEREX) has also been developed and widely used to identify and characterize the tumor antigens [105] in various types of cancers, including breast cancer [106], hepatocellular carcinoma [107], and gastric cancer [108], as well as pancreatic cancer [109]. The interaction of tumor antigens with antibodies in the autologous and allogeneic sera of cancer patients allows the identification of respective tumor antigens in the recombinant cDNA library [105]. The application of SEREX in identifying tumor antigens has also helped to define factors involved in tumorigenesis and further identify targets for diagnosis and vaccine-based therapy [107]. It allows a rapid identification of multiple tumor antigens and does not require the generation of tumor cell lines and pre-established CTL clones [110]. Furthermore, a proteome-based approach has recently been implemented in the prediction and identification of tumor neoantigens in cancer patients; this allows the screening of large number of patient sera and autoantigens [98]. In the future, the therapeutic efficacy of cancer vaccines can be enhanced by developing customized treatments based on the genomic and transcriptomic features of each patient, administered in conjunction with anti-immunosuppressive agents. Future directions also include strategies to increase the accuracy of choosing the right neoepitopes for personalized cancer vaccines and to overcome the occurrence of immune tolerance [96]. A continued focus on scientifically driven clinical trials is required to develop more potent and specific vaccines for the treatment of pancreatic cancer.
-
(3)
Immune checkpoint therapy with agents that enhance T cell immunity
CD40 is a member of the TNF receptor family and is constitutively expressed on APCs. The binding of CD40 with its ligand (CD154), which is expressed on activated T cells, results in APCs activation, leading to activation of adaptive immunity. Ligation of CD40 on DC can increase the expression of MHC and co-stimulatory molecules, the production of pro-inflammatory cytokines, and enhanced T cell immunity [111]. In another preclinical trial, it was reported that CD40 activation itself was insufficient to induce a productive antitumor immune response, and required macrophages to rapidly infiltrate the tumor lesions, become tumoricidal and facilitate stroma depletion [112]. However, the use of a CD40 agonist was shown to promote maturation of macrophages and DCs, as well as cross-presentation of tumor antigens to CD8+ T cells, and to facilitate macrophage tumoricidal activity [113]. The administration of an agonist CD40 Ab with gemcitabine and nab-paclitaxel to mice resulted in macrophage-independent T cell immunity. This demonstrated that while the combination of gemcitabine and albumin- bound paclitaxel did not induce regression in established tumors, the use of an agonist CD40 Ab together with the chemotherapeutic agents significantly reduced tumor growth and improved survival compared to those receiving chemotherapeutic agents only.
Furthermore, the combination of CD40 mAb with chemotherapy was shown to enhance the efficacy of immune checkpoint therapy by priming the T cell response; treatment of tumor-bearing mice with this combination therapy resulted in reduced tumor progression and prolonged survival [63].CD40 mAb/chemotherapy was found to transform the TME of pancreatic cancer, resulting in reduced level of Tregs and increased infiltration of CD8+ T cells to the tumor site; this could further enhance the induction of a T cell response, and consequently augment the antitumor effects of anti-PD-1 in PDAC [63]. The induction of T cell immunity could transform pancreatic tumors that are completely resistant to immune checkpoint inhibitors into those in which tumor growth can be controlled with immune checkpoint blockade [63]. Luheshi and coworkers also demonstrated that the combination of an agonist CD40 mAb with PD-L1 blockade significantly delayed tumor growth and increased the overall survival in a murine model. All these data showed that CD40 mAbs play a role in stromal remodeling which transforms the immunosuppressive TME of pancreatic cancer, increases the infiltration of functional CD8+ T cells, enhances the expression of IL-2 and Th1 chemokines, and upregulates both the tumor and systemic PD-L1 expression; this could help to improve the sensitivity towards immune checkpoint therapy. In summary, while immune checkpoint monotherapy alone has minimal effects against PDAC, the combination of a CD40 mAb and PD-L1 blockade can improve the overall survival in comparison to either therapy alone.
Currently, a phase Ib clinical trial (NCT02304393) is assessing the safety, pharmacokinetics, pharmacodynamics and activity of a combination treatment of a CD40 mAb (RO7009789)) in conjunction with atezolizumab (anti-PD-L1) in patients with metastatic or locally advanced solid tumors. Another ongoing phase Ib/II study (NCT03214250) aims to investigate the safety and efficacy of a CD40 mAb (APX005M) administered with gemcitabine and nab-paclitaxel with or withoutanti-PD-1 mAb (nivolumab) in metastatic pancreatic cancer patients.
-
(4)
Immune checkpoint therapy with adoptive T cell transfer
Other than CD40 mAbs, adoptive T cell transfer (ACT) also represents a promising immunotherapy approach for cancer. ACT uses genetically modified T cells to express chimeric antigen receptors (CAR), and has shown impressive activity in treating acute lymphoblastic leukemia [114, 115]. CARs are artificial receptors that are engineered to target specific antigens that are expressed in tumors but are not expressed, or expressed only at low levels, in normal tissues. By combining the antigen binding properties of mAb with lytic capacity of T cells, ‘off target’ effects and unspecific cytotoxicity can be minimized. CAR-T cells can also target cells in an MHC-independent fashion, bypassing tumor cell resistance towards MHC-restricted T-cell recognition [116, 117]. The efficacy of CAR T cells was found to be limited in solid tumors, particularly in pancreatic cancer, as there is a lack of an ideal target analogous to CD19 (B cell activation receptor), as well as the immunosuppressive environment of pancreatic cancer [118]. As a result, to treat pancreatic cancer, CAR is engineered to recognize mesothelin (membrane protein antigen) which is overexpressed in pancreatic cancer and other common solid tumors (Fig. 2) but is not expressed on T cells [118, 119] Mesothelin is an attractive target for immunotherapy due to its limited expression in normal tissues, its overexpression in malignant tissues, and its high immunogenicity [120]. Mesothelin might not be essential for the growth and reproduction of both wild type and mesothelin knockdown mice [121], although its aberrant or overexpression in preclinical and clinical studies showed that it plays an active role in both tumor malignancy and aggressiveness by promoting tumor proliferation, leading to invasion, metastasis, and conferring resistance towards cytotoxic agents [119].

Frequency of mesothelin protein expression in various common solid malignancies
In one of the completed phase I clinical trials (NCT01897415), T cells were engineered to express anti-mesothelin CAR and administered into patients with chemotherapy refractory metastatic pancreatic cancer. Of the six patients treated, two patients achieved stable disease in that study. Currently, an ongoing non-randomized phase I/II clinical trial (NCT01583686) is being conducted to determine the safety and efficacy of administering the engineered tumor fighting cells (anti-mesothelin cells) in metastatic cancer patients (ovarian, lung, cervical, mesothelioma and pancreatic cancer), followed by the administration of a preparative regimen (fludarabine, cyclophosphamide and aldeslekin). Furthermore, an ongoing non-randomized phase I study (NCT03323944) is evaluating the safety and feasibility of transducing meso-cells in unresectable or metastatic pancreatic cancer patients, either given as a single agent or in combination with cyclophosphamide. Several clinical trials (Table 1) and ongoing clinical trials (Table 2) using CAR T cell mesothelin-targeted immunotherapies in various solid tumors.
Targeting of other tumor antigens such as CEA (carcinoembryonic antigen), FAP, and mucin (MUC)-1 also has marked activity in murine models of pancreatic cancer. CEA is a glycoprotein that is highly expressed on the surface of the majority of pancreatic cancer cells. Targeting CEA by CAR T cells has been shown to induce tumor regression and produce long-term tumor eradication in 67% of pancreatic tumor-bearing mice without autoimmune effects. Interestingly, engineered T cells with a dual-receptor CAR (dCAR-T) exert high cytotoxicity against pancreatic tumor cells expressing tumor antigens, CEA and mesothelin, resulting in 80% apoptosis of tumor cell [96]. Pancreatic tumor-bearing mice receiving dCAR-T cells released higher levels of cytokines, including IL-2, IL-6, IFN-λ and TNF-α, and showed a marked reduction in tumor growth compared to controls receiving CAR-T cells alone [96]. However, a CEA-targeted CAR T cell clinical study was halted early, after several treated patients suffered from shortness of breath, highlighting the risks and caution needed when pursuing these studies [122]. Several clinical trials are ongoing to better understand the safety, adverse events and potential effectiveness of CEA-targeted CAR T cells in liver metastases (NCT02850536), colorectal cancer (NCT02959151), and CEA positive cancer, including gastric cancer, lung cancer, pancreatic cancer, breast cancer and colorectal cancer (NCT02349724).
MUC-1 has recently become an interesting target in cancer immunotherapy and it is overexpressed in nearly 90% of pancreatic cancers [123]. Overexpression of MUC-1 has been observed to play a role in tumor progression, invasion, metastasis and therapy resistance. MUC-1 may protect tumor cells from immune surveillance by inhibiting antigen recognition by T cells, thereby, inhibiting the effector function of T cells and promoting an anti-inflammatory TME [124]. Generation of CAR T cells targeting MUC-1 and tested in a MUC-1-expressing breast cancer xenograft mouse model showed that MUC-1-CAR-T cells significantly reduced tumor growth compared to untreated control mice by promoting T cell proliferation and increasing production of inflammatory cytokines such as IFN-γ, resulting in enhanced antitumor immunity and the killing of MUC-1 positive tumor cells [125]. An early phase I study evaluating the therapeutic efficacy and safety profile of CAR T cells targeting MUC-1 in seminal vesicle cancer, revealed no adverse effects. This study showed increased levels of CD4+ and CD8+ T cells and significant tumor necrosis in treated patients, suggesting that CAR-T cell therapy could be a promising treatment strategy for patients with solid tumors [126]. Clinical trials are ongoing to assess the efficacy and safety of MUC-1-targeted CAR-T cell therapy in patients with relapsed or refractory solid tumors, including pancreatic adenocarcinoma (NCT02587689 & NCT02617134).
