
Abstract
Transactive response DNA binding protein of 43 kDa (TDP-43) is an intranuclear protein encoded by the TARDBP gene that is involved in RNA splicing, trafficking, stabilization, and thus, the regulation of gene expression. Cytoplasmic inclusion bodies containing phosphorylated and truncated forms of TDP-43 are hallmarks of amyotrophic lateral sclerosis (ALS) and a subset of frontotemporal lobar degeneration (FTLD). Additionally, TDP-43 inclusions have been found in up to 57% of Alzheimer’s disease (AD) cases, most often in a limbic distribution, with or without hippocampal sclerosis. In some cases, TDP-43 deposits are also found in neurons with neurofibrillary tangles. AD patients with TDP-43 pathology have increased severity of cognitive impairment compared to those without TDP-43 pathology. Furthermore, the most common genetic risk factor for AD, apolipoprotein E4 (APOE4), is associated with increased frequency of TDP-43 pathology. These findings provide strong evidence that TDP-43 pathology is an integral part of multiple neurodegenerative conditions, including AD. Here, we review the biology and pathobiology of TDP-43 with a focus on its role in AD. We emphasize the need for studies on the mechanisms that lead to TDP-43 pathology, especially in the setting of age-related disorders such as AD.
Similar content being viewed by others

Background
Alzheimer’s disease (AD), the leading cause of dementia, is a heterogeneous neurodegenerative disorder in terms of clinical presentations and the density and distribution of the cardinal neuropathologic lesions. The neuropathologic hallmarks of AD are senile plaques composed of extracellular deposits of amyloid-β (Aβ) and neurofibrillary tangles composed of intracellular aggregates of tau protein with multiple post-translational modifications including phosphorylation. Senile plaques are complicated and heterogeneous lesions that contain not only amyloid deposits and tau positive neurites, but also neurites with degenerating pre- and post-synaptic elements (so-called dystrophic neurites), as well as activated microglia and reactive astrocytes [1]. Aβ deposits within the walls of blood vessels in the form of amyloid angiopathy are found in many patients with AD, but it is also found in other neurologic disorders [2]. Tau deposits are also found in neuronal cell processes (“neuropil threads”) and in dystrophic neurites within senile plaques [1, 3]. Neurofibrillary tangles are not exclusive to AD, but are found in a wide range of neurological disorders [4], as hereditary disorders [5] or secondary pathologic processes [6], due to environmental or genetic factors. Based on the density of neurofibrillary tangles in the hippocampus relative to those in the neocortex, AD can be classified into three clinicopathologic subtypes: typical AD, hippocampal sparing AD, and limbic predominant AD [7]. The clinicopathologic classification of AD subtypes has recently been confirmed and extended in living patients with neuroimaging methods [8, 9], identifying additional subtypes, including minimal change AD and AD with asymmetrical neocortical involvement.
Clinically, the two major presentations of AD can be classified as amnestic and non-amnestic. The former is characterized by deficiencies in short-term memory, recall and learning, which are the most common clinical presentations of typical and limbic predominant subtypes of AD. The latter shows impairment in other cognitive domains, such as language, visuospatial skills, or executive functioning. This is often associated with hippocampal sparing AD.
In addition to senile plaques and neurofibrillary tangles, many AD brains have other pathological lesions, such as cerebrovascular pathology, Lewy bodies, argyrophilic grain disease, hippocampal sclerosis, cerebral amyloid angiopathy, and transactive response DNA binding protein of 43 kDa (TDP-43) pathology [10, 11]. Importantly, these additional pathologies significantly increase the risk for dementia compared to patients with only one pathology [12]. The mixed pathologies also lower the threshold and accelerate the progression for clinical diagnosis of AD [13]. More recently, Spina and coworkers systematically investigated co-pathologies in early-onset and late-onset AD patients and found that the number of co-pathologies was associated with worse cognitive performance [11]. In this review, we focus on TDP-43 in aging and AD from clinical, pathological, and basic research perspectives.
Biology of TDP-43
TDP-43 is a 43 kDa heterogeneous nuclear ribonuclear protein (hnRNP) composed of 414 amino acids and is encoded by the TARDBP gene located on chromosome 1 (1p36.22) [14]. TDP-43 is synthesized in the cytoplasm and shuttled into the nucleus where it primarily resides to perform its physiological functions.
Biological function of TDP-43
The function of TDP-43, much like other hnRNPs, is to regulate gene expression and other aspects of RNA processing including RNA splicing, mRNA turnover, RNA trafficking, and microRNA (miRNA) biogenesis [15,16,17,18,19,20,21,22]. TDP-43 targets over 4,000 different mRNA transcripts [23], ranging from disease-associated transcripts [18], to its own mRNA transcript [17]. Disruption of the proper regulation of TDP-43 may contribute to its pathogenesis. Studies have shown that TDP-43 self-regulates through a negative feedback loop where TDP-43 destabilizes its mRNA transcript by binding to the 3’ untranslated region [17]. Interestingly, TDP-43 has been shown to down-regulate tau expression by destabilizing its mRNA transcripts [18]. Furthermore, TDP-43 might regulate the ratio of 4-repeat tau and 3-repeat tau via alternative splicing of tau exon 10 [24]. However, the regulation of tau expression by TDP-43 was not replicated in another independent study of AD [25]. Thus, the relationship between TDP-43 and the expression of tau remains unclear and needs to be further investigated.
Additionally, TDP-43 plays a role in the cellular stress response [15, 26,27,28]. If a cell is exposed to certain stressors (i.e., heat shock, oxidative stress, or viral infection), it can regulate levels of mRNA to conserve energy and prioritize cell survival [29, 30]. Stress granules are cytoplasmic foci in response to cellular stress that contain non-essential RNA. TDP-43 associates with ribosomes in stress granules to temporarily halt translation and promote cytoprotective protein synthesis [15, 31].
TDP-43 has been reported to regularly shuttle between the cytoplasm and nucleus depending on transcriptional needs [32]. Interestingly, low levels of TDP-43 have even been found to reside inside of mitochondria in human motor and cortical neurons; however, age-matched neurons from amyotrophic lateral sclerosis (ALS) and frontotemporal lobar degeneration (FTLD) patients expressed a significantly higher amount of mitochondrial TDP-43, reportedly altering their morphology and impairing mitochondrial function [33].
Protein structure of TDP-43
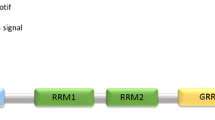
The structure of TDP-43 is composed of an N-terminal domain, a nuclear localization sequence (NLS), two RNA binding domains (RBD1 and RBD2), a nuclear export signal (NES), and a C-terminal glycine rich domain (GRD) (Fig 1) [34]. The protein also has an amyloidogenic core region (residues 311-360) with two alpha-helices that convert into beta sheets in TDP-43 aggregates [35]. The NLS is critical for physiological function, as mutations or deletions of the NLS result in mislocalization and aggregation of TDP-43 that are characteristic of disease models [36,37,38]. Importin-α facilitates the transport of TDP-43 into the nucleus by binding to the NLS. The role of the NES in TDP-43 remains controversial. The export of TDP-43 from nucleus to cytoplasm is thought to be mediated by exportin XPO1 binding to the NES in the second RBD [39, 40]; however, recent data suggests that the export of TDP-43 from the nucleus does not require either XPO1 or the NES, but instead is exported through passive diffusion [37, 41,42,43,44]. The function of the N-terminus is to regulate the homodimerization of TDP-43 to ensure proper folding and mRNA splicing [45]. The C-terminus is important for mRNA splicing and hnRNP interactions, and it is also thought to play a role in the formation of TDP-43 inclusions [46]. Additionally, this portion of the protein has been referred to as a prion-like domain due to its low complexity and high proclivity for aggregation, as well as being the site for over 50 sporadic and familial ALS-associated mutations [34, 47,48,49].

Protein structure of transactive response DNA binding protein of 43 kDa (TDP-43). TDP-43 is a 414 amino acid protein with a nuclear localization sequence (NLS) followed by two RNA binding domains (RBD1 and RBD2), a nuclear export sequence (NES), and a glycine rich prion-like (GRD) C-terminus. The mutations reported to increase the risk of ALS (red), FTLD (blue), and AD (orange) are indicated
Polymorphisms of TARDBP gene and disease risks
Mutations in the TARDBP gene are mainly associated with ALS and located along the glycine rich C-terminal domain (Fig 1). In particular, TARDBP mutants, such as Q331K and M337V, have been well studied for their associations to ALS [50]. Fewer TARDBP mutations, including P112H [51] and G295S [52], have been linked to FTLD. A reported mutant, I383V, has been implicated in both ALS and FTLD [52,53,54,55]. In general, most of the disease-associated mutations in the TARDBP gene are associated with an increase in TDP-43 aggregation and toxicity [48]. Interestingly, there are reports of a rare missense mutation in the NLS region of TDP-43, A90V, which is speculated to increase the risk of AD through a loss-of-function mechanism [56,57,58].
