
Abstract
Mitochondrial dysfunction is strongly implicated in the etiology of idiopathic and genetic Parkinson’s disease (PD). However, strategies aimed at ameliorating mitochondrial dysfunction, including antioxidants, antidiabetic drugs, and iron chelators, have failed in disease-modification clinical trials. In this review, we summarize the cellular determinants of mitochondrial dysfunction, including impairment of electron transport chain complex 1, increased oxidative stress, disturbed mitochondrial quality control mechanisms, and cellular bioenergetic deficiency. In addition, we outline mitochondrial pathways to neurodegeneration in the current context of PD pathogenesis, and review past and current treatment strategies in an attempt to better understand why translational efforts thus far have been unsuccessful.
Similar content being viewed by others


Background
Parkinson’s disease (PD) is the most prevalent neurodegenerative movement disorder affecting up to 2 % of those aged 60 years and older [1]. Clinically, PD is defined by presence of the levodopa-responsive motor symptoms bradykinesia with resting tremor or rigidity [2]. These motor symptoms are frequently accompanied by non-motor symptoms, including but not limited to sleep disturbances, depression, autonomic dysfunction, and hyposmia [3, 4]. Today, there are still no proven strategies for slowing the progression of PD. This unmet medical need reflects our incomplete grasp of disease mechanisms.
Neuropathologically, PD is characterized by two imperfectly aligned features: selective neuronal degeneration of vulnerable cell-types within particular brain regions (e.g., midbrain dopaminergic (DA) substantia nigra pars compacta (SNc) neurons [5, 6]), and the presence of eosinophilic alpha-synuclein (aSYN) positive inclusion bodies, termed Lewy pathology (LP). Systematic cross-sectional characterization of human postmortem PD brains revealed that even in late-stage disease LP is not globally distributed in the brain of PD patients, but is restricted to certain vulnerable nuclei, thereby showing a patch-like distribution [7, 8]. While there is clear evidence that some regions (SNc, olfactory bulb, dorsal motor nucleus of vagus, locus coeruleus, pedunculopontine nucleus, amygdala) are more susceptible to LP than others, it has been difficult to establish the sequence and extent in which they develop LP. In addition to brain pathology, LP also affects many structures of the peripheral nervous system (nerve fibers within e.g., skin, heart, esophagus) [7, 9]. The observation that misfolded, fibrillar forms of aSYN can propagate from one cell to another in PD animal models [10], has fueled the thought that also in humans toxic aSYN species might spread between synaptically coupled brain regions, thereby driving the development of brain-wide LP formation [11].
In contrast to the relatively well-mapped distribution of LP, the spatio-temporal development of cell-loss within affected regions remains largely elusive. While loss of dopaminergic SNc neurons has been well-documented and clearly linked to the onset of PD motor symptomatology, there is no brain-wide assessment of neurodegeneration, and the available studies investigating cell loss show notable heterogeneity [12]. Given the absence of a clear correlation between LP formation and neuronal cell loss, it is crucial to disentangle the cell-intrinsic factors which render neurons susceptible to LP formation and those who drive neurodegeneration. So far, several core pathogenetic factors have been identified. Among those are impaired cellular protein homeostasis, dysfunctional proteasomal and lysosomal clearance systems, impaired protein and membrane trafficking, synaptic dysfunction including disturbed neurotransmission, neuroinflammation, and mitochondrial dysfunction [3, 13,14,15,16].
Mitochondrial dysfunction has long been implicated as a key pathological hallmark in PD. Since mitochondria are highly multifunctional organelles, their integrity is essential for neuronal function and survival. This review summarizes the evidence for mitochondrial dysfunction in genetic and idiopathic PD, discusses the bidirectional interaction between mitochondrial stress and aSYN aggregation, and points out potential mitochondrial pathways to neurodegeneration in the current context of PD pathogenesis. Further, we review current and past therapeutic strategies targeting mitochondrial dysfunction in an attempt to modify disease progression, and outline current gaps in our understanding.
Main text
Importance of mitochondrial health in PD at-risk neurons
Neurons possess a complex network of mitochondria stretching from dendrites that receive synaptic contacts to the synaptic terminals that communicate with neighboring neurons. Mitochondria perform a variety of tasks, including generation of adenosine triphosphate (ATP), Ca2+ buffering and epigenetic signaling [17,18,19]. Two central tenets of the mitochondrial theory of pathogenesis are that neurons have a high bioenergetic demand and that neurons rely heavily on mitochondria for ATP production. Indeed, all cells rely upon ATP to drive basic cellular processes. Neurons differ from many other cell types in ways that increase their bioenergetic needs. In particular, they need ATP to maintain ionic homeostasis which is being constantly challenged by 1) their reliance upon electrical signals generated by transmembrane ion fluxes, 2) their sequestration of transmitter into vesicles, fusion of these vesicles during synaptic activity and reuptake of membrane during vesicular recycling, and 3) the need to maintain and repair an often massive transmitter release machinery [20]. The ATP necessary for these processes can be derived both from glycolysis and mitochondrial oxidative phosphorylation (OXPHOS). While glycolytic mechanisms are fast, they are relatively inefficient and generate roughly one tenth the ATP from glucose that mitochondria can extract. It has been hypothesized that neurons rely exclusively upon mitochondrial OXPHOS for ATP generation (using lactate shuttled from astrocytes), but more recent direct measurements have shown that neurons use both glycolysis and OXPHOS to generate ATP [21].
Despite the clear importance of mitochondria to neuronal bioenergetics, they also play a variety of other roles. One of these is Ca2+ buffering. This may be particularly important in axons of some neurons [22]. Another important function is metabolic signaling [19]. For example, mitochondria are critical sources of citrate, which is important to the production of acetyl-coenzyme A and acetylation of proteins and DNA.
Compromised mitochondrial function may have a disproportionate impact on those neurons that are at-risk in PD. The best studied example of this phenotype is the SNc dopaminergic neuron. These neurons are constantly active and have extensive axonal arbors with as many as 1–2 million transmitter release sites per axon in humans [23]. Many (if not all) of the other neurons at greatest risk in PD have a similar phenotype: locus coeruleus noradrenergic neurons, dorsal motor nucleus of the vagus cholinergic neurons, and pedunculopontine nucleus cholinergic neurons [20, 24,25,26]. These neurons play a key role in organismal survival, particularly during times of crisis when sustained, efficient function is critical.
To meet this bioenergetic demand, many at-risk neurons engage a feed-forward control mechanism that utilizes plasma membrane L-type Ca2+ channels to drive mitochondrial OXPHOS [27,28,29,30,31,32]. While this feed-forward control helps to ensure that ATP levels do not fall during times of high demand, it also increases the production of damaging reactive oxygen species (ROS) and basal mitochondrial oxidant stress. ROS and mitochondrial oxidant stress damages lipids, proteins and DNA [33]. This can not only compromise cellular function but leads to an increased demand on catabolic processes in neurons, most importantly lysosomal degradation. This increased demand should in principle decrease spare capacity, providing a linkage between mitochondrial stress and genetic mutations linked to familial cases of PD involving mitochondrial quality control (DJ1, PINK1, parkin) and lysosomal function (GBA1, LRRK2, VPS35, others).
Evidence for mitochondrial impairment in PD patients
A key piece of evidence that mitochondrial dysfunction is implicated in PD pathogenesis stems from the observation in 1983 that several recreational drug users which intravenously administered the new synthetic heroin drug MPPP (1-methyl-4-phenyl-4-propionoxy-piperidine) developed acute-onset but levodopa (L-DOPA) responsive parkinsonian motor symptoms shortly after drug administration [34]. Subsequently, the mitochondrial ETC inhibitor MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) was identified as a byproduct of poor MPPP synthesis. Following absorption, MPTP crosses the blood-brain barrier and is converted to MPP+ within astroglia by monoaminoxidase B (Fig. 1). Extracellularly released MPP+ is then actively taken up via the DA transporter and accumulates within mitochondria of DA neurons where it inhibits mitochondrial complex I (CI) of the ETC [35,36,37]. Since its first discovery, MPTP induced toxicity has been established and validated many times as a reliable approach to model neurodegeneration and development of motor symptoms in rodents and primates [38, 39]. From a translational standpoint, the MPTP studies have taught us that mitochondrial CI inhibition in DA SNc neurons can cause a disease phenotype that resembles many features of idiopathic PD, e.g. all cardinal motor symptoms (bradykinesia, rigidity, tremor), some non-motor symptoms (dribbling of saliva, urinary disturbances), and L-DOPA responsiveness.

Mechanism of action of neurotoxins inducing PD. MPTP readily crosses the blood-brain barrier and is taken up by nearby astroglia which subsequently convert it to MPP+ via MAO-B. Extracellularly released MPP+ is then actively taken up via DAT and accumulates within mitochondria of DA neurons where it inhibits mitochondrial CI of the ETC resulting in ROS production and energetic deficiency. Similarly, the pesticide rotenone (Rot), due to its high lipophilicity, readily crosses biological membranes and reaches the inner mitochondrial membrane where it inhibits CI. In contrast, paraquat (PQ2+) relies on the LAT1 to cross the blood-brain barrier. Hereafter, it is taken up by DAT or OCT3 into DA neurons and generates ROS by redox cycling at CI and CIII of the ETC. Abbreviations: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP); 1-methyl-4-phenylpyridinium (MPP+); coenzyme Q (CoQ); dopamine (DA); dopamine transporter (DAT); L-amino acid transporter (LAT1); mitochondrial Complex I (CI); mitochondrial Complex II (CII); mitochondrial Complex III (CIII); mitochondrial Complex IV (CIV); mitochondrial Complex V (CV); monoamino oxidase B (MAO-B); organic cation transporter 3 (OCT3); paraquat (PQ2+); reactive oxygen species (ROS); rotenone (Rot); vesicular monoamino transporter (VMAT). Created with BioRender.com
The observation that CI blockade can induce PD-like symptoms is further substantiated by the finding that the chemically related substance paraquat, as well as the CI inhibitor rotenone (Fig. 1), are not only shown by epidemiology to be risk factors for the development of PD, but also induce PD-like symptomatology in animal experiments [40,41,42]. More recently, genetic approaches have shown that disruption of CI function specifically in dopaminergic neurons is sufficient to produce a progressive, L-DOPA-responsive parkinsonism [43].
But is mitochondrial dysfunction also a constant and reliable feature of idiopathic PD, meaning in the absence of mitochondrial toxins or genetic disease drivers? Important aspects can be derived from the analyses of brain tissue from deceased idiopathic PD patients. In several studies, tissue samples of the SNc but also of other brain regions, as well as lymphocytes and platelets were analyzed for the presence of ETC alterations by immunoblotting, immunohistochemistry, or enzyme activity analysis. The most pronounced and consistently reported finding is a decreased activity of CI of the ETC in SNc tissue homogenates [44,45,46,47]. Some studies even observed a decreased activity of CI in tissue samples from the frontal as well as prefrontal cortices and striatum, but not peripheral tissues [48,49,50]. In contrast, deficiency of ETC CII, CIII or CIV was only sporadically observed, and when ETC function was either assessed by immunohistochemistry or other peripheral specimens (e.g. lymphocytes, platelets, or muscle) were analyzed, CI dysfunction was only inconsistently reported [47].
Regarding the lack of concordance in some studies it is important to consider that most experiments either used mixed tissue homogenates (neuronal and non-neuronal cells), or investigated peripheral tissue, which from our current point of view is likely not the main manifestation place of PD pathology. Future studies investigating ETC dysfunction in human postmortem tissue using now available cell-type specific approaches might therefore possess great potential to further enhance our understanding of mitochondrial dysfunction in idiopathic PD [43].
Another line of evidence pointing to mitochondrial dysfunction in PD is based on the observation of increased mitochondrial DNA (mtDNA) aberrations in tissue samples of deceased patients with idiopathic PD. While initial approaches investigating mtDNA deletions produced conflicting results [51,52,53], more recent studies confirmed an increased amount of mtDNA deletions specifically in postmortem SNc tissue of PD patients [54,55,56]. In addition, patients carrying a mutation of the polymerase gamma gene, the only polymerase present in human mitochondria, develop rare genetic syndromes including parkinsonian symptoms and loss of SNc neurons [57]. Taken together, there is mounting clear evidence implicating mitochondrial dysfunction as a key disease hallmark in idiopathic PD.