New generations of CAR are under investigation to further enhance their activities and specificities, while also decreasing off-target toxicity. It should be noted that the immunosuppressive environment, as well as the effect of upregulation of immune checkpoint inhibitors on CAR T cells, can significantly restrict the full potential of CAR T cell therapy [127, 128]. Therefore, the new generations of CAR constructs incorporate the ability of antigen-redirected T cells to produce immuno-modulatory cytokines such as IL-12 or immune checkpoint inhibitors; this approach enhances infiltration of immune effector cells via the expression of chemokine receptors, and converts immunosuppressive signals into antitumor immune response within the TME [129]. We believe that the efficacy of CAR T cell therapy can be enhanced by combining it with the other chemotherapeutic agents such as cyclophosphamide or immune checkpoint inhibitors such as CTLA-4 and anti-PD-1 mAb [130]. Strategies that combine CAR T cell therapy with immune checkpoint blockade have been studied in murine models. The combination of CAR T cells with PD-1 blockade significantly enhanced tumor regression in comparison with either treatment alone in transgenic mice with lung cancer [131]. Surprisingly, mice treated with combination therapy also showed decreased levels of MDSCs at the tumor site compared with non-treated control mice. Importantly, administration of the combination treatment was well tolerated, with no signs of autoimmunity [131]. Another study demonstrated that the killing activity of CAR T cells was significantly restored upon the addition of anti-PD-L1, suggesting that the efficacy of CAR T cells can be markedly enhanced by blocking PD-L1 immunosuppression. Furthermore, the engineering of CAR T cells to produce immune checkpoint antibodies within the host was shown to be more effective compared with CAR T cells alone, or CAR T cells combined with anti–PD-1 antibody treatment, resulting in enhanced antitumor immunity of CAR T cells and prolonged overall survival of xenograft mouse models [132]. The expression of PD-1 was significantly reduced in anti-PD-1-secreting CAR T cells compared with CAR T cells. Compared with both CAR T cell monotherapy alone and combination therapy, mice treated with anti-PD-1-secreting CAR T cells had a significantly higher ratio of CD8+ versus CD4+ T cells at the tumor site [133]. Similar results were observed in a renal cell carcinoma mouse model, where CAR T cells secreting anti-PD-L1 antibodies were shown to greatly diminish the exhaustion of T cells and further enhance tumor regression compared with CAR T cells alone [134]. Altogether, these data indicated that this combinatorial strategy could enhance effector function of T cells in the presence of immune checkpoint inhibitors, resulting in tumor regression and improved survival [128]. Ongoing novel clinical studies (NCT03182803 and NCT03030001) are designed to evaluate the efficacy and safety of CAR T cells engineered to express immune checkpoint antibodies in advanced recurrent or refractory malignant solid tumors. Another clinical trial (NCT03179007) is evaluating the safety and efficacy of a novel combination of autologous MUC-1-CAR T cells which express immune checkpoint antibodies in patients with MUC1 positive, advanced solid tumors. Although CAR T cell therapy could produce large populations of T cells specific to tumor antigens, it is time-consuming and expensive compared to vaccine-based therapy [116].
Targeting the immunosuppressive environment
Targeting immunosuppressive environment enriched with immunosuppressive cells such as TAMs and MDSCs, is an important strategy to the success of immunotherapy in pancreatic cancer. Preclinical studies have elucidated the critical role of TAMs not only in tumor progression and metastasis but also in conferring resistance to chemotherapy and radiotherapy [135]. Furthermore, a higher number of tumor infiltrating immunosuppressive cells always correlate with local or metastatic relapse, leading to reduced survival in pancreatic cancer patients [136]. Thus, Table 3 summarizes intervention strategies using CSF1R blockers, JAK/STAT inhibitors, BTK inhibitors and radiotherapy with or without other checkpoint inhibitors, to target the immunosuppressive environment in tumors.
-
(1)
Colony-stimulating factor 1 receptor (CSF1R)
In the TME, CSF1R is expressed on TAMs and MDSCs, which play an important role in suppressing cytotoxic immunity. Upon binding to its ligands, CSF-1 or IL-34, CSF1R undergoes oligomerization and autophosphorylation, leading to the activation of signal transduction, and consequently promoting the proliferation, differentiation and survival of macrophages [137]. CSF-1/CSF1R acts a key regulator of the differentiation, recruitment and survival of TAMs. Importantly, TAMs were shown to promote tumor proliferation, angiogenesis, invasion, and metastasis, as well as resistance to therapies [138]. Infiltration of TAMs was shown to be associated with poor survival in cancer patients [139, 140], as a consequence of the overexpression of CSF-1 and immunosuppressive cytokines such as IL-4 and IL-10 in the TME [141].
CSF1R inhibition is associated with reduced immune suppression, enhanced tumor regression and activation of antitumor immune cells as a consequence of a reduced percentage of TAMs to support antigen presentation and T cell activation within the TME [136]. CSF1R was shown to be involved in the recruitment of macrophages in murine models of pancreatic cancer [142]. The administration of the CSF1R inhibitor (AZD7507) caused a reduction in tumor burden and was associated with improved overall survival in murine models. Pro-tumor cytokines such as IL-6 and IL10 levels were reduced in the tumors of mice treated with AZD7507 [142]. Selective depletion of TAMs via inhibition of CSF1R activity in a mouse model resulted in increased CTL infiltration, decreased Treg infiltration [136] and significantly improved efficacy of chemotherapy-induced antitumor immunity, leading to the hypothesis that targeting the CSF1R/CSF1 interaction in combination with immune checkpoint blockade could produce a synergistic response [135]. Zhu and coworkers [136] demonstrated that the efficacy of anti-PD-1 or anti-CTLA4-based immunotherapy was enhanced via CSF1R/CSF1 blockade. It is important to highlight that the tumor growth reduced more than 90% when CSF1R blockade was combined with either anti-CTLA-4 or anti-PD-1 compared to the mice treated only with anti-CTLA-4 or anti-PD-1 alone. Building on these results, a phase I clinical trial (NCT03153410) using the combination of IMC-CS4 (CSF1R mAb) with GVAX and anti-PD-1 is ongoing for patients with borderline resectable pancreatic cancer. Another anti-CSF1R agent, PLX-3397 (pexidartinib) in combination with anti-PD-L1 Ab (durvalumab) is currently in phase I clinical trial (NCT02777710) for patients with advanced pancreatic and colorectal cancers.
-
(2)
Janus Kinase (JAK) inhibitors
The JAK/STAT signaling pathways are essential for a wide range of cytokines and growth factors, leading to critical cellular events such as hematopoiesis and the development of the immune system [143]. Both type I (IFN-α and IFN-β) and type II (IFN-γ) IFNs are potent activators of the JAK/STAT pathway and play a crucial role in cancer immune surveillance and tumor suppression by regulating the expression of PD-L1 through that pathway. It is observed that tumors grow significantly faster in IFN-γ knockout mice than in wild-type animals. However, the level of IFN-γ is higher in pancreatic tumor tissue compared to normal pancreatic tissue [133, 144]. This means that sustained IFN-γ-STAT1 signaling could lead to chronic inflammation and inflammation-mediated tumor development.
Therefore, the over-activation of JAK/STAT signaling is often associated with inflammatory diseases and malignancies [145], the production of inflammatory cytokines and angiogenic factors, and expansion of MDSCs which promote an immunosuppressive TME [146]. Moreover, the JAK/STAT pathway was found to upregulate PD-L1 expression in pancreatic cancer via the activity of IFNs. In an in vivo study of pancreatic tumor cells, flow cytometric analysis revealed that IFNs can upregulate PD-L1 expression, while the expression of PD-L1 induced by IFNs can be diminished by ruxolitinib, a JAK/STAT inhibitor. The administration of ruxolitinib to pancreatic tumor-bearing mice also resulted in significantly reduced tumor growth [147]. Long-term treatment with ruxolitinib is known to decrease the levels of STAT1 and STAT3 phosphorylation, reverse dysregulated development of Th1 and T follicular helper cells (Tfh), and enhance Th17 responses [148]. The inhibition of STAT1 phosphorylation represses upregulation of PD-L1 by IFN-γ, whereas the inhibition of STAT3 phosphorylation decreases the production of immunosuppressive cytokines by tumor cells, resulting in the conversion of tumor-mediated immune suppression to activation of T cells, as well as increased infiltration of CD8+ T cells and expression of T-bet, IL-21, perforin, and FasL [147]. In addition, the activation of IL-21 by ruxolitinib is known to act as a costimulatory signal that enhances the effector function of immune cells and activation of T cells, thereby suppressing tumor growth. Consequently, ruxolitinib may be effective in overcoming pancreatic cancer resistance to immune checkpoint therapy. The combination of ruxolitinib with anti-PD-1 exhibited significantly greater efficacy in reducing tumor growth compared with ruxolitinib or PD-1 blockade monotherapy. Additionally, the levels of IFN-γ, CD8+ T cells and FasL within TME were significantly higher in tumor-bearing mice treated with combined therapy [144]. All these data indicate that ruxolitinib is effective in facilitating the infiltration and activation of CTLs, thus enhancing the efficacy of immune checkpoint therapy in pancreatic cancer.
In a randomized double-blind, phase II study, ruxolitinib plus capecitabine improved survival in patients with metastatic pancreatic cancer who failed to respond to gemcitabine chemotherapy [149]. Unfortunately, subsequent phase III studies (NCT02117479 & NCT02119663) have closed to enrolment as there was no significant improvement in patient survival. The efficacy of ruxolitinib therapy could be enhanced by using it as an adjunct agent to suppress chronic inflammation and facilitate infiltration of CD8+ T cells, rather than as a monotherapeutic agent for overcoming anti-PD-L1 immunotherapy resistance in pancreatic cancer patients [144].
-
(3)
Bruton’s Tyrosine Kinase (BTK) inhibitors
BTK is a member of Tec tyrosine kinase family which is important for B cell development, differentiation and signaling. Activation of BTK has been implicated in the pathogenesis of B cell malignancies as it can trigger downstream signaling events such as proliferation and differentiation mediated through transcription factors such as NFκB, as well as survival signaling cascades such as RAS/RAF/MEK/ERK and PI3K/AKT/mTOR [150]. BTK also leads to T cell suppression by regulating interactions between B cells and macrophages, promoting pancreatic cancer progression [151]. In pancreatic cancer, the infiltration of mast cells was found to correlate with high tumor grade and poor survival [152].
Ibrutinib, a BTK inhibitor, was shown to exhibit anti-fibrotic effects in PDAC by effectively inhibiting infiltration of mast cells in both transgenic mice and patient-derived xenograft models. Ibrutinib reduces stromal fibrosis and inhibits tumor progression, indicating that it may have the potential to sensitize tumors to checkpoint blockade. The administration of ibrutinib in a mouse model of insulinoma, resulted in vascular collapse and tumor regression [153]. Ibrutinib is also known to inhibit interleukin-2-inducible T-cell kinase (ITK), a type of enzyme which is essential for the survival of Th2 cells. As a result, it may shift from Th2 cell protumor response to Th1 cell antitumor response, and augment the deposition of CD8+ T cells in tumors [154].
Surprisingly, the combination therapies with ibrutinib and immune checkpoint blockade showed impressive therapeutic effects not only in mouse models of lymphoma that are resistant to ibrutinib, but also in animal models of breast and colon cancers [154]. The depletion of CD4+ and CD8+ T cells abrogated the treatment efficacy of anti-PD-L1, confirming the role of T cells in activating antitumor activity. Conversely, the antitumor T cell response could be enhanced via the addition of ibrutinib to anti-PD-L1, resulting in tumor regression and prolonged survival of mice with lymphoma [154]. Treatment with ibrutinib or anti-PD-L1 monotherapy exhibited neither delayed tumor growth nor enhanced survival of tumor-bearing mice. In contrast, the combination of ibrutinib and anti-PD-L1 delayed tumor growth, improved survival, and reduced lung metastasis in both breast tumor-bearing mice and colon tumor-bearing mice [154]. Additionally, mice cured by the combination therapies also displayed long term immune memory, as they were resistant to the respective tumors upon tumor re-challenge [154]. A phase I/II clinical trial (NCT02403271) was conducted to assess the safety and efficacy of ibrutinib in combination with anti-PD-L1 antibody (durvalumab) in patients with relapsed or refractory solid tumors, including pancreatic cancer. Another phase II clinical trial (NCT02940301) is recruiting patients with Hodgkin lymphoma to determine the efficacy of ibrutinib in combination with an anti-PD-1 antibody (nivolumab).
-
(4)
Immune checkpoint therapy with radiotherapy (RT)
RT can convert the TME from a ‘cold’ state that lacks infiltration of antitumor immune cells and is resistant to immunotherapy to a ‘hot’ state, which can activate the immune system in triggering an antitumor response, leading to cytotoxicity and the release of stimulatory agents that could enhance the recruitment of T cells to the tumor site. For instance, antitumor T cells that are generated spontaneously or via vaccination may be prevented from entry into the tumor site due to the presence of the desmoplastic stroma of cancer [155]. In contrast, by inducing tumor cell death coupled with the release of danger signals, radiation can stimulate activation and migration of DCs to the tumor-draining lymph nodes where the activation of antitumor T cells will be stimulated. The activated T cells then migrate to the established tumor, kill tumor cells and secrete cytokines, which further enhance activation of DCs and conversion of TAMs to the antitumor M1 phenotype; this indicates that RT converts cold tumor to hot tumors [155].
RT has emerged as a front-runner strategy, where previous studies using murine models of breast cancer showed that it can convert tumors to become responsive to immune checkpoint therapy [156, 157] Interestingly, Azad and coworkers [158] reported that PD-L1 was upregulated after RT and chemotherapy in a JAK/STAT dependent manner, while the intratumoral milieu was shifted away from infiltration of immunosuppressive MDSCs and Tregs towards the infiltration of activated CD8+ cells. It is important to note that the tumor response was significantly improved in the Pan02 murine model receiving only higher RT doses (12 Gy or 5 × 3 Gy) plus PD-L1 blockade, whereas anti-PD-L1 alone did not affect tumor growth. PD-L1 blockade did, however, sensitize pancreatic allografts to high RT doses. Flow cytometric analysis revealed that there was a significant increase in infiltration of both CD45+CD4+ T cells and CD45+CD8+ T cells upon tumor irradiation, which was further enhanced by blockade of PD-L1. As a result, the authors concluded that the efficacy of RT in delaying tumor growth can be augmented via the blockade of PD-L1, and that the infiltration of MDSCs and Tregs into the tumor site can be significantly decreased by treatment with higher RT doses in combination with PD-L1 blockade. PD-L1 blockade also showed potential in enhancing the anti-metastatic effect of RT in murine liver cancer models, supporting the use of this combination strategy in future clinical studies [158]. These findings were consistent with those of Deng and coworkers [159], who revealed that RT plus anti-PD-L1 could synergistically reduce the infiltration of MDSCs which normally suppress the antitumor T cells response, thus altering the immune response in the TME of breast cancer-bearing mouse models. Although the molecular mechanisms have yet to be fully elucidated, the central message generated by Azad and coworkers [158] clearly provides an important insight on the potential of immune checkpoint inhibitors to radio-sensitize a large group of RT-resistant tumors, including pancreatic cancer that traditionally has been classified as non-immunogenic.
Clinical research on combination strategies is rapidly progressing. There are numerous clinical studies that examine concurrent treatment, in combination with RT, with either PD-L1 blockade or anti-PD-L1 mAbs in various types of solid tumors. Interestingly, unresectable, non-metastatic pancreatic cancer is also being investigated in a phase Ib clinical study (NCT02868632) to evaluate the efficacy of RT plus either anti-CTLA-4 mAb alone, anti-PD-L1 mAb alone, or the combination of both immune checkpoint inhibitors. Recently, a randomized phase II study (NCT02866383) in metastatic pancreatic cancer patients who are intolerant to chemotherapeutic agents is ongoing to determine the efficacy and safety profile of nivolumab or nivolumab plus ipilimumab administered simultaneously with high dose RT; this is estimated to end in 2019. Another similar pilot study (NCT02311361) is also investigating the efficacy of immune checkpoint inhibition (tremelimumab and/or MEDI4736) with RT in unresectable metastatic pancreatic cancer patients.
Targeting the desmoplastic barrier
Pancreatic cancers are highly desmoplastic, with low vascular perfusion leading to hypoxia, and impeded delivery and efficacy of drugs. The desmoplastic reaction is known to be driven by the production of hyaluronan (HA) by fibroblasts. The accumulation of HA in the TME results in an increase in tumor interstitial fluid pressure (IFP), which significantly compresses blood vessels and impedes blood flow [160]. This in turn leads to the hypoxic environment in the tumor. In the hypoxic microenvironment, tumor cells can achieve tumor escape by upregulating the activation of immunosuppressive cells such as Tregs and MDSCs, which may further lead to the dysfunction of infiltrating CD8+ T cells, and consequently, the facilitation of tumor invasion, and metastasis, as well as resistance to therapy [161]. Furthermore, the secretion of immunosuppressive cytokines by the hypoxic TME can induce apoptosis of CD8+ T cells and the production of Tregs, along with inhibition of the activation of APCs; this, in turn, results in the failure of DCs to present tumor antigens to T cells for the activation of an antitumor response [161].
Therefore, the treatment strategies which target tumor hypoxia and excessive fibrosis are likely to shift the TME from being immunosuppressive to one that facilitates the activation of T cell immune response and sensitizes pancreatic cancer to immune checkpoint therapy. Focal adhesion kinase (FAK) represents one of the stromal targets and plays a role in cancer cell proliferation, progression and survival [162]. It has been recognized as one of the key factors in regulating the fibrotic TME of PDAC. The overexpression of FAK in many solid tumors is inversely associated with survival [163, 164]. Inhibition of FAK expression with a FAK inhibitor (PF-562,271) in mouse models of pancreatic cancer decreased tumor proliferation and reduced tumor fibrosis; it also reduced the recruitment of MDSCs, Tregs and TAMs into the tumor site. The reduction of myeloid cells was significantly associated with increased levels of CD8+ T cells [162, 165]. FAK inhibition also dramatically reduced the secretion of both pro-inflammatory and pro-fibrotic cytokines, such as IL-1α, IL-1β, E-selectin, MMP3, and CCL6 which play a role in the recruitment of myeloid cells [165]. These data suggested that the expression of FAK in pancreatic cancer could facilitate the creation of a fibrotic and immunosuppressive TME that protects tumor cells from CTL-mediated antitumor activity. Additionally, increased infiltration of CD8+ T cells, reduced tumor burden and improved overall survival were observed in tumor-bearing mice treated with combination therapy comprising FAK inhibition and PD-1 blockade, compared to PD-1 blockade alone; this suggests that the efficacy of anti-PD-1 can be enhanced by inhibiting the expression of FAK [165]. More conclusive evidence will be available when the phase I/II clinical trial (NCT02758587) is completed. This trial involves patients with advanced solid tumors, including pancreatic cancers, who receive a FAK inhibitor (defactinib) in combination with an anti-PD-1 mAb (pembrolizumab).
Another potential therapeutic target within the stroma of pancreatic cancer is hyaluronan (HA). Accumulation of HA or HA combined with its binding proteins (HABPs) forms a size selective barrier to antitumor immune cells and the efficient delivery of therapeutic drugs; this creates an immunosuppressive environment which prevents infiltration of T cells, as well as preventing chemotherapeutic agents and mAbs from entering the tumor and reaching their site of action. Its accumulation is thus always associated with poor prognosis in cancer patients [160]. A novel investigational agent, PEGPH20 (pegvorhyaluronidase alfa), which is a PEGylated form of recombinant human hyaluronidase, PH20, has been identified as an enzyme that can degrade HA [160, 166]. PEGPH20-induced HA depletion resulted in increased recruitment of NK cells and antibody delivery into the high HA tumor site, greatly enhancing cetuximab or trastuzumab ADCC [167]. The increased infiltration of CD8+ T cells as a consequence of the degradation of HA by PEGPH20 also improved the efficacy of anti-PD-L1 in HA-rich breast cancer tumor models, resulting in inhibition of tumor growth, in comparison with PEGPH20 or anti-PD-L1 alone [168]. The combination of PEGPH20 and anti-PD-L1 also rendered HA-rich tumors sensitive to anti-PD-L1. Imaging studies showed that there was an accumulation of anti-PD-L1 within the TME which was thought to be enhanced by PEGPH20 [168]. These data suggested that removal of HA by PEGPH20 enhanced the efficacy of immune checkpoint therapy by increasing infiltration of CD4+ T cells and NK cells, while decreasing the percentage of MDSCs; at the same time, it also enhanced the accumulation of immunotherapeutic antibody in HA-rich tumors.
PEGPH20 has also been shown to delay tumor growth and metastasis and to enhance chemotherapy efficacy in HA-rich tumor models, including pancreatic cancer. This is likely due to the enhanced perfusion of therapeutic agents into the tumor. In the PEGPH20-treated KPC mouse model, the diameter of vessels was significantly increased, resulting in improved blood flow. Surprisingly, when PEGPH20 was administered together with gemcitabine, the combination regimen significantly suppressed tumor growth and increased apoptosis resulting in an increased overall survival compared to gemcitabine alone [166, 169]. Moreover, a preclinical studydemonstrated that in mice treated with a combination of PEGPH20 and shIDO-ST, a Salmonella-based therapy which targets the immunosuppressive molecule indoleamine 2,3-dioxygenase (IDO), there was a significant reduction in tumor burden, an increased infiltration of immune cells into the desmoplastic stroma of pancreatic cancer, and an enhancement of FasL-mediated apoptosis of tumor cells [170]. These results suggested that stromal barriers to infiltration of antitumor immune cells can be overcome with PEGPH20, with a consequent enhancement of the antitumor activity of immunotherapy. However, the actual mechanism of HA depletion induced by PEGPH20 is still under investigation [171].
To date, there are no clinical studies investigating PEGPH20 as monotherapy or combinatory treatment in cancers [172]. Numerous clinical trials are currently enrolling patients; these trials will evaluate the efficacy of PEGPH20 in combination with other therapeutic agents (Table 4). A non-randomized pilot study (NCT02921022) investigating the effects of PEGPH20 plus a combination regimen (gemcitabine, nab-paclitaxel and rivaroxaban) in patients with/without prior thromboembolic events is currently ongoing. A Phase I clinical trial (NCT03481920) is currently underway to evaluate the pharmacodynamics and efficacy of PEGPH20 plus avelumab (anti-PD-L1 mAb) in chemotherapy-resistant pancreatic cancer patients [173]. Another clinical trial (NCT03267940) is ongoing to evaluate the safety, tolerability and antitumor activity of PEGPH20 with anti-PD-L1 in patients with cholangiocarcinoma/gallbladder cancer. The safety and tolerability of a combination of PEGPH20 and anti-PD-1 is also under investigation in HA-high patients with lung cancer and gastric cancer (NCT02563548). Targeting the tumor stroma could modulate its immunosuppressive conditions by facilitating the normalization of blood vessels and weakening the immunosuppressive effects of cancer-associated fibroblasts; this will effectively enhance the transportation of oxygen and therapeutic agents as well as the infiltration of effector T cells to the tumor tissues, thereby, enhancing the efficacy of the immune checkpoint therapies [174].
Combinations of mismatch repair deficiency and PD-1 blockade in tumors
Surprisingly, the effectiveness of immunotherapy can be predicted based on the presence of mismatch repair deficiency. Mismatch repair deficient (dMMR) cancers are hypothesized to have large numbers of mutation-associated neoantigens (MANAs) which can be recognized by the host antitumor immune cells [175, 176]. dMMR is associated with frameshift mutations that take place within the coding sequences, resulting in the production of functionally inactive proteins that can be presented via MHC molecules to CD8+ T cells as tumor neoantigens, consequently eliciting an antitumor immune response as well as the infiltration of T cells into the TME [177]. An early clinical trial (NCT02060188) showed that colorectal cancer with mismatch repair deficiency was associated with an increased tumor neoantigen load and infiltration of immune effector cells, consequently enhancing tumor sensitivity to immune checkpoint blockade, particularly anti-PD-1 [178]. A phase II clinical trial (NCT01876511) is ongoing to evaluate the clinical activity of pembrolizumab (anti-PD-1) in patients with dMMR tumors. The objective response rate (ORR) and progrssion-free survival were significantly higher (40 and 78% respectively) in dMMR cancer patients compared with MMR-proficient colorectal cancer patients ((0 and 11%, respectively) [179], supporting the hypothesis that dMMR tumors are more responsive to immune checkpoint blockade than MMR-proficient tumors.
Future prospects
Pancreatic cancer has been recognized as one of the most aggressive malignancies and is usually diagnosed at an advanced level, with limited or no effective therapeutic options thus far. However, preclinical and clinical trials have shown promising results in therapies targeting immune checkpoint molecules. Of note, immune checkpoint therapies is largely ineffective in pancreatic cancer due to the low mutational load along with the hypoxic pancreatic TME that is filled with immunosuppressive cells, which acts as a selective barrier to drug penetration and infiltration of immune effector cells, significantly limiting the efficacy of immunotherapy. Many efforts have been made to gain insights into patients who exhibit resistance towards immune checkpoint therapy and to find ways to maximize treatment efficacy via combination therapies. Rational combinations of immunotherapy may represent one potential strategy to synergistically overcome the immunosuppressive microenvironment of pancreatic cancer, as well as to induce long-lasting antitumor activity within the TME. The optimal dose, schedule, and ideal sequence, for example, when to combine immunotherapy with other therapies such as chemotherapy, radiation therapy, or targeted agents, must be determined, as these therapies have different mode of actions [180]. In view of the increasing number of novel compounds being synthesized [181, 182] or isolated from natural products [183,184,185,186,187,188,189,190], more pre-clinical studies are required to test whether these new classes of compounds can also target the tumor microenvironment of pancreatic cancer. These new compounds may act synergistically with chemotherapy, radiation therapy, immunotherapy or other targeted therapies.
Additionally, several studies are being conducted to develop (1) vaccine-based therapy with immune checkpoint blockade to increase infiltration of T cells; (2) co-delivery of stromal targeting agents with immune checkpoint inhibitor; and (3) co-delivery of T cells priming agents with immunotherapeutic antibodies [191]. Interestingly, the current focus has shifted to characterization of tumor neoantigens. In pancreatic cancer, assessment of the quality of tumor neoantigens has opened a new avenue for investigating tumor progression. Advancement in neoantigen discovery may unlock the potential of personalized cancer vaccines to work alone or in combination with other therapies to enhance the strength of antitumor effects and improve clinical outcomes. Future efforts can be anticipated to harness neoantigen-specific antitumor immunity to treat patients with immune checkpoint inhibitor-resistant cancers such as pancreatic cancer and to identify immunogenic hotspots for directed neoantigen targeting [192]. Personalized immunotherapy based on individual genetic information, molecular biology and immune profiling is hypothesized to result in the greatest clinical outcomes for cancer patients [193], leading either to the conversion of untreatable cancer into a controllable chronic disease or long term tumor eradication [194]. In addition, more efforts should be made in the development of databases and bioinformatics platforms, as an enormous amount of genomics and biomarkers data have been generated; at the same time, this could empower the practice of precision immune-oncology. More validation studies and well-designed clinical trials are warranted to provide evidence to support personalized immunotherapy for patients with pancreatic cancer.
Conclusion
The development of combination strategies that act to stimulate the immune response and break down the barriers of TME in conjunction with immunotherapies, hold promises for improving the overall survival of pancreatic cancer patients. To achieve this, researchers and clinicians need to advance their understandings of tumor immunology, identify novel biomarkers, optimize the timing of immunotherapy, and implement novel preclinical models to better predict the therapeutic efficacy; there also needs to be an improvement in clinical trial designs to achieve a better understanding of the mechanism of action and resistance of pancreatic cancer towards immunotherapy. We believe that combination immunotherapies represent a promising modality in pancreatic cancer; these are advancing from bench to bedside at a rapid pace. Translation of immunotherapies into clinical practice will bring a new hope for patients suffering from this aggressive, silent, asymptomatic killer.
Abbreviations
- ACT:
-
Adoptive T cell transfer
- APC:
-
Antigen presenting cells
- Arg:
-
Arginine
- Arg-1:
-
Arginase-1
- BTK:
-
Bruton’s Tyrosine Kinase
- CAR:
-
Chimeric antigen receptor
- Cox-2:
-
Cyclooxygenase-2
- CSF1R:
-
Colony-stimulating factor 1 receptor
- CT-011:
-
Pidilizumab
- CTLA-4:
-
Cyototoxic T-lymphocyte antigen-4
- DC:
-
Dendritic cells
- ECM:
-
Extracellular matrix
- FAP:
-
Fibroblast activation protein
- FDA:
-
Food and Drug Administration
- GM-CSF:
-
Granulocyte macrophage-colony stimulating factor
- HA:
-
Hyaluronan
- HABP:
-
Hyaluronan binding proteins
- IFN:
-
Interferon
- IFP:
-
Interstitial fluid pressure
- iNOS:
-
Inducible nitric oxide synthase
- ITK:
-
Inducible T-cell kinase
- JAK:
-
Janus kinase
- mAb:
-
Monoclonal antibody
- Mb:
-
Megabase
- MDSC:
-
Myeloid-derived suppressor cells
- MIP-3α:
-
Macrophage inflammatory protein-3 alpha
- MMPs:
-
Matrix metalloproteinases
- MUC-1:
-
Mucin-1
- NK:
-
Natural killer
- NSCLC:
-
Non-small cell lung cancer
- ORR:
-
Objective response rate
- OS:
-
Overall survival
- PD-1:
-
Programmed death 1
- PDAC:
-
Pancreatic ductal adenocarcinoma
- PD-L1:
-
Programmed death-ligand1
- PEGPH20:
-
Pegvorhyaluronidase alfa
- PFS:
-
Progression free survival
- PNT:
-
Peroxynitrite
- ROS:
-
Reactive oxygen species
- RT:
-
Radiotherapy
- SD:
-
Stable disease
- TAA:
-
Tumor associated antigen
- TAM:
-
Tumor associated macrophages
- TCR:
-
T cell receptors
- TGF-α:
-
Transforming growth factor-α
- TGF-β:
-
Transforming growth factor-β
- TIL:
-
Tumor infiltrating lymphocytes
- TME:
-
Tumor microenvironment
- TNF-α:
-
Tumor necrosis factor-α
- TP:
-
Thymidine phosphorylase
- Treg:
-
Regulatory T-cells
- VEGF:
-
Vascular endothelial growth factor
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68(6):394–424.
Ilic M, Ilic I. Epidemiology of pancreatic cancer. World J Gastroenterol. 2016;22(44):9694–705.
Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74(11):2913–21.
Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol. 1997;15(6):2403–13.
Sun C, Ansari D, Andersson R, Wu DQ. Does gemcitabine-based combination therapy improve the prognosis of unresectable pancreatic cancer? World J Gastroenterol. 2012;18(35):4944–58.
Conroy T, Desseigne F, Ychou M, Bouche O, Guimbaud R, Becouarn Y, et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N Engl J Med. 2011;364(19):1817–25.
Von Hoff DD, Ervin T, Arena FP, Chiorean EG, Infante J, Moore M, et al. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N Engl J Med. 2013;369(18):1691–703.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32(10):1020–30.
Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin's lymphoma. N Engl J Med. 2015;372(4):311–9.
Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, et al. Nivolumab versus docetaxel in advanced nonsquamous non-small-cell lung Cancer. N Engl J Med. 2015 Oct 22;373(17):1627–39.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Er JL, Goh PN, Lee CY, Tan YJ, Hii LW, Mai CW, et al. Identification of inhibitors synergizing gemcitabine sensitivity in the squamous subtype of pancreatic ductal adenocarcinoma (PDAC). Apoptosis. 2018;23(5-6):343-55.
Beatty GL, Gladney WL. Immune escape mechanisms as a guide for cancer immunotherapy. Clin Cancer Res. 2015;21(4):687–92.
Chung FF, Mai CW, Ng PY, Leong CO. Cytochrome P450 2W1 (CYP2W1) in colorectal cancers. Curr Cancer Drug Targets. 2016;16(1):71–8.
Mai CW, Chung FF, Leong CO. Targeting Legumain as a novel therapeutic strategy in cancers. Curr Drug Targets. 2017;18(11):1259–68.
Mai CW, Kang YB, Pichika MR. Should a toll-like receptor 4 (TLR-4) agonist or antagonist be designed to treat cancer? TLR-4: its expression and effects in the ten most common cancers. Onco Targets Ther. 2013;6:1573–87.
Maniam G, Mai CW, Zulkefeli M, Dufes C, Tan DM, Fu JY. Challenges and opportunities of nanotechnology as delivery platform for Tocotrienols in Cancer therapy. Front Pharmacol. 2018;9:1358.
Feig C, Gopinathan A, Neesse A, Chan DS, Cook N, Tuveson DA. The pancreas cancer microenvironment. Clin Cancer Res. 2012;18(16):4266–76.
Waghray M, Yalamanchili M, di Magliano MP, Simeone DM. Deciphering the role of stroma in pancreatic cancer. Curr Opin Gastroenterol. 2013;29(5):537–43.
Parente P, Parcesepe P, Covelli C, Olivieri N, Remo A, Pancione M, et al. Crosstalk between the tumor microenvironment and immune system in pancreatic ductal adenocarcinoma: potential targets for new therapeutic approaches. Gastroenterol Res Pract. 2018;2018:15.
Haque I, Subramanian A, Banerjee S, Banerjee SK. Current and emerging perspectives on immunotherapy for pancreatic cancer. Transl Cancer Res 2017;6(Suppl 2):S331-S336.
Liu Q, Liao Q, Zhao Y. Chemotherapy and tumor microenvironment of pancreatic cancer. Cancer Cell Int. 2017;17(68):017–0437.
von Ahrens D, Bhagat TD, Nagrath D, Maitra A, Verma A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J Hematol Oncol. 2017;10(1):76.
Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16(9):582–98.
Qu C, Wang Q, Meng Z, Wang P. Cancer-associated fibroblasts in pancreatic Cancer: should they be deleted or reeducated? Integr Cancer Ther. 2018;17(4):1016–9.
Özdemir BC, Pentcheva-Hoang T, Carstens JL, Zheng X, Wu C-C, Simpson TR, et al. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 2014;25(6):719–34.
Cheng JD, Dunbrack RL Jr, Valianou M, Rogatko A, Alpaugh RK, Weiner LM. Promotion of tumor growth by murine fibroblast activation protein, a serine protease, in an animal model. Cancer Res. 2002;62(16):4767–72.
Joyce JA, Fearon DT. T cell exclusion, immune privilege, and the tumor microenvironment. Science. 2015;348(6230):74–80.
Cheng JD, Valianou M, Canutescu AA, Jaffe EK, Lee HO, Wang H, et al. Abrogation of fibroblast activation protein enzymatic activity attenuates tumor growth. Mol Cancer Ther. 2005;4(3):351–60.
Lo A, Li CP, Buza EL, Blomberg R, Govindaraju P, Avery D, et al. Fibroblast activation protein augments progression and metastasis of pancreatic ductal adenocarcinoma. JCI Insight. 2017;2(19).
Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119(12):3613–25.
Cremasco V, Astarita JL, Grauel AL, Keerthivasan S, MacIsaac K, Woodruff MC, et al. FAP delineates heterogeneous and functionally divergent stromal cells in immune-excluded breast tumors. Cancer Immunol Res. 2018;6(12):1472-85.
Lo A, Wang L-CS, Scholler J, Monslow J, Avery D, Newick K, et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer Res. 2015;75(14):2800-810. https://doi.org/10.1158/0008-5472.CAN-14-3041.
Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein–α. Science. 2010;330(6005):827–30.
Foucher ED, Ghigo C, Chouaib S, Galon J, Iovanna J, Olive D. Pancreatic ductal adenocarcinoma: a strong imbalance of good and bad immunological cops in the tumor microenvironment. Front Immunol. 2018;9:1044.
Fu Y, Liu S, Zeng S, Shen H. The critical roles of activated stellate cells-mediated paracrine signaling, metabolism and onco-immunology in pancreatic ductal adenocarcinoma. Mol Cancer. 2018;17(1):62.
Ren B, Cui M, Yang G, Wang H, Feng M, You L, et al. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol Cancer. 2018;17(1):108.
Ene-Obong A, Clear AJ, Watt J, Wang J, Fatah R, Riches JC, et al. Activated pancreatic stellate cells sequester CD8+ T cells to reduce their infiltration of the juxtatumoral compartment of pancreatic ductal adenocarcinoma. Gastroenterology. 2013;145(5):1121–32.
Tang D, Yuan Z, Xue X, Lu Z, Zhang Y, Wang H, et al. High expression of Galectin-1 in pancreatic stellate cells plays a role in the development and maintenance of an immunosuppressive microenvironment in pancreatic cancer. Int J Cancer. 2012;130(10):2337–48.
Chang JH, Jiang Y, Pillarisetty VG. Role of immune cells in pancreatic cancer from bench to clinical application: an updated review. Medicine. 2016;95(49):0000000000005541.
Tan MC, Goedegebuure PS, Belt BA, Flaherty B, Sankpal N, Gillanders WE, et al. Disruption of CCR5-dependent homing of regulatory T cells inhibits tumor growth in a murine model of pancreatic cancer. J Immunol. 2009;182(3):1746–55.
Liyanage UK, Goedegebuure PS, Moore TT, Viehl CT, Moo-Young TA, Larson JW, et al. Increased prevalence of regulatory T cells (Treg) is induced by pancreas adenocarcinoma. J Immunother. 2006;29(4):416–24.
Moo-Young TA, Larson JW, Belt BA, Tan MC, Hawkins WG, Eberlein TJ, et al. Tumor-derived TGF-beta mediates conversion of CD4+Foxp3+ regulatory T cells in a murine model of pancreas cancer. J Immunother. 2009;32(1):12–21.
Tang Y, Xu X, Guo S, Zhang C, Tang Y, Tian Y, et al. An increased abundance of tumor-infiltrating regulatory T cells is correlated with the progression and prognosis of pancreatic ductal adenocarcinoma. PLoS One. 2014;9(3):e91551.
Keenan BP, Saenger Y, Kafrouni MI, Leubner A, Lauer P, Maitra A, et al. A Listeria vaccine and depletion of T-regulatory cells activate immunity against early stage pancreatic intraepithelial neoplasms and prolong survival of mice. Gastroenterology. 2014;146(7):1784–94 e6.
Leao IC, Ganesan P, Armstrong TD, Jaffee EM. Effective depletion of regulatory T cells allows the recruitment of mesothelin-specific CD8 T cells to the antitumor immune response against a mesothelin-expressing mouse pancreatic adenocarcinoma. Clin Transl Sci. 2008;1(3):228–39.
Gabitass RF, Annels NE, Stocken DD, Pandha HA, Middleton GW. Elevated myeloid-derived suppressor cells in pancreatic, esophageal and gastric cancer are an independent prognostic factor and are associated with significant elevation of the Th2 cytokine interleukin-13. Cancer Immunol Immunother. 2011;60(10):1419–30.
Sinha P, Clements VK, Bunt SK, Albelda SM, Ostrand-Rosenberg S. Cross-talk between myeloid-derived suppressor cells and macrophages subverts tumor immunity toward a type 2 response. J Immunol. 2007;179(2):977–83.
Liu C, Yu S, Kappes J, Wang J, Grizzle WE, Zinn KR, et al. Expansion of spleen myeloid suppressor cells represses NK cell cytotoxicity in tumor-bearing host. Blood. 2007;109(10):4336–42.
Raber P, Ochoa AC, Rodriguez PC. Metabolism of L-arginine by myeloid-derived suppressor cells in cancer: mechanisms of T cell suppression and therapeutic perspectives. Immunol Investig. 2012;41(6–7):614–34.
Kusmartsev S, Nefedova Y, Yoder D, Gabrilovich DI. Antigen-specific inhibition of CD8+ T cell response by immature myeloid cells in cancer is mediated by reactive oxygen species. J Immunol. 2004;172(2):989–99.
Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S. Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8+ T cells. J Immunol. 2009;183(2):937–44.
Ostrand-Rosenberg S. Myeloid-derived suppressor cells: more mechanisms for inhibiting antitumor immunity. Cancer Immunol Immunother. 2010;59(10):1593–600.
Mielgo A, Schmid MC. Impact of tumour associated macrophages in pancreatic cancer. BMB Rep. 2013;46(3):131–8.
Clark CE, Hingorani SR, Mick R, Combs C, Tuveson DA, Vonderheide RH. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007;67(19):9518–27.
Menen RS, Hassanein MK, Momiyama M, Suetsugu A, Moossa AR, Hoffman RM, et al. Tumor-educated macrophages promote tumor growth and peritoneal metastasis in an orthotopic nude mouse model of human pancreatic cancer. In Vivo. 2012;26(4):565–9.
Cui R, Yue W, Lattime EC, Stein MN, Xu Q, Tan XL. Targeting tumor-associated macrophages to combat pancreatic cancer. Oncotarget. 2016;7(31):50735–54.
Campbell AS, Albo D, Kimsey TF, White SL, Wang TN. Macrophage inflammatory protein-3α promotes pancreatic cancer cell invasion. J Surg Res. 2005;123(1):96–101.
Guo S, Contratto M, Miller G, Leichman L, Wu J. Immunotherapy in pancreatic cancer: unleash its potential through novel combinations. World J Clin Oncol. 2017;8(3):230–40.
Hilmi M, Bartholin L, Neuzillet C. Immune therapies in pancreatic ductal adenocarcinoma: where are we now? World J Gastroenterol. 2018;24(20):2137–51.
Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature. 2013;500(7463):415–21.
Winograd R, Byrne KT, Evans RA, Odorizzi PM, Meyer AR, Bajor DL, et al. Induction of T-cell immunity overcomes complete resistance to PD-1 and CTLA-4 blockade and improves survival in pancreatic carcinoma. Cancer Immunol Res. 2015;3(4):399–411.
Elnemr A, Ohta T, Yachie A, Kayahara M, Kitagawa H, Ninomiya I, et al. Human pancreatic cancer cells express non-functional Fas receptors and counterattack lymphocytes by expressing Fas ligand; a potential mechanism for immune escape. Int J Oncol. 2001;18(1):33–9.
Gong J, Hendifar A, Tuli R, Chuang J, Cho M, Chung V, et al. Combination systemic therapies with immune checkpoint inhibitors in pancreatic cancer: overcoming resistance to single-agent checkpoint blockade. Clin Transl Med. 2018;7(1):32.
Schober M, Jesenofsky R, Faissner R, Weidenauer C, Hagmann W, Michl P, et al. Desmoplasia and chemoresistance in pancreatic cancer. Cancers (Basel). 2014;6(4):2137–54.
Li J, Wientjes MG, Au JL. Pancreatic cancer: pathobiology, treatment options, and drug delivery. AAPS J. 2010;12(2):223–32.
Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med. 2011;364(26):2517–26.
Johansson H, Andersson R, Bauden M, Hammes S, Holdenrieder S, Ansari D. Immune checkpoint therapy for pancreatic cancer. World J Gastroenterol. 2016;22(43):9457–76.
Barnhart C. Pembrolizumab: first in class for treatment of metastatic melanoma. J Adv Pract Oncol. 2015;6(3):234–8.
Burns MC, O'Donnell A, Puzanov I. Pembrolizumab for the treatment of advanced melanoma. Expert Opin Orphan Drugs. 2016;4(8):867–73.
Atkins MB, Clark JI, Quinn DI. Immune checkpoint inhibitors in advanced renal cell carcinoma: experience to date and future directions. Ann Oncol. 2017;28(7):1484–94.
McDermott DF, Sosman JA, Sznol M, Massard C, Gordon MS, Hamid O, et al. Atezolizumab, an anti-programmed death-ligand 1 antibody, in metastatic renal cell carcinoma: long-term safety, clinical activity, and immune correlates from a phase Ia study. J Clin Oncol. 2016;34(8):833–42.
Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after Chemoradiotherapy in stage III non–small-cell lung Cancer. N Engl J Med. 2017;377(20):1919–29.
Palla AR, Doll D. Immunotherapy in Merkel cell carcinoma: role of Avelumab. Immunotargets Ther. 2018;7:15–9.
Sun D, Ma J, Wang J, Zhang F, Wang L, Zhang S, et al. Clinical observation of immune checkpoint inhibitors in the treatment of advanced pancreatic cancer: a real-world study in Chinese cohort. Ther Clin Risk Manag. 2018;14:1691–700.
Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–33.
Vonderheide RH. The immune revolution: a case for priming, not checkpoint. Cancer Cell. 2018;33(4):563–9.
Chang JH, Jiang Y, Pillarisetty VG. Role of immune cells in pancreatic cancer from bench to clinical application: an updated review. Medicine (Baltimore). 2016;95(49):e5541.
Er JL, Goh PN, Lee CY, Tan YJ, Hii LW, Mai CW, et al. Identification of inhibitors synergizing gemcitabine sensitivity in the squamous subtype of pancreatic ductal adenocarcinoma (PDAC). Apoptosis. 2018;23(5–6):343–55.
McDonnell AM, Lesterhuis WJ, Khong A, Nowak AK, Lake RA, Currie AJ, et al. Tumor-infiltrating dendritic cells exhibit defective cross-presentation of tumor antigens, but is reversed by chemotherapy. Eur J Immunol. 2015;45(1):49–59.
Haynes NM, van der Most RG, Lake RA, Smyth MJ. Immunogenic anti-cancer chemotherapy as an emerging concept. Curr OpinImmunol. 2008;20(5):545–57.
Ma Y, Adjemian S, Mattarollo SR, Yamazaki T, Aymeric L, Yang H, et al. Anticancer chemotherapy-induced intratumoral recruitment and differentiation of antigen-presenting cells. Immunity. 2013;38(4):729–41.
Nowak AK, Robinson BW, Lake RA. Synergy between chemotherapy and immunotherapy in the treatment of established murine solid tumors. Cancer Res. 2003;63(15):4490–6.
Aglietta M, Barone C, Sawyer MB, Moore MJ, Miller WH Jr, Bagala C, et al. A phase I dose escalation trial of tremelimumab (CP-675,206) in combination with gemcitabine in chemotherapy-naive patients with metastatic pancreatic cancer. Ann Oncol. 2014;25(9):1750–5.
Nomi T, Sho M, Akahori T, Hamada K, Kubo A, Kanehiro H, et al. Clinical significance and therapeutic potential of the programmed death-1 ligand/programmed death-1 pathway in human pancreatic cancer. Clin Cancer Res. 2007;13(7):2151–7.
Thind K, Padrnos LJ, Ramanathan RK, Borad MJ. Immunotherapy in pancreatic cancer treatment: a new frontier. Ther Adv Gastroenterol. 2017;10(1):168–94.
Lutz E, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma. A phase II trial of safety, efficacy, and immune activation. Ann Surg. 2011;253(2):328–35.
Lutz ER, Kinkead H, Jaffee EM, Zheng L. Priming the pancreatic cancer tumor microenvironment for checkpoint-inhibitor immunotherapy. Oncoimmunology. 2014;3(11):e962401.
van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–66.
Le DT, Lutz E, Uram JN, Sugar EA, Onners B, Solt S, et al. Evaluation of ipilimumab in combination with allogeneic pancreatic tumor cells transfected with a GM-CSF gene in previously treated pancreatic cancer. J Immunother. 2013;36(7):382–9.
Soares KC, Rucki AA, Wu AA, Olino K, Xiao Q, Chai Y, et al. PD-1/PD-L1 blockade together with vaccine therapy facilitates effector T-cell infiltration into pancreatic tumors. J Immunother. 2015;38(1):1–11.
Kleponis J, Skelton R, Zheng L. Fueling the engine and releasing the break: combinational therapy of cancer vaccines and immune checkpoint inhibitors. Cancer Biol Med. 2015;12(3):201–8.
Pejawar-Gaddy S, Finn OJ. Cancer vaccines: accomplishments and challenges. Crit Rev Oncol Hematol. 2008;67(2):93–102.
Lu YC, Robbins PF. Cancer immunotherapy targeting neoantigens. Semin Immunol. 2016 Feb;28(1):22–7.
Zhang J, Wolfgang CL, Zheng L. Precision Immuno-oncology: prospects of individualized immunotherapy for pancreatic Cancer. Cancers (Basel). 2018;10(2).
Dodson LF, Hawkins WG, Goedegebuure P. Potential targets for pancreatic cancer immunotherapeutics. Immunotherapy. 2011;3(4):517–37.
Zhang JY, Looi KS, Tan EM. Identification of tumor-associated antigens as diagnostic and predictive biomarkers in cancer. Methods Mol Biol. 2009;520:1–10.
