
Abstract
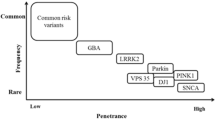
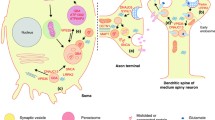
There has been tremendous progress toward understanding the genetic basis of Parkinson’s disease and related movement disorders. We summarize the genetic, clinical and pathological findings of autosomal dominant disease linked to mutations in SNCA, LRRK2, ATXN2, ATXN3, MAPT, GCH1, DCTN1 and VPS35. We then discuss the identification of mutations in PARK2, PARK7, PINK1, ATP13A2, FBXO7, PANK2 and PLA2G6 genes. In particular we discuss the clinical and pathological characterization of these forms of disease, where neuropathology has been important in the likely coalescence of pathways highly relevant to typical PD. In addition to the identification of the causes of monogenic forms of PD, significant progress has been made in defining genetic risk loci for PD; we discuss these here, including both risk variants at LRRK2 and GBA, in addition to discussing the results of recent genome-wide association studies and their implications for PD. Finally, we discuss the likely path of genetic discovery in PD over the coming period and the implications of these findings from a clinical and etiological perspective.

Similar content being viewed by others

References
Abou-Sleiman PM, Healy DG, Quinn N, Lees AJ, Wood NW (2003) The role of pathogenic DJ-1 mutations in Parkinson’s disease. Ann Neurol 54:283–286. doi:10.1002/ana.10675
Aharon-Peretz J, Rosenbaum H, Gershoni-Baruch R (2004) Mutations in the glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N Engl J Med 351:1972–1977
Al-Chalabi A, Durr A, Wood NW et al (2009) Genetic variants of the alpha-synuclein gene SNCA are associated with multiple system atrophy. PLoS One 4:e7114. doi:10.1371/journal.pone.0007114
Ballana E, Govea N, de Cid R et al (2008) Detection of unrecognized low-level mtDNA heteroplasmy may explain the variable phenotypic expressivity of apparently homoplasmic mtDNA mutations. Hum Mutat 29:248–257. doi:10.1002/humu.20639
Bardien S, Lesage S, Brice A, Carr J (2011) Genetic characteristics of leucine-rich repeat kinase 2 (LRRK2) associated Parkinson’s disease. Parkinsonism Relat Disord 17:501–508. pii: S1353-8020(10)00287-7
Bonifati V, Rizzu P, van Baren MJ et al (2003) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299:256–259. doi:10.1126/science.1077209
Bonifati V, Rohe CF, Breedveld GJ et al (2005) Early-onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 65:87–95
Bras JM, Guerreiro RJ, Ribeiro MH et al (2005) G2019S dardarin substitution is a common cause of Parkinson’s disease in a Portuguese cohort. Mov Disord : Official J Mov Disord Soc 20:1653–1655. doi:10.1002/mds.20682
Chartier-Harlin MC, Dachsel JC, Vilarino-Guell C et al (2011) Translation initiator EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet 89:398–406. doi:10.1016/j.ajhg.2011.08.009
Chartier-Harlin MC, Kachergus J, Roumier C et al (2004) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364:1167–1169. doi:10.1016/S0140-6736(04)17103-1
Choi JM, Woo MS, Ma HI et al (2008) Analysis of PARK genes in a Korean cohort of early-onset Parkinson disease. Neurogenetics 9:263–269. doi:10.1007/s10048-008-0138-0
Clark IE, Dodson MW, Jiang C et al. (2006) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166
Consortium UKPsD, Wellcome Trust Case Control Consortium, Spencer CC et al (2011) Dissection of the genetics of Parkinson’s disease identifies an additional association 5′ of SNCA and multiple associated haplotypes at 17q21. Hum Mol Genet 20:345–353. pii: ddq469
Criscuolo C, Volpe G, De Rosa A et al (2006) PINK1 homozygous W437X mutation in a patient with apparent dominant transmission of parkinsonism. Mov Disord: Off J Mov Disord Soc 21:1265–1267. doi:10.1002/mds.20933