Mitochondrial dysfunction is tightly linked to genetic PD
Although only roughly 10% of PD cases are associated with defined genetic alterations, the study of these familial PD (PARK) genes has led to major advances in our understanding of PD etiopathogenesis. While numerous PARK genes have been identified, several of these are directly linked to impaired mitochondrial function and integrity (Table 1).
Mutations of the genes coding for PINK1 (PARK6) and Parkin (PARK2) are the most frequent causes of autosomal recessive early-onset PD. Their clinical manifestation is characterized by relatively pure motor symptomatology and L-DOPA responsiveness, which can be accompanied by dopamimetica associated dyskinesia, hyperreflexia, and sometimes psychiatric symptoms. Interestingly, histopathological examination of postmortem tissue indicates loss of SNc dopaminergic neurons and neurons in other brain regions normally vulnerable in idiopathic PD (e.g., locus coeruleus, nucleus basalis meynert). However, presence of aSYN inclusions, a hallmark of idiopathic PD, is not a consistent feature of these PD cases [111,112,113,114,115,116]. At the cellular level, PINK1 and Parkin play key roles in mitochondrial quality control mechanisms and signaling cascades in response to mitochondrial damage [67]. PINK1/Parkin can not only initiate mitophagy, but also control fission and fusion of mitochondria, promote the generation of mitochondria derived vesicles and induce mitochondrial biogenesis [70, 117,118,119,120,121]. In fibroblasts from PINK1 and Parkin familial PD cases, loss of protein function leads to ETC impairment with reduced ATP production and high levels of ROS [122,123,124]. While experimental studies using Parkin-KO mice revealed lower levels of mitochondrial respiratory capacity [125,126,127], PINK1-KO mice additionally exhibited defects in CI function, reduced Ca2+ buffering capacity, and impairments in mitochondrial membrane potential [128,129,130,131]. Comparable findings have also been reported in Drosophila Parkin and PINK1 models [71,72,73, 132,133,134]. As the underlying pathophysiological event, increased mitochondrial fission has been identified in Parkin and PINK1 mutant mice and Drosophila models [117, 135]. This is supported by the fact that inhibition of mitochondrial fission via mdivi-1 treatment, was able to rescue mitochondrial function by normalizing the balance between mitochondrial fission and fusion [136]. Apart from increased fission, defects in mitochondrial biogenesis have been shown to contribute to mitochondrial dysfunction in Parkin deficient human dopaminergic neurons [121].
Interestingly, there is additional evidence for accumulation of insoluble Parkin within idiopathic PD patients. While previous studies observed that accumulating Parkin is S-nitrosylated [137,138,139,140], a more recent study discovered that Parkin itself functions as a redox molecule by providing antioxidant capacity for human midbrain neurons. Subsequent oxidizing posttranslational modifications then contribute to the decrease in Parkin solubility [141].
Another example indicating mitochondrial driven parkinsonism, are mutations in the gene coding for DJ1 (PARK7). Resulting loss of function leads to an autosomal recessive form of PD which is less common than PINK1 or Parkin familial PD. The clinical presentation of individuals with DJ1 mutations is characterized by early onset slow progressing parkinsonism, which is frequently accompanied by non-motor symptomatology (e.g., anxiety, cognitive decline, and psychotic symptoms), and good L-DOPA responsiveness [142]. Notably, postmortem histopathological analysis revealed widespread cortical and subcortical LP and neurodegeneration [77]. DJ1 is involved in counteracting oxidative stress and subsequent mitochondrial dysfunction under physiological conditions. In the experimental setting, DJ1 depletion leads to impaired mitochondrial respiration, high levels of intracellular ROS, compromised mitochondrial membrane potential, and altered mitochondrial morphology [78, 143,144,145]. Furthermore, mutated DJ1 is translocated from the cytosol into the mitochondrial matrix where it gets degraded [146]. Despite the increasing interest in DJ1’s function, the molecular mechanisms remain incompletely understood. Several lines of evidence suggest that DJ1 is a redox-sensitive protein which relies on cysteine oxidation to sense oxidative stress and then counteract this stress through activation of different signaling pathways [147,148,149]. Other reports suggest that DJ1 may additionally possess chaperone activity [150, 151], supported by data showing that DJ1 is able to attenuate aSYN aggregation [152], and the observation that human induced pluripotent stem cells (IPSCs) derived from fibroblasts of DJ1 PD patients exhibit increased aSYN pathology [153]. However, further evidence highlights DJ1’s enzymatic functions, including glyoxalase and deglycase activities, showing that DJ1 can decrease reactive carbonyl products and repair glycated nucleic acids [154, 155]. Albeit the exact biological interplay of these processes is still debated, DJ1 clearly links antioxidant pathways, mitochondrial dysfunction, and aSYN aggregation.
More recently mutations affecting vacuolar protein sorting 35 (VPS35 = PARK17) have been linked to late-onset autosomal dominant PD, and VPS13C (PARK23) to early onset rapid progressing autosomal recessive PD [100, 156, 157]. Although the exact pathophysiological mechanisms are still intensively debated, experimental studies on VPS35 mutant fibroblasts, mice, or cell culture systems reported increased mitochondrial fragmentation, disturbed mitochondrial fission and fusion dynamics, and abnormal configuration of ETC CI [101,102,103, 158]. Mechanistically, VPS35 is a part of the retromer complex and thereby plays an important role in endosomal sorting and trafficking of proteins. VPS35 mutations have been shown to lead to an enhanced interaction of VPS35 with DLP1, which subsequently causes increased turnover of mitochondrial DLP1 complex, thereby fueling excessive mitochondrial fission, finally culminating in mitochondrial dysfunction and fragmentation [101, 158]. Further, VPS13C mutations have been shown to decrease mitochondrial membrane potential, promote mitochondrial fragmentation, and elevate mitophagy [100]. In addition, mutations in FBXO7 (PARK15), causing a rare syndrome of juvenile parkinsonism with pyramidal signs, have been linked to impaired mitophagy and decreased CI function [94].
Taken together, the familial PD cases not only show us that there is a clear link between genetic PD and mitochondrial dysfunction, they also highlight that multiple mitochondrial pathways may be impaired, including CI function, mitophagy, fission and fusion, and mitochondrial biogenesis.
Causal link between α-synuclein pathology and mitochondrial dysfunction
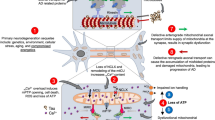
While for a portion of PD patients, the occurrence of mitochondrial dysfunction can be explained by PARK genes, the etiology of idiopathic PD is still a matter of intensive debate. However, broad experimental evidence stemming from observations in isolated mitochondria [59,60,61, 159], and rodents [62, 63], suggests aSYN pathology as a major source of mitochondrial dysfunction (Fig. 2). Under physiological conditions, monomeric aSYN was shown to modulate the function of the mitochondrial ATP synthase subunit alpha, as aSYN knock-out mice showed reduced ATP synthase efficiency and reduced ATP levels [64]. Similarly, another study employing aSYN deficient mice observed an altered neuronal mitochondrial membrane structure and CI deficiency [65].

Synucleinopathy-driven mechanisms of cellular dysfunction and death in PD. Abbreviations: mitochondrial permeability transition pore (mPTP); reactive oxygen species (ROS). Created with BioRender.com
In the presence of aSYN pathology, meaning excessive amounts of overexpressed monomeric aSYN or existence of oligomeric and fibrillar aSYN, several studies reported decreased mitochondrial CI activity, alterations of mitochondrial membrane potential, and elevated oxidative stress levels [59, 63, 66, 160,161,162]. The effect on CI is further substantiated by another study which reported a dose-dependent effect of aSYN pathology on CI inhibition [163]. Based on the observation that aSYN knock-out mice were resistant to MPTP induced toxicity, it has been hypothesized that aSYN directly influences CI function [35, 164]. This view is supported by studies which reported that overexpression of human aSYN in wildtype mice or use of SNCA A30P mutated transgenic mice worsened MPTP induced toxicity [165, 166]. Similar findings have also been observed for the CI inhibitor rotenone [167, 168]. However, CI does not seem to be the only engagement point for aSYN pathology. More recently, interaction of pathological aSYN oligomers with the ATP synthase subunit alpha in combination with mitochondrial permeability transition pore opening has been suggested as a mediator of aSYN induced mitochondrial dysfunction [169]. Further, it has been shown that aSYN oligomers interact with the outer mitochondrial membrane protein TOM20 [170, 171]. As a consequence of aSYN binding to TOM20, mitochondrial protein import is impaired causing ETC malfunction, accumulation of ROS and loss of mitochondrial membrane potential [170]. aSYN induced loosening of contacts between mitochondria and the endoplasmic reticulum (ER), which are considered essential for proper Ca2+ exchange between those two organelles, has been reported as another possible cause of reduced mitochondrial respiration, primarily by dysregulated intracellular Ca2+ levels [172, 173].
Taken together, these studies not only show that aSYN pathology can trigger mitochondrial dysfunction, they reveal that there are several independent pathways how aSYN pathology affects mitochondrial function (Fig. 2). Notably, many of those pathways converge to a shared pathological phenotype exhibiting increased cellular and mitochondrial ROS, impairment of mitochondrial membrane potential, and reduced mitochondrial respiration.
Pathways linking mitochondrial dysfunction to neurodegeneration
Does mitochondrial dysfunction cause neurodegeneration in PD, or is it simply a disease tombstone?
While this question is difficult to answer for idiopathic PD, important information can be gleaned again from familial PD cases by looking at those few histopathological postmortem reports which are available. Notably, PINK1 as well as Parkin, and DJ1 mutation carriers, all familial PD cases where PD is thought to be majorly driven by mitochondrial dysfunction, exhibit marked neuronal cell loss within the SNc and other susceptible brain regions [77, 111, 112]. This clearly indicates that at least genetically driven mitochondrial dysfunction is causative of neuronal cell loss in these individuals. This is supported by the finding that targeted disruption of mitochondrial CI in mice leads to dopaminergic degeneration culminating in a human-like type of parkinsonism [43]. However, what is less clear is whether mitochondrial dysfunction is necessary for PD.
As mentioned above, mitochondrial dysfunction and damage can contribute to several pathological cascades implicated in PD [67, 174, 175]. As shown by using direct ratiometric probes, many at-risk neurons have been found to manifest elevated levels of mitochondrial oxidant stress [30, 31, 176]. Sustained oxidant stress damages membranes, proteins, and DNA. This damage elevates mitophagy in SNc dopaminergic neurons [176], thereby diminishing the overall autophagic capacity. Cytosolic ROS can further damage proteins of the mitophagy pathway [138] and increase mitochondrial dysfunction. Mitochondrially-generated ROS also compromises lysosomal and proteasomal function and increases the accumulation of misfolded forms of aSYN [153, 177]. Further, intracellular ROS triggers induction of parthanatos, an apoptosis independent pathway of neurodegeneration [178]. In parallel, damaged mitochondria or excessive mitochondrial stress can induce mtDNA release into the cytosol and subsequent increases in the production of proinflammatory cytokines [179, 180], as shown in Parkin-KO mice which also exhibited a POLG mutation [181]. Mitochondrial dysfunction is further connected to neuroinflammation by the observation that loss of PINK1 and Parkin function results in increased mitochondrial antigen presentation and subsequent activation of cytotoxic T-cells [182]. Intestinal infection with Gram-negative bacteria in PINK1 mice enhanced mitochondrial antigen presentation which was followed by elevated levels of CD8+ T-cells in the brain and periphery [183].