Srivatsan S, Patel JM, Bozeman EN, Imasuen IE, He S, Daniels D, et al. Allogeneic tumor cell vaccines: the promise and limitations in clinical trials. Hum Vaccin Immunother. 2014;10(1):52–63.
Giannakis M, Mu XJ, Shukla SA, Qian ZR, Cohen O, Nishihara R, et al. Genomic correlates of immune-cell infiltrates in colorectal carcinoma. Cell Rep. 2016 Apr 26;15(4):857–65.
Howitt BE, Shukla SA, Sholl LM, Ritterhouse LL, Watkins JC, Rodig S, et al. Association of Polymerase e-mutated and microsatellite-instable endometrial cancers with Neoantigen load, number of tumor-infiltrating lymphocytes, and expression of PD-1 and PD-L1. JAMA Oncol. 2015;1(9):1319–23.
Aurisicchio L, Pallocca M, Ciliberto G, Palombo F. The perfect personalized cancer therapy: cancer vaccines against neoantigens. J Exp Clin Cancer Res. 2018;37(1):86.
Viatte S, Alves PM, Romero P. Reverse immunology approach for the identification of CD8 T-cell-defined antigens: advantages and hurdles. Immunol Cell Biol. 2006;84(3):318–30.
Vigneron N. Human tumor antigens and Cancer immunotherapy. Biomed Res Int. 2015;2015:948501.
Even-Desrumeaux K, Baty D, Chames P. State of the art in tumor antigen and biomarker discovery. Cancers (Basel). 2011;3(2):2554–96.
Lu H, Knutson KL, Gad E, Disis ML. The tumor antigen repertoire identified in tumor-bearing neu transgenic mice predicts human tumor antigens. Cancer Res. 2006;66(19):9754–61.
Wang Y, Han KJ, Pang XW, Vaughan HA, Qu W, Dong XY, et al. Large scale identification of human hepatocellular carcinoma-associated antigens by autoantibodies. J Immunol. 2002;169(2):1102–9.
Obata Y, Takahashi T, Sakamoto J, Tamaki H, Tominaga S, Hamajima N, et al. SEREX analysis of gastric cancer antigens. Cancer Chemother Pharmacol. 2000;46(Suppl):S37–42.
Okada T, Akada M, Fujita T, Iwata T, Goto Y, Kido K, et al. A novel cancer testis antigen that is frequently expressed in pancreatic, lung, and endometrial cancers. Clin Cancer Res. 2006;12(1):191–7.
Preuss KD, Zwick C, Bormann C, Neumann F, Pfreundschuh M. Analysis of the B-cell repertoire against antigens expressed by human neoplasms. Immunol Rev. 2002;188:43–50.
Vonderheide RH, Glennie MJ. Agonistic CD40 antibodies and cancer therapy. Clin Cancer Res. 2013;19(5):1035–43.
Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6.
Luheshi NM, Coates-Ulrichsen J, Harper J, Mullins S, Sulikowski MG, Martin P, et al. Transformation of the tumour microenvironment by a CD40 agonist antibody correlates with improved responses to PD-L1 blockade in a mouse orthotopic pancreatic tumour model. Oncotarget. 2016;7(14):18508–20.
Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368(16):1509–18.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17.
Brunet LR, Hagemann T, Andrew G, Mudan S, Marabelle A. Have lessons from past failures brought us closer to the success of immunotherapy in metastatic pancreatic cancer? Oncoimmunology. 2016;5(4):e1112942.
Vera JF, Brenner MK, Dotti G. Immunotherapy of human cancers using gene modified T lymphocytes. Current gene therapy. 2009;9(5):396–408.
Watanabe K, Luo Y, Da T, Guedan S, Ruella M, Scholler J, et al. Pancreatic cancer therapy with combined mesothelin-redirected chimeric antigen receptor T cells and cytokine-armed oncolytic adenoviruses. JCI Insight. 2018;3(7).
Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov. 2016;6(2):133–46.
Hassan R, Ho M. Mesothelin targeted cancer immunotherapy. Eur J Cancer. 2008;44(1):46–53.
Bera TK, Pastan I. Mesothelin is not required for normal mouse development or reproduction. Mol Cell Biol. 2000;20(8):2902–6.
Gilham DE, Anderson J, Bridgeman JS, Hawkins RE, Exley MA, Stauss H, et al. Adoptive T-cell therapy for cancer in the United Kingdom: a review of activity for the British Society of Gene and Cell Therapy annual meeting 2015. Hum Gene Ther. 2015;26(5):276–85.
DeSelm CJ, Tano ZE, Varghese AM, Adusumilli PS. CAR T-cell therapy for pancreatic cancer. J Surg Oncol. 2017;116(1):63–74.
Townsend MH, Shrestha G, Robison RA, O'Neill KL. The expansion of targetable biomarkers for CAR T cell therapy. J Exp Clin Cancer Res. 2018;37(1):163.
Wilkie S, Picco G, Foster J, Davies DM, Julien S, Cooper L, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180(7):4901–9.
You F, Jiang L, Zhang B, Lu Q, Zhou Q, Liao X, et al. Phase 1 clinical trial demonstrated that MUC1 positive metastatic seminal vesicle cancer can be effectively eradicated bymodified anti-MUC1 chimeric antigen receptor transduced T cells. Sci China Life Sci. 2016;59(4):386–97.
Akce M, Zaidi MY, Waller EK, El-Rayes BF, Lesinski GB. The potential of CAR T cell therapy in pancreatic Cancer. Front Immunol. 2018;9:2166.
Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126(8):3130–44.
Knochelmann HM, Smith AS, Dwyer CJ, Wyatt MM, Mehrotra S, Paulos CM. CAR T cells in solid tumors: blueprints for building effective therapies. Front Immunol. 2018;9:1740.
Kunk PR, Bauer TW, Slingluff CL, Rahma OE. From bench to bedside a comprehensive review of pancreatic cancer immunotherapy. J Immunother Cancer. 2016;4:14.
John LB, Devaud C, Duong CP, Yong CS, Beavis PA, Haynes NM, et al. Anti-PD-1 antibody therapy potently enhances the eradication of established tumors by gene-modified T cells. Clin Cancer Res. 2013;19(20):5636–46.
Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res. 2014;20(16):4262–73.
Li S, Siriwon N, Zhang X, Yang S, Jin T, He F, et al. Enhanced Cancer immunotherapy by chimeric antigen receptor-modified T cells engineered to secrete checkpoint inhibitors. Clin Cancer Res. 2017;23(22):6982–92.
Suarez ER, Chang de K, Sun J, Sui J, Freeman GJ, Signoretti S, et al. Chimeric antigen receptor T cells secreting anti-PD-L1 antibodies more effectively regress renal cell carcinoma in a humanized mouse model. Oncotarget. 2016;7(23):34341–55.
Torphy RJ, Zhu Y, Schulick RD. Immunotherapy for pancreatic cancer: barriers and breakthroughs. Ann Gastroenterol Surg. 2018;2(4):274–81.
Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014;74(18):5057–69.
Achkova D, Maher J. Role of the colony-stimulating factor (CSF)/CSF-1 receptor axis in cancer. Biochem Soc Trans. 2016;44(2):333–41.
Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41(1):49–61.
Pedersen MB, Danielsen AV, Hamilton-Dutoit SJ, Bendix K, Norgaard P, Moller MB, et al. High intratumoral macrophage content is an adverse prognostic feature in anaplastic large cell lymphoma. Histopathology. 2014;65(4):490–500.
Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One. 2012;7(12):28.
Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J Immunother Cancer. 2017;5(1):53.
Candido JB, Morton JP, Bailey P, Campbell AD, Karim SA, Jamieson T, et al. CSF1R+ macrophages sustain pancreatic tumor growth through T cell suppression and maintenance of key gene programs that define the squamous subtype. Cell Rep. 2018;23(5):1448–60.
Seif F, Khoshmirsafa M, Aazami H, Mohsenzadegan M, Sedighi G, Bahar M. The role of JAK-STAT signaling pathway and its regulators in the fate of T helper cells. Cell Commun Signal. 2017;15(1):23.
Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, et al. PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci. 2018;109(1):43-53.
Quintas-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19(8):1933–40.
Yu H, Pardoll D, Jove R. STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer. 2009;9(11):798–809.
Lu C, Talukder A, Savage NM, Singh N, Liu K. JAK-STAT-mediated chronic inflammation impairs cytotoxic T lymphocyte activation to decrease anti-PD-1 immunotherapy efficacy in pancreatic cancer. Oncoimmunology. 2017;6(3):e1291106.
Weinacht KG, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol. 2017;139(5):1629–40 e2.
Hurwitz HI, Uppal N, Wagner SA, Bendell JC, Beck JT, Wade SM 3rd, et al. Randomized, double-blind, phase II study of Ruxolitinib or placebo in combination with Capecitabine in patients with metastatic pancreatic Cancer for whom therapy with gemcitabine has failed. J Clin Oncol. 2015;33(34):4039–47.
Campbell R, Chong G, Hawkes EA. Novel indications for Bruton's tyrosine kinase inhibitors, beyond hematological malignancies. J Clin Med. 2018;7(4).
Gunderson AJ, Kaneda MM, Tsujikawa T, Nguyen AV, Affara NI, Ruffell B, et al. Bruton tyrosine kinase-dependent immune cell cross-talk drives pancreas Cancer. Cancer Discov. 2016;6(3):270–85.
Strouch MJ, Cheon EC, Salabat MR, Krantz SB, Gounaris E, Melstrom LG, et al. Crosstalk between mast cells and pancreatic cancer cells contributes to pancreatic tumor progression. Clin Cancer Res. 2010;16(8):2257–65.
Masso-Valles D, Jauset T, Serrano E, Sodir NM, Pedersen K, Affara NI, et al. Ibrutinib exerts potent antifibrotic and antitumor activities in mouse models of pancreatic adenocarcinoma. Cancer Res. 2015;75(8):1675–81.
Sagiv-Barfi I, Kohrt HE, Czerwinski DK, Ng PP, Chang BY, Levy R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc Natl Acad Sci U S A. 2015;112(9):17.
Demaria S, Coleman CN, Formenti SC. Radiotherapy: changing the game in immunotherapy. Trends Cancer. 2016;2(6):286–94.
Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005;11(2 Pt 1):728–34.
Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, et al. Fractionated but not single-dose radiotherapy induces an immune-mediated abscopal effect when combined with anti-CTLA-4 antibody. Clin Cancer Res. 2009;15(17):5379–88.
Azad A, Yin Lim S, D'Costa Z, Jones K, Diana A, Sansom OJ, et al. PD-L1 blockade enhances response of pancreatic ductal adenocarcinoma to radiotherapy. EMBO Mol Med. 2017;9(2):167–80.
Deng L, Liang H, Burnette B, Beckett M, Darga T, Weichselbaum RR, et al. Irradiation and anti-PD-L1 treatment synergistically promote antitumor immunity in mice. J Clin Invest. 2014;124(2):687–95.
Kultti A, Zhao C, Singha NC, Zimmerman S, Osgood RJ, Symons R, et al. Accumulation of extracellular hyaluronan by hyaluronan synthase 3 promotes tumor growth and modulates the pancreatic cancer microenvironment. Biomed Res Int. 2014;2014:817613.
Lee CT, Mace T, Repasky EA. Hypoxia-driven immunosuppression: a new reason to use thermal therapy in the treatment of cancer? Int J Hyperth. 2010;26(3):232–46.
Stokes JB, Adair SJ, Slack-Davis JK, Walters DM, Tilghman RW, Hershey ED, et al. Inhibition of focal adhesion kinase by PF-562,271 inhibits the growth and metastasis of pancreatic cancer concomitant with altering the tumor microenvironment. Mol Cancer Ther. 2011;10(11):2135–45.
Miyazaki T, Kato H, Nakajima M, Sohda M, Fukai Y, Masuda N, et al. FAK overexpression is correlated with tumour invasiveness and lymph node metastasis in oesophageal squamous cell carcinoma. Br J Cancer. 2003;89(1):140–5.
Itoh S, Maeda T, Shimada M, Aishima S, Shirabe K, Tanaka S, et al. Role of expression of focal adhesion kinase in progression of hepatocellular carcinoma. Clin Cancer Res. 2004;10(8):2812–7.
Jiang H, Hegde S, Knolhoff BL, Zhu Y, Herndon JM, Meyer MA, et al. Targeting focal adhesion kinase renders pancreatic cancers responsive to checkpoint immunotherapy. Nat Med. 2016;22(8):851–60.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. 2012;21(3):418–29.
Singha NC, Nekoroski T, Zhao C, Symons R, Jiang P, Frost GI, et al. Tumor-associated hyaluronan limits efficacy of monoclonal antibody therapy. Mol Cancer Ther. 2015;14(2):523–32.
Clift R, Li X, Blouw B, Thompson CB, Huang Y. Abstract 2740: PEGylated recombinant hyaluronidase PH20 (pegvorhyaluronidase alfa PEGPH20) converts HA-rich tumors from resistant to sensitive to anti-PD-L1 immunotherapy in murine syngeneic breast cancer models. Cancer Res. 2018;78(13 Supplement):2740.
Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–20.
Manuel ER, Chen J, D'Apuzzo M, Lampa MG, Kaltcheva TI, Thompson CB, et al. Salmonella-based therapy targeting Indoleamine 2,3-dioxygenase coupled with enzymatic depletion of tumor Hyaluronan induces complete regression of aggressive pancreatic tumors. Cancer Immunol Res. 2015;3(9):1096–107.
Shepard HM. Breaching the castle walls: Hyaluronan depletion as a therapeutic approach to Cancer therapy. Front Oncol. 2015;5:192.
Hingorani SR, Harris WP, Beck JT, Berdov BA, Wagner SA, Pshevlotsky EM, et al. Phase Ib study of PEGylated recombinant human hyaluronidase and gemcitabine in patients with advanced pancreatic Cancer. Clin Cancer Res. 2016;22(12):2848–54.
Bullock AJ, Hingorani SR, Wu XW, Jiang P, Chondros D, Khelifa S, et al. Final analysis of stage 1 data from a randomized phase II study of PEGPH20 plus nab-paclitaxel/gemcitabine in stage IV previously untreated pancreatic cancer patients (pts), utilizing Ventana companion diagnostic assay. J Clin Oncol. 2016;34(15_suppl):4104.
Yu Y, Cui J. Present and future of cancer immunotherapy: a tumor microenvironmental perspective. Oncol Lett. 2018;16(4):4105–13.
Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science. 2017;357(6349):409–13.
Antonarakis ES. A new molecular taxonomy to predict immune checkpoint inhibitor sensitivity in prostate Cancer. Oncologist. 2019;9(10):2018–0819.
Lee V, Murphy A, Le DT, Diaz LA Jr. Mismatch repair deficiency and response to immune checkpoint blockade. Oncologist. 2016;21(10):1200–11.
Overman MJ, McDermott R, Leach JL, Lonardi S, Lenz HJ, Morse MA, et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. Lancet Oncol. 2017;18(9):1182–91.
Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med. 2015;372(26):2509–20.
Antonia SJ, Larkin J, Ascierto PA. Immuno-oncology combinations: a review of clinical experience and future prospects. Clin Cancer Res. 2014;20(24):6258–68.
Mai CW, Yaeghoobi M, Abd-Rahman N, Kang YB, Pichika MR. Chalcones with electron-withdrawing and electron-donating substituents: anticancer activity against TRAIL resistant cancer cells, structure-activity relationship analysis and regulation of apoptotic proteins. Eur J Med Chem. 2014;77:378–87.
Yeong KY, Tan SC, Mai CW, Leong CO, Chung FF, Lee YK, et al. Contrasting sirtuin and poly(ADP-ribose)polymerase activities of selected 2,4,6-trisubstituted benzimidazoles. Chem Biol Drug Des. 2018;91(1):213–9.
Soo HC, Chung FF, Lim KH, Yap VA, Bradshaw TD, Hii LW, et al. Cudraflavone C induces tumor-specific apoptosis in colorectal Cancer cells through inhibition of the phosphoinositide 3-kinase (PI3K)-AKT pathway. PLoS One. 2017;12(1):e0170551.
Mai CW, Kang YB, Nadarajah VD, Hamzah AS, Pichika MR. Drug-like dietary vanilloids induce anticancer activity through proliferation inhibition and regulation of bcl-related apoptotic proteins. Phytother Res. 2018;32(6):1108–18.
Mai CW, Kang YB, Hamzah AS, Pichika MR. Comparative efficacy of vanilloids in inhibiting toll-like receptor-4 (TLR-4)/myeloid differentiation factor (MD-2) homodimerisation. Food Funct. 2018;9(6):3344–50.
Krishnan P, Lee FK, Chong KW, Mai CW, Muhamad A, Lim SH, et al. Alstoscholactine and Alstolaxepine, Monoterpenoid indole alkaloids with gamma-lactone-bridged Cycloheptane and Oxepane moieties from Alstonia scholaris. Org Lett. 2018.
Chung FF, Tan PF, Raja VJ, Tan BS, Lim KH, Kam TS, et al. Jerantinine a induces tumor-specific cell death through modulation of splicing factor 3b subunit 1 (SF3B1). Sci Rep. 2017;7:42504.
Al-Khdhairawi AAQ, Krishnan P, Mai CW, Chung FF, Leong CO, Yong KT, et al. A Bis-benzopyrroloisoquinoline alkaloid incorporating a Cyclobutane Core and a Chlorophenanthroindolizidine alkaloid with cytotoxic activity from Ficus fistulosa var. tengerensis. J Nat Prod. 2017;80(10):2734–40.
Mai CW, Yap KS, Kho MT, Ismail NH, Yusoff K, Shaari K, et al. Mechanisms underlying the anti-inflammatory effects of Clinacanthus nutans Lindau extracts: inhibition of cytokine production and toll-like Receptor-4 activation. Front Pharmacol. 2016;7:7.
Tan BS, Kang O, Mai CW, Tiong KH, Khoo AS, Pichika MR, et al. 6-Shogaol inhibits breast and colon cancer cell proliferation through activation of peroxisomal proliferator activated receptor gamma (PPARgamma). Cancer Lett. 2013;336(1):127–39.
Blair AB, Zheng L. Rational combinations of immunotherapy for pancreatic ductal adenocarcinoma. Chin Clin Oncol. 2017;6(3):31.
Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, et al. Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512.
Whiteside TL, Demaria S, Rodriguez-Ruiz ME, Zarour HM, Melero I. Emerging opportunities and challenges in Cancer immunotherapy. Clin Cancer Res. 2016;22(8):1845–55.
Tsiatas M, Mountzios G, Curigliano G. Future perspectives in cancer immunotherapy. Ann Transl Med. 2016;4(14):273.
Acknowledgments
The authors wish to acknowledge the researchers at the Institute of Research, Development and Innovation, International Medical University for the critical comments on the manuscripts. The authors also appreciate the critical review and elaborative language editing by Emeritus Professor Brian L Furman, University of Strathclyde, United Kingdom.
Funding
The review was a collective effort of researchers receiving a pancreatic cancer research grant from the Malaysia Ministry of Education Fundamental Research Grant Scheme (FRGS/1/2017/SKK08/IMU/03/1).
Availability of data and materials
Not applicable.
Author information
Authors and Affiliations
Contributions
CKL, FFLC, COL, SFW, RR, CWM participated in drafting, writing, and reviewing the manuscript. CKL and CWM drafted and finalised all figures and tables. All authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
This review does not contain any individual personal data and thus consent for publication is not applicable.
Competing interests
The authors declare that the review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Looi, CK., Chung, F.FL., Leong, CO. et al. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J Exp Clin Cancer Res 38, 162 (2019). https://doi.org/10.1186/s13046-019-1153-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13046-019-1153-8