Dekker MC, van Swieten JC, Houwing-Duistermaat JJ et al (2003) A clinical-genetic study of Parkinson’s disease in a genetically isolated community. J Neurol 250:1056–1062. doi:10.1007/s00415-003-0151-z
Di Fonzo A, Chien HF, Socal M et al (2007) ATP13A2 missense mutations in juvenile parkinsonism and young onset Parkinson disease. Neurology 68:1557–1562. pii: 68/19/1557
Di Fonzo A, Dekker MC, Montagna P et al (2009) FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome. Neurology 72:240–245. doi:10.1212/01.wnl.0000338144.10967.2b
Di Fonzo A, Rohe CF, Ferreira J et al (2005) A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson’s disease. Lancet 365:412–415. doi:S0140-6736(05)17829-5
Dick KJ, Eckhardt M, Paisan-Ruiz C et al (2010) Mutation of FA2H underlies a complicated form of hereditary spastic paraplegia (SPG35). Hum Mutat 31:E1251–E1260. doi:10.1002/humu.21205
Djarmati A, Hagenah J, Reetz K et al (2009) ATP13A2 variants in early-onset Parkinson’s disease patients and controls. Mov Disord: Off J Mov Disord Soc 24:2104–2111. doi:10.1002/mds.22728
Do CB, Tung JY, Dorfman E et al (2011) Web-based genome-wide association study identifies two novel loci and a substantial genetic component for Parkinson’s disease. PLoS Genet 7:e1002141. doi:10.1371/journal.pgen.1002141
Duda JE, Giasson BI, Mabon ME et al (2002) Concurrence of alpha-synuclein and tau brain pathology in the Contursi kindred. Acta Neuropathol 104:7–11. doi:10.1007/s00401-002-0563-3
Edwards TL, Scott WK, Almonte C et al (2010) Genome-wide association study confirms SNPs in SNCA and the MAPT region as common risk factors for Parkinson disease. Ann Hum Genet 74:97–109
Farrer M, Chan P, Chen R et al (2001) Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 50:293–300
Farrer M, Kachergus J, Forno L et al (2004) Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol 55:174–179. doi:10.1002/ana.10846
Farrer MJ, Hulihan MM, Kachergus JM et al (2009) DCTN1 mutations in Perry syndrome. Nat Genet 41:163–165. doi:10.1038/ng.293
Funayama M, Hasegawa K, Kowa H, Saito M, Tsuji S, Obata F (2002) A new locus for Parkinson’s disease (PARK8) maps to chromosome 12p11.2-q13.1. Ann Neurol 51:296–301. doi:10.1002/ana.10113
Fung HC, Scholz S, Matarin M et al (2006) Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol 5:911–916. pii: S1474-4422(06)70578-6
Furtado S, Payami H, Lockhart PJ et al (2004) Profile of families with parkinsonism-predominant spinocerebellar ataxia type 2 (SCA2). Mov Disord : Off J Mov Disord Soc 19:622–629. doi:10.1002/mds.20074
Gaig C, Marti MJ, Ezquerra M, Rey MJ, Cardozo A, Tolosa E (2007) G2019S LRRK2 mutation causing Parkinson’s disease without Lewy bodies. J Neurol Neurosurg Psychiatry 78:626–628. doi:jnnp.2006.107904
Geisler S, Holmstrom KM, Skujat D et al (2010) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12:119–131. doi:10.1038/ncb2012
Giasson BI, Van Deerlin VM (2008) Mutations in LRRK2 as a cause of Parkinson’s disease. Neurosignals 16:99–105. doi:10.1159/000109764
Gilks WP, Abou-Sleiman PM, Gandhi S et al (2005) A common LRRK2 mutation in idiopathic Parkinson’s disease. Lancet 365:415–416. doi:10.1016/S0140-6736(05)17830-1
Goker-Alpan O, Lopez G, Vithayathil J, Davis J, Hallett M, Sidransky E (2008) The spectrum of parkinsonian manifestations associated with glucocerebrosidase mutations. Arch Neurol 65:1353–1357
Golbe LI, Di Iorio G, Bonavita V, Miller DC, Duvoisin RC (1990) A large kindred with autosomal dominant Parkinson’s disease. Ann Neurol 27:276–282. doi:10.1002/ana.410270309
Goldwurm S, Tunesi S, Tesei S et al (2011) Kin-cohort analysis of LRRK2-G2019S penetrance in Parkinson’s disease. Mov Disord: Off J Mov Disord Soc. doi:10.1002/mds.23807
Gouider-Khouja N, Larnaout A, Amouri R et al (2003) Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat Disord 9:247–251 pii: S1353802003000166
Gwinn-Hardy K, Chen JY, Liu HC et al (2000) Spinocerebellar ataxia type 2 with parkinsonism in ethnic Chinese. Neurology 55:800–805
Gwinn-Hardy K, Singleton A, O’Suilleabhain P et al (2001) Spinocerebellar ataxia type 3 phenotypically resembling parkinson disease in a black family. Arch Neurol 58:296–299
Gwinn K, Devine MJ, Jin LW et al (2011) Clinical features, with video documentation, of the original familial Lewy body parkinsonism caused by alpha-synuclein triplication (Iowa kindred). Mov Disord : Off J Mov Disord Soc 26:2134–2136
Hamza TH, Chen H, Hill-Burns EM et al (2011) Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson’s disease modifier gene via interaction with coffee. PLoS Genet 7:e1002237. doi:10.1371/journal.pgen.1002237
Hasegawa K, Stoessl AJ, Yokoyama T, Kowa H, Wszolek ZK, Yagishita S (2009) Familial parkinsonism: study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord 15:300–306 pii: S1353-8020(08)00233-2
Hayashi S, Wakabayashi K, Ishikawa A et al (2000) An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord: Off J Mov Disord Soc 15:884–888
Hayflick SJ, Westaway SK, Levinson B et al (2003) Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 348:33–40. doi:10.1056/NEJMoa020817
Healy DG, Falchi M, O’Sullivan SS et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case–control study. Lancet Neurol 7:583–590. doi:10.1016/S1474-4422(08)70117-0
Hernandez D, Paisan Ruiz C, Crawley A et al (2005) The dardarin G 2019 S mutation is a common cause of Parkinson’s disease but not other neurodegenerative diseases. Neurosci Lett 389:137–139. pii:S0304-3940(05)00861-X
Hernandez DG, Paisan-Ruiz C, McInerney-Leo A et al (2005) Clinical and positron emission tomography of Parkinson’s disease caused by LRRK2. Ann Neurol 57:453–456. doi:10.1002/ana.20401
Hjermind LE, Johannsen LG, Blau N et al (2006) Dopa-responsive dystonia and early-onset Parkinson’s disease in a patient with GTP cyclohydrolase I deficiency? Mov Disord : Off J Mov Disord Soc 21:679–682. doi:10.1002/mds.20773
Hutton M, Lendon CL, Rizzu P et al (1998) Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 393:702–705. doi:10.1038/31508
Ibanez P, Bonnet AM, Debarges B et al (2004) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364:1169–1171. doi:10.1016/S0140-6736(04)17104-3
International Parkinson’s Disease Genomics Consortium, Wellcome Trust Case Control Consortium (2011) A two-stage meta-analysis identifies several new loci for Parkinson’s disease. PLoS Genet 7: e1002142. doi:10.1371/journal.pgen.1002142
International Parkinson Disease Genomics Consortium, Nalls MA, Plagnol V et al (2011) Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet 377:641–649. [pii:S0140-6736(10)62345-8]
Kachergus J, Mata IF, Hulihan M et al (2005) Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 76:672–680. pii:S0002-9297(07)62878-X
Kann M, Jacobs H, Mohrmann K et al (2002) Role of parkin mutations in 111 community-based patients with early-onset parkinsonism. Ann Neurol 51:621–625. doi:10.1002/ana.10179
Kawaguchi Y, Okamoto T, Taniwaki M et al (1994) CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat Genet 8:221–228. doi:10.1038/ng1194-221
Kay DM, Moran D, Moses L et al (2007) Heterozygous parkin point mutations are as common in control subjects as in Parkinson’s patients. Ann Neurol 61:47–54. doi:10.1002/ana.21039