As indicated above, failure of mitochondrial quality control mechanisms defines another pathway to neurodegeneration in PD. Substantial evidence shows that the concerted interplay of PINK1 and Parkin is essential for maintaining mitochondrial health. Loss of function mutations result in disruption of cellular mitophagy, as well as impaired fusion and fission of mitochondria, and reduced generation of mitochondrial derived vesicles [67]. As a consequence, damaged mitochondria accumulate, cytochrome c and other proapoptotic proteins are released into the cytosol, and apoptosis might be induced. Damaged mitochondria due to loss of mitochondrial quality control mechanisms also contribute to the generation of oxidative stress and mtDNA mutations. Importantly, in idiopathic PD, LP also directly inactivates Parkin and thereby contributes to failure of mitochondrial quality control even in the absence of genetic mutations [138, 139].
Intracellular Ca2+ signaling also may contribute to pathogenesis [5]. At-risk neurons have low intrinsic Ca2+ buffering capacity and strong engagement of both plasma membrane and ER-dependent Ca2+ signaling, leading to large cytosolic oscillations in intracellular Ca2+ concentration [176]. Elevated intracellular Ca2+ can promote aSYN misfolding and aggregation [184, 185] thereby linking aSYN and Ca2+ in a vicious cycle.
Another key hallmark of PD is impaired cellular proteasomal and lysosomal mechanisms [153, 186, 187]. Proteasomal degradation as well as lysosomal function are energy consuming processes. It is easy to infer that compromised ATP production by mitochondria will reduce their functional capacity. Thus, elevated mitochondrial ROS production – and the resulting cellular damage – not only increases the burden on these systems, but with declining mitochondrial capacity it will likely diminish their capacity. As a consequence, not only aSYN aggregation is promoted but clearance of oxidized proteins is reduced, leading to further generation of ROS and oxidative damage in terms of a feedforward mechanism. Moreover, there is evidence for dynamic mitochondria-lysosome contacts which allow inter-organelle crosstalk. Interestingly, patient derived neurons harboring a heterozygous mutation within the gene coding for β-glucocerebrosidase (GBA1) show disturbed loosening of these contact sites which resulted in prolonged tethering and disruption of intracellular mitochondrial distribution [188].
Taken together, current evidence indicates that there are several mitochondrial pathways which are tightly linked to other pathogenic mechanisms of PD. While some of these pathways are highly interdependent, others act in parallel to each other. From a translational standpoint, this suggests that, as in cancer, new therapeutic approaches will either need to target several of these pathways at once or be tailored to pathological endpoints shared by these pathways.
Therapeutic approaches targeting mitochondrial dysfunction in PD
One of the greatest challenges facing the biomedical community is the development of a disease-modifying therapy for PD. Several clinical trials have been attempted to address this challenge, but none have succeeded. Several have targeted mitochondrial function either directly or indirectly.
Given the recognition that mitochondrial oxidant stress is a potential driver of pathogenesis, some of the earliest trials aimed at reducing it (Table 2). For example, the antioxidant coenzyme Q10 (CoQ10) was tested in several trials, as was minocycline; they all failed [252, 253]. Mitochondrially-targeted antioxidants, like MitoQ, MitoVitE, MitoApocynin and MitoTEMPOL were developed to achieve better target engagement and showed promise in pre-clinical experiments, but this general strategy has not shown a clear benefit in PD patients [207]. One of the key issues with these trials is that it is difficult to demonstrate adequate target engagement and biological efficacy of these compounds in humans. So, it is unclear whether they are testing the core hypothesis or not.
A related approach is to try and boost brain concentrations of glutathione (Table 2). Nigral levels of glutathione are lower in PD patients, possibly because of an increased reliance upon glycolysis for ATP production in PD patients [210]. Elevating glutathione has been proposed and explored in preclinical and clinical trials [213]. However, it is unclear whether this is simply an effect of mitochondrial dysfunction and whether adequate brain concentrations can be achieved with oral dosing. N-Acetyl cysteine (NAC), an approved drug to treat acetaminophen induced liver failure [254], increases cellular glutathione levels in vivo. Notably, weekly intravenous administration of NAC over 3 months in idiopathic PD patients revealed a significant clinical improvement which was paralleled by increased dopamine transporter binding during ioflupane imaging (DaTSCAN) [217].
Another consequence of mitochondrial dysfunction is a lowering of nicotinamide adenine dinucleotide (NAD) [255]. Mitochondrial CI metabolizes NADH to NAD+. Boosting cellular NAD levels by dietary supplements of the precursor nicotinamide (vitamin B3) has neuroprotective effects in some preclinical models of PD [220]. The recent phase I study NADPARK in which drug naïve de novo PD patients received 1000 mg of nicotinamide riboside over 30 days achieved some desired metabolic outcomes and a mild clinical benefit [223].
A related approach is based upon epidemiological studies showing reduced risk of developing PD when using antidiabetic drugs like exenatide or pioglitazone [256]. Both drugs have been studied intensively in preclinical animal models and clinical PD trials (Table 3). Exenatide appears to exert its neuroprotective effects by dampening neuroinflammatory pathways, reduction of ROS, lowering intracellular Ca2+ levels, restoring mitophagy, and improving overall bioenergetic efficiency [258]. In a randomized double-blind placebo-controlled trial on PD patients under symptomatic dopamine replacement therapy, 48 weeks of exenatide, slightly although significantly, improved motor symptoms [260]. Currently, a phase III trial [261] is investigating the effects of a two-year exenatide treatment on motor symptoms in PD patients, which are again also receiving symptomatic dopamine replacement therapy. In contrast, 52 weeks long treatment with liraglutide, also a glucagon-like peptide 1 (GLP-1) agonist, resulted in improvement of non-motor symptoms and activities of daily living while motor symptoms were unchanged [262]. While preclinical models suggest a mitochondria-based mechanism of action, there is no robust data from clinical studies regarding GLP-1 agonist’s cellular mechanism of action.
Pioglitazone, a peroxisome proliferator-activated receptor gamma (PPARγ) agonist, also has been considerably studied in PD. In animal studies, it reduced neuroinflammation, suppressed nitric oxide synthase activity, improved proteasomal clearance, and enhanced mitochondrial biogenesis [264, 279]. However, a phase II clinical trial in early PD patients found no clinical benefit of 44 weeks treatment with pioglitazone on disease progression [265].
As outlined above, mitochondria are highly dynamic organelles that form a complex network within the cell soma, axon and down to the synaptic buttons. Maintaining this network in a viable state relies on constant spatial redistribution via mitochondrial trafficking, as well as balanced mitochondrial fusion and fission, to keep a pool of healthy mitochondria at any time. However, many of these mitochondrial quality control processes appear to be disrupted in PD patients [67]. Based on that, several preclinical approaches have been developed to correct this putative defect in mitochondrial dynamics (Table 3). Inhibition of mitochondrial fission via the mitochondrial division inhibitor 1 (mdivi-1) has been reported to be neuroprotective in an aSYN overexpression rat model. Treatment with mdivi-1, reduced mitochondrial fragmentation and was simultaneously associated with reduced oxidative stress and improved mitochondrial health [267]. Further, accumulation of the mitochondrial adaptor protein Miro on the outer mitochondrial membrane has been identified in PD and linked to delayed mitophagy in experimental PD models [271]. Pharmacological reduction of Miro in cellular and PD Drosophila fly models was able to restore mitophagy and decrease neuronal cell loss [272].
Further, gene therapy approaches targeting PINK1 and Parkin deficiencies have been explored (Table 3). PINK1 overexpression not only ameliorated mitochondrial dysfunction resulting from prior induced PINK1 deficiency in PINK1 mutant Drosophila models [71, 280], but also was protective in an aSYN induced phenotype in aSYN transgenic Drosophila PD model [277], and protected against neuronal loss and mitochondrial dysfunction in in vitro and in vivo MPTP models [274]. Overexpression of parkin has similar effects [275, 276]. A protein-based therapy using a cell-permeable Parkin was protective in 6-hydroxydopamine (6-OHDA) and adeno-associated viral vector (AAV) mouse models, presumably by enhancing mitochondrial quality control via facilitating mitochondrial biogenesis, and promoting mitophagy [278]. It should be noted however that the predictive validity of both the 6-OHDA and MPTP models of PD is questionable, as all of the failed drugs have passed this test in preclinical work.
Limiting mitochondrial stimulation as a new therapeutic approach
As outlined above, most of the mitochondrially-targeted, disease-modifying strategies that have moved to clinical trials, or are in the planning stages, are aimed at either limiting the consequences of mitochondrial damage (e.g., CoQ10), enhancing the clearance of damaged mitochondria (e.g., Miro targeting) or blunting the inflammatory consequences of mitochondrial dysfunction (e.g., exenatide) [252, 253]. An alternative strategy is to first diminish mitochondrial damage. The mechanistic studies focusing on the origins of mitochondrial oxidant stress in at-risk neurons (like SNc dopaminergic neurons) point to their feedforward stimulation by plasma membrane L-type Ca2+ channels. Inhibiting L-type channels with dihydropyridine negative allosteric modulators lowered mitochondrial oxidant stress and mitophagy in at-risk dopaminergic neurons in animal models [145, 176]. They also diminished mitochondrial oxidant stress in a model of recessive PD [145], and showed neuroprotective effects in the MPTP and 6-OHDA models of PD [281, 282]. More importantly, epidemiological studies have shown that use of dihydropyridines is associated with a reduced risk of developing PD [283, 284]. These observations motivated two clinical trials with the dihydropyridine isradipine. Isradipine was chosen for these trials because it has the highest relative affinity for the sub-class of L-type channel thought to be the most important in driving mitochondrial stress in SNc dopaminergic neurons (channels with a pore-forming Cav1.3 subunit). While initial reports stated that there was no evidence of efficacy in modifying disease progression [285], a subsequent re-analysis reopened the discussion on an extended release formulation of isradipine, suggesting that there may be a disease modifying effect based on the UPDRS assessed progression in patients given 10 mg isradipine per day [286].
Current gaps in our understanding
Based on our current knowledge of mitochondrial dysfunction in PD, there are at least four major gaps in our understanding.
First, the chain of events arising from mitochondrial dysfunction needs to be more rigorously characterized. As in modern cancer treatment, this would allow combination therapies that maximize biological efficacy and minimize unwanted side-effects of treatment (see Tables 2 and 3).
Second, there need to be more objective, and quantitative measures of disease progression. The reliance upon highly variable clinical rating scales adds an enormous amount of noise to clinical trial outcomes and prevents modest disease-modifying effects to be resolved. These biomarkers should include ones that assess mitochondrial function and dysfunction [253]. Current strategies are mainly focused on improving neuroimaging of cellular bioenergetics (e.g., magnetic resonance spectroscopy). However, studies should also implement blood- or CSF-based biomarkers as recently demonstrated [223].
Third, we need to have a better understanding of the mitochondrial pathways leading to neurodegeneration in the different PD subtypes [287]. This could allow personalized disease-modification therapies and better target engagement.
Fourth, we need to know whether the mechanisms driving disease progression in PD are time invariant or not. It could be that mitochondrial dysfunction is important in the early stages of PD pathogenesis, but not in later stages. For example, the later stages of cell loss in PD could be driven by network dysfunction caused by less than complete disruption of at-risk neuron function. A clear understanding of these mechanisms would allow disease-modifying treatments to be tailored to the respective disease stage.
Conclusions
Mitochondrial dysfunction is a core hallmark of PD. Preclinical, epidemiological, histopathological, and clinical trial data point towards mitochondrial dysfunction as being a significant disease driving factor in idiopathic and familial PD. On the cellular level, core features are CI impairment, increased oxidative stress, disturbed mitochondrial quality control mechanisms, and bioenergetic deficiency. Current experimental evidence indicates that there are several mitochondrial pathways that contribute to PD pathogenesis. Targeting more than one of these pathways at the same time may be a more effective strategy than trying to affect just one. Moreover, given that the pathology in PD is largely in the brain, drug delivery strategies that optimize brain delivery and target engagement need to be pursued. So, while no treatment has been unequivocally shown to slow disease progression in the early stage of PD, there remains optimism that this situation will change soon.
Availability of data and materials
N/A.
Abbreviations
- 6-OHDA:
-
6-hydroxydopamine
- AAV:
-
Adeno-associated viral vector
- aSYN:
-
Alpha-synuclein
- ATP:
-
Adenosine triphosphate
- ATP13A2:
-
ATPase type 13A2
- CI:
-
Mitochondrial complex I
- CII:
-
Mitochondrial complex II
- CIII:
-
Mitochondrial complex III
- CIV:
-
Mitochondrial complex IV
- CHCHD2:
-
Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing protein 2
- CoQ10:
-
Coenzyme Q10
- DA:
-
Dopamine
- DaTSCAN:
-
Dopamine transporter ioflupane imaging
- DLP1:
-
Dynamin-1-like protein
- DJ1:
-
Protein deglycase DJ1
- ER:
-
Endoplasmic reticulum
- ETC:
-
Electron transport chain
- FBXO7:
-
F-box protein 7
- GBA1:
-
β-glucocerebrosidase
- GLP-1:
-
Glucagon-like peptide 1
- HSP31:
-
Heat shock protein 31
- IPSCs:
-
Induced pluripotent stem cells
- L-DOPA:
-
Levodopa
- LP:
-
Lewy pathology
- LRRK2:
-
Leucine-rich repeat kinase 2
- mdivi-1:
-
Mitochondrial division inhibitor 1
- MPP+ :
-
1-methyl-4-phenylpyridinium
- MPPP:
-
1-methyl-4-phenyl-4-propionoxy-piperidine
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- mtDNA:
-
Mitochondrial DNA
- NAC:
-
N-Acetyl cysteine
- NAD:
-
Nicotinamide adenine dinucleotide
- OXPHOS:
-
Oxidative phosphorylation
- PARK:
-
Familial PD genes
- Parkin:
-
E3-Ubiquitin-protein-Ligase Parkin
- PD:
-
Parkinson’s disease
- PINK1:
-
PTEN-induced kinase-1
- PPARγ:
-
Peroxisome proliferator-activated receptor gamma
- POLG:
-
Gene coding for DNA polymerase gamma
- ROS:
-
Reactive oxygen species
- SNc:
-
Substantia nigra pars compacta
- SNCA:
-
Gene coding for alpha-synuclein
- TOM:
-
Translocase of outer mitochondrial membrane
- UDCA:
-
Ursodeoxycholic acid
- VPS35:
-
Vacuolar protein sorting 35
- VPS13C:
-
Vacuolar protein sorting 13 homolog C
References
Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet. 2021;397:2284–303.
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W, et al. MDS clinical diagnostic criteria for Parkinson's disease. Mov Disord. 2015;30:1591–601.
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, et al. Parkinson disease. Nat Rev Dis Primers. 2017;3:17013.
Chaudhuri KR, Healy DG, Schapira AH. National Institute for clinical E: non-motor symptoms of Parkinson's disease: diagnosis and management. Lancet Neurol. 2006;5:235–45.
Surmeier DJ, Obeso JA, Halliday GM. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 2017;18:101–13.
Koga S, Sekiya H, Kondru N, Ross OA, Dickson DW. Neuropathology and molecular diagnosis of Synucleinopathies. Mol Neurodegener. 2021;16:83.
Beach TG, Adler CH, Sue LI, Vedders L, Lue L, White Iii CL, et al. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010;119:689–702.
Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging. 2003;24:197–211.
Doppler K, Jentschke HM, Schulmeyer L, Vadasz D, Janzen A, Luster M, et al. Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson's disease. Acta Neuropathol. 2017;133:535–45.
Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, et al. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science. 2012;338:949–53.
Henderson MX, Henrich MT, Geibl FF, Oertel WH, Brundin P, Surmeier DJ. The roles of connectivity and neuronal phenotype in determining the pattern of alpha-synuclein pathology in Parkinson's disease. Neurobiol Dis. 2022;168:105687.
Giguere N, Burke Nanni S, Trudeau LE. On cell loss and selective vulnerability of neuronal populations in Parkinson's disease. Front Neurol. 2018;9:455.
Kalia LV, Lang AE. Parkinson's disease. Lancet. 2015;386:896–912.
Hirsch EC, Standaert DG. Ten unsolved questions about Neuroinflammation in Parkinson's disease. Mov Disord. 2021;36:16–24.
Sahoo S, Padhy AA, Kumari V, Mishra P. Role of ubiquitin-proteasome and autophagy-lysosome pathways in alpha-Synuclein aggregate clearance. Mol Neurobiol. 2022;59:5379–407.
Wong YC, Luk K, Purtell K, Burke Nanni S, Stoessl AJ, Trudeau LE, et al. Neuronal vulnerability in Parkinson disease: should the focus be on axons and synaptic terminals? Mov Disord. 2019;34:1406–22.
Yellen G. Fueling thought: management of glycolysis and oxidative phosphorylation in neuronal metabolism. J Cell Biol. 2018;217:2235–46.
Spinelli JB, Haigis MC. The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol. 2018;20:745–54.
Chakrabarty RP, Chandel NS. Mitochondria as signaling organelles control mammalian stem cell fate. Cell Stem Cell. 2021;28:394–408.
Matsuda W, Furuta T, Nakamura KC, Hioki H, Fujiyama F, Arai R, et al. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J Neurosci. 2009;29:444–53.
Area-Gomez E, Guardia-Laguarta C, Schon EA, Przedborski S. Mitochondria, OxPhos, and neurodegeneration: cells are not just running out of gas. J Clin Invest. 2019;129:34–45.
Lewis TL Jr, Kwon SK, Lee A, Shaw R, Polleux F. MFF-dependent mitochondrial fission regulates presynaptic release and axon branching by limiting axonal mitochondria size. Nat Commun. 2018;9:5008.
Bolam JP, Pissadaki EK. Living on the edge with too many mouths to feed: why dopamine neurons die. Mov Disord. 2012;27:1478–83.
Pacelli C, Giguere N, Bourque MJ, Levesque M, Slack RS, Trudeau LE. Elevated mitochondrial bioenergetics and axonal Arborization size are key contributors to the vulnerability of dopamine neurons. Curr Biol. 2015;25:2349–60.
Martinez-Gonzalez C, Bolam JP, Mena-Segovia J. Topographical organization of the pedunculopontine nucleus. Front Neuroanat. 2011;5:22.
Aston-Jones G, Waterhouse B. Locus coeruleus: from global projection system to adaptive regulation of behavior. Brain Res. 2016;1645:75–8.
Guzman JN, Sanchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–9.
Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, et al. Rejuvenation' protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–6.
Surmeier DJ, Guzman JN, Sanchez J, Schumacker PT. Physiological phenotype and vulnerability in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009290.
Sanchez-Padilla J, Guzman JN, Ilijic E, Kondapalli J, Galtieri DJ, Yang B, et al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat Neurosci. 2014;17:832–40.
Goldberg JA, Guzman JN, Estep CM, Ilijic E, Kondapalli J, Sanchez-Padilla J, et al. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson's disease. Nat Neurosci. 2012;15:1414–21.
Zampese E, Wokosin DL, Gonzalez-Rodriguez P, Guzman JN, Tkatch T, Kondapalli J, et al. Ca(2+) channels couple spiking to mitochondrial metabolism in substantia nigra dopaminergic neurons. Sci Adv. 2022;8:eabp8701.
Angelova PR, Abramov AY. Role of mitochondrial ROS in the brain: from physiology to neurodegeneration. FEBS Lett. 2018;592:692–702.
Langston JW, Ballard P, Tetrud JW, Irwin I. Chronic parkinsonism in humans due to a product of meperidine-analog synthesis. Science. 1983;219:979–80.
Dauer W, Przedborski S. Parkinson's disease: mechanisms and models. Neuron. 2003;39:889–909.
Desai VG, Feuers RJ, Hart RW, Ali SF. MPP(+)-induced neurotoxicity in mouse is age-dependent: evidenced by the selective inhibition of complexes of electron transport. Brain Res. 1996;715:1–8.
Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503–8.
Burns RS, Chiueh CC, Markey SP, Ebert MH, Jacobowitz DM, Kopin IJ. A primate model of parkinsonism: selective destruction of dopaminergic neurons in the pars compacta of the substantia nigra by N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A. 1983;80:4546–50.
Meredith GE, Rademacher DJ. MPTP mouse models of Parkinson's disease: an update. J Parkinsons Dis. 2011;1:19–33.
Hoglinger GU, Feger J, Prigent A, Michel PP, Parain K, Champy P, et al. Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J Neurochem. 2003;84:491–502.
Pan-Montojo F, Schwarz M, Winkler C, Arnhold M, O'Sullivan GA, Pal A, et al. Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep. 2012;2:898.
Martinez TN, Greenamyre JT. Toxin models of mitochondrial dysfunction in Parkinson's disease. Antioxid Redox Signal. 2012;16:920–34.
Gonzalez-Rodriguez P, Zampese E, Stout KA, Guzman JN, Ilijic E, Yang B, et al. Disruption of mitochondrial complex I induces progressive parkinsonism. Nature. 2021;599:650–6.
Schapira AH, Cooper JM, Dexter D, Jenner P, Clark JB, Marsden CD. Mitochondrial complex I deficiency in Parkinson's disease. Lancet. 1989;1:1269.
Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD. Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease. Brain. 1992;115(Pt 2):333–42.
Janetzky B, Hauck S, Youdim MB, Riederer P, Jellinger K, Pantucek F, et al. Unaltered aconitase activity, but decreased complex I activity in substantia nigra pars compacta of patients with Parkinson's disease. Neurosci Lett. 1994;169:126–8.
Subrahmanian N, LaVoie MJ. Is there a special relationship between complex I activity and nigral neuronal loss in Parkinson's disease? A critical reappraisal. Brain Res. 2021;1767:147434.
Flones IH, Fernandez-Vizarra E, Lykouri M, Brakedal B, Skeie GO, Miletic H, et al. Neuronal complex I deficiency occurs throughout the Parkinson's disease brain, but is not associated with neurodegeneration or mitochondrial DNA damage. Acta Neuropathol. 2018;135:409–25.
Mizuno Y, Ohta S, Tanaka M, Takamiya S, Suzuki K, Sato T, et al. Deficiencies in complex I subunits of the respiratory chain in Parkinson's disease. Biochem Biophys Res Commun. 1989;163:1450–5.
Hattori N, Tanaka M, Ozawa T, Mizuno Y. Immunohistochemical studies on complexes I, II, III, and IV of mitochondria in Parkinson's disease. Ann Neurol. 1991;30:563–71.
Ozawa T, Tanaka M, Ikebe S, Ohno K, Kondo T, Mizuno Y. Quantitative determination of deleted mitochondrial DNA relative to normal DNA in parkinsonian striatum by a kinetic PCR analysis. Biochem Biophys Res Commun. 1990;172:483–9.
Lestienne P, Nelson J, Riederer P, Jellinger K, Reichmann H. Normal mitochondrial genome in brain from patients with Parkinson's disease and complex I defect. J Neurochem. 1990;55:1810–2.
Mann VM, Cooper JM, Schapira AH. Quantitation of a mitochondrial DNA deletion in Parkinson's disease. FEBS Lett. 1992;299:218–22.
Gu G, Reyes PE, Golden GT, Woltjer RL, Hulette C, Montine TJ, et al. Mitochondrial DNA deletions/rearrangements in parkinson disease and related neurodegenerative disorders. J Neuropathol Exp Neurol. 2002;61:634–9.
Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. 2006;38:515–7.
Dolle C, Flones I, Nido GS, Miletic H, Osuagwu N, Kristoffersen S, et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat Commun. 2016;7:13548.
Rahman S, Copeland WC. POLG-related disorders and their neurological manifestations. Nat Rev Neurol. 2019;15:40–52.
Sulzer D, Edwards RH. The physiological role of alpha-synuclein and its relationship to Parkinson's disease. J Neurochem. 2019;150:475–86.
Devi L, Raghavendran V, Prabhu BM, Avadhani NG, Anandatheerthavarada HK. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J Biol Chem. 2008;283:9089–100.
Li L, Nadanaciva S, Berger Z, Shen W, Paumier K, Schwartz J, et al. Human A53T alpha-synuclein causes reversible deficits in mitochondrial function and dynamics in primary mouse cortical neurons. PLoS One. 2013;8:e85815.
Arias-Fuenzalida J, Jarazo J, Qing X, Walter J, Gomez-Giro G, Nickels SL, et al. FACS-assisted CRISPR-Cas9 genome editing facilitates Parkinson's disease modeling. Stem Cell Rep. 2017;9:1423–31.
Subramaniam SR, Vergnes L, Franich NR, Reue K, Chesselet MF. Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol Dis. 2014;70:204–13.
Tapias V, Hu X, Luk KC, Sanders LH, Lee VM, Greenamyre JT. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell Mol Life Sci. 2017;74:2851–74.
Ludtmann MH, Angelova PR, Ninkina NN, Gandhi S, Buchman VL, Abramov AY. Monomeric alpha-Synuclein exerts a physiological role on brain ATP synthase. J Neurosci. 2016;36:10510–21.
Ellis CE, Murphy EJ, Mitchell DC, Golovko MY, Scaglia F, Barcelo-Coblijn GC, et al. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol Cell Biol. 2005;25:10190–201.
Hsu LJ, Sagara Y, Arroyo A, Rockenstein E, Sisk A, Mallory M, et al. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am J Pathol. 2000;157:401–10.
Ge P, Dawson VL, Dawson TM. PINK1 and Parkin mitochondrial quality control: a source of regional vulnerability in Parkinson's disease. Mol Neurodegener. 2020;15:20.
Kamienieva I, Duszynski J, Szczepanowska J. Multitasking guardian of mitochondrial quality: Parkin function and Parkinson's disease. Transl Neurodegener. 2021;10:5.
Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson's disease. Neuron. 2015;85:257–73.
Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015;524:309–14.
Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, et al. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature. 2006;441:1162–6.
Gautier CA, Kitada T, Shen J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc Natl Acad Sci U S A. 2008;105:11364–9.
Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, et al. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–38.
Thomas KJ, McCoy MK, Blackinton J, Beilina A, van der Brug M, Sandebring A, et al. DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet. 2011;20:40–50.
Liu Y, Ma X, Fujioka H, Liu J, Chen S, Zhu X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc Natl Acad Sci U S A. 2019;116:25322–8.
Shtifman A, Zhong N, Lopez JR, Shen J, Xu J. Altered Ca2+ homeostasis in the skeletal muscle of DJ-1 null mice. Neurobiol Aging. 2011;32:125–32.
Taipa R, Pereira C, Reis I, Alonso I, Bastos-Lima A, Melo-Pires M, et al. DJ-1 linked parkinsonism (PARK7) is associated with Lewy body pathology. Brain. 2016;139:1680–7.
Irrcher I, Aleyasin H, Seifert EL, Hewitt SJ, Chhabra S, Phillips M, et al. Loss of the Parkinson's disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum Mol Genet. 2010;19:3734–46.
Wang X, Petrie TG, Liu Y, Liu J, Fujioka H, Zhu X. Parkinson's disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J Neurochem. 2012;121:830–9.
Giaime E, Yamaguchi H, Gautier CA, Kitada T, Shen J. Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLoS One. 2012;7:e40501.
Kuhlmann N, Milnerwood AJ. A critical LRRK at the synapse? The neurobiological function and pathophysiological dysfunction of LRRK2. Front Mol Neurosci. 2020;13:153.
Bonet-Ponce L, Cookson MR. LRRK2 recruitment, activity, and function in organelles. FEBS J. 2022;289:6871–90.
Rocha EM, Keeney MT, Di Maio R, De Miranda BR, Greenamyre JT. LRRK2 and idiopathic Parkinson's disease. Trends Neurosci. 2022;45:224–36.
Tolosa E, Vila M, Klein C, Rascol O. LRRK2 in Parkinson disease: challenges of clinical trials. Nat Rev Neurol. 2020;16:97–107.
Heo HY, Park JM, Kim CH, Han BS, Kim KS, Seol W. LRRK2 enhances oxidative stress-induced neurotoxicity via its kinase activity. Exp Cell Res. 2010;316:649–56.
Singh A, Zhi L, Zhang H. LRRK2 and mitochondria: recent advances and current views. Brain Res. 2019;1702:96–104.
Xiao B, Deng X, Ng EY, Allen JC Jr, Lim SY, Ahmad-Annuar A, et al. Association of LRRK2 haplotype with age at onset in Parkinson disease. JAMA Neurol. 2018;75:127–8.
Luth T, Konig IR, Grunewald A, Kasten M, Klein C, Hentati F, et al. Age at onset of LRRK2 p.Gly2019Ser is related to environmental and lifestyle factors. Mov Disord. 2020;35:1854–8.
van Veen S, Martin S, Van den Haute C, Benoy V, Lyons J, Vanhoutte R, et al. ATP13A2 deficiency disrupts lysosomal polyamine export. Nature. 2020;578:419–24.
Fujii T, Nagamori S, Wiriyasermkul P, Zheng S, Yago A, Shimizu T, Tabuchi Y, Okumura T, Fujii T, Takeshima H, Sakai H. Parkinson’s disease-associated ATP13A2/PARK9 functions as a lysosomal H(+),K(+)-ATPase. Nat Commun. 2023;14:2174.
Ramirez A, Heimbach A, Grundemann J, Stiller B, Hampshire D, Cid LP, et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet. 2006;38:1184–91.
Vrijsen S, Besora-Casals L, van Veen S, Zielich J, Van den Haute C, Hamouda NN, et al. ATP13A2-mediated endo-lysosomal polyamine export counters mitochondrial oxidative stress. Proc Natl Acad Sci U S A. 2020;117:31198–207.
Dehay B, Ramirez A, Martinez-Vicente M, Perier C, Canron MH, Doudnikoff E, et al. Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A. 2012;109:9611–6.
Joseph S, Schulz JB, Stegmuller J. Mechanistic contributions of FBXO7 to Parkinson disease. J Neurochem. 2018;144:118–27.
Burchell VS, Nelson DE, Sanchez-Martinez A, Delgado-Camprubi M, Ivatt RM, Pogson JH, et al. The Parkinson's disease-linked proteins Fbxo7 and Parkin interact to mediate mitophagy. Nat Neurosci. 2013;16:1257–65.
Zhou ZD, Xie SP, Sathiyamoorthy S, Saw WT, Sing TY, Ng SH, et al. F-box protein 7 mutations promote protein aggregation in mitochondria and inhibit mitophagy. Hum Mol Genet. 2015;24:6314–30.
Delgado-Camprubi M, Esteras N, Soutar MP, Plun-Favreau H, Abramov AY. Deficiency of Parkinson's disease-related gene Fbxo7 is associated with impaired mitochondrial metabolism by PARP activation. Cell Death Differ. 2017;24:2210.
Sassone J, Reale C, Dati G, Regoni M, Pellecchia MT, Garavaglia B. The role of VPS35 in the pathobiology of Parkinson's disease. Cell Mol Neurobiol. 2021;41:199–227.
Williams ET, Chen X, Otero PA, Moore DJ. Understanding the contributions of VPS35 and the retromer in neurodegenerative disease. Neurobiol Dis. 2022;170:105768.
Lesage S, Drouet V, Majounie E, Deramecourt V, Jacoupy M, Nicolas A, et al. Loss of VPS13C function in autosomal-recessive parkinsonism causes mitochondrial dysfunction and increases PINK1/Parkin-dependent Mitophagy. Am J Hum Genet. 2016;98:500–13.
Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, et al. Parkinson's disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med. 2016;22:54–63.
Tang FL, Liu W, Hu JX, Erion JR, Ye J, Mei L, et al. VPS35 deficiency or mutation causes dopaminergic neuronal loss by impairing mitochondrial fusion and function. Cell Rep. 2015;12:1631–43.
Zhou L, Wang W, Hoppel C, Liu J, Zhu X. Parkinson's disease-associated pathogenic VPS35 mutation causes complex I deficits. Biochim Biophys Acta Mol basis Dis. 2017;1863:2791–5.
Ruan Y, Hu J, Che Y, Liu Y, Luo Z, Cheng J, et al. CHCHD2 and CHCHD10 regulate mitochondrial dynamics and integrated stress response. Cell Death Dis. 2022;13:156.
Kee TR, Espinoza Gonzalez P, Wehinger JL, Bukhari MZ, Ermekbaeva A, Sista A, et al. Mitochondrial CHCHD2: disease-associated mutations, physiological functions, and current animal models. Front Aging Neurosci. 2021;13:660843.
Ikeda A, Nishioka K, Meng H, Takanashi M, Hasegawa I, Inoshita T, et al. Mutations in CHCHD2 cause alpha-synuclein aggregation. Hum Mol Genet. 2019;28:3895–911.
Liu YT, Huang X, Nguyen D, Shammas MK, Wu BP, Dombi E, et al. Loss of CHCHD2 and CHCHD10 activates OMA1 peptidase to disrupt mitochondrial cristae phenocopying patient mutations. Hum Mol Genet. 2020;29:1547–67.
Sato S, Noda S, Torii S, Amo T, Ikeda A, Funayama M, et al. Homeostatic p62 levels and inclusion body formation in CHCHD2 knockout mice. Hum Mol Genet. 2021;30:443–53.
Kumar N, Leonzino M, Hancock-Cerutti W, Horenkamp FA, Li P, Lees JA, et al. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J Cell Biol. 2018;217:3625–39.
Hancock-Cerutti W, Wu Z, Xu P, Yadavalli N, Leonzino M, Tharkeshwar AK, et al. ER-lysosome lipid transfer protein VPS13C/PARK23 prevents aberrant mtDNA-dependent STING signaling. J Cell Biol. 2022;221(7):e202106046
Samaranch L, Lorenzo-Betancor O, Arbelo JM, Ferrer I, Lorenzo E, Irigoyen J, et al. PINK1-linked parkinsonism is associated with Lewy body pathology. Brain. 2010;133:1128–42.
Pramstaller PP, Schlossmacher MG, Jacques TS, Scaravilli F, Eskelson C, Pepivani I, et al. Lewy body Parkinson's disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol. 2005;58:411–22.
Steele JC, Guella I, Szu-Tu C, Lin MK, Thompson C, Evans DM, et al. Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol. 2015;77:458–68.
Takanashi M, Li Y, Hattori N. Absence of Lewy pathology associated with PINK1 homozygous mutation. Neurology. 2016;86:2212–3.
Houlden H, Singleton AB. The genetics and neuropathology of Parkinson's disease. Acta Neuropathol. 2012;124:325–38.
Schneider SA, Alcalay RN. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov Disord. 2017;32:1504–23.
Deng H, Dodson MW, Huang H, Guo M. The Parkinson's disease genes pink1 and parkin promote mitochondrial fission and/or inhibit fusion in Drosophila. Proc Natl Acad Sci U S A. 2008;105:14503–8.
Yang Y, Ouyang Y, Yang L, Beal MF, McQuibban A, Vogel H, et al. Pink1 regulates mitochondrial dynamics through interaction with the fission/fusion machinery. Proc Natl Acad Sci U S A. 2008;105:7070–5.
Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, et al. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol. 2012;22:135–41.
McLelland GL, Lee SA, McBride HM, Fon EA. Syntaxin-17 delivers PINK1/parkin-dependent mitochondrial vesicles to the endolysosomal system. J Cell Biol. 2016;214:275–91.
Kumar M, Acevedo-Cintron J, Jhaldiyal A, Wang H, Andrabi SA, Eacker S, et al. Defects in mitochondrial biogenesis drive mitochondrial alterations in PARKIN-deficient human dopamine neurons. Stem Cell Rep. 2020;15:629–45.
Abramov AY, Smulders-Srinivasan TK, Kirby DM, Acin-Perez R, Enriquez JA, Lightowlers RN, et al. Mechanism of neurodegeneration of neurons with mitochondrial DNA mutations. Brain. 2010;133:797–807.
Rakovic A, Grunewald A, Seibler P, Ramirez A, Kock N, Orolicki S, et al. Effect of endogenous mutant and wild-type PINK1 on Parkin in fibroblasts from Parkinson disease patients. Hum Mol Genet. 2010;19:3124–37.
Mortiboys H, Thomas KJ, Koopman WJ, Klaffke S, Abou-Sleiman P, Olpin S, et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann Neurol. 2008;64:555–65.
Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, et al. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–22.
Periquet M, Corti O, Jacquier S, Brice A. Proteomic analysis of parkin knockout mice: alterations in energy metabolism, protein handling and synaptic function. J Neurochem. 2005;95:1259–76.
Stichel CC, Zhu XR, Bader V, Linnartz B, Schmidt S, Lubbert H. Mono- and double-mutant mouse models of Parkinson's disease display severe mitochondrial damage. Hum Mol Genet. 2007;16:2377–93.
Gautier CA, Giaime E, Caballero E, Nunez L, Song Z, Chan D, et al. Regulation of mitochondrial permeability transition pore by PINK1. Mol Neurodegener. 2012;7:22.
Heeman B, Van den Haute C, Aelvoet SA, Valsecchi F, Rodenburg RJ, Reumers V, et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J Cell Sci. 2011;124:1115–25.
Morais VA, Verstreken P, Roethig A, Smet J, Snellinx A, Vanbrabant M, et al. Parkinson's disease mutations in PINK1 result in decreased complex I activity and deficient synaptic function. EMBO Mol Med. 2009;1:99–111.
Akundi RS, Huang Z, Eason J, Pandya JD, Zhi L, Cass WA, et al. Increased mitochondrial calcium sensitivity and abnormal expression of innate immunity genes precede dopaminergic defects in Pink1-deficient mice. PLoS One. 2011;6:e16038.
Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–83.
Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature. 2006;441:1157–61.
Whitworth AJ, Theodore DA, Greene JC, Benes H, Wes PD, Pallanck LJ. Increased glutathione S-transferase activity rescues dopaminergic neuron loss in a Drosophila model of Parkinson's disease. Proc Natl Acad Sci U S A. 2005;102:8024–9.
Gao Q, Tian R, Han H, Slone J, Wang C, Ke X, et al. PINK1-mediated Drp1(S616) phosphorylation modulates synaptic development and plasticity via promoting mitochondrial fission. Signal Transduct Target Ther. 2022;7:103.
Cui M, Tang X, Christian WV, Yoon Y, Tieu K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J Biol Chem. 2010;285:11740–52.
Schlossmacher MG, Frosch MP, Gai WP, Medina M, Sharma N, Forno L, et al. Parkin localizes to the Lewy bodies of Parkinson disease and dementia with Lewy bodies. Am J Pathol. 2002;160:1655–67.
LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–21.
Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, et al. S-nitrosylation of parkin regulates ubiquitination and compromises parkin's protective function. Science. 2004;304:1328–31.
Sunico CR, Nakamura T, Rockenstein E, Mante M, Adame A, Chan SF, et al. S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson's disease. Mol Neurodegener. 2013;8:29.
Tokarew JM, El-Kodsi DN, Lengacher NA, Fehr TK, Nguyen AP, Shutinoski B, et al. Age-associated insolubility of parkin in human midbrain is linked to redox balance and sequestration of reactive dopamine metabolites. Acta Neuropathol. 2021;141:725–54.
Bonifati V, Rizzu P, van Baren MJ, Schaap O, Breedveld GJ, Krieger E, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science. 2003;299:256–9.
Krebiehl G, Ruckerbauer S, Burbulla LF, Kieper N, Maurer B, Waak J, et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson's disease-associated protein DJ-1. PLoS One. 2010;5:e9367.
Andres-Mateos E, Perier C, Zhang L, Blanchard-Fillion B, Greco TM, Thomas B, et al. DJ-1 gene deletion reveals that DJ-1 is an atypical peroxiredoxin-like peroxidase. Proc Natl Acad Sci U S A. 2007;104:14807–12.
Guzman JN, Sanchez-Padilla J, Wokosin D, Kondapalli J, Ilijic E, Schumacker PT, et al. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature. 2010;468:696–700.
Queliconi BB, Kojima W, Kimura M, Imai K, Udagawa C, Motono C, et al. Unfolding is the driving force for mitochondrial import and degradation of the Parkinson's disease-related protein DJ-1. J Cell Sci. 2021;134(22):jcs258653.
Kim RH, Peters M, Jang Y, Shi W, Pintilie M, Fletcher GC, et al. DJ-1, a novel regulator of the tumor suppressor PTEN. Cancer Cell. 2005;7:263–73.
Bahmed K, Messier EM, Zhou W, Tuder RM, Freed CR, Chu HW, et al. DJ-1 modulates nuclear erythroid 2-related Factor-2-mediated protection in human primary alveolar type II cells in smokers. Am J Respir Cell Mol Biol. 2016;55:439–49.
Cao J, Ying M, Xie N, Lin G, Dong R, Zhang J, et al. The oxidation states of DJ-1 dictate the cell fate in response to oxidative stress triggered by 4-hpr: autophagy or apoptosis? Antioxid Redox Signal. 2014;21:1443–59.
Lee SJ, Kim SJ, Kim IK, Ko J, Jeong CS, Kim GH, et al. Crystal structures of human DJ-1 and Escherichia coli Hsp31, which share an evolutionarily conserved domain. J Biol Chem. 2003;278:44552–9.
Shendelman S, Jonason A, Martinat C, Leete T, Abeliovich A. DJ-1 is a redox-dependent molecular chaperone that inhibits alpha-synuclein aggregate formation. PLoS Biol. 2004;2:e362.
Zondler L, Miller-Fleming L, Repici M, Goncalves S, Tenreiro S, Rosado-Ramos R, et al. DJ-1 interactions with alpha-synuclein attenuate aggregation and cellular toxicity in models of Parkinson's disease. Cell Death Dis. 2014;5:e1350.
Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson's disease. Science. 2017;357:1255–61.
Richarme G, Liu C, Mihoub M, Abdallah J, Leger T, Joly N, et al. Guanine glycation repair by DJ-1/Park7 and its bacterial homologs. Science. 2017;357:208–11.
Mazza MC, Shuck SC, Lin J, Moxley MA, Termini J, Cookson MR, et al. DJ-1 is not a deglycase and makes a modest contribution to cellular defense against methylglyoxal damage in neurons. J Neurochem. 2022;162:245–61.
Vilarino-Guell C, Wider C, Ross OA, Dachsel JC, Kachergus JM, Lincoln SJ, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet. 2011;89:162–7.
Zimprich A, Benet-Pages A, Struhal W, Graf E, Eck SH, Offman MN, et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet. 2011;89:168–75.
Wang W, Ma X, Zhou L, Liu J, Zhu X. A conserved retromer sorting motif is essential for mitochondrial DLP1 recycling by VPS35 in Parkinson's disease model. Hum Mol Genet. 2017;26:781–9.
Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ. Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+−induced mitochondrial dysfunction. J Biol Chem. 2014;289:21490–507.
Chinta SJ, Mallajosyula JK, Rane A, Andersen JK. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci Lett. 2010;486:235–9.
Loeb V, Yakunin E, Saada A, Sharon R. The transgenic overexpression of alpha-synuclein and not its related pathology associates with complex I inhibition. J Biol Chem. 2010;285:7334–43.
Reeve AK, Ludtmann MH, Angelova PR, Simcox EM, Horrocks MH, Klenerman D, et al. Aggregated alpha-synuclein and complex I deficiency: exploration of their relationship in differentiated neurons. Cell Death Dis. 2015;6:e1820.
Liu G, Zhang C, Yin J, Li X, Cheng F, Li Y, et al. Alpha-Synuclein is differentially expressed in mitochondria from different rat brain regions and dose-dependently down-regulates complex I activity. Neurosci Lett. 2009;454:187–92.
Klivenyi P, Siwek D, Gardian G, Yang L, Starkov A, Cleren C, et al. Mice lacking alpha-synuclein are resistant to mitochondrial toxins. Neurobiol Dis. 2006;21:541–8.
Song DD, Shults CW, Sisk A, Rockenstein E, Masliah E. Enhanced substantia nigra mitochondrial pathology in human alpha-synuclein transgenic mice after treatment with MPTP. Exp Neurol. 2004;186:158–72.
Song LK, Ma KL, Yuan YH, Mu Z, Song XY, Niu F, et al. Targeted overexpression of alpha-Synuclein by rAAV2/1 vectors induces progressive Nigrostriatal degeneration and increases vulnerability to MPTP in mouse. PLoS One. 2015;10:e0131281.
Cannon JR, Geghman KD, Tapias V, Sew T, Dail MK, Li C, et al. Expression of human E46K-mutated alpha-synuclein in BAC-transgenic rats replicates early-stage Parkinson's disease features and enhances vulnerability to mitochondrial impairment. Exp Neurol. 2013;240:44–56.
Shavali S, Brown-Borg HM, Ebadi M, Porter J. Mitochondrial localization of alpha-synuclein protein in alpha-synuclein overexpressing cells. Neurosci Lett. 2008;439:125–8.
Ludtmann MHR, Angelova PR, Horrocks MH, Choi ML, Rodrigues M, Baev AY, et al. Alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson's disease. Nat Commun. 2018;9:2293.
Di Maio R, Barrett PJ, Hoffman EK, Barrett CW, Zharikov A, Borah A, et al. Alpha-Synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson's disease. Sci Transl Med. 2016;8:342ra378.
Martinez JH, Fuentes F, Vanasco V, Alvarez S, Alaimo A, Cassina A, et al. Alpha-synuclein mitochondrial interaction leads to irreversible translocation and complex I impairment. Arch Biochem Biophys. 2018;651:1–12.
Paillusson S, Gomez-Suaga P, Stoica R, Little D, Gissen P, Devine MJ, et al. Alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017;134:129–49.
Erustes AG, D'Eletto M, Guarache GC, Ureshino RP, Bincoletto C, da Silva Pereira GJ, et al. Overexpression of alpha-synuclein inhibits mitochondrial ca(2+) trafficking between the endoplasmic reticulum and mitochondria through MAMs by altering the GRP75-IP3R interaction. J Neurosci Res. 2021;99:2932–47.
Bose A, Beal MF. Mitochondrial dysfunction in Parkinson's disease. J Neurochem. 2016;139(Suppl 1):216–31.
Nicoletti V, Palermo G, Del Prete E, Mancuso M, Ceravolo R. Understanding the multiple role of mitochondria in Parkinson's disease and related disorders: lesson from genetics and protein-interaction network. Front Cell Dev Biol. 2021;9:636506.
Guzman JN, Ilijic E, Yang B, Sanchez-Padilla J, Wokosin D, Galtieri D, et al. Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J Clin Invest. 2018;128:2266–80.
Perfeito R, Lazaro DF, Outeiro TF, Rego AC. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol Cell Neurosci. 2014;62:51–9.
Kam TI, Mao X, Park H, Chou SC, Karuppagounder SS, Umanah GE, et al. Poly(ADP-ribose) drives pathologic alpha-synuclein neurodegeneration in Parkinson's disease. Science. 2018;362(6414):eaat8407.
Oka T, Hikoso S, Yamaguchi O, Taneike M, Takeda T, Tamai T, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature. 2012;485:251–5.
West AP, Khoury-Hanold W, Staron M, Tal MC, Pineda CM, Lang SM, et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–7.
Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, et al. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–62.
Matheoud D, Sugiura A, Bellemare-Pelletier A, Laplante A, Rondeau C, Chemali M, et al. Parkinson's disease-related proteins PINK1 and Parkin repress mitochondrial antigen presentation. Cell. 2016;166:314–27.
Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, et al. Intestinal infection triggers Parkinson's disease-like symptoms in Pink1(−/−) mice. Nature. 2019;571:565–9.
Nath S, Goodwin J, Engelborghs Y, Pountney DL. Raised calcium promotes alpha-synuclein aggregate formation. Mol Cell Neurosci. 2011;46:516–26.
Han JY, Choi TS, Kim HI. Molecular role of ca(2+) and hard divalent metal cations on accelerated fibrillation and Interfibrillar aggregation of alpha-Synuclein. Sci Rep. 1895;2018:8.
McNaught KS, Belizaire R, Isacson O, Jenner P, Olanow CW. Altered proteasomal function in sporadic Parkinson's disease. Exp Neurol. 2003;179:38–46.
Bukhatwa S, Zeng BY, Rose S, Jenner P. A comparison of changes in proteasomal subunit expression in the substantia nigra in Parkinson's disease, multiple system atrophy and progressive supranuclear palsy. Brain Res. 2010;1326:174–83.
Kim S, Wong YC, Gao F, Krainc D. Dysregulation of mitochondria-lysosome contacts by GBA1 dysfunction in dopaminergic neuronal models of Parkinson's disease. Nat Commun. 1807;2021:12.
Hargreaves IP, Lane A, Sleiman PM. The coenzyme Q10 status of the brain regions of Parkinson's disease patients. Neurosci Lett. 2008;447:17–9.
Younes-Mhenni S, Frih-Ayed M, Kerkeni A, Bost M, Chazot G. Peripheral blood markers of oxidative stress in Parkinson's disease. Eur Neurol. 2007;58:78–83.
Ilic TV, Jovanovic M, Jovicic A, Tomovic M. Oxidative stress indicators are elevated in de novo Parkinson's disease patients. Funct Neurol. 1999;14:141–7.
Littarru GP, Tiano L. Bioenergetic and antioxidant properties of coenzyme Q10: recent developments. Mol Biotechnol. 2007;37:31–7.
Moon Y, Lee KH, Park JH, Geum D, Kim K. Mitochondrial membrane depolarization and the selective death of dopaminergic neurons by rotenone: protective effect of coenzyme Q10. J Neurochem. 2005;93:1199–208.
Park HW, Park CG, Park M, Lee SH, Park HR, Lim J, et al. Intrastriatal administration of coenzyme Q10 enhances neuroprotection in a Parkinson's disease rat model. Sci Rep. 2020;10:9572.
Abdin AA, Hamouda HE. Mechanism of the neuroprotective role of coenzyme Q10 with or without L-dopa in rotenone-induced parkinsonism. Neuropharmacology. 2008;55:1340–6.
Beal MF, Matthews RT, Tieleman A, Shults CW. Coenzyme Q10 attenuates the 1-methyl-4-phenyl-1,2,3,tetrahydropyridine (MPTP) induced loss of striatal dopamine and dopaminergic axons in aged mice. Brain Res. 1998;783:109–14.
Cleren C, Yang L, Lorenzo B, Calingasan NY, Schomer A, Sireci A, et al. Therapeutic effects of coenzyme Q10 (CoQ10) and reduced CoQ10 in the MPTP model of parkinsonism. J Neurochem. 2008;104:1613–21.
Shults CW, Oakes D, Kieburtz K, Beal MF, Haas R, Plumb S, et al. Effects of coenzyme Q10 in early Parkinson disease: evidence of slowing of the functional decline. Arch Neurol. 2002;59:1541–50.
Parkinson Study Group QEI, Beal MF, Oakes D, Shoulson I, Henchcliffe C, Galpern WR, et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: no evidence of benefit. JAMA Neurol. 2014;71:543–52.
Negida A, Menshawy A, El Ashal G, Elfouly Y, Hani Y, Hegazy Y, et al. Coenzyme Q10 for patients with Parkinson's disease: a systematic review and Meta-analysis. CNS Neurol Disord Drug Targets. 2016;15:45–53.
Noda N, Hamatani T, Shibue Y, Takagaki T, Kitamura A, Haranaka M, et al. Phase 1 study to investigate the safety, tolerability, and pharmacokinetics of EPI-589 in healthy participants. Clin Pharmacol Drug Dev. 2022;11:1136–46.
Prasuhn J, Kasten M, Vos M, Konig IR, Schmid SM, Wilms B, et al. The use of vitamin K2 in patients with Parkinson's disease and mitochondrial dysfunction (PD-K2): a Theranostic pilot study in a placebo-controlled parallel group design. Front Neurol. 2020;11:592104.
Banerjee P, Saha I, Sarkar D, Maiti AK. Contributions and limitations of mitochondria-targeted and non-targeted antioxidants in the treatment of parkinsonism: an updated review. Neurotox Res. 2022;40:847–73.
Ghosh A, Chandran K, Kalivendi SV, Joseph J, Antholine WE, Hillard CJ, et al. Neuroprotection by a mitochondria-targeted drug in a Parkinson's disease model. Free Radic Biol Med. 2010;49:1674–84.
Langley M, Ghosh A, Charli A, Sarkar S, Ay M, Luo J, et al. Mito-Apocynin prevents mitochondrial dysfunction, microglial activation, oxidative damage, and progressive neurodegeneration in MitoPark transgenic mice. Antioxid Redox Signal. 2017;27:1048–66.
Ghosh A, Langley MR, Harischandra DS, Neal ML, Jin H, Anantharam V, et al. Mitoapocynin treatment protects against Neuroinflammation and dopaminergic neurodegeneration in a preclinical animal model of Parkinson's disease. J NeuroImmune Pharmacol. 2016;11:259–78.
Snow BJ, Rolfe FL, Lockhart MM, Frampton CM, O'Sullivan JD, Fung V, et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov Disord. 2010;25:1670–4.
Poulter MO, Payne KB, Steiner JP. Neuroimmunophilins: a novel drug therapy for the reversal of neurodegenerative disease? Neuroscience. 2004;128:1–6.
Investigators. NN-P: a randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology. 2007;68:20–8.
Charisis S, Ntanasi E, Stamelou M, Xiromerisiou G, Maraki M, Veskoukis AS, et al. Plasma glutathione and prodromal Parkinson's disease probability. Mov Disord. 2022;37:200–5.
Meister A. On the antioxidant effects of ascorbic acid and glutathione. Biochem Pharmacol. 1992;44:1905–15.
Zeevalk GD, Bernard LP, Guilford FT. Liposomal-glutathione provides maintenance of intracellular glutathione and neuroprotection in mesencephalic neuronal cells. Neurochem Res. 2010;35:1575–87.
Mischley LK, Lau RC, Shankland EG, Wilbur TK, Padowski JM. Phase IIb study of intranasal glutathione in Parkinson's disease. J Parkinsons Dis. 2017;7:289–99.
Rahimmi A, Khosrobakhsh F, Izadpanah E, Moloudi MR, Hassanzadeh K. N-acetylcysteine prevents rotenone-induced Parkinson's disease in rat: an investigation into the interaction of parkin and Drp1 proteins. Brain Res Bull. 2015;113:34–40.
Caridade-Silva R, Araujo B, Martins-Macedo J, Teixeira FG. N-Acetylcysteine treatment may compensate motor impairments through dopaminergic transmission modulation in a striatal 6-Hydroxydopamine Parkinson's disease rat model. Antioxidants (Basel). 2023:12(6):1257.
Clark J, Clore EL, Zheng K, Adame A, Masliah E, Simon DK. Oral N-acetyl-cysteine attenuates loss of dopaminergic terminals in alpha-synuclein overexpressing mice. PLoS One. 2010;5:e12333.
Monti DA, Zabrecky G, Kremens D, Liang TW, Wintering NA, Bazzan AJ, et al. N-acetyl cysteine is associated with dopaminergic improvement in Parkinson's disease. Clin Pharmacol Ther. 2019;106:884–90.
Lautrup S, Sinclair DA, Mattson MP, Fang EF. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 2019;30:630–55.
Shan C, Gong YL, Zhuang QQ, Hou YF, Wang SM, Zhu Q, et al. Protective effects of beta- nicotinamide adenine dinucleotide against motor deficits and dopaminergic neuronal damage in a mouse model of Parkinson's disease. Prog Neuro-Psychopharmacol Biol Psychiatry. 2019;94:109670.
Schondorf DC, Ivanyuk D, Baden P, Sanchez-Martinez A, De Cicco S, Yu C, et al. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and Fly models of Parkinson's disease. Cell Rep. 2018;23:2976–88.
Conze D, Brenner C, Kruger CL. Safety and metabolism of long-term administration of NIAGEN (nicotinamide riboside chloride) in a randomized, double-blind, placebo-controlled clinical trial of healthy overweight adults. Sci Rep. 2019;9:9772.
Martens CR, Denman BA, Mazzo MR, Armstrong ML, Reisdorph N, McQueen MB, et al. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat Commun. 2018;9:1286.
Brakedal B, Dolle C, Riemer F, Ma Y, Nido GS, Skeie GO, et al. The NADPARK study: a randomized phase I trial of nicotinamide riboside supplementation in Parkinson's disease. Cell Metab. 2022;34(396–407):e396.
Schlesinger I, Schlesinger N. Uric acid in Parkinson's disease. Mov Disord. 2008;23:1653–7.
Guerreiro S, Ponceau A, Toulorge D, Martin E, Alvarez-Fischer D, Hirsch EC, et al. Protection of midbrain dopaminergic neurons by the end-product of purine metabolism uric acid: potentiation by low-level depolarization. J Neurochem. 2009;109:1118–28.
Chen X, Burdett TC, Desjardins CA, Logan R, Cipriani S, Xu Y, et al. Disrupted and transgenic urate oxidase alter urate and dopaminergic neurodegeneration. Proc Natl Acad Sci U S A. 2013;110:300–5.
Cipriani S, Bakshi R, Schwarzschild MA. Protection by inosine in a cellular model of Parkinson's disease. Neuroscience. 2014;274:242–9.
El-Shamarka MEA, Kozman MR, Messiha BAS. The protective effect of inosine against rotenone-induced Parkinson's disease in mice; role of oxido-nitrosative stress, ERK phosphorylation, and A2AR expression. Naunyn Schmiedeberg's Arch Pharmacol. 2020;393:1041–53.
Khanal S, Bok E, Kim J, Park GH, Choi DY. Dopaminergic neuroprotective effects of inosine in MPTP-induced parkinsonian mice via brain-derived neurotrophic factor upregulation. Neuropharmacology. 2023;238:109652.
El-Latif AMA, Rabie MA, Sayed RH, Fattah M, Kenawy SA. Inosine attenuates rotenone-induced Parkinson's disease in rats by alleviating the imbalance between autophagy and apoptosis. Drug Dev Res. 2023;84:1159–74.
Parkinson Study Group S-PDI, Schwarzschild MA, Ascherio A, Beal MF, Cudkowicz ME, Curhan GC, et al. Inosine to increase serum and cerebrospinal fluid urate in Parkinson disease: a randomized clinical trial. JAMA Neurol. 2014;71:141–50.
Parkinson Study Group S-PDI, Schwarzschild MA, Ascherio A, Casaceli C, Curhan GC, Fitzgerald R, et al. Effect of urate-elevating inosine on early Parkinson disease progression: the SURE-PD3 randomized clinical trial. JAMA. 2021;326:926–39.
Abdelkader NF, Safar MM, Salem HA. Ursodeoxycholic acid ameliorates apoptotic Cascade in the rotenone model of Parkinson's disease: modulation of mitochondrial perturbations. Mol Neurobiol. 2016;53:810–7.
Mortiboys H, Furmston R, Bronstad G, Aasly J, Elliott C, Bandmann O. UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2(G2019S) carriers and in vivo. Neurology. 2015;85:846–52.
Jiang C, Shen D, Li K, Wang H, Sang W, Qi H. Protective effects of Ursodeoxycholic acid against oxidative stress and Neuroinflammation through mitogen-activated protein kinases pathway in MPTP-induced Parkinson disease. Clin Neuropharmacol. 2022;45:168–74.
Qi H, Shen D, Jiang C, Wang H, Chang M. Ursodeoxycholic acid protects dopaminergic neurons from oxidative stress via regulating mitochondrial function, autophagy, and apoptosis in MPTP/MPP(+)-induced Parkinson's disease. Neurosci Lett. 2021;741:135493.
Sathe AG, Tuite P, Chen C, Ma Y, Chen W, Cloyd J, et al. Pharmacokinetics, safety, and tolerability of orally administered Ursodeoxycholic acid in patients with Parkinson's disease-a pilot study. J Clin Pharmacol. 2020;60:744–50.
Klopstock T, Elstner M, Bender A. Creatine in mouse models of neurodegeneration and aging. Amino Acids. 2011;40:1297–303.