TDP-43 pathology in ALS and FTLD
Pathological forms of TDP-43 were first identified in 2006 when ALS and FTLD patients were found to have tau-negative, ubiquitin-positive cytoplasmic inclusion bodies [59,60,61]. The pathogenic mechanisms in these brains ultimately result in TDP-43 depletion from the nucleus, TDP-43 mislocalization into the cytoplasm, and the formation of insoluble aggregates that contain TDP-43 with multiple posttranslational modifications including ubiquitination, phosphorylation, and truncation [59,60,61,62,63]. These TDP-43 inclusion bodies found in neurons, neuronal cell processes, and glia are now characteristic of the pathology in the most common forms of ALS and FTLD [60, 63, 64].
Subtypes of TDP-43 pathology in ALS and FTLD
Based on the morphology, cell type, and distribution of TDP-43 pathology, FTLD-TDP can be classified into four main subtypes [65,66,67,68,69] (Fig 2). Type A is characterized by compact neuronal cytoplasmic inclusions (NCIs) and short dystrophic neurites (DNs) with occasional neuronal intranuclear inclusions (“cat-eye” inclusions) (NIIs) distributed preferentially in upper neocortical layers. Type B is characterized by diffuse granular NCIs and sparse DNs with inclusions showing no preference for superficial or deep neocortical layers. Oligodendroglial cytoplasmic inclusions are common in affected cortices and subcortical white matter, especially Type B cases associated with motor neuron disease. Type C is characterized by numerous DNs predominantly in superficial and deep neocortical layers, which are longer and thicker than those seen in Type A. Sparse NCIs are detected in the neocortex, but dense, compact, and round NCIs (“Pick body-like”) are frequent in the hippocampal dentate gyrus and in the basal ganglia, especially the putamen. The most distinctive feature of Type D is the presence of numerous NIIs, including both round inclusions and “cat-eye” type inclusions. Type D has variable DNs and NCIs. A fifth subtype, Type E, has been proposed [68], but it is less widely accepted. The characteristic features of Type E are granulofilamentous neuronal inclusions, abundant grains, and oligodendroglial inclusions that affect all layers of the neocortex. Among the TDP-43 subtypes, Type A is the most common type, followed by Type B. This pathologic subtyping has a good correlation with clinical phenotypes and genetics. Type A is most often associated with behavioral variant frontotemporal dementia (bvFTD) or progressive non-fluent aphasia (PNFA), while Type B is associated with bvFTD with or without motor neuron disease (MND). Most cases of FTLD due to GRN mutations have Type A; many, but not all cases of FTLD with C9ORF72 mutations have Type B. Semantic dementia (SD) and bvFTD are common clinical phenotypes in Type C, but no genetic association has been reported. Type D is associated with frontotemporal dementia (FTD) and Paget’s disease of bone caused by mutations of VCP gene [70]. This classification has been demonstrated to be supported by clinical, biochemical, and genetic correlational studies for FTLD-TDP [69], but needs to be further examined in AD cases to evaluate its pathological significance.

Representative images of TDP-43 pathology subtypes in FTLD-TDP brains. (A) Immunohistochemistry with an anti-phosphorylated-TDP-43 antibody (pSer409/pSer410) shows numerous neuronal cytoplasmic inclusions, short dystrophic neurites, and neuronal intranuclear inclusion (inset) in Type A; diffuse granular neuronal cytoplasmic inclusions in Type B; and numerous thick and long dystrophic neurites in Type C, in the superficial layer of the midfrontal gyrus (upper panel). In the dentate gyrus (lower panel), Type A shows compact neuronal cytoplasmic inclusions; Type B shows diffuse granular neuronal cytoplasmic inclusions; and Type C shows Pick body-like neuronal cytoplasmic inclusion. Scale bar = 50 μm. (B) A summary of clinical, pathological, and genetic features of TDP-43 pathology subtypes. NCI: Neuronal cytoplasmic inclusion; NII: Neuronal intranuclear inclusion; DN: Dystrophic neurite; GCI: Glia cytoplasmic inclusion; DG: Dentate gyrus; FTD: Frontotemporal dementia; bvFTD: Behavioral variant frontotemporal dementia; PNFA: Progressive non-fluent aphasia; MND: Motor neuron disease; and SD: Semantic dementia
Progression pattern of TDP-43 pathology in ALS and FTLD
Pathological progression of TDP-43 varies depending upon the underlying neurodegenerative disease with different progression patterns proposed for FTLD and ALS by Brettschneider et al [71, 72]. In bvFTD, stage 1 is associated with the lowest level of TDP-43 pathology in the basal and anterior portions of the prefrontal cortex, the pathology then invades other regions of the prefrontal cortex including the middle frontal gyrus and insular cortex as stage 2, leading into the motor cortex and parietal cortical areas as stage 3, and finally reaches stage 4, the most advanced stage, with widespread and high density TDP-43 pathology involved in the occipital cortex [71]. The staging scheme for ALS includes early involvement of the motor cortex, brainstem and spinal cord (stage 1), prefrontal cortex (stage 2), postcentral cortex and striatum (stage 3), and finally, TDP-43 pathology infiltrates the anteromedial temporal lobe (stage 4) [72].
Pathogenesis of TDP-43 in ALS and FTLD
Ubiquitination, phosphorylation, and truncation modifies the conformation of TDP-43, as well as its size and charge, contributing to the decreased shuttling into the nucleus [73,74,75]. Ubiquitin commonly binds to proteins to target them for eventual degradation. Lys-84, one of the multiple TDP-43 ubiquitination sites, is reported to be involved in the nuclear import of TDP-43 [76]. TDP-43 is phosphorylated most often at serine residues but can also be phosphorylated at tyrosine or threonine residues. The serine residues most often affected are serines 403, 404, 409 and 410; with serines 409 and 410 being the most common [77, 78]. Cytoplasmic TDP-43 can be cleaved by calpains and caspases into N-terminal fragments and C-terminal fragments (CTFs) with molecular weights of 35 and 20-25 kDa, respectively [79,80,81]. These fragments, in particular the CTFs, have been found to induce formation of ubiquitinated and phosphorylated cytoplasmic TDP-43 aggregates in vitro [82]. It is possible that neither phosphorylation nor ubiquitination is necessary for TDP-43 aggregation. Early-stage inclusions are neither ubiquitinated nor phosphorylated, and ubiquitination is usually associated with late stages in the aggregation process of in vitro neuronal cell culture models [79, 83]. Additionally, the potential lack of the NLS, precluding TDP-43 from shuttling back to the nucleus, may contribute to formation of aggregates [73].
An impairment in the clearance of TDP-43 may also contribute to the pathogenic process. An in vitro study has indicated that soluble TDP-43 is degraded by the ubiquitin-proteasome system, while insoluble TDP-43 aggregates are degraded via the autophagy system [84]. Other investigators determined that TDP-43 has a KFERQ-like sequence in the RBD1 domain, specifically QVKKD (amino acids 134 to 138), that allows Hsc70 binding and degradation of soluble, non-aggregated TDP-43 by chaperone-mediated autophagy [85]. Interestingly, degradation of TDP-43 species, particularly the CTFs, were much higher by the ubiquitin-proteasome system than by autophagy [85], suggesting that TDP-43 can be cleared through both mechanisms depending on its specific form.
In a non-diseased state, a balance between soluble and insoluble forms of RNA binding proteins (including TDP-43) and cell stress granules is maintained in the cytoplasm primarily due to their reversibility during cellular stress response [31]. In ALS and FTLD, this balance is possibly compromised due to the increased presence of aggregated TDP-43 within the cytoplasm, which in turn may increase cellular stress that leads to the formation of additional stress granules and the aggregation of RNA binding proteins, acting as seeds for TDP-43 aggregation [31]. TDP-43 can also be found within stress granules themselves depending on the conditions used to induced stress. For example, stress induced by sodium arsenite produces increased TDP-43 in stress granules [15, 86]. It has also been reported that TDP-43 inclusion bodies co-localize with markers of stress granules [26, 78, 86,87,88,89]. Interestingly, only the full length TDP-43 species, but not the CTFs, are recruited into stress granules, which requires both the RBD1 and GRD domains [90]. On the other hand, some investigators suggested that co-localization of TDP-43 with stress granules depends on RNA-bound forms of TDP-43. RNA-bound TDP-43 in stress granules is soluble, while free TDP-43 can form insoluble aggregates independent of stress granules [15, 91]. Together, the relationship between stress granules and TDP-43 pathology is a research focus that needs further investigation.
Gain of toxic and loss of normal function of TDP-43 in ALS and FTLD
Neuronal death associated with pathological TDP-43 is thought to be caused by a combination of both a toxic gain of function, as well as a loss of physiological function associated with depletion of TDP-43 from the nucleus [73]. Oligomeric and cytoplasmic aggregates of TDP-43 have been shown to be cytotoxic both in vitro and in vivo [92,93,94,95]. Additionally, mislocalized and aggregated TDP-43 can enhance mislocalization of nuclear TDP-43 and hinder intracellular transport [20, 47, 96,97,98]. Cytoplasmic mislocalization of TDP-43 may predispose the cell to stress since this has been shown to be associated with markers of stress response in cell culture model systems [47, 91, 99, 100].
Loss of function of TDP-43 is another mechanism implicated in neuronal loss in ALS and FTLD. Studies in mouse models rarely detect TDP-43 cytoplasmic inclusion bodies; however, neurodegeneration associated with loss nuclear TDP-43 can be evident [101]. In humans with C9ORF72-linked FTLD, there is loss of nuclear TDP-43 at pre-symptomatic stages [102]. Furthermore, the mere lack of nuclear TDP-43 is sufficient to cause neuronal atrophy [103]. This observation suggests that loss of nuclear TDP-43 is an early pathological event that might drive neurodegeneration. Additionally, loss of nuclear TDP-43 may modify chromatin accessibility leading to altered gene expression [20, 27, 47, 97, 104,105,106,107,108,109,110,111].
Interestingly, nuclear TDP-43 suppresses splicing of non-conserved cryptic exons, reducing the number of frameshift or nonsense mutations in mRNA transcripts [104, 112]. Patients with ALS or FTLD have impairments in non-conserved cryptic exon suppression function leading to the decay of mutated transcripts and disturbance in translation [110, 111, 113]. Cryptic exon splicing has also been noted in AD with TDP-43 pathology, including those with cytoplasmic inclusion bodies and those with only nuclear depletion of TDP-43, suggesting that impairments of TDP-43 cryptic exon repression may be an early event in TDP-43 pathogenesis in FTLD, ALS and a subset of patients with AD [114].
TDP-43 pathology in AD
TDP-43 pathology is frequently detected in pathologically confirmed AD brains in up to 57% of AD cases [10, 115,116,117,118,119,120,121,122,123], where it has been associated with worse brain atrophy and greater memory loss in AD patients [116]. TDP-43 species have been shown to colocalize with senile plaques and neurofibrillary tangles, with experimental evidence suggesting a direct interaction between TDP-43 and Aβ or tau [122, 124,125,126,127,128]. Furthermore, TDP-43 pathology in AD is associated with the severity of AD pathology, including higher Braak neurofibrillary tangle stages and Thal amyloid phases [129]. Additionally, TDP-43 NCIs in AD cases exhibit a variety of TDP-43 species with distinct patterns in terms of TDP-43 phosphorylation sites and the presence or absence of non-phosphorylated, N-terminal and C-terminal epitopes [130]. Altogether, it suggests that TDP-43 pathology could play a role in AD progression or be secondary to reactive changes that occur in advanced AD (Fig 3).

Illustration of the involvement of TDP-43 in the progression of Alzheimer’s disease. In the brain of Alzheimer’s disease (AD), the amyloid-β (Aβ) peptide is produced through the proteolytic processing of a transmembrane protein, amyloid precursor protein (APP) by β- and γ-secretases. The accumulation of soluble Aβ monomers in the brain parenchyma leads to the formation of Aβ oligomers, fibrils, and eventually Aβ plaques, due to overproduction and/or impaired Aβ clearance pathways contributing to the development of AD. The pathological changes of tau protein decrease its microtubule binding capacity and disrupts microtubule stability causing microtubule disintegration. The intracellular aggregates of tau protein form the neurofibrillary tangles. Tau deposits are also found in neuronal cell processes (“neuropil threads”) and in dystrophic neurites within Aβ plaques. Aβ plaques are heterogeneous lesions containing not only amyloid deposits and tau-positive neurites, but also neurites with degenerating pre- and post-synaptic elements (neurite dystrophy), as well as activated microglia, reactive astrocytes, and dysfunction of oligodendrocytes causing demyelination. TDP-43 is synthesized in the cytoplasm and retains the ability to shuttle from the cytoplasm into the nucleus where it primarily resides to perform its physiological functions such as RNA splicing. During the progression of AD, the pathogenic events lead to TDP-43 depletion from the nucleus, TDP-43 mislocalization into the cytoplasm, and the formation of insoluble TDP-43 aggregates. The neurodegeneration brought about by pathological TDP-43 can be caused by a potential combination of both a loss of physiological function and a gain of toxic functions
Clinical significance of TDP-43 in AD
TDP-43 has been reported to influence the clinical features of dementia, including cognitive deficits and the likelihood of dementia. Josephs and coworkers sought to determine the frequency of TDP-43 pathology across AD subtypes and its effects on cognition [119]. They found that deposition of TDP-43 was frequent in limbic predominant (67%) and typical AD subtypes (59%), but less frequent in the hippocampal sparing subtype (21%) [119]. Although the frequency of TDP-43 deposition in AD varies by pathological subtype, the observed effects of TDP-43 on clinical features, such as exacerbating cognitive decline, were consistent across pathological subtypes [119]. Another study investigated TDP-43, mixed pathologies, and clinical AD type dementia in the Religious Orders Study and the Rush Memory and Aging Project (ROSMAP) cohort with 946 old-age adults (89.3 ± 6.5 years) [115]. TDP-43 pathology was present in 52% of the participants; 65% in individuals with Alzheimer’s-type dementia and 44% in cognitively normal individuals. Additionally, coexistence of both TDP-43 and AD pathology was more common in those with Alzheimer’s-type dementia (54%) than those without dementia (25%) [115]. After using a logistic regression model and accounting for age, sex, and education, the investigators discovered that not only mixed AD and TDP-43 pathology, but also TDP-43 pathology, alone, was associated with Alzheimer’s-type dementia with an odds ratio of 6.73 and 1.51, respectively [115]. Similarly, McAleese and coworkers investigated the frequency of TDP-43 pathology in 119 individuals with autopsy-confirmed AD, dementia with Lewy bodies (DLB), mixed AD/DLB, and non-demented elderly controls [120]. TDP-43 pathology was present in all groups but was the highest in AD (73.9%) and mixed AD/DLB (52.6%) groups.
Overall, these results suggest that TDP-43 pathology is common in AD, especially in the limbic predominant subtype. These results also suggest TDP-43 pathology is a risk factor for developing dementia of the Alzheimer type independent of pathological subtypes, and TDP-43 pathology increases the rate of hippocampal atrophy in AD.
Progression pattern of TDP-43 pathology in AD
Interestingly, TDP-43 presenting as a secondary comorbid pathology in AD follows its own distinct pathological distribution pattern compared to that of ALS and FTLD. Josephs et al proposed that the progression of TDP-43 pathology in AD occurs in six stages, with stage 1 being characterized by TDP-43 pathology present within the amygdala (Fig 4) [117]. Progression into the entorhinal cortex and subiculum of the hippocampus defines stage 2, while stage 3 involves the hippocampal dentate gyrus and occipitotemporal cortex. In a subset of cases, the hippocampus has neuronal loss and gliosis consistent with hippocampal sclerosis [118, 123], but in other cases TDP-43 pathology is associated with Alzheimer type lesions, in particular neurofibrillary tangles [123]. The phenomenon of TDP-43 colocalization in neurons with neurofibrillary tangles has been termed Type β [131], to distinguish it from genuine NCI in Type B cellular pathology. As the pathology progresses into stage 4, the insular cortex, ventral striatum, basal forebrain, and inferior temporal cortex become affected. In stage 5, TDP-43 pathology now involves the brainstem nuclei, including the substantia nigra, inferior olivary nucleus, and midbrain tectum. The final stage, stage 6, is associated with involvement of basal ganglia and middle frontal cortex [117]. The TDP-43 stage was not affected by the age at onset, nor the time from onset to death in these AD patients [117]. This staging scheme is supported by assessment of clinical behavior, pathological characteristics, neuroimaging, and genetics; however, the underlying mechanisms driving distribution of TDP-43 in AD is unclear.

Distribution pattern of TDP-43 pathology in AD. (Upper panel) Illustration of TDP-43 stage in AD. An anterior coronal section depicts TDP-43 pathology progression from the amygdala (Stage 1), into the subiculum and entorhinal cortex (Stage 2), and then leads into the occipitotemporal cortex and dentate gyrus (Stage 3) of the hippocampus, followed by the insular cortex and the inferior temporal cortex (Stage 4). After progressing into the substantia nigra (Stage 5), the pathology reaches its final stage at the basal ganglia (putamen and globus pallidus) and middle frontal cortex (Stage 6). (Lower panel) Immunohistochemistry with an anti-phosphorylated-TDP-43 antibody shows representative images of TDP-43 pathology in different brain regions of each stage. Stage 1, amygdala; Stage 2, entorhinal cortex; Stage 3, dentate gyrus; Stage 4, inferior temporal gyrus; Stage 5, substantia nigra; Stage 6, middle frontal gyrus. Scale bar = 50 μm
TDP-43 and Aβ
In vitro and in vivo data have indicated that pathologic processes leading to AD and those leading to TDP-43 aggregation may influence one another. One study found that full length recombinant TDP-43 can form stable and spherical oligomers that can be recognized and bound by A11, an anti-amyloid oligomer specific antibody [92]. TDP-43 oligomers found in AD and FTLD brains [92, 127] are toxic to neurons both in vitro and in vivo through mechanisms that include reducing the DNA binding capacity of TDP-43, suggesting that oligomerization of TDP-43 may lead to gain of toxic function, as well as loss of physiological function [92]. The investigators also noted that soluble Aβ is converted to Aβ oligomers in the presence of TDP-43 oligomers due to their ability to cross-seed [92, 132]. This suggests that TDP-43 and Aβ have structurally similar domains that could contribute to the formation of Aβ-TDP-43 complexes. The frequent detection of TDP-43 positive inclusion bodies in AD could be due in part to this potential cross-seeding capacity of Aβ with TDP-43 [132]. Interestingly, full length TDP-43, as well as truncated N-terminal and C-terminal variants, were found to reduce Aβ fibrillization in a dose-dependent manner at oligomeric and other pre-fibril stages [92, 125]. Analogous to the most significant deficits seen in humans with AD and TDP-43 pathologies, mice with recombinant TDP-43 oligomers injected into the hippocampus had exacerbation of neuroinflammation and memory deficits [125].
A study investigating the relationship between TDP-43 and AD found that late stage AD patients have increased pathological cortical TDP-43 [122], which is consistent with the finding that TDP-43 pathology is associated with severe AD pathology [129]. Similar to late-stage AD, the investigators also noted an increase in TDP-43 pathology after Aβ (1-42) expressing lentiviral injections into the cortices of rats, as well as co-localization of intracellular Aβ with TDP-43, and association between phospho-TDP-43 and Aβ [122]. These data suggest a direct relationship between pathological TDP-43 and expression of Aβ in cells [122].
Another study revealed that the overexpression of TDP-43 in the cortex and hippocampus of an APP/PS1 mouse model (carrying mutant APP and PSEN1 genes) resulted in a decrease in Aβ plaque burden [124]. In this TDP-43 overexpression model there was also increased formation of TDP-43 oligomers [124]. In addition, there were increased levels of the amyloid precursor protein (APP) in the lysosomes, which might be the explanation for reduced Aβ plaques rather than inhibition of amyloid fibrilization by direct interaction with Aβ and extracellular TDP-43.
In another model system, there was increased neurodegeneration in the hippocampus of an APP/PS1 mouse model with conditional TDP-43 knockout in the forebrain [133], suggesting that TDP-43 depletion may contribute to neurodegeneration. Perhaps loss of TDP-43 function due to pathological modifications and mislocalization in a background of AD pathology may function in a similar way to TDP-43 depletion, possibly exacerbating neurodegeneration similar to results observed in AD [116]. Interestingly, the APP/PS1 TDP-43 knockout mice had a decrease in Aβ burden but increased oligomeric Aβ levels [133], suggesting that both overexpression and depletion of TDP-43 result in similar Aβ outcomes. Similarly, microglial-specific inducible conditional TDP-43 knockout in an APP mouse line was found to increase phagocytic activity of microglia, which resulted in increased amyloid clearance and reduction in Aβ plaque burden [134]. Additionally, microglial-specific TDP-43 depletion induced synaptic loss, even in the absence of amyloid, which may contribute to downstream neurodegeneration possibly due to synaptic pruning by overactive microglia [134]. These data suggest that microglial phagocytic activity, and thus Aβ clearance, may be at least in part regulated through TDP-43.
TDP-43 and Tau
Cytoplasmic inclusions in FTLD are typically immunoreactive for either Tau or TDP-43, respectively, thus the clarification of two subtypes FTLD-Tau and FTLD-TDP [135]. However, there has been studies that investigated the relationship between tau and TDP-43 outside the context of FTLD. For example, an in vitro study revealed that tau oligomer treatment increased nuclear levels of both phosphorylated and non-phosphorylated TDP-43 monomers in a dose-dependent manner [127]. Additionally, as the concentration of tau oligomers increased, the levels of phosphorylated TDP-43 oligomers in the cytoplasm increased as well, resulting in accumulations of phosphorylated TDP-43 oligomers that were also immunoreactive for tau oligomers [127], suggesting that the presence of tau oligomers induces the mislocalization and polymerization of TDP-43 species into oligomers and aggregates, and that tau oligomers may be able to cross-seed with TDP-43. Furthermore, TDP-43 oligomers were found to co-localize with tau and Aβ in AD and FTLD post-mortem brains [127].
The previously discussed APP/PS1 mouse model with TDP-43 overexpression was associated with increased pathological tau, which suggests that TDP-43 could play a role in neurofibrillary tangle development [124]. Furthermore, phosphorylated tau was present within mouse neuronal extensions in APP/PS1 transgenic mice with TDP-43 overexpression. In addition, colocalization of TDP-43 and phosphorylated tau has been detected in AD brains, with distinct tau and TDP-43 filaments within the same neuron [123, 136]. These data suggest that depending on the context, TDP-43 and tau may influence one another’s pathological progression; TDP-43 can promote pathological tau accumulation, or vice versa. However, an inverse association between TDP-43 and tau within post-mortem AD brains was also reported, possibly due to the negative regulation of tau transcripts by TDP-43 [18]. Therefore, additional studies are required to elucidate the relationship between TDP-43 and tau in AD development.
TDP-43 and APOE in AD
Apolipoprotein E (apoE), a glycoprotein present within the central nervous system and periphery, is an important lipid transporter, especially for cholesterol [137]. The human APOE gene has three alleles: APOE2, APOE3, and APOE4, with APOE2 being associated with the reduced risk for late-onset AD, while APOE4 is a major risk factor for late-onset AD [137,138,139]. ApoE has been well-known to influence Aβ pathology, as well as other neurodegenerative disease pathologies, including α-synuclein, in an isoform-dependent manner [118, 140,141,142,143,144]. Associations between APOE4 and TDP-43 pathology have also been reported [116, 118, 145]. A case study suggested that apoE and TDP-43 can form complexes based on co-immunoprecipitation data, and that APOE genotype can affect the severity of the complex burden with the APOE4/4 individual suffering from a higher burden compared to APOE3/3 [146]. Using a cohort from Mayo Clinic’s brain bank, Josephs and coworkers determined that pathologically confirmed AD patients with TDP-43 co-pathology were also more likely to carry the APOE4 allele when compared to TDP-43 negative AD cases [116]. Additionally, these individual’s scores on multiple cognitive impairment tests were decreased and cognitive impairment was more likely to present itself before death [116]. Similarly, another study based upon the ROSMAP cohort has reported that the stage and burden of TDP-43 pathology are positively correlated with the number of APOE4 alleles, even after controlling for amyloid, tau, and Lewy body pathologies [118]. Wennberg and coworkers analyzed a cohort of 751 pathologically confirmed AD cases for TDP-43 status, APOE genotype, tau neurofibrillary tangle stage, and Aβ status and found a direct association between APOE4 and TDP-43; the association was mediated by Aβ and tau [145]. Overall, these data suggest that APOE4 increases TDP-43 burden and likely increases the risk of TDP-43 pathology in AD by processes linked to Alzheimer type pathology and also processes independent of Aβ, thus contributing to detrimental effects of APOE4 on cognition later in life.
TDP-43 pathology in aging and hippocampal sclerosis (HS) of the elderly
Age-dependent demethylation of the TARDBP 3′ untranslated region has been reported to increase TARDBP mRNA expression in the motor cortex in ALS [147]. Besides ALS, aging is considered a risk factor for developing TDP-43 pathology even in neurologically normal individuals [148,149,150]. From 286 consecutive autopsy brains, Uchino and coworkers reported that 40% of control elderly individuals (78.5 ± 9.7 years) with minimal senile plaques had TDP-43 pathology [151]. Additionally, TDP-43-positive individuals were reported to be significantly older than those without TDP-43 pathology from a study investigating TDP-43 in the anterior temporal pole cortex [152]. These data suggest that TDP-43 pathology in the anterior temporal pole cortex is an important early neocortical stage of TDP-43 progression in aging and AD while extension of TDP-43 pathology to the midfrontal cortex is a late stage associated with more severe and global cognitive impairment [152]. Similarly, a study exploring age-related interneuron degeneration discovered that aged TDP-43 transgenic mice suffered from a significantly higher amount of TDP-43 positive inclusions than did non-transgenic aged mice as well as worse degeneration [153].
Hippocampal sclerosis (HS) increases in frequency with age and is a distinct process from AD, even though they both are associated with an amnestic clinical syndrome [154]. About 10-25% of individuals over the age of 85 are affected by HS-aging with the pathological feature of TDP-43 pathology in the hippocampus [150]. Neuronal loss in HS overlaps with that seen in epilepsy and hypoxic-ischemia, but the latter are not associated with TDP-43 pathology [155]. The discovery of TDP-43 pathology in HS of the elderly was the first evidence that this was a unique disease process that is associated with advanced age. Common genetic variants in GRN and TMEM106B are risk factors for FTLD [156, 157] and subsequent studies have also shown that they are risk factors for HS of the elderly [158, 159], linking this old age pathology to a similar disease process associated with FTLD. The GRN and TMEM106B genetic associations have also been observed in HS in the setting to Lewy body dementia, most of whom have at least some co-existing Alzheimer type pathology [160].
Given the fact that HS can be associated with degenerative, toxic, and selective hippocampal neuronal loss associated with anoxic-ischemic injury or epilepsy, the term HS has fallen out of favor. An international group of experts proposed a new name for TDP-43 pathology in the elderly, often associated with HS, “limbic-predominant age-related TDP-43 encephalopathy” (LATE) [149]. LATE neuropathological change (LATE-NC) is the term to refer to the pathology to distinguish it from the clinical syndrome, LATE, which remains to be defined, but is clearly associated with at least an amnestic syndrome. LATE-NC is characterized by TDP-43 neuronal and glial inclusions, with or without neuronal loss. TDP-43 pathology in LATE is concentrated in the limbic regions, including the amygdala, hippocampus, and anterior cingulate gyrus. According to a simplified staging scheme of LATE-NC, TDP-43 pathology initially forms in the amygdala (stage 1) and then extends to the hippocampus (stage 2) and the middle frontal gyrus (stage 3). Although it remains controversial [161], LATE can be differentiated from FTLD-TDP based on its epidemiology and severity of cortical TDP-43 pathology. LATE usually affects much older adults (present in 20-50% of individuals past 80 years old) than FTLD-TDP [149, 162]. TDP-43 pathology in the middle frontal gyrus in LATE-NC stage 3 is less severe than that of FTLD-TDP [163]. LATE is commonly found with co-pathologies including Aβ and tau [149]. Indeed, AD and LATE are often comorbid processes. LATE has been linked with robust disease-specific cognitive impairment, and it is one of the common age-related diseases that can imitate AD [149]. In the ROSMAP cohort, 15-20 percent of clinically diagnosed AD dementia patients at 80 years of age or older are associated with LATE [149].
TDP-43 and other neurodegenerative disorders
TDP-43 has been reported as a co-pathology in other neurodegenerative disorders besides AD, including Huntington’s disease, progressive supranuclear palsy (PSP), corticobasal degeneration (CBD), argyrophilic grain disease, DLB, and multiple system atrophy (MSA) [10, 120, 126, 128, 164,165,166,167,168,169,170]. In most cases, phosphorylated or truncated TDP-43 is a component of the cytoplasmic inclusions in these disorders, occasionally co-localizing with the primary pathology [120, 125,126,127]. The prevalence of the co-pathology depends on the primary pathology. For instance, over 57% of AD patients or 45% of CBD patients had TDP-43 pathology, while less than 6% of PSP or MSA patients had TDP-43 pathology [10, 116, 164, 168, 169].
An in vivo study using transgenic mice expressing human TDP-43 mutants found that administration of an autophagy-inducing drug could ameliorate TDP-43 pathology in the brain and spinal cord of the transgenic animals [171]. Given the fact that tau and α-synuclein pathologies also implicate disruption of autophagic pathways [172,173,174], developing active pharmacological agents to enhance autophagy flux may alleviate intracellular aggregation-prone proteins. Due to the ubiquitous nature of TDP-43 expression, it may not be a viable therapeutic approach to target TDP-43 in a generalized manner; however, strategies to modify the TDP-43 toxicity and to reduce TDP-43 aggregation may not only benefit FTLD and ALS patients [175], but also be relevant to more common age-related neurodegenerative disorders such as AD, Lewy body dementia, and LATE.
Conclusions
Significant efforts in the past decade have been placed in finding and testing new treatment methods for AD in hopes to prevent or cure this devastating disease. TDP-43 pathology, commonly found in AD brains, has been shown to influence AD pathology and neurodegeneration, whether it be decreasing senile plaque load through overexpression, or increasing amyloid oligomers and synapse loss through depletion. It also shares an important genetic risk factor with AD, the APOE4 gene. The mere presence of TDP-43 pathology increases the likelihood of developing Alzheimer-type dementia. These findings provide strong evidence for TDP-43 being an integral part of multiple neurodegenerative conditions, emphasizing the need to better understand the mechanisms of TDP-43 pathogenesis in AD and other age-related disorders.
Availability of data and materials
Not applicable.
Abbreviations
- Aβ :
-
Amyloid-β
- AD :
-
Alzheimer’s disease
- ALS :
-
Amyotrophic lateral sclerosis
- APOE :
-
Apolipoprotein E
- CBD :
-
Corticobasal degeneration
- CTF :
-
C-terminal fragment
- DG :
-
Dentate gyrus
- DLB :
-
Dementia with Lewy bodies
- DN :
-
Dystrophic neurite
- FTD :
-
Frontotemporal dementia
- bvFTD :
-
Behavioral variant frontotemporal dementia
- FTLD :
-
Frontotemporal lobar degeneration
- GCI :
-
Glia cytoplasmic inclusion
- GRD :
-
Glycine rich domain
- hnRNP :
-
Heterogeneous nuclear ribonuclear protein
- HS :
-
Hippocampal sclerosis
- LATE :
-
Limbic-predominant age related TDP-43 encephalopathy
- LATE-NC :
-
LATE neuropathological change
- MND :
-
Motor neuron disease
- MSA :
-
Multiple system atrophy
- NCI :
-
Neuronal cytoplasmic inclusion
- NES :
-
Nuclear export signal
- NII :
-
Neuronal intranuclear inclusion
- NLS :
-
Nuclear localization sequence
- PNFA :
-
Progressive non-fluent aphasia
- PSP :
-
Progressive supranuclear palsy
- RBD :
-
RNA binding domain
- ROSMAP :
-
Religious Orders Study and Rush Memory and Aging Project
- SD :
-
Semantic dementia
- SOD1 :
-
Superoxide dismutase
- TDP-43 :
-
Transactive response DNA binding protein of 43 kDa
References
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer's disease. Mol Neurodegener. 2019;14:32.
Ghiso J, Frangione B. Cerebral amyloidosis, amyloid angiopathy, and their relationship to stroke and dementia. J Alzheimers Dis. 2001;3:65–73.
Guo T, Zhang D, Zeng Y, Huang TY, Xu H, Zhao Y. Molecular and cellular mechanisms underlying the pathogenesis of Alzheimer's disease. Mol Neurodegener. 2020;15:40.
Spillantini MG, Goedert M. Tau protein pathology in neurodegenerative diseases. Trends Neurosci. 1998;21:428–33.
Spillantini MG, Murrell JR, Goedert M, Farlow M, Klug A, Ghetti B. Mutations in the tau gene (MAPT) in FTDP-17: the family with Multiple System Tauopathy with Presenile Dementia (MSTD). J Alzheimers Dis. 2006;9:373–80.
McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy. Brain Pathol. 2015;25:350–64.
Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol. 2011;10:785–96.
Levin F, Ferreira D, Lange C, Dyrba M, Westman E, Buchert R, et al. Alzheimer's Disease Neuroimaging I: Data-driven FDG-PET subtypes of Alzheimer's disease-related neurodegeneration. Alzheimers Res Ther. 2021;13:49.
Vogel JW, Young AL, Oxtoby NP, Smith R, Ossenkoppele R, Strandberg OT, et al. Four distinct trajectories of tau deposition identified in Alzheimer's disease. Nat Med. 2021;27:871–81.
Robinson JL, Lee EB, Xie SX, Rennert L, Suh E, Bredenberg C, et al. Neurodegenerative disease concomitant proteinopathies are prevalent, age-related and APOE4-associated. Brain. 2018;141:2181–93.
Spina S, La Joie R, Petersen C, Nolan AL, Cuevas D, Cosme C, et al. Comorbid neuropathological diagnoses in early versus late-onset Alzheimer's disease. Brain. 2021;144:2186–98.
Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204.
Kapasi A, DeCarli C, Schneider JA. Impact of multiple pathologies on the threshold for clinically overt dementia. Acta Neuropathol. 2017;134:171–86.
Stover CM, Lynch NJ, Hanson SJ, Windbichler M, Gregory SG, Schwaeble WJ. Organization of the MASP2 locus and its expression profile in mouse and rat. Mamm Genome. 2004;15:887–900.
Higashi S, Kabuta T, Nagai Y, Tsuchiya Y, Akiyama H, Wada K. TDP-43 associates with stalled ribosomes and contributes to cell survival during cellular stress. J Neurochem. 2013;126:288–300.
De Conti L, Akinyi MV, Mendoza-Maldonado R, Romano M, Baralle M, Buratti E. TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic Acids Res. 2015;43:8990–9005.
Ayala YM, De Conti L, Avendano-Vazquez SE, Dhir A, Romano M, D'Ambrogio A, et al. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011;30:277–88.
Gu J, Wu F, Xu W, Shi J, Hu W, Jin N, et al. TDP-43 suppresses tau expression via promoting its mRNA instability. Nucleic Acids Res. 2017;45:6177–93.
Fukushima M, Hosoda N, Chifu K, Hoshino SI. TDP-43 accelerates deadenylation of target mRNAs by recruiting Caf1 deadenylase. FEBS Lett. 2019;593:277–87.
Alami NH, Smith RB, Carrasco MA, Williams LA, Winborn CS, Han SSW, et al. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron. 2014;81:536–43.
Kawahara Y, Mieda-Sato A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc Natl Acad Sci U S A. 2012;109:3347–52.
Chew J, Cook C, Gendron TF, Jansen-West K, Del Rosso G, Daughrity LM, et al. Aberrant deposition of stress granule-resident proteins linked to C9orf72-associated TDP-43 proteinopathy. Mol Neurodegener. 2019;14:9.
Sephton CF, Cenik C, Kucukural A, Dammer EB, Cenik B, Han Y, et al. Identification of neuronal RNA targets of TDP-43-containing ribonucleoprotein complexes. J Biol Chem. 2011;286:1204–15.
Gu J, Chen F, Iqbal K, Gong CX, Wang X, Liu F. Transactive response DNA-binding protein 43 (TDP-43) regulates alternative splicing of tau exon 10: Implications for the pathogenesis of tauopathies. J Biol Chem. 2017;292:10600–12.
Niblock M, Hortobagyi T, Troakes C, Al-Sarraj S, Spickett C, Jones R, et al. Lack of association between TDP-43 pathology and tau mis-splicing in Alzheimer's disease. Neurobiol Aging. 2016;37:45–6.
Dewey CM, Cenik B, Sephton CF, Johnson BA, Herz J, Yu G. TDP-43 aggregation in neurodegeneration: are stress granules the key? Brain Res. 2012;1462:16–25.
Khalfallah Y, Kuta R, Grasmuck C, Prat A, Durham HD, Vande Velde C. TDP-43 regulation of stress granule dynamics in neurodegenerative disease-relevant cell types. Sci Rep. 2018;8:7551.
Kim HJ, Raphael AR, LaDow ES, McGurk L, Weber RA, Trojanowski JQ, et al. Therapeutic modulation of eIF2alpha phosphorylation rescues TDP-43 toxicity in amyotrophic lateral sclerosis disease models. Nat Genet. 2014;46:152–60.
Anderson P, Kedersha N. Stress granules: the Tao of RNA triage. Trends Biochem Sci. 2008;33:141–50.
Kedersha N, Chen S, Gilks N, Li W, Miller IJ, Stahl J, et al. Evidence that ternary complex (eIF2-GTP-tRNA(i)(Met))-deficient preinitiation complexes are core constituents of mammalian stress granules. Mol Biol Cell. 2002;13:195–210.
Baradaran-Heravi Y, Van Broeckhoven C, van der Zee J. Stress granule mediated protein aggregation and underlying gene defects in the FTD-ALS spectrum. Neurobiol Dis. 2020;134:104639.
Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, et al. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121:3778–85.
Wang W, Wang L, Lu J, Siedlak SL, Fujioka H, Liang J, et al. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat Med. 2016;22:869–78.
Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010;6:211–20.
Zhuo XF, Wang J, Zhang J, Jiang LL, Hu HY, Lu JX. Solid-State NMR Reveals the Structural Transformation of the TDP-43 Amyloidogenic Region upon Fibrillation. J Am Chem Soc. 2020;142:3412–21.
Walker AK, Spiller KJ, Ge G, Zheng A, Xu Y, Zhou M, et al. Functional recovery in new mouse models of ALS/FTLD after clearance of pathological cytoplasmic TDP-43. Acta Neuropathol. 2015;130:643–60.
Pinarbasi ES, Cagatay T, Fung HYJ, Li YC, Chook YM, Thomas PJ. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci Rep. 2018;8:7083.
Besnard-Guerin C. Cytoplasmic localization of amyotrophic lateral sclerosis-related TDP-43 proteins modulates stress granule formation. Eur J Neurosci. 2020;52:3995–4008.
Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, et al. The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature. 2015;525:56–61.
Chou CC, Zhang Y, Umoh ME, Vaughan SW, Lorenzini I, Liu F, et al. TDP-43 pathology disrupts nuclear pore complexes and nucleocytoplasmic transport in ALS/FTD. Nat Neurosci. 2018;21:228–39.
Nishimura AL, Zupunski V, Troakes C, Kathe C, Fratta P, Howell M, et al. Nuclear import impairment causes cytoplasmic trans-activation response DNA-binding protein accumulation and is associated with frontotemporal lobar degeneration. Brain. 2010;133:1763–71.
Winton MJ, Igaz LM, Wong MM, Kwong LK, Trojanowski JQ, Lee VM. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J Biol Chem. 2008;283:13302–9.
Archbold HC, Jackson KL, Arora A, Weskamp K, Tank EM, Li X, et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Sci Rep. 2018;8:4606.
Ederle H, Funk C, Abou-Ajram C, Hutten S, Funk EBE, Kehlenbach RH, et al. Nuclear egress of TDP-43 and FUS occurs independently of Exportin-1/CRM1. Sci Rep. 2018;8:7084.
Zhang YJ, Caulfield T, Xu YF, Gendron TF, Hubbard J, Stetler C, et al. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum Mol Genet. 2013;22:3112–22.
Conicella AE, Zerze GH, Mittal J, Fawzi NL. ALS Mutations Disrupt Phase Separation Mediated by alpha-Helical Structure in the TDP-43 Low-Complexity C-Terminal Domain. Structure. 2016;24:1537–49.
Suk TR, Rousseaux MWC. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol Neurodegener. 2020;15:45.
Jo M, Lee S, Jeon YM, Kim S, Kwon Y, Kim HJ. The role of TDP-43 propagation in neurodegenerative diseases: integrating insights from clinical and experimental studies. Exp Mol Med. 2020;52:1652–62.
de Boer EMJ, Orie VK, Williams T, Baker MR, De Oliveira HM, Polvikoski T, et al. TDP-43 proteinopathies: a new wave of neurodegenerative diseases. J Neurol Neurosurg Psychiatry. 2020;92(1):86–95.
Arnold ES, Ling SC, Huelga SC, Lagier-Tourenne C, Polymenidou M, Ditsworth D, et al. ALS-linked TDP-43 mutations produce aberrant RNA splicing and adult-onset motor neuron disease without aggregation or loss of nuclear TDP-43. Proc Natl Acad Sci U S A. 2013;110:E736–45.
Moreno F, Rabinovici GD, Karydas A, Miller Z, Hsu SC, Legati A, et al. A novel mutation P112H in the TARDBP gene associated with frontotemporal lobar degeneration without motor neuron disease and abundant neuritic amyloid plaques. Acta Neuropathol Commun. 2015;3:19.
Caroppo P, Camuzat A, Guillot-Noel L, Thomas-Anterion C, Couratier P, Wong TH, et al. Defining the spectrum of frontotemporal dementias associated with TARDBP mutations. Neurol Genet. 2016;2:e80.
Charoniti E, Papastefanopoulou V, Florou-Hatziyiannidou C, Koros C, Stanitsa E, Papatriantafyllou JD, et al. TARDBP p.I383V, a recurrent alteration in Greek FTD patients. J Neurol Sci. 2021;428:117566.
Rutherford NJ, Zhang YJ, Baker M, Gass JM, Finch NA, Xu YF, et al. Novel mutations in TARDBP (TDP-43) in patients with familial amyotrophic lateral sclerosis. PLoS Genet. 2008;4:e1000193.
Feng F, Wang H, Liu J, Wang Z, Xu B, Zhao K, et al. Genetic and clinical features of Chinese sporadic amyotrophic lateral sclerosis patients with TARDBP mutations. Brain Behav. 2021;11:e2312.
Vanden Broeck L, Kleinberger G, Chapuis J, Gistelinck M, Amouyel P, Van Broeckhoven C, et al. Functional complementation in Drosophila to predict the pathogenicity of TARDBP variants: evidence for a loss-of-function mechanism. Neurobiol Aging. 2015;36:1121–9.
Chang XL, Tan MS, Tan L, Yu JT. The Role of TDP-43 in Alzheimer's Disease. Mol Neurobiol. 2016;53:3349–59.
Brouwers N, Bettens K, Gijselinck I, Engelborghs S, Pickut BA, Van Miegroet H, et al. Contribution of TARDBP to Alzheimer's disease genetic etiology. J Alzheimers Dis. 2010;21:423–30.
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–11.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–3.
Mackenzie IR, Bigio EH, Ince PG, Geser F, Neumann M, Cairns NJ, et al. Pathological TDP-43 distinguishes sporadic amyotrophic lateral sclerosis from amyotrophic lateral sclerosis with SOD1 mutations. Ann Neurol. 2007;61:427–34.
Feneberg E, Gray E, Ansorge O, Talbot K, Turner MR. Towards a TDP-43-Based Biomarker for ALS and FTLD. Mol Neurobiol. 2018;55:7789–801.
Neumann M, Kwong LK, Lee EB, Kremmer E, Flatley A, Xu Y, et al. Phosphorylation of S409/410 of TDP-43 is a consistent feature in all sporadic and familial forms of TDP-43 proteinopathies. Acta Neuropathol. 2009;117:137–49.
Sreedharan J, Blair IP, Tripathi VB, Hu X, Vance C, Rogelj B, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–72.
Mackenzie IR, Neumann M, Baborie A, Sampathu DM, Du Plessis D, Jaros E, et al. A harmonized classification system for FTLD-TDP pathology. Acta Neuropathol. 2011;122:111–3.
Mackenzie IR, Neumann M. Subcortical TDP-43 pathology patterns validate cortical FTLD-TDP subtypes and demonstrate unique aspects of C9orf72 mutation cases. Acta Neuropathol. 2020;139:83–98.
Sampathu DM, Neumann M, Kwong LK, Chou TT, Micsenyi M, Truax A, et al. Pathological heterogeneity of frontotemporal lobar degeneration with ubiquitin-positive inclusions delineated by ubiquitin immunohistochemistry and novel monoclonal antibodies. Am J Pathol. 2006;169:1343–52.
Lee EB, Porta S, Michael Baer G, Xu Y, Suh E, Kwong LK, et al. Expansion of the classification of FTLD-TDP: distinct pathology associated with rapidly progressive frontotemporal degeneration. Acta Neuropathol. 2017;134:65–78.
Tan RH, Shepherd CE, Kril JJ, McCann H, McGeachie A, McGinley C, et al. Classification of FTLD-TDP cases into pathological subtypes using antibodies against phosphorylated and non-phosphorylated TDP43. Acta Neuropathol Commun. 2013;1:33.
Forman MS, Mackenzie IR, Cairns NJ, Swanson E, Boyer PJ, Drachman DA, et al. Novel ubiquitin neuropathology in frontotemporal dementia with valosin-containing protein gene mutations. J Neuropathol Exp Neurol. 2006;65:571–81.
Brettschneider J, Del Tredici K, Irwin DJ, Grossman M, Robinson JL, Toledo JB, et al. Sequential distribution of pTDP-43 pathology in behavioral variant frontotemporal dementia (bvFTD). Acta Neuropathol. 2014;127:423–39.
Brettschneider J, Del Tredici K, Toledo JB, Robinson JL, Irwin DJ, Grossman M, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38.
Gendron TF, Petrucelli L. Rodent models of TDP-43 proteinopathy: investigating the mechanisms of TDP-43-mediated neurodegeneration. J Mol Neurosci. 2011;45:486–99.
Guedes ACB, Santin R, Costa ASR, Reiter KC, Hilbig A, Fernandez LL. Distinct Phospho-TDP-43 brain distribution in two cases of FTD, one associated with ALS. Dement Neuropsychol. 2017;11:249–54.
Martinez-Gonzalez L, Rodriguez-Cueto C, Cabezudo D, Bartolome F, Andres-Benito P, Ferrer I, et al. Motor neuron preservation and decrease of in vivo TDP-43 phosphorylation by protein CK-1delta kinase inhibitor treatment. Sci Rep. 2020;10:4449.
Hans F, Eckert M, von Zweydorf F, Gloeckner CJ, Kahle PJ. Identification and characterization of ubiquitinylation sites in TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem. 2018;293:16083–99.
Gendron TF, Josephs KA, Petrucelli L. Review: transactive response DNA-binding protein 43 (TDP-43): mechanisms of neurodegeneration. Neuropathol Appl Neurobiol. 2010;36:97–112.
Gao J, Wang L, Huntley ML, Perry G, Wang X. Pathomechanisms of TDP-43 in neurodegeneration. J Neurochem. 2018;146(1):7–20.
Dormann D, Capell A, Carlson AM, Shankaran SS, Rodde R, Neumann M, et al. Proteolytic processing of TAR DNA binding protein-43 by caspases produces C-terminal fragments with disease defining properties independent of progranulin. J Neurochem. 2009;110:1082–94.
Berning BA, Walker AK. The Pathobiology of TDP-43 C-Terminal Fragments in ALS and FTLD. Front Neurosci. 2019;13:335.
Li Q, Yokoshi M, Okada H, Kawahara Y. The cleavage pattern of TDP-43 determines its rate of clearance and cytotoxicity. Nat Commun. 2015;6:6183.
Igaz LM, Kwong LK, Chen-Plotkin A, Winton MJ, Unger TL, Xu Y, et al. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J Biol Chem. 2009;284:8516–24.
Farrawell NE, Lambert-Smith IA, Warraich ST, Blair IP, Saunders DN, Hatters DM, et al. Distinct partitioning of ALS associated TDP-43, FUS and SOD1 mutants into cellular inclusions. Sci Rep. 2015;5:13416.
Scotter EL, Vance C, Nishimura AL, Lee YB, Chen HJ, Urwin H, et al. Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J Cell Sci. 2014;127:1263–78.
Huang CC, Bose JK, Majumder P, Lee KH, Huang JT, Huang JK, et al. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J Cell Sci. 2014;127:3024–38.
Ratti A, Gumina V, Lenzi P, Bossolasco P, Fulceri F, Volpe C, et al. Chronic stress induces formation of stress granules and pathological TDP-43 aggregates in human ALS fibroblasts and iPSC-motoneurons. Neurobiol Dis. 2020;145:105051.
Harrison AF, Shorter J. RNA-binding proteins with prion-like domains in health and disease. Biochem J. 2017;474:1417–38.
Zhang K, Daigle JG, Cunningham KM, Coyne AN, Ruan K, Grima JC, et al. Stress Granule Assembly Disrupts Nucleocytoplasmic Transport. Cell. 2018;173:958–71 e917.
Parker SJ, Meyerowitz J, James JL, Liddell JR, Crouch PJ, Kanninen KM, et al. Endogenous TDP-43 localized to stress granules can subsequently form protein aggregates. Neurochem Int. 2012;60:415–24.
Bentmann E, Neumann M, Tahirovic S, Rodde R, Dormann D, Haass C. Requirements for stress granule recruitment of fused in sarcoma (FUS) and TAR DNA-binding protein of 43 kDa (TDP-43). J Biol Chem. 2012;287:23079–94.
Mann JR, Gleixner AM, Mauna JC, Gomes E, DeChellis-Marks MR, Needham PG, et al. RNA Binding Antagonizes Neurotoxic Phase Transitions of TDP-43. Neuron. 2019;102:321–38 e328.
Fang YS, Tsai KJ, Chang YJ, Kao P, Woods R, Kuo PH, et al. Full-length TDP-43 forms toxic amyloid oligomers that are present in frontotemporal lobar dementia-TDP patients. Nat Commun. 2014;5:4824.
Capitini C, Conti S, Perni M, Guidi F, Cascella R, De Poli A, et al. TDP-43 inclusion bodies formed in bacteria are structurally amorphous, non-amyloid and inherently toxic to neuroblastoma cells. PLoS One. 2014;9:e86720.
Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity. J Biol Chem. 2009;284:20329–39.
Zhang T, Mullane PC, Periz G, Wang J. TDP-43 neurotoxicity and protein aggregation modulated by heat shock factor and insulin/IGF-1 signaling. Hum Mol Genet. 2011;20:1952–65.
Cascella R, Capitini C, Fani G, Dobson CM, Cecchi C, Chiti F. Quantification of the Relative Contributions of Loss-of-function and Gain-of-function Mechanisms in TAR DNA-binding Protein 43 (TDP-43) Proteinopathies. J Biol Chem. 2016;291:19437–48.
Liu-Yesucevitz L, Lin AY, Ebata A, Boon JY, Reid W, Xu YF, et al. ALS-linked mutations enlarge TDP-43-enriched neuronal RNA granules in the dendritic arbor. J Neurosci. 2014;34:4167–74.
Leibiger C, Deisel J, Aufschnaiter A, Ambros S, Tereshchenko M, Verheijen BM, et al. TDP-43 controls lysosomal pathways thereby determining its own clearance and cytotoxicity. Hum Mol Genet. 2018;27:1593–607.
Gasset-Rosa F, Lu S, Yu H, Chen C, Melamed Z, Guo L, et al. Cytoplasmic TDP-43 De-mixing Independent of Stress Granules Drives Inhibition of Nuclear Import, Loss of Nuclear TDP-43, and Cell Death. Neuron. 2019;102:339–57 e337.
Asakawa K, Handa H, Kawakami K. Optogenetic modulation of TDP-43 oligomerization accelerates ALS-related pathologies in the spinal motor neurons. Nat Commun. 2020;11:1004.
Igaz LM, Kwong LK, Lee EB, Chen-Plotkin A, Swanson E, Unger T, et al. Dysregulation of the ALS-associated gene TDP-43 leads to neuronal death and degeneration in mice. J Clin Invest. 2011;121:726–38.
Vatsavayai SC, Yoon SJ, Gardner RC, Gendron TF, Vargas JN, Trujillo A, et al. Timing and significance of pathological features in C9orf72 expansion-associated frontotemporal dementia. Brain. 2016;139:3202–16.
Nana AL, Sidhu M, Gaus SE, Hwang JL, Li L, Park Y, et al. Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 2019;137:27–46.
Torres P, Ramirez-Nunez O, Romero-Guevara R, Bares G, Granado-Serrano AB, Ayala V, et al. Cryptic exon splicing function of TARDBP interacts with autophagy in nervous tissue. Autophagy. 2018;14:1398–403.
Sasaguri H, Chew J, Xu YF, Gendron TF, Garrett A, Lee CW, et al. The extreme N-terminus of TDP-43 mediates the cytoplasmic aggregation of TDP-43 and associated toxicity in vivo. Brain Res. 1647;2016:57–64.
McDonald KK, Aulas A, Destroismaisons L, Pickles S, Beleac E, Camu W, et al. TAR DNA-binding protein 43 (TDP-43) regulates stress granule dynamics via differential regulation of G3BP and TIA-1. Hum Mol Genet. 2011;20:1400–10.
Tibshirani M, Zhao B, Gentil BJ, Minotti S, Marques C, Keith J, et al. Dysregulation of chromatin remodelling complexes in amyotrophic lateral sclerosis. Hum Mol Genet. 2017;26:4142–52.
Liu EY, Russ J, Cali CP, Phan JM, Amlie-Wolf A, Lee EB. Loss of Nuclear TDP-43 Is Associated with Decondensation of LINE Retrotransposons. Cell Rep. 2019;27:1409–21 e1406.
Briese M, Saal-Bauernschubert L, Luningschror P, Moradi M, Dombert B, Surrey V, et al. Loss of Tdp-43 disrupts the axonal transcriptome of motoneurons accompanied by impaired axonal translation and mitochondria function. Acta Neuropathol Commun. 2020;8:116.
Melamed Z, Lopez-Erauskin J, Baughn MW, Zhang O, Drenner K, Sun Y, et al. Premature polyadenylation-mediated loss of stathmin-2 is a hallmark of TDP-43-dependent neurodegeneration. Nat Neurosci. 2019;22:180–90.
Klim JR, Williams LA, Limone F. Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, et al: ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019;22:167–79.
Donde A, Sun M, Ling JP, Braunstein KE, Pang B, Wen X, et al. Splicing repression is a major function of TDP-43 in motor neurons. Acta Neuropathol. 2019;138:813–26.
Ling JP, Pletnikova O, Troncoso JC, Wong PC. TDP-43 repression of nonconserved cryptic exons is compromised in ALS-FTD. Science. 2015;349:650–5.
Sun M, Bell W, LaClair KD, Ling JP, Han H, Kageyama Y, et al. Cryptic exon incorporation occurs in Alzheimer's brain lacking TDP-43 inclusion but exhibiting nuclear clearance of TDP-43. Acta Neuropathol. 2017;133:923–31.
James BD, Wilson RS, Boyle PA, Trojanowski JQ, Bennett DA, Schneider JA. TDP-43 stage, mixed pathologies, and clinical Alzheimer's-type dementia. Brain. 2016;139:2983–93.
Josephs KA, Whitwell JL, Weigand SD, Murray ME, Tosakulwong N, Liesinger AM, et al. TDP-43 is a key player in the clinical features associated with Alzheimer's disease. Acta Neuropathol. 2014;127:811–24.
Josephs KA, Murray ME, Whitwell JL, Tosakulwong N, Weigand SD, Petrucelli L, et al. Updated TDP-43 in Alzheimer's disease staging scheme. Acta Neuropathol. 2016;131:571–85.
Yang HS, Yu L, White CC, Chibnik LB, Chhatwal JP, Sperling RA, et al. Evaluation of TDP-43 proteinopathy and hippocampal sclerosis in relation to APOE epsilon4 haplotype status: a community-based cohort study. Lancet Neurol. 2018;17:773–81.
Josephs KA, Whitwell JL, Tosakulwong N, Weigand SD, Murray ME, Liesinger AM, et al. TAR DNA-binding protein 43 and pathological subtype of Alzheimer's disease impact clinical features. Ann Neurol. 2015;78:697–709.
McAleese KE, Walker L, Erskine D, Thomas AJ, McKeith IG, Attems J. TDP-43 pathology in Alzheimer's disease, dementia with Lewy bodies and ageing. Brain Pathol. 2017;27:472–9.
Josephs KA, Dickson DW, Tosakulwong N, Weigand SD, Murray ME, Petrucelli L, et al. Rates of hippocampal atrophy and presence of post-mortem TDP-43 in patients with Alzheimer's disease: a longitudinal retrospective study. Lancet Neurol. 2017;16:917–24.
Herman AM, Khandelwal PJ, Stanczyk BB, Rebeck GW. Moussa CE: beta-amyloid triggers ALS-associated TDP-43 pathology in AD models. Brain Res. 2011;1386:191–9.
Amador-Ortiz C, Lin WL, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer's disease. Ann Neurol. 2007;61:435–45.
Davis SA, Gan KA, Dowell JA, Cairns NJ, Gitcho MA. TDP-43 expression influences amyloidbeta plaque deposition and tau aggregation. Neurobiol Dis. 2017;103:154–62.
Shih YH, Tu LH, Chang TY, Ganesan K, Chang WW, Chang PS, et al. TDP-43 interacts with amyloid-beta, inhibits fibrillization, and worsens pathology in a model of Alzheimer's disease. Nat Commun. 2020;11:5950.
Higashi S, Iseki E, Yamamoto R, Minegishi M, Hino H, Fujisawa K, et al. Concurrence of TDP-43, tau and alpha-synuclein pathology in brains of Alzheimer's disease and dementia with Lewy bodies. Brain Res. 2007;1184:284–94.
Montalbano M, McAllen S, Cascio FL, Sengupta U, Garcia S, Bhatt N, et al. TDP-43 and Tau Oligomers in Alzheimer's Disease, Amyotrophic Lateral Sclerosis, and Frontotemporal Dementia. Neurobiol Dis. 2020;146:105130.
Yokota O, Davidson Y, Bigio EH, Ishizu H, Terada S, Arai T, et al. Phosphorylated TDP-43 pathology and hippocampal sclerosis in progressive supranuclear palsy. Acta Neuropathol. 2010;120:55–66.
Katsumata Y, Fardo DW, Kukull WA, Nelson PT. Dichotomous scoring of TDP-43 proteinopathy from specific brain regions in 27 academic research centers: associations with Alzheimer's disease and cerebrovascular disease pathologies. Acta Neuropathol Commun. 2018;6:142.
Tome SO, Vandenberghe R, Ospitalieri S, Van Schoor E, Tousseyn T, Otto M, et al. Distinct molecular patterns of TDP-43 pathology in Alzheimer's disease: relationship with clinical phenotypes. Acta Neuropathol Commun. 2020;8:61.
Josephs KA, Murray ME, Tosakulwong N, Weigand SD, Serie AM, Perkerson RB, et al. Pathological, imaging and genetic characteristics support the existence of distinct TDP-43 types in non-FTLD brains. Acta Neuropathologica. 2019;137:227–38.
Guerrero-Munoz MJ, Castillo-Carranza DL, Krishnamurthy S, Paulucci-Holthauzen AA, Sengupta U, Lasagna-Reeves CA, et al. Amyloid-beta oligomers as a template for secondary amyloidosis in Alzheimer's disease. Neurobiol Dis. 2014;71:14–23.
LaClair KD, Donde A, Ling JP, Jeong YH, Chhabra R, Martin LJ, et al. Depletion of TDP-43 decreases fibril and plaque beta-amyloid and exacerbates neurodegeneration in an Alzheimer's mouse model. Acta Neuropathol. 2016;132:859–73.
Paolicelli RC, Jawaid A, Henstridge CM, Valeri A, Merlini M, Robinson JL, et al. TDP-43 Depletion in Microglia Promotes Amyloid Clearance but Also Induces Synapse Loss. Neuron. 2017;95:297–308 e296.
Bahia VS, Takada LT, Deramecourt V. Neuropathology of frontotemporal lobar degeneration: a review. Dement Neuropsychol. 2013;7:19–26.
Lin WL, Dickson DW. Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 2008;116:205–13.
Zhao N, Liu CC, Qiao W, Bu G. Apolipoprotein E, Receptors, and Modulation of Alzheimer's Disease. Biol Psychiatry. 2018;83:347–57.
Yamazaki Y, Zhao N, Caulfield TR, Liu CC, Bu G. