
Abstract
Spleen tyrosine kinase (Syk) is a cytosolic non-receptor protein tyrosine kinase (PTK) and is mainly expressed in hematopoietic cells. Syk was recognized as a critical element in the B-cell receptor signaling pathway. Syk is also a key component in signal transduction from other immune receptors like Fc receptors and adhesion receptors. Several oral Syk inhibitors including fostamatinib (R788), entospletinib (GS-9973), cerdulatinib (PRT062070), and TAK-659 are being assessed in clinical trials. The second generation compound, entospletinib, showed promising results in clinical trials against B-cell malignancies, mainly chronic lymphoid leukemia. Syk inhibitors are being evaluated in combination regimens in multiple malignancies.
Similar content being viewed by others
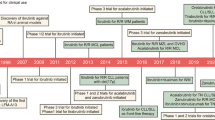


Background
Spleen tyrosine kinase (Syk) is a cytosolic non-receptor protein tyrosine kinase (PTK) which was discovered in 1990 [1]. The full-length syk cDNA was first cloned from porcine spleen cells in 1991 [2]. The human syk cDNA contains an open reading frame of 1908 base pairs. The human syk gene encodes a 635-aa polypeptide with an estimated molecular weight of 72 kDa [3, 4]. The syk gene was found to be localized on chromosome 9q22. Syk is mainly expressed in hematopoietic cells. Syk belongs to the Src family of non-receptor type PTKs and is highly homologous to ZAP-70, which is thought to be the Syk counterpart in T cells [4, 5].
Syk contains two N-terminal SH2 domains and one C-terminal tyrosine kinase domain. The SH2 domains of Syk bind to immunoreceptor tyrosine-based activation motifs (ITAMs), leading to Syk activation. Syk protein lacks myristoylation site, therefore does not attach directly to the cell membrane [2, 4, 5].
Syk has an autophosphorylation site at Tyr-518. Following receptor engagement such as antigen binding or sIgM ligation in B cells, tyrosine residues are phosphorylated by Lyn, another Src-family non-receptor PTK. The phosphorylation on the tyrosine residues in Syk creates binding sites for CBL, VAV1, and phospholipase C-gamma, the regulators of B-cell receptor (BCR) signaling pathways. These lead to an increase in second messenger IP3 which stimulates calcium ion mobilization.
Syk functions
Syk was recognized as a critical element in the BCR signaling pathway [6, 7]. Syk is also a key component in signal transduction from other immune receptors like Fc receptors and adhesion receptors. Syk along with other BCR signaling molecules, Bruton tyrosine kinase (BTK), PI3K delta (PI3Kδ), and tumor necrosis factor (TNF) superfamily receptors was also found to be involved in signal transduction independent from the BCR [8,9,10]. Syk is expressed primarily in hematopoietic cells like B-cells, monocytes, macrophages, mast cells, and neutrophils. Syk was recognized to be a potential target for the treatment of various hematologic cancers, autoimmune disorders, and other inflammatory states [11,12,13,14,15,16]. Under resting conditions, Syk remains in unphosphorylated state. Activation of the BCR leads to oligomerization and phosphorylation of the Igα and ß (immunoreceptor tyrosine-based activation motifs, ITAMs), the transmembrane signaling proteins CD79a and CD79b. The latter results in activation of the Syk tyrosine kinase, which in turn initiates downstream signaling through PI3K and BTK leading to amplification of the original BCR signal. In animal models Syk was found to be a critical point in B-cell antibody responses, differentiation into germinal center or plasma cells and memory B-cells [17,18,19].
Syk inhibitors
Several oral Syk inhibitors including fostamatinib (R788), entospletinib (GS-9973), cerdulatinib (PRT062070), and TAK-659 are being assessed in clinical trials [9]. Preclinical studies of a few Syk inhibitors have been reported [20,21,22,23].
Fostamatinib
Fostamatinib is the first oral Syk inhibitor (previously known as R788). It is rapidly metabolized in vivo to R406 [24, 25]. Fostamatinib can selectively abrogate the BCR signaling pathway. Fostamatinib has potent anti-inflammatory effects [18, 26]. It was first demonstrated to have activities in rheumatoid arthritis and immune thrombocytopenia [11, 27,28,29,30,31]. In a murine model of chronic lymphoid leukemia (CLL) fostamatinib was found to induce an early and transient mobilization of both normal and malignant B cells, but selectively inhibited the growth of the malignant B-cell population [32]. This effect on mobilization of B cells is similar to that of BTK inhibitors [10, 33,34,35,36,37,38,39].
Fostamatinib was the first clinically available Syk inhibitor which was tested in a phase I/II study in patients with refractory B-cell lymphomas [40]. The dose-limiting toxicity (DLT) was diarrhea, neutropenia, and thrombocytopenia. The highest response (n = 6, 55%) was seen in patients with CLL/SLL (n = 11). The overall response rate (ORR) among the 23 patients with diffuse large B-cell lymphoma (DLBCL) in the phase II part was 22%. Among the five responders, there were one complete response (CR) and four partial responses (PR). Median progression-free survival (PFS) for the patients in the phase II group was 4.2 months, and for DLBCL patients, the median PFS was 2.7 months. This trial established the phase II dose at 200 mg BID.
Fostamatinib was studied in patients with relapsed or refractory DLBCL in a phase II, two-arm, randomized, double-blinded trial at either 100 mg twice a day (BID) or 200 mg BID. The ORR was the primary endpoint. Due to limited efficacy at the lower dosage, all subsequent patients were treated at 200 mg BID. At the time of the report [41], 68 patients were treated (47 at 200 mg BID, 21 at 100 mg BID). Of these patients, 58% were germinal B-cell (GCB) origin, 30% activated B-cell (ABC) origin. Diarrhea, nausea, and fatigue were the most common treatment-related adverse events (TEAE). The ORR was 3%. Patients with the ABC genotype failed to respond. Off-target effects on VEGFR and other tyrosine kinases were thought to be responsible for the most common TEAEs [24, 42].
Entospletinib (GS-9973)
The search for more kinase-selective oral Syk inhibitor resulted in the discovery of GS-9973, entospletinib [43]. This second generation compound demonstrated favorable in vitro and in vivo selectivity profile with fewer dose-limiting adverse effects. Entospletinib in comparison to fostamatinib revealed increased selectivity for Janus kinase 2 (JAK-2), c-KIT, FMS-like tyrosine kinase 3 (FLT 3), RET, and VEGFR2 [43].
Preclinical studies in CLL cell lines demonstrated that inhibition of Syk activity leads to decreased secretion of CCL3 and CCL4 resulting in cell redistribution. The disruption of the microenvironment of B-cells and surrounding stroma by Syk inhibitors mitigates the chemokine- and integrin-mediated protective effects of CLL cells [44,45,46]. Clinically this effect manifests as lymph node reduction with associated transient marked lymphocytosis. These effects make entospletinib an ideal agent for refractory CLL and other B cell non-Hodgkin lymphomas (NHL) [47, 48].
Entospletinib (GS-9973) was evaluated in a multicenter, phase 2 study on subjects with relapsed or refractory CLL (n = 41) and non-Hodgkin lymphoma (n = 145) [47]. Patients received 800 mg entospletinib twice daily. This report provided results of efficacy in CLL and safety in all patients (n = 186). The prior treatment for CLL must have included at least 2 cycles of therapeutic antibody or cytotoxic therapy. The median progression-free survival (PFS) for CLL was 13.8 months (95% CI, 7.7 months to not reached). The ORR was 61.0% (95% CI, 44.5–75.8%), with 25 partial responses (PR), 13 patients with stable disease, and no complete responses (CR). 94.9% of the patients in the CLL cohort achieved reduction in adenopathy. There were three patients who had nodal reduction with persistent lymphocytosis. The rate of serious adverse events (SAEs) was 29%. The most common TEAEs included dyspnea, pneumonia, febrile neutropenia, and pyrexia. The study showed that entospletinib had clinical activity in subjects with relapsed or refractory CLL [49]. The toxicity was acceptable in the whole patient population with CLL and NHL.
In updated report of 69 patients with indolent NHL (41 follicular lymphoma (FL), 11 lymphoplasmacytoid lymphoma (LPL), 17 marginal zone lymphoma (MZL)), entospletinib 800 mg BID was evaluated in a multicenter phase II study as mentioned above [48]. Median age of the cohorts was 66 years (range 41–89). The subjects received a median number of 3 prior treatment regimens (range 1–14). Entospletinib was generally well tolerated. The most common TEAEs were fatigue and GI toxicities. Common laboratory abnormalities were elevation of ALT /AST and bilirubin as well as anemia and neutropenia. The ORR was 13.0% (95% CI: 6.1% to 23.3%), with 7 PR, and one CR. The median PFS was 5.5 months (95% CI: 4.4 to 8.2 months). Therefore, entospletinib monotherapy 800 mg BID was well tolerated and showed clinical activity in patients with advanced indolent NHL. Entospletinib was shown recently in a phase II study to have modest activity in 39 patients with relapsed or refractory mantle cell lymphoma (MCL) [50]. Further development of entospletinib in NHL will likely focus on the combination regimens [50].
Recently, the interim results of the entospletinib monotherapy and in combination with chemotherapy in patients with AML were presented. In this ongoing phase 1b/2 study (NCT02343939), entospletinib was well tolerated [51]. At the time of this report, the study enrolled 12 patients with a median age of 54 (range, 18–69) years. Three patients were treated at 200 mg BID, and six were dosed at 400 mg BID. The most common non-hematologic AEs included febrile neutropenia, nausea, and diarrhea. Taken all the data together, there was lack of benefit to escalate dose pass 400 mg BID. Therefore, 400 mg BID was selected as the recommended phase 2 dose. Surprisingly, 9/9 patients (100%) treated at both levels achieved CR. One patient with 11q23-rearranged AML achieved morphologic and cytogenetic CR after 14 days on entospletinib monotherapy, indicating unique sensitivity to the drug in this patient with poor prognosis [51].
Entospletinib in combination regimens
PI3Kδ inhibitors and BTK inhibitors as well as immune check point inhibitors are novel agents that have changed the clinical management of CLL and NHLs [8, 10, 35, 52,53,54,55,56]. Prolonged administration of these agents is, however, needed, and resistance is already emerging in a proportion of patients [57, 58]. Therefore, combination of entospletinib and idelalisib was evaluated in an attempt to enhance the efficacy and reduce resistance [59, 60].
An open-label phase II study evaluated the safety and efficacy of the combination of entospletinib and idelalisib [60]. The study included 66 patients in five cohorts of indolent NHL, CLL, MCL, and DLBCL. The primary end point was ORR. The patients underwent intra-patient dose escalation of idelalisib and entospletinib (from 100 to 150 mg and 200 to 800 mg twice daily, respectively) every 2 or 4 weeks. After treatment for a median of 10 weeks, the ORR reached 60% in the CLL cohort, 36% in follicular lymphoma, and 17% in DLBCL. The most common AEs were diarrhea, rash, and transaminitis, which improved after treatment discontinuation. However, there were 12 patients who developed pneumonitis, 11 of grade 3 or more, resulting in two fatalities. The study was therefore terminated. Such a severe pro-inflammatory response was attributed to the unexpected potent inhibition of the mammalian target of rapamycin (mTOR) observed with the combination of the two study drugs. Increased T helper 1 (Th-1) response mediated by interferon-γ, IL- 6, 7, 8 was reported in patients with pneumonitis [60].
Events from the idelalisib and entospletinib phase II study prompt researchers to exert caution in planning and conducting further trials with Syk inhibitors in conjunction with other BCR inhibitors or in combination with chemotherapy (Table 1).
Despite recent advances in molecular diagnosis and clinical therapy, relapsed and refractory acute lymphoid leukemia (R/R ALL) remains a significant challenge [61,62,63,64,65,66,67,68]. Novel agents are being developed to improve the outcome of R/R ALL [64, 65, 69,70,71,72,73,74]. Entospletinib was studied in combination with vincristine and shown to have synergistic activity in vitro in 19 hematological cancer cell lines including lymphoma, multiple myeloma, ALL, and AML [75]. The in vivo efficacy of entospletinib and vincristine as singe agents and in combination was further evaluated in a lymphoma xenograft model using the SU-DHL-10 cell line. The combination of vincristine with entospletinib showed synergistic activity in ALL in the mouse model and thus supported the further clinical development of entospletinib in combination with vincristine [76]. A phase 1b trial (NCT02404220) is evaluating entospletinib in combination with chemotherapy (vincristine and dexamethasone) for the treatment of R/R Pre B-ALL. In the last update, three patients have been enrolled [77].
Cerdulatinib (PRT062070)
Cerdulatinib is a dual inhibitor of Syk and JAK 1/3 and has been shown to have activity against DLBCL in in vitro studies [78]. Cerdulatinib is an oral kinase inhibitor against Syk and JAK. It has been shown in vitro to have specific inhibitory activity against Syk and JAK1/3 in a subset of B-cell lymphoma cell lines. Cerdulatinib suppressed inflammation and autoantibody generation and blocked B-cell activation in a rat model and splenomegaly in a mouse model of chronic B-cell antigen receptor stimulation. In both ABC and GCB lymphoma cell lines, cerdulatinib induced apoptosis and cell-cycle arrest. Cerdulatinib blocked JAK/STAT and BCR signaling in primary GCB and non-GCB DLBCL tumor cells [79]. Primary CLL cells from patients were treated in vitro with cerdulatinib alone or in combination with venetoclax. Cerdulatinib inhibited BCR- and IL4-induced downstream signaling in CLL cells and reduced CCL3/CCL4 production. Cerdulatinib and venetoclax have synergistic activity in primary CLL cells [80]. Interestingly, cerdulatinib was shown to be better than ibrutinib in overcoming the stromal support and blocking proliferation of ibrutinib-resistant primary CLL cells and of BTKC481S-transfected/ibrutinib-resistant lymphoma cells [81]. Phase I/II dose escalation study in CLL/SLL/NHL is underway (NCT01994382) [78, 82].
TAK-659
Syk directly binds to and activates FMS-like tyrosine kinase 3 (FLT-3) [17]. Through structure-based drug design, a novel series of heteroaromatic Syk inhibitors was screened and optimized, resulting in the discovery of TAK-659, a dual Syk and FLT3 inhibitor [83]. TAK-659 inhibited the pro-survival, proliferative, chemoresistant, and activation effects promoted by the microenvironment. Combination of TAK-659 with other BCR inhibitors showed synergistic effect in inducing apoptosis. Combination of TAK-659 and ibrutinib induced significantly higher cytotoxicity toward CLL cells [84].
TAK-659 suppressed splenomegaly and tumor development in a LMP2A/Myc mouse model in nanomolar concentrations. In addition, TAK-659 also blocked metastasis of tumor cells into bone marrow. Interestingly, TAK-659 killed tumor cells without damaging host cells in spleen and tumors [85].
In a phase 1 first-in-human study of TAK-659 (NCT02000934), patients (pts) aged ≥18 years with advanced solid tumors or lymphoma received oral TAK-659 daily (QD, 60–120 mg) [86]. At the data cutoff on June 15, 2015, 24 pts had been enrolled (14 solid tumor pts; 10 lymphoma pts) at four dose levels of TAK-659 (60, 80, 100, 120 mg). Among the 10 lymphoma pts, 7 had DLBCL (5 GCB and 2 ABC subtypes) and 3 had follicular lymphoma (FL). Among all the pts, the most common AEs were fatigue, elevated AST, anemia, and diarrhea. Eight patients (33%) had grade ≥3 drug-related AEs. Of 7 response-evaluable DLBCL pts, 3 achieved PR. Therefore, TAK-659 at dose 60–100 mg QD appears to be acceptable for further studies.
In the updated report of this phase I first-in-human study, 54 adult patients with advanced solid tumors/lymphoma received oral TAK-659 QD at doses of 60, 80, 100, and 120 mg during dose escalation (n = 35) or 100 mg during dose expansion (n = 19) [87]. There were five dose-limiting toxicities (DLTs): increased lipase levels, generalized edema, mucositis, and increased AST. MTD (maximal tolerated dose) was 100 mg daily. In this updated report, 24 DLBCL pts were evaluable for responses. Among these 24 pts, 11 (46%) achieved an objective response; (7 (29%) CR, 4 PR); all 3 response-evaluable FL pts achieved PR. No solid tumor patients achieved an objective response.
A phase Ib/II study of TAK-659 is underway in pts with relapsed or refractory acute myelogenous leukemia (R/R AML) [88]. During the phase Ib dose escalation study, adult pts with R/R AML received oral TAK-659 daily at doses of 60, 100, 120, and 160 mg. The pharmacodynamic effect of TAK-659 was assessed using flow cytometry by measuring the phosphorylation of ribosomal protein S6 (pS6) in peripheral AML blasts. DLT has not been observed in the latest update. The median age of the patients was 67 years (range 25–86), and 38% had received ≥4 prior lines of therapy. TEAEs were seen in 12 (92%) pts overall; the most common AEs were elevated AST (31%), ALT (23%), and amylase levels (23%). For PD studies, pS6 was detected at baseline and reduced after dosing in four patients (2 FLT-3-ITD; 2 FLT-3-WT). Inhibition of FLT-3-ITD phosphorylation and early activity with decreases in peripheral myeloblasts was observed. The unique mechanism of action with dual inhibition of Syk and FLT-3 appears to warrant further studies in R/R AML patients with FLT-3 mutations.
Conclusion
Syk inhibitors are a new group of small molecule inhibitors targeting BCR-mediated signaling pathways. The second generation compound, entospletinib, showed promising results in clinical trials against B-cell malignancies, mainly CLL. There is an unmet need in hematological malignancies for the new non-chemotherapy drugs. Multidrug regimens in combination with other small molecule inhibitors and new antibodies may have the potential of overcoming or delaying the development of drug resistance [89,90,91,92,93,94,95]. The off-target effects of the small molecule inhibitors are a limiting factor. Caution should be exerted especially in testing drug combinations [60].
Abbreviations
- ALK:
-
Anaplastic lymphoma kinase
- BTK:
-
Bruton tyrosine kinase
- cfDNA:
-
Cell free DNA
- EGFR:
-
Epidermal growth factor receptor
References
Kobayashi T, Nakamura S, Taniguchi T, Yamamura H. Purification and characterization of a cytosolic protein-tyrosine kinase from porcine spleen. Eur J Biochem. 1990;188(3):535–40.
Taniguchi T, Kobayashi T, Kondo J, Takahashi K, Nakamura H, Suzuki J, Nagai K, Yamada T, Nakamura S, Yamamura H. Molecular cloning of a porcine gene syk that encodes a 72-kDa protein-tyrosine kinase showing high susceptibility to proteolysis. J Biol Chem. 1991;266(24):15790–6.
Ku G, Malissen B, Mattei MG. Chromosomal location of the Syk and ZAP-70 tyrosine kinase genes in mice and humans. Immunogenetics. 1994;40(4):300–2.
Law CL, Sidorenko SP, Chandran KA, Draves KE, Chan AC, Weiss A, Edelhoff S, Disteche CM, Clark EA. Molecular cloning of human Syk. A B cell protein-tyrosine kinase associated with the surface immunoglobulin M-B cell receptor complex. J Biol Chem. 1994;269(16):12310–9.
Yanagi S, Kurosaki T, Yamamura H. The structure and function of nonreceptor tyrosine kinase p72syk expressed in hematopoietic cells. Cell Signal. 1995;7(3):185–93.
Tsang E, Giannetti AM, Shaw D, Dinh M, Tse JK, Gandhi S, Ho H, Wang S, Papp E, Bradshaw JM. Molecular mechanism of the Syk activation switch. J Biol Chem. 2008;283(47):32650–9.
Uckun FM, Qazi S. SYK as a new therapeutic target in B-cell precursor acute lymphoblastic leukemia. J Cancer Ther. 2014;5(1):124–31.
Rai KR. Therapeutic potential of new B cell-targeted agents in the treatment of elderly and unfit patients with chronic lymphocytic leukemia. J Hematol Oncol. 2015;8:85.
Singh R, Masuda ES, Payan DG. Discovery and development of spleen tyrosine kinase (SYK) inhibitors. J Med Chem. 2012;55(8):3614–43.
Wu J, Liu C, Tsui ST, Liu D. Second-generation inhibitors of Bruton tyrosine kinase. J Hematol Oncol. 2016;9:80.
Gomez-Puerta JA, Bosch X. Therapy: spleen tyrosine kinase inhibitors—novel therapies for RA? Nat Rev Rheumatol. 2011;7(3):134–6.
Ma TK, McAdoo SP, Tam FW. Spleen tyrosinekinase: a crucial player and potential therapeutic target in renal disease. Nephron. 2016;133(4):261–9.
Pritchard EM, Stewart E, Zhu F, Bradley C, Griffiths L, Yang L, Suryadevara PK, Zhang J, Freeman BB 3rd, Guy RK, Dyer MA. Pharmacokinetics and efficacy of the spleen tyrosine kinase inhibitor r406 after ocular delivery for retinoblastoma. Pharm Res. 2014;31(11):3060–72.
Masuda ES, Schmitz J. Syk inhibitors as treatment for allergic rhinitis. Pulm Pharmacol Ther. 2008;21(3):461–7.
Geahlen RL. Getting Syk: spleen tyrosine kinase as a therapeutic target. Trends Pharmacol Sci. 2014;35(8):414–22.
Braegelmann C, Holzel M, Ludbrook V, Dickson M, Turan N, Ferring-Schmitt S, Sternberg S, Bieber T, Kuhn A, Wenzel J. Spleen tyrosine kinase (SYK) is a potential target for the treatment of cutaneous lupus erythematosus patients. Exp Dermatol. 2016;25(5):375–9.
Puissant A, Fenouille N, Alexe G, Pikman Y, Bassil CF, Mehta S, Du J, Kazi JU, Luciano F, Ronnstrand L, Kung AL, Aster JC, Galinsky I, Stone RM, DeAngelo DJ, Hemann MT, Stegmaier K. SYK is a critical regulator of FLT3 in acute myeloid leukemia. Cancer Cell. 2014;25(2):226–42.
Riccaboni M, Bianchi I, Petrillo P. Spleen tyrosine kinases: biology, therapeutic targets and drugs. Drug Discov Today. 2010;15(13–14):517–30.
Chen Z, Shojaee S, Buchner M, Geng H, Lee JW, Klemm L, Titz B, Graeber TG, Park E, Tan YX, Satterthwaite A, Paietta E, Hunger SP, Willman CL, Melnick A, Loh ML, Jung JU, Coligan JE, Bolland S, Mak TW, Limnander A, Jumaa H, Reth M, Weiss A, Lowell CA, Muschen M. Signalling thresholds and negative B-cell selection in acute lymphoblastic leukaemia. Nature. 2015;521(7552):357–61.
Koerber RM, Held SA, Heine A, Kotthoff P, Daecke SN, Bringmann A, Brossart P. Analysis of the anti-proliferative and the pro-apoptotic efficacy of Syk inhibition in multiple myeloma. Exp Hematol Oncol. 2015;4:21.
Yamamoto N, Takeshita K, Shichijo M, Kokubo T, Sato M, Nakashima K, Ishimori M, Nagai H, Li YF, Yura T, Bacon KB. The orally available spleen tyrosine kinase inhibitor 2-[7-(3,4-dimethoxyphenyl)-imidazo[1,2-c]pyrimidin-5-ylamino]nicotinamide dihydrochloride (BAY 61-3606) blocks antigen-induced airway inflammation in rodents. J Pharmacol Exp Ther. 2003;306(3):1174–81.
Bhavaraju K, Kim S, Daniel JL, Kunapuli SP. Evaluation of [3-(1-methyl-1H-indol-3-yl-methylene)-2-oxo-2, 3-dihydro-1H-indole-5-sulfonamide] (OXSI-2), as a Syk-selective inhibitor in platelets. Eur J Pharmacol. 2008;580(3):285–90.
Chen KH, Hsu HH, Yang HY, Tian YC, Ko YC, Yang CW, Hung CC. Inhibition of spleen tyrosine kinase (syk) suppresses renal fibrosis through anti-inflammatory effects and down regulation of the MAPK-p38 pathway. Int J Biochem Cell Biol. 2016;74:135–44.
Rolf MG, Curwen JO, Veldman-Jones M, Eberlein C, Wang J, Harmer A, Hellawell CJ, Braddock M. In vitro pharmacological profiling of R406 identifies molecular targets underlying the clinical effects of fostamatinib. Pharmacol Res Perspect. 2015;3(5):e00175.
Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, Qu K, Herlaar E, Lau A, Young C, Wong BR, Lovell S, Sun T, Park G, Argade A, Jurcevic S, Pine P, Singh R, Grossbard EB, Payan DG, Masuda ES. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. 2006;319(3):998–1008.
Can G, Ayvaz S, Can H, Demirtas S, Aksit H, Yilmaz B, Korkmaz U, Kurt M, Karaca T. The Syk inhibitor fostamatinib decreases the severity of colonic mucosal damage in a rodent model of colitis. J Crohns Colitis. 2015;9(10):907–17.
Weinblatt ME, Genovese MC, Ho M, Hollis S, Rosiak-Jedrychowicz K, Kavanaugh A, Millson DS, Leon G, Wang M, van der Heijde D. Effects of fostamatinib, an oral spleen tyrosine kinase inhibitor, in rheumatoid arthritis patients with an inadequate response to methotrexate: results from a phase III, multicenter, randomized, double-blind, placebo-controlled, parallel-group study. Arthritis Rheum. 2014;66(12):3255–64.
Weinblatt ME, Kavanaugh A, Burgos-Vargas R, Dikranian AH, Medrano-Ramirez G, Morales-Torres JL, Murphy FT, Musser TK, Straniero N, Vicente-Gonzales AV, Grossbard E. Treatment of rheumatoid arthritis with a Syk kinase inhibitor: a twelve-week, randomized, placebo-controlled trial. Arthritis Rheum. 2008;58(11):3309–18.
Roark JH, Bussel JB, Cines DB, Siegel DL. Genetic analysis of autoantibodies in idiopathic thrombocytopenic purpura reveals evidence of clonal expansion and somatic mutation. Blood. 2002;100(4):1388–98.
Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med. 2002;346(13):995–1008.
Baluom M, Samara E, Grossbard EB, Lau DT. Fostamatinib, a Syk-kinase inhibitor, does not affect methotrexate pharmacokinetics in patients with rheumatoid arthritis. J Clin Pharmacol. 2011;51(9):1310–8.
Suljagic M, Longo PG, Bennardo S, Perlas E, Leone G, Laurenti L, Efremov DG. The Syk inhibitor fostamatinib disodium (R788) inhibits tumor growth in the Emu- TCL1 transgenic mouse model of CLL by blocking antigen-dependent B-cell receptor signaling. Blood. 2010;116(23):4894–905.
Wang ML, Lee H, Chuang H, Wagner-Bartak N, Hagemeister F, Westin J, Fayad L, Samaniego F, Turturro F, Oki Y, Chen W, Badillo M, Nomie K, DeLa RM, Zhao D, Lam L, Addison A, Zhang H, Young KH, Li S, Santos D, Medeiros LJ, Champlin R, Romaguera J, Zhang L. Ibrutinib in combination with rituximab in relapsed or refractory mantle cell lymphoma: a single-centre, open-label, phase 2 trial. Lancet Oncol. 2016;17(1):48–56.
Furtado M, Wang ML, Munneke B, McGreivy J, Beaupre DM, Rule S. Ibrutinib-associated lymphocytosis corresponds to bone marrow involvement in mantle cell lymphoma. Br J Haematol. 2015;170(1):131–4.
Wu J, Zhang M, Liu D. Acalabrutinib (ACP-196): a selective second-generation BTK inhibitor. J Hematol Oncol. 2016;9(1):21.
Byrd JC, Harrington B, O'Brien S, Jones JA, Schuh A, Devereux S, Chaves J, Wierda WG, Awan FT, Brown JR, Hillmen P, Stephens DM, Ghia P, Barrientos JC, Pagel JM, Woyach J, Johnson D, Huang J, Wang X, Kaptein A, Lannutti BJ, Covey T, Fardis M, McGreivy J, Hamdy A, Rothbaum W, Izumi R, Diacovo TG, Johnson AJ, Furman RR. Acalabrutinib (ACP-196) in relapsed chronic lymphocytic leukemia. N Engl J Med. 2016;374(4):323–32.
Byrd JC, O'Brien S, James DF. Ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(13):1278–9.
Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, Grant B, Sharman JP, Coleman M, Wierda WG, Jones JA, Zhao W, Heerema NA, Johnson AJ, Sukbuntherng J, Chang BY, Clow F, Hedrick E, Buggy JJ, James DF, O'Brien S. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32–42.
Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–16.
Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, Schaefer-Cutillo J, De Vos S, Sinha R, Leonard JP, Cripe LD, Gregory SA, Sterba MP, Lowe AM, Levy R, Shipp MA. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 2010;115(13):2578–85.
Flinn IW, Bartlett NL, Blum KA, Ardeshna KM, LaCasce AS, Flowers CR, Shustov AR, Thress KS, Mitchell P, Zheng F, Skolnik JM, Friedberg JW. A phase II trial to evaluate the efficacy of fostamatinib in patients with relapsed or refractory diffuse large B-cell lymphoma (DLBCL). Eur J Cancer. 2016;54:11–7.
Skinner M, Philp K, Lengel D, Coverley L, Lamm Bergstrom E, Glaves P, Musgrove H, Prior H, Braddock M, Huby R, Curwen JO, Duffy P, Harmer AR. The contribution of VEGF signalling to fostamatinib-induced blood pressure elevation. Br J Pharmacol. 2014;171(9):2308–20.
Currie KS, Kropf JE, Lee T, Blomgren P, Xu J, Zhao Z, Gallion S, Whitney JA, Maclin D, Lansdon EB, Maciejewski P, Rossi AM, Rong H, Macaluso J, Barbosa J, Di Paolo JA, Mitchell SA. Discovery of GS-9973, a selective and orally efficacious inhibitor of spleen tyrosine kinase. J Med Chem. 2014;57(9):3856–73.
Buchner M, Baer C, Prinz G, Dierks C, Burger M, Zenz T, Stilgenbauer S, Jumaa H, Veelken H, Zirlik K. Spleen tyrosine kinase inhibition prevents chemokine- and integrin-mediated stromal protective effects in chronic lymphocytic leukemia. Blood. 2010;115(22):4497–506.
Buchner M, Fuchs S, Prinz G, Pfeifer D, Bartholome K, Burger M, Chevalier N, Vallat L, Timmer J, Gribben JG, Jumaa H, Veelken H, Dierks C, Zirlik K. Spleen tyrosine kinase is overexpressed and represents a potential therapeutic target in chronic lymphocytic leukemia. Cancer Res. 2009;69(13):5424–32.
Sharman J, Di Paolo J. Targeting B-cell receptor signaling kinases in chronic lymphocytic leukemia: the promise of entospletinib. Ther Adv Hematol. 2016;7(3):157–70.
Sharman J, Hawkins M, Kolibaba K, Boxer M, Klein L, Wu M, Hu J, Abella S, Yasenchak C. An open-label phase 2 trial of entospletinib (GS-9973), a selective spleen tyrosine kinase inhibitor, in chronic lymphocytic leukemia. Blood. 2015;125(15):2336–43.
Sharman JP, Klein LM, Boxer M, Kolibaba KS, Abella S, Eng C, He J, Hu J, Yasenchak CA. Phase 2 trial of entospletinib (GS-9973), a selective Syk inhibitor, in indolent non-Hodgkin's lymphoma (iNHL). Blood. 2015;126(23):1545.
Sharman JP, Shustov AR, Smith MR, Hagenstad CT, Kolibaba KS, Abella-Dominicis E, Zhang D, Mitra S, Yasenchak CA, Awan FT. Updated results on the clinical activity of entospletinib (GS-9973), a selective Syk inhibitor, in patients with CLL previously treated with an inhibitor of the B-cell receptor signaling pathway. Blood. 2016;128(22):3225.
Sharman JP, Kolibaba KS, Shustov AR, Giever TA, Klein LM, Nay D, Rado TA, Saleh A, Zhang D, Shi W, Forero A, Assouline S. Results of a phase 2 trial evaluating efficacy and safety of entospletinib (GS-9973) in patients with mantle cell lymphoma. Blood. 2016;128(22):2963.
Walker AR, Bhatnagar B, Marcondes AMQ, DiPaolo J, Vasu S, Mims AS, Klisovic RB, Walsh KJ, Canning R, Behbehani GK, Garzon R, Blachly JS, Johnson AJ, Abella-Dominicis E, Byrd JC, Blum W. Interim results of a phase 1b/2 study of entospletinib (GS-9973) monotherapy and in combination with chemotherapy in patients with acute myeloid leukemia. Blood. 2016;128(22):2831.
Seiter K, Stiefel MF, Barrientos J, Shaikh A, Ahmed N, Baskind P, Liu D. Successful treatment of ibrutinib-associated central nervous system hemorrhage with platelet transfusion support. Stem Cell Invest. 2016;3:27.
Wu J, Zhang M, Liu D. Bruton tyrosine kinase inhibitor ONO/GS-4059: from bench to bedside. Oncotarget. 2017;8:7201–7.
Maffei R, Fiorcari S, Martinelli S, Potenza L, Luppi M, Marasca R. Targeting neoplastic B cells and harnessing microenvironment: the “double face” of ibrutinib and idelalisib. J Hematol Oncol. 2015;8:60.
Mathur R, Sehgal L, Braun FK, Berkova Z, Romaguerra J, Wang M, Rodriguez MA, Fayad L, Neelapu SS, Samaniego F. Targeting Wnt pathway in mantle cell lymphoma-initiating cells. J Hematol Oncol. 2015;8(1):63.
Cheson BD, Leonard JP. Monoclonal antibody therapy for B-cell non-Hodgkin’s lymphoma. N Engl J Med. 2008;359(6):613–26.
Furman RR, Cheng S, Lu P, Setty M, Perez AR, Guo A, Racchumi J, Xu G, Wu H, Ma J, Steggerda SM, Coleman M, Leslie C, Wang YL. Ibrutinib resistance in chronic lymphocytic leukemia. N Engl J Med. 2014;370(24):2352–4.
Hamadani M, Balasubramanian S, Hari PN. Ibrutinib in refractory classic Hodgkin’s lymphoma. N Engl J Med. 2015;373(14):1381–2.
Burke RT, Meadows S, Loriaux MM, Currie KS, Mitchell SA, Maciejewski P, Clarke AS, Dipaolo JA, Druker BJ, Lannutti BJ, Spurgeon SE. A potential therapeutic strategy for chronic lymphocytic leukemia by combining Idelalisib and GS-9973, a novel spleen tyrosine kinase (Syk) inhibitor. Oncotarget. 2014;5(4):908–15.
Barr PM, Saylors GB, Spurgeon SE, Cheson BD, Greenwald DR, O’Brien SM, Liem AKD, Mclntyre RE, Joshi A, Abella-Dominicis E, Hawkins MJ, Reddy A, Di Paolo J, Lee H, He J, Hu J, Dreiling LK, Friedberg JW. Phase 2 study of idelalisib and entospletinib: pneumonitis limits combination therapy in relapsed refractory CLL and NHL. Blood. 2016;127(20):2411–5.
Cai B, Guo M, Wang Y, Zhang Y, Yang J, Guo Y, Dai H, Yu C, Sun Q, Qiao J, Hu K, Zuo H, Dong Z, Zhang Z, Feng M, Li B, Sun Y, Liu T, Liu Z, Wang Y, Huang Y, Yao B, Han W, Ai H. Co-infusion of haplo-identical CD19-chimeric antigen receptor T cells and stem cells achieved full donor engraftment in refractory acute lymphoblastic leukemia. J Hematol Oncol. 2016;9(1):131.
Fan D, Li W, Yang Y, Zhang X, Zhang Q, Yan Y, Yang M, Wang J, Xiong D. Redirection of CD4+ and CD8+ T lymphocytes via an anti-CD3xanti-CD19 bi-specific antibody combined with cytosine arabinoside and the efficient lysis of patient-derived B-ALL cells. J Hematol Oncol. 2015;8:108.
Fan G, Wang Z, Hao M, Li J. Bispecific antibodies and their applications. J Hematol Oncol. 2015;8:130.
Kantarjian H, Stein A, Gökbuget N, Fielding AK, Schuh AC, Ribera J-M, Wei A, Dombret H, Foà R, Bassan R, Arslan Ö, Sanz MA, Bergeron J, Demirkan F, Lech-Maranda E, Rambaldi A, Thomas X, Horst H-A, Brüggemann M, Klapper W, Wood BL, Fleishman A, Nagorsen D, Holland C, Zimmerman Z, Topp MS. Blinatumomab versus chemotherapy for advanced acute lymphoblastic leukemia. N Engl J Med. 2017;376(9):836–47.
Kantarjian HM, DeAngelo DJ, Stelljes M, Martinelli G, Liedtke M, Stock W, Gokbuget N, O'Brien S, Wang K, Wang T, Paccagnella ML, Sleight B, Vandendries E, Advani AS. Inotuzumab ozogamicin versus standard therapy for acute lymphoblastic leukemia. N Engl J Med. 2016;375(8):740–53.
Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, Chew A, Gonzalez VE, Zheng Z, Lacey SF, Mahnke YD, Melenhorst JJ, Rheingold SR, Shen A, Teachey DT, Levine BL, June CH, Porter DL, Grupp SA. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371(16):1507–17.
Topp MS, Gokbuget N, Stein AS, Zugmaier G, O'Brien S, Bargou RC, Dombret H, Fielding AK, Heffner L, Larson RA, Neumann S, Foa R, Litzow M, Ribera JM, Rambaldi A, Schiller G, Bruggemann M, Horst HA, Holland C, Jia C, Maniar T, Huber B, Nagorsen D, Forman SJ, Kantarjian HM. Safety and activity of blinatumomab for adult patients with relapsed or refractory B-precursor acute lymphoblastic leukaemia: a multicentre, single-arm, phase 2 study. Lancet Oncol. 2014;16(1):57–66.
Chao M-W, Lai M-J, Liou J-P, Chang Y-L, Wang J-C, Pan S-L, Teng C-M. The synergic effect of vincristine and vorinostat in leukemia in vitro and in vivo. J Hematol Oncol. 2015;8(1):82.
Sidaway P. Haematological cancer: Treg predict responsiveness to blinatumomab. Nat Rev Clin Oncol. 2017;14(5):262.
Barlev A, Lin VW, Katz A, Hu K, Cong Z, Barber B. Estimating long-term survival of adults with Philadelphia chromosome-negative relapsed/refractory B-precursor acute lymphoblastic leukemia treated with blinatumomab using historical data. Adv Ther. 2017;34(1):148–55.
Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature. 2017;545(7655):423–31.
Riviere I, Sadelain M. Chimeric antigen receptors: a cell and gene therapy perspective. Mol Ther. 2017;25(5):1117–24.
Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJ, Hamieh M, Cunanan KM, Odak A, Gonen M, Sadelain M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543(7643):113–7.
Ghosh A, Smith M, James SE, Davila ML, Velardi E, Argyropoulos KV, Gunset G, Perna F, Kreines FM, Levy ER, Lieberman S, Jay HV, Tuckett AZ, Zakrzewski JL, Tan L, Young LF, Takvorian K, Dudakov JA, Jenq RR, Hanash AM, Motta AC, Murphy GF, Liu C, Schietinger A, Sadelain M, van den Brink MR. Donor CD19 CAR T cells exert potent graft-versus-lymphoma activity with diminished graft-versus-host activity. Nat Med. 2017;23(2):242–9.
Perova T, Grandal I, Nutter LM, Papp E, Matei IR, Beyene J, Kowalski PE, Hitzler JK, Minden MD, Guidos CJ, Danska JS. Therapeutic potential of spleen tyrosine kinase inhibition for treating high-risk precursor B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6(236):236ra262.
Axelrod MJ, Fowles P, Silverman J, Clarke A, Tang J, Rousseau E, Webb HK, Di Paolo J. The combination of entospletinib and vincristine demonstrates synergistic activity in a broad panel of hematological cancer cell lines and anti-tumor efficacy in a DLBCL xenograft model. Blood. 2015;126(23):5123.
Minden MD, Stock W, Crosswell H, Siegel RD, Chun P, Abella S, He J, Eng C, Douer D. A phase 1b, open-label, dose escalation and expansion study evaluating the safety and efficacy of entospletinib (GS-9973) with vincristine and dexamethasone in adult subjects with relapsed or refractory acute lymphoid leukemia (ALL). Blood. 2015;126(23):4866.
Coffey G, Betz A, DeGuzman F, Pak Y, Inagaki M, Baker DC, Hollenbach SJ, Pandey A, Sinha U. The novel kinase inhibitor PRT062070 (Cerdulatinib) demonstrates efficacy in models of autoimmunity and B-cell cancer. J Pharmacol Exp Ther. 2014;351(3):538–48.
Ma J, Xing W, Coffey G, Dresser K, Lu K, Guo A, Raca G, Pandey A, Conley P, Yu H, Wang YL. Cerdulatinib, a novel dual SYK/JAK kinase inhibitor, has broad anti-tumor activity in both ABC and GCB types of diffuse large B cell lymphoma. Oncotarget. 2015;6(41):43881–96.
Blunt MD, Koehrer S, Dobson RC, Larrayoz M, Wilmore S, Hayman A, Parnell J, Smith LD, Davies A, Johnson PW, Conley PB, Pandey A, Strefford JC, Stevenson FK, Packham G, Forconi F, Coffey GP, Burger JA, Steele AJ. The dual Syk/JAK inhibitor cerdulatinib antagonizes B-cell receptor and microenvironmental signaling in chronic lymphocytic leukemia. Clin Cancer Res. 2017;23:2313. doi:10.1158/1078-0432.CCR-16-1662.
Guo A, Lu P, Coffey G, Conley P, Pandey A, Wang YL. Dual SYK/JAK inhibition overcomes ibrutinib resistance in chronic lymphocytic leukemia: cerdulatinib, but not ibrutinib, induces apoptosis of tumor cells protected by the microenvironment. Oncotarget. 2017;8:12953–67.
Coffey G, Rani A, Betz A, Pak Y, Haberstock-Debic H, Pandey A, Hollenbach S, Gretler DD, Mant T, Jurcevic S, Sinha U. PRT062607 achieves complete inhibition of the spleen tyrosine kinase at tolerated exposures following oral dosing in healthy volunteers. Clin Pharmacol 2017;57(2):194–210.
Lam B, Arikawa Y, Cramlett J, Dong Q, de Jong R, Feher V, Grimshaw CE, Farrell PJ, Hoffman ID, Jennings A, Jones B, Matuszkiewicz J, Miura J, Miyake H, Natala SR, Shi L, Takahashi M, Taylor E, Wyrick C, Yano J, Zalevsky J, Nie Z. Discovery of TAK-659 an orally available investigational inhibitor of Spleen Tyrosine Kinase (SYK). Bioorg Med Chem Lett. 2016;26(24):5947–50.
Purroy N, Carabia J, Abrisqueta P, Egia L, Aguilo M, Carpio C, Palacio C, Crespo M, Bosch F. Inhibition of BCR signaling using the Syk inhibitor TAK-659 prevents stroma-mediated signaling in chronic lymphocytic leukemia cells. Oncotarget. 2017;8(1):742–56.
Cen O, Kannan K, Huck JJ, Yu J, Zhang M, Gordon LI, Longnecker R. The Syk inhibitor TAK-659 prevents splenomegaly and tumor development in a murine model of EBV-associated lymphoma. Blood. 2016;128(22):4179.
Petrich AM, Gordon LI, Infante JR, Madan S, Giles FJ, Nimeiri HS, Kaplan JB, Stumpo K, Zhang B, Faucette S, Shou Y, Shih KC. Phase 1 dose-escalation study of TAK-659, an investigational SYK inhibitor, in patients (Pts) with advanced solid tumor or lymphoma malignancies. Blood. 2015;126(23):2693.
Kaplan JB, Gordon LI, Infante JR, Popat R, Rambaldi A, Madan S, Patel MR, Gritti G, Ng C-H, Chau I, Radford JA, de Oteyza JP, Zinzani PL, Faucette S, Sheldon-Waniga E, Williams M, Stumpo K, Shou Y, Carpio C, Bosch F. Updated results from a phase 1 study of TAK-659, an investigational and reversible SYK inhibitor, in patients (Pts) with advanced solid tumor or lymphoma malignancies. Blood. 2016;128(22):624.
Kaplan JB, Bixby DL, Morris JC, Frankfurt O, Altman J, Wise-Draper T, Burke PW, Collins S, Kannan K, Wang L, Faucette S, Lee B, Shou Y, Levis MJ, Pratz KW. A phase 1b/2 study of TAK-659, an investigational dual SYK and FLT-3 inhibitor, in patients (Pts) with relapsed or refractory acute myelogenous leukemia (R/R AML). Blood. 2016;128(22):2834.
Cang S, Iragavarapu C, Savooji J, Song Y, Liu D. ABT-199 (venetoclax) and BCL-2 inhibitors in clinical development. J Hematol Oncol. 2015;8(1):129.
Wu J, Fu J, Zhang M, Liu D. Blinatumomab: a bispecific T cell engager (BiTE) antibody against CD19/CD3 for refractory acute lymphoid leukemia. J Hematol Oncol. 2015;8:104.
Wu J, Fu J, Zhang M, Liu D. AFM13: a first-in-class tetravalent bispecific anti-CD30/CD16A antibody for NK cell-mediated immunotherapy. J Hematol Oncol. 2015;8(1):96.
Linder K, Gandhiraj D, Hanmantgad M, Seiter K, Liu D. Complete remission after single agent blinatumomab in a patient with pre-B acute lymphoid leukemia relapsed and refractory to three prior regimens: hyperCVAD, high dose cytarabine mitoxantrone and CLAG. Exp Hematol Oncol. 2016;5:20.
Ma W, Gilligan BM, Yuan J, Li T. Current status and perspectives in translational biomarker research for PD-1/PD-L1 immune checkpoint blockade therapy. J Hematol Oncol. 2016;9:47.
Xu KY, Wu S. Update on the treatment of metastatic clear cell and non-clear cell renal cell carcinoma. Biomarker Res. 2015;3:5.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61.
Funding
This study was partly funded by The first Affiliated Hospital of Zhengzhou University, Zhengzhou, China.
Availability of data and materials
The material supporting the conclusion of this review has been included within the article.
Author information
Authors and Affiliations
Contributions
DL designed the study. AMD and DL drafted the manuscript and finalized the table. Both authors read and approved final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This is not applicable for this review.
Consent for publication
This is not applicable for this review.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Liu, D., Mamorska-Dyga, A. Syk inhibitors in clinical development for hematological malignancies. J Hematol Oncol 10, 145 (2017). https://doi.org/10.1186/s13045-017-0512-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-017-0512-1