Cankaya S, Cankaya B, Kilic U, Kilic E, Yulug B. The therapeutic role of minocycline in Parkinson's disease. Drugs Context. 2019;8:212553.
Matthews RT, Ferrante RJ, Klivenyi P, Yang L, Klein AM, Mueller G, et al. Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp Neurol. 1999;157:142–9.
Investigators. NN-P: a randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology. 2006;66:664–71.
Dodel R, Spottke A, Gerhard A, Reuss A, Reinecker S, Schimke N, et al. Minocycline 1-year therapy in multiple-system-atrophy: effect on clinical symptoms and [(11)C] (R)-PK11195 PET (MEMSA-trial). Mov Disord. 2010;25:97–107.
Sofic E, Riederer P, Heinsen H, Beckmann H, Reynolds GP, Hebenstreit G, et al. Increased iron (III) and total iron content in post mortem substantia nigra of parkinsonian brain. J Neural Transm. 1988;74:199–205.
Wang JY, Zhuang QQ, Zhu LB, Zhu H, Li T, Li R, et al. Meta-analysis of brain iron levels of Parkinson's disease patients determined by postmortem and MRI measurements. Sci Rep. 2016;6:36669.
Dexter DT, Statton SA, Whitmore C, Freinbichler W, Weinberger P, Tipton KF, et al. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson's disease after peripheral administration. J Neural Transm (Vienna). 2011;118:223–31.
Kaur D, Yantiri F, Rajagopalan S, Kumar J, Mo JQ, Boonplueang R, et al. Genetic or pharmacological iron chelation prevents MPTP-induced neurotoxicity in vivo: a novel therapy for Parkinson's disease. Neuron. 2003;37:899–909.
Workman DG, Tsatsanis A, Lewis FW, Boyle JP, Mousadoust M, Hettiarachchi NT, et al. Protection from neurodegeneration in the 6-hydroxydopamine (6-OHDA) model of Parkinson's with novel 1-hydroxypyridin-2-one metal chelators. Metallomics. 2015;7:867–76.
Molina-Holgado F, Gaeta A, Francis PT, Williams RJ, Hider RC. Neuroprotective actions of deferiprone in cultured cortical neurones and SHSY-5Y cells. J Neurochem. 2008;105:2466–76.
Martin-Bastida A, Ward RJ, Newbould R, Piccini P, Sharp D, Kabba C, et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson's disease. Sci Rep. 2017;7:1398.
Devos D, Moreau C, Devedjian JC, Kluza J, Petrault M, Laloux C, et al. Targeting chelatable iron as a therapeutic modality in Parkinson's disease. Antioxid Redox Signal. 2014;21:195–210.
Devos D, Labreuche J, Rascol O, Corvol JC, Duhamel A, Guyon Delannoy P, et al. Trial of Deferiprone in Parkinson's disease. N Engl J Med. 2022;387:2045–55.
Elkouzi A, Vedam-Mai V, Eisinger RS, Okun MS. Emerging therapies in Parkinson disease - repurposed drugs and new approaches. Nat Rev Neurol. 2019;15:204–23.
Prasuhn J, Davis RL, Kumar KR. Targeting mitochondrial impairment in Parkinson's disease: challenges and opportunities. Front Cell Dev Biol. 2020;8:615461.
Raghu G, Berk M, Campochiaro PA, Jaeschke H, Marenzi G, Richeldi L, et al. The multifaceted therapeutic role of N-Acetylcysteine (NAC) in disorders characterized by oxidative stress. Curr Neuropharmacol. 2021;19:1202–24.
Katsyuba E, Romani M, Hofer D, Auwerx J. NAD(+) homeostasis in health and disease. Nat Metab. 2020;2:9–31.
Chen L, Tao Y, Li J, Kang M. Pioglitazone use is associated with reduced risk of Parkinson's disease in patients with diabetes: a systematic review and meta-analysis. J Clin Neurosci. 2022;106:154–8.
Dahiya S, Tisch S, Greenfield J. The effect of GLP-1 receptor agonists in pre-clinical rodent models of Parkinson's disease: a systematic review and meta-analysis. Clin Park Relat Disord. 2022;6:100133.
Reich N, Holscher C. The neuroprotective effects of glucagon-like peptide 1 in Alzheimer's and Parkinson's disease: an in-depth review. Front Neurosci. 2022;16:970925.
Bergkvist L, Johnson ME, Mercado G, Steiner JA, Meyerdirk L, Schulz E, et al. An extended release GLP-1 analogue increases alpha-synuclein accumulation in a mouse model of prodromal Parkinson's disease. Exp Neurol. 2021;341:113693.
Athauda D, Maclagan K, Skene SS, Bajwa-Joseph M, Letchford D, Chowdhury K, et al. Exenatide once weekly versus placebo in Parkinson's disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–75.
Vijiaratnam N, Girges C, Auld G, Chau M, Maclagan K, King A, et al. Exenatide once weekly over 2 years as a potential disease-modifying treatment for Parkinson's disease: protocol for a multicentre, randomised, double blind, parallel group, placebo controlled, phase 3 trial: the 'Exenatide-PD3' study. BMJ Open. 2021;11:e047993.
Malatt C, Wu T, Bresee C, Hogg E, Wertheimer J, Tan E, et al. Liraglutide improves non-motor function and activities of daily living in patients with Parkinson’s disease: a randomized, double-blind, placebo-controlled trial (P9-11.005). Neurology. 2022;98:3068.
Dehmer T, Heneka MT, Sastre M, Dichgans J, Schulz JB. Protection by pioglitazone in the MPTP model of Parkinson's disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem. 2004;88:494–501.
Corona JC, Duchen MR. PPARgamma and PGC-1alpha as therapeutic targets in Parkinson's. Neurochem Res. 2015;40:308–16.
Investigators. NETiPDF-Z: pioglitazone in early Parkinson’s disease: a phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015;14:795–803.
Oliver D, Reddy PH. Dynamics of dynamin-related protein 1 in Alzheimer's disease and other neurodegenerative diseases. Cells. 2019:823;8(9):961.
Bido S, Soria FN, Fan RZ, Bezard E, Tieu K. Mitochondrial division inhibitor-1 is neuroprotective in the A53T-alpha-synuclein rat model of Parkinson's disease. Sci Rep. 2017;7:7495.
Fan RZ, Guo M, Luo S, Cui M, Tieu K. Exosome release and neuropathology induced by alpha-synuclein: new insights into protective mechanisms of Drp1 inhibition. Acta Neuropathol Commun. 2019;7:184.
Zhang XL, Huang WM, Tang PC, Sun Y, Zhang X, Qiu L, et al. Anti-inflammatory and neuroprotective effects of natural cordycepin in rotenone-induced PD models through inhibiting Drp1-mediated mitochondrial fission. Neurotoxicology. 2021;84:1–13.
Rappold PM, Cui M, Grima JC, Fan RZ, de Mesy-Bentley KL, Chen L, et al. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat Commun. 2014;5:5244.
Hsieh CH, Shaltouki A, Gonzalez AE. Bettencourt da Cruz a, Burbulla LF, St Lawrence E, Schule B, Krainc D, palmer TD, Wang X: functional impairment in Miro degradation and Mitophagy is a shared feature in familial and sporadic Parkinson's disease. Cell Stem Cell. 2016;19:709–24.
Shaltouki A, Hsieh CH, Kim MJ, Wang X. Alpha-synuclein delays mitophagy and targeting Miro rescues neuron loss in Parkinson's models. Acta Neuropathol. 2018;136:607–20.
Hsieh CH, Li L, Vanhauwaert R, Nguyen KT, Davis MD, Bu G, et al. Miro1 Marks Parkinson's disease subset and Miro1 reducer rescues neuron loss in Parkinson's models. Cell Metab. 2019;30(1131–1140):e1137.
Haque ME, Thomas KJ, D'Souza C, Callaghan S, Kitada T, Slack RS, et al. Cytoplasmic Pink1 activity protects neurons from dopaminergic neurotoxin MPTP. Proc Natl Acad Sci U S A. 2008;105:1716–21.
Yasuda T, Hayakawa H, Nihira T, Ren YR, Nakata Y, Nagai M, et al. Parkin-mediated protection of dopaminergic neurons in a chronic MPTP-minipump mouse model of Parkinson disease. J Neuropathol Exp Neurol. 2011;70:686–97.
Bian M, Liu J, Hong X, Yu M, Huang Y, Sheng Z, et al. Overexpression of parkin ameliorates dopaminergic neurodegeneration induced by 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. PLoS One. 2012;7:e39953.
Todd AM, Staveley BE. Pink1 suppresses alpha-synuclein-induced phenotypes in a Drosophila model of Parkinson's disease. Genome. 2008;51:1040–6.
Chung E, Choi Y, Park J, Nah W, Park J, Jung Y, et al. Intracellular delivery of Parkin rescues neurons from accumulation of damaged mitochondria and pathological alpha-synuclein. Sci Adv. 2020;6:eaba1193.
Pinto M, Nissanka N, Peralta S, Brambilla R, Diaz F, Moraes CT. Pioglitazone ameliorates the phenotype of a novel Parkinson's disease mouse model by reducing neuroinflammation. Mol Neurodegener. 2016;11:25.
Yang Y, Gehrke S, Imai Y, Huang Z, Ouyang Y, Wang JW, et al. Mitochondrial pathology and muscle and dopaminergic neuron degeneration caused by inactivation of Drosophila Pink1 is rescued by Parkin. Proc Natl Acad Sci U S A. 2006;103:10793–8.
Ilijic E, Guzman JN, Surmeier DJ. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson's disease. Neurobiol Dis. 2011;43:364–71.
Singh A, Verma P, Balaji G, Samantaray S, Mohanakumar KP. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced parkinsonism in mice. Neurochem Int. 2016;99:221–32.
Pasternak B, Svanstrom H, Nielsen NM, Fugger L, Melbye M, Hviid A. Use of calcium channel blockers and Parkinson's disease. Am J Epidemiol. 2012;175:627–35.
Tseng YF, Lin HC, Chao JC, Hsu CY, Lin HL. Calcium Channel blockers are associated with reduced risk of Parkinson's disease in patients with hypertension: a population-based retrospective cohort study. J Neurol Sci. 2021;424:117412.
Parkinson Study Group S-PDIIII. Isradipine versus placebo in early Parkinson disease: a randomized trial. Ann Intern Med. 2020;172:591–8.
Surmeier DJ, Nguyen JT, Lancki N, Venuto CS, Oakes D, Simuni T, et al. Re-analysis of the STEADY-PD II trial-evidence for slowing the progression of Parkinson's disease. Mov Disord. 2022;37:334–42.
Fereshtehnejad SM, Zeighami Y, Dagher A, Postuma RB. Clinical criteria for subtyping Parkinson's disease: biomarkers and longitudinal progression. Brain. 2017;140:1959–76.
Acknowledgements
N/A
Funding
Open Access funding enabled and organized by Projekt DEAL. FFG received grants from the ParkinsonFonds Deutschland/Stichting ParkinsonFonds, the Netherlands, and the von Behring-Röntgen-Stiftung, Germany. WHO is a Hertie-Senior Research Professor, supported by the charitable Hertie Foundation, Frankfurt/Main, Germany. WHO received grants from the MJFF, the DFG (IRTG-GRK 1901), and the ParkinsonFonds Deutschland/Stichting ParkinsonFonds, the Netherlands, outside of this work.
Author information
Authors and Affiliations
Contributions
MTH and FFG wrote the first draft of the manuscript and prepared the figures. WHO and DJS revised and edited the manuscript. MTH prepared the final version of this manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
N/A
Consent for publication
All authors consented to publish this manuscript.
Competing interests
The authors report no competing interests. WHO received honoraria for educational and scientific presentations at symposia from AbbVie and Stada Pharma unrelated to the work presented in this manuscript.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Henrich, M.T., Oertel, W.H., Surmeier, D.J. et al. Mitochondrial dysfunction in Parkinson’s disease – a key disease hallmark with therapeutic potential. Mol Neurodegeneration 18, 83 (2023). https://doi.org/10.1186/s13024-023-00676-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-023-00676-7