Khan NL, Brooks DJ, Pavese N et al (2002) Progression of nigrostriatal dysfunction in a parkin kindred: an [18F]dopa PET and clinical study. Brain 125:2248–2256
Khan NL, Jain S, Lynch JM et al (2005) Mutations in the gene LRRK2 encoding dardarin (PARK8) cause familial Parkinson’s disease: clinical, pathological, olfactory and functional imaging and genetic data. Brain 128:2786–2796. doi:awh667
Kitada T, Asakawa S, Hattori N et al (1998) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392:605–608. doi:10.1038/33416
Kruer MC, Hiken M, Gregory A et al (2011) Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 134:947–958
Kruer MC, Paisan-Ruiz C, Boddaert N et al (2010) Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann Neurol 68:611–618. doi:10.1002/ana.22122
Kruger R, Kuhn W, Muller T et al (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18:106–108. doi:10.1038/ng0298-106
Kruger R, Vieira-Saecker AM, Kuhn W et al (1999) Increased susceptibility to sporadic Parkinson’s disease by a certain combined alpha-synuclein/apolipoprotein E genotype. Ann Neurol 45:611–617
Kurian MA, Morgan NV, MacPherson L et al (2008) Phenotypic spectrum of neurodegeneration associated with mutations in the PLA2G6 gene (PLAN). Neurology 70:1623–1629
Latourelle JC, Sun M, Lew MF et al (2008) The Gly2019Ser mutation in LRRK2 is not fully penetrant in familial Parkinson’s disease: the GenePD study. BMC Med 6:32. doi:10.1186/1741-7015-6-32
Lesage S, Durr A, Tazir M et al (2006) LRRK2 G2019S as a cause of Parkinson’s disease in North African Arabs. N Engl J Med 354:422–423
Lesage S, Leutenegger AL, Ibanez P et al (2005) LRRK2 haplotype analyses in European and North African families with Parkinson disease: a common founder for the G2019S mutation dating from the 13th century. Am J Hum Genet 77:330–332 pii: S0002-9297(07)62924-3
Lill CM, Roehr JT, McQueen MB et al (2012) Comprehensive research synopsis and systematic meta-analyses in Parkinson’s disease genetics: the PDGene database. PLoS Genet 8:e1002548. doi:10.1371/journal.pgen.1002548
Lincoln SJ, Maraganore DM, Lesnick TG et al (2003) Parkin variants in North American Parkinson’s disease: cases and controls. Mov Disord : Off J Mov Disord Soc 18:1306–1311. doi:10.1002/mds.10601
Lohmann E, Periquet M, Bonifati V et al (2003) How much phenotypic variation can be attributed to parkin genotype? Ann Neurol 54:176–185. doi:10.1002/ana.10613
Lu CS, Chang HC, Kuo PC et al (2004) The parkinsonian phenotype of spinocerebellar ataxia type 3 in a Taiwanese family. Parkinsonism Relat Disord 10:369–373. doi:10.1016/j.parkreldis.2004.03.009
Lynch T, Sano M, Marder KS et al (1994) Clinical characteristics of a family with chromosome 17-linked disinhibition-dementia-parkinsonism-amyotrophy complex. Neurology 44:1878–1884
Machaczka M, Rucinska M, Skotnicki AB, Jurczak W (1999) Parkinson’s syndrome preceding clinical manifestation of Gaucher’s disease. Am J Hematol 61:216–217. doi:10.1002/(SICI)1096-8652(199907)61:3<216:AID-AJH11>3.0.CO;2-E
Maraganore DM, de Andrade M, Elbaz A et al (2006) Collaborative analysis of alpha-synuclein gene promoter variability and Parkinson disease. JAMA 296:661–670 pii: 296/6/661
Maraganore DM, de Andrade M, Lesnick TG et al (2005) High-resolution whole-genome association study of Parkinson disease. Am J Hum Genet 77:685–693 pii: S0002-9297(07)63354-0
Marongiu R, Brancati F, Antonini A et al (2007) Whole gene deletion and splicing mutations expand the PINK1 genotypic spectrum. Hum Mutat 28:98. doi:10.1002/humu.9472
Marti-Masso JF, Ruiz-Martinez J, Bolano MJ et al (2009) Neuropathology of Parkinson’s disease with the R1441G mutation in LRRK2. Mov Disord: Off J Mov Disord Soc 24:1998–2001. doi:10.1002/mds.22677
Matilla T, McCall A, Subramony SH, Zoghbi HY (1995) Molecular and clinical correlations in spinocerebellar ataxia type 3 and Machado-Joseph disease. Ann Neurol 38:68–72. doi:10.1002/ana.410380113
Matsuda N, Sato S, Shiba K et al (2010) PINK1 stabilized by mitochondrial depolarization recruits parkin to damaged mitochondria and activates latent parkin for mitophagy. J Cell Biol 189:211–221. doi:10.1083/jcb.200910140
Mellick GD, Siebert GA, Funayama M et al (2009) Screening PARK genes for mutations in early-onset Parkinson’s disease patients from Queensland, Australia. Parkinsonism Relat Disord 15:105–109. [pii:S1353-8020(08)00108-9]
Miller DW, Hague SM, Clarimon J et al (2004) Alpha-synuclein in blood and brain from familial Parkinson disease with SNCA locus triplication. Neurology 62:1835–1838
Morgan NV, Westaway SK, Morton JE et al (2006) PLA2G6, encoding a phospholipase A2, is mutated in neurodegenerative disorders with high brain iron. Nat Genet 38:752–754
Mori H, Kondo T, Yokochi M et al (1998) Pathologic and biochemical studies of juvenile parkinsonism linked to chromosome 6q. Neurology 51:890–892
Muenter MD, Forno LS, Hornykiewicz O et al (1998) Hereditary form of parkinsonism–dementia. Ann Neurol 43:768–781. doi:10.1002/ana.410430612
Neudorfer O, Giladi N, Elstein D et al (1996) Occurrence of Parkinson’s syndrome in type I Gaucher disease. QJM 89:691–694
Nichols WC, Pankratz N, Hernandez D et al (2005) Genetic screening for a single common LRRK2 mutation in familial Parkinson’s disease. Lancet 365:410–412. doi:10.1016/S0140-6736(05)17828-3
Ning YP, Kanai K, Tomiyama H et al (2008) PARK9-linked parkinsonism in eastern Asia: mutation detection in ATP13A2 and clinical phenotype. Neurology 70:1491–1493
Nishioka K, Ross OA, Ishii K et al (2009) Expanding the clinical phenotype of SNCA duplication carriers. Mov Disord : Off J Mov Disord Soc 24:1811–1819. doi:10.1002/mds.22682
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31:763–780. doi:10.1002/humu.21277
Obi T, Nishioka K, Ross OA et al (2008) Clinicopathologic study of a SNCA gene duplication patient with Parkinson disease and dementia. Neurology 70:238–241
Ozelius LJ, Senthil G, Saunders-Pullman R et al (2006) LRRK2 G2019S as a cause of Parkinson’s disease in Ashkenazi Jews. N Engl J Med 354:424–425
Paisan-Ruiz C, Bhatia KP, Li A et al (2009) Characterization of PLA2G6 as a locus for dystonia-parkinsonism. Ann Neurol 65:19–23. doi:10.1002/ana.21415
Paisan-Ruiz C, Dogu O, Yilmaz A, Houlden H, Singleton A (2008) SPG11 mutations are common in familial cases of complicated hereditary spastic paraplegia. Neurology 70:1384–1389. doi:10.1212/01.wnl.0000294327.66106.3d
Paisan-Ruiz C, Guevara R, Federoff M et al (2010) Early-onset l-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations. Mov Disord: Off J Mov Disord Soc 25:1791–1800. doi:10.1002/mds.23221
Paisan-Ruiz C, Jain S, Evans EW et al (2004) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44:595–600. doi:10.1016/S0896627304006890
Paisan-Ruiz C, Nath P, Wood NW, Singleton A, Houlden H (2008) Clinical heterogeneity and genotype-phenotype correlations in hereditary spastic paraplegia because of Spatacsin mutations (SPG11). Eur J Neurol: Off J Eur Fed Neurol Soc 15:1065–1070. doi:10.1111/j.1468-1331.2008.02247.x
Paisan-Ruiz C, Scopes G, Lee P, Houlden H (2009) Homozygosity mapping through whole genome analysis identifies a COL18A1 mutation in an Indian family presenting with an autosomal recessive neurological disorder. Am J Med Genet Part B, Neuropsychiatric Genet: Off Publ Int Soc Psychiatric Genet 150B:993–997. doi:10.1002/ajmg.b.30929
Pankratz N, Wilk JB, Latourelle JC et al (2009) Genomewide association study for susceptibility genes contributing to familial Parkinson disease. Hum Genet 124:593–605. doi:10.1007/s00439-008-0582-9
Park J, Lee SB, Lee S et al (2006) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441:1157–1161
Perry TL, Bratty PJ, Hansen S, Kennedy J, Urquhart N, Dolman CL (1975) Hereditary mental depression and Parkinsonism with taurine deficiency. Arch Neurol 32:108–113
Polymeropoulos MH, Higgins JJ, Golbe LI et al (1996) Mapping of a gene for Parkinson’s disease to chromosome 4q21-q23. Science 274:1197–1199
Polymeropoulos MH, Lavedan C, Leroy E et al (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276:2045–2047
Pramstaller PP, Schlossmacher MG, Jacques TS et al (2005) Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 58:411–422. doi:10.1002/ana.20587
Ramirez A, Heimbach A, Grundemann J et al (2006) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38:1184–1191
Rieley MB, Stevenson DA, Viskochil DH, Tinkle BT, Martin LJ, Schorry EK (2011) Variable expression of neurofibromatosis 1 in monozygotic twins. Am J Med Genet Part A 155A:478–485. doi:10.1002/ajmg.a.33851
Ross OA, Toft M, Whittle AJ et al (2006) Lrrk2 and Lewy body disease. Ann Neurol 59:388–393. doi:10.1002/ana.20731
Samaranch L, Lorenzo-Betancor O, Arbelo JM et al (2010) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133:1128–1142
Sasaki S, Shirata A, Yamane K, Iwata M (2004) Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 63:678–682
Satake W, Nakabayashi Y, Mizuta I et al (2009) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41:1303–1307
Schneider SA, Paisan-Ruiz C, Quinn NP et al (2010) ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov Disord : Off J Mov Disord Soc 25:979–984. doi:10.1002/mds.22947
Scholz SW, Houlden H, Schulte C et al (2009) SNCA variants are associated with increased risk for multiple system atrophy. Ann Neurol 65:610–614. doi:10.1002/ana.21685
Shojaee S, Sina F, Banihosseini SS et al (2008) Genome-wide linkage analysis of a Parkinsonian-pyramidal syndrome pedigree by 500 K SNP arrays. Am J Hum Genet 82:1375–1384. [pii:S0002-9297(08)00314-5]
Sidransky E, Nalls MA, Aasly JO et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661
Simon-Sanchez J, Schulte C, Bras JM et al (2009) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41:1308–1312
Simon-Sanchez J, van Hilten JJ, van de Warrenburg B et al (2011) Genome-wide association study confirms extant PD risk loci among the Dutch. Eur J Hum Genet 19:655–661
Singleton A, Gwinn-Hardy K (2004) Parkinson’s disease and dementia with Lewy bodies: a difference in dose? Lancet 364:1105–1107. doi:10.1016/S0140-6736(04)17117-1
Singleton AB, Farrer M, Johnson J et al (2003) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302:841. doi:10.1126/science.1090278
Spillantini MG, Crowther RA, Jakes R, Cairns NJ, Lantos PL, Goedert M (1998) Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251:205–208
Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95:6469–6473
Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M (1997) Alpha-synuclein in Lewy bodies. Nature 388:839–840. doi:10.1038/42166
Spira PJ, Sharpe DM, Halliday G, Cavanagh J, Nicholson GA (2001) Clinical and pathological features of a Parkinsonian syndrome in a family with an Ala53Thr alpha-synuclein mutation. Ann Neurol 49:313–319
Subramony SH, Hernandez D, Adam A et al (2002) Ethnic differences in the expression of neurodegenerative disease: Machado-Joseph disease in Africans and Caucasians. Mov Disord: Off J Mov Disord Soc 17:1068–1071. doi:10.1002/mds.10241
Tanner CM, Ottman R, Goldman SM et al (1999) Parkinson disease in twins: an etiologic study. JAMA 281:341–346
Tayebi N, Callahan M, Madike V et al (2001) Gaucher disease and parkinsonism: a phenotypic and genotypic characterization. Mol Genet Metab 73:313–321. doi:10.1006/mgme.2001.3201
Tayebi N, Walker J, Stubblefield B et al (2003) Gaucher disease with parkinsonian manifestations: does glucocerebrosidase deficiency contribute to a vulnerability to parkinsonism? Mol Genet Metab 79:104–109. doi:10.1016/S1096719203000714
Thomas M, Hayflick SJ, Jankovic J (2004) Clinical heterogeneity of neurodegeneration with brain iron accumulation (Hallervorden-Spatz syndrome) and pantothenate kinase-associated neurodegeneration. Mov Disord: Off J Mov Disord Soc 19:36–42. doi:10.1002/mds.10650
Valente EM, Abou-Sleiman PM, Caputo V et al (2004) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304:1158–1160. doi:10.1126/science.1096284
van de Warrenburg BP, Lammens M, Lucking CB et al (2001) Clinical and pathologic abnormalities in a family with parkinsonism and parkin gene mutations. Neurology 56:555–557
Vilarino-Guell C, Wider C, Ross OA et al (2011) VPS35 mutations in Parkinson disease. Am J Hum Genet 89:162–167 pii:S0002-9297(11)00242-4
Weng YH, Chou YH, Wu WS et al (2007) PINK1 mutation in Taiwanese early-onset parkinsonism: clinical, genetic, and dopamine transporter studies. J Neurol 254:1347–1355. doi:10.1007/s00415-007-0534-7
Wider C, Dickson DW, Wszolek ZK (2010) Leucine-rich repeat kinase 2 gene-associated disease: redefining genotype-phenotype correlation. Neuro-degenerative diseases 7:175–179. doi:10.1159/000289232
Wiest V, Eisenbarth I, Schmegner C, Krone W, Assum G (2003) Somatic NF1 mutation spectra in a family with neurofibromatosis type 1: toward a theory of genetic modifiers. Hum Mutat 22:423–427. doi:10.1002/humu.10272
Wilhelmsen KC, Lynch T, Pavlou E, Higgins M, Nygaard TG (1994) Localization of disinhibition–dementia–parkinsonism–amyotrophy complex to 17q21-22. Am J Hum Genet 55:1159–1165
Wirdefeldt K, Gatz M, Reynolds CA, Prescott CA, Pedersen NL (2011) Heritability of Parkinson disease in Swedish twins: a longitudinal study. Neurobiol Aging 32(1923):e1921–e1928. doi:10.1016/j.neurobiolaging.2011.02.017
Wszolek ZK, Pfeiffer RF, Bhatt MH et al (1992) Rapidly progressive autosomal dominant parkinsonism and dementia with pallido-ponto-nigral degeneration. Ann Neurol 32:312–320. doi:10.1002/ana.410320303
Zarranz JJ, Alegre J, Gomez-Esteban JC et al (2004) The new mutation, E46 K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. doi:10.1002/ana.10795
Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ (2001) A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 28:345–349. doi:10.1038/ng572
Zimprich A, Benet-Pages A, Struhal W et al (2011) A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89:168–175. [pii:S0002-9297(11)00261-8]
Zimprich A, Biskup S, Leitner P et al (2004) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44:601–607. [pii:S0896627304007202]
Zuk O, Hechter E, Sunyaev SR, Lander ES (2012) The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci USA. doi:10.1073/pnas.1119675109
Acknowledgments
This work was supported in part by the Intramural Research Program of the National Institute on Aging, National Institutes of Health, Department of Health and Human Services; projects number Z01 AG000957-08 and AG000958-08. We are also grateful to the Medical Research Council (MRC), NORD, the Parkinson’s Disease Foundation (PDF), the DMRF and The Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Houlden, H., Singleton, A.B. The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol 124, 325–338 (2012). https://doi.org/10.1007/s00401-012-1013-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-012-1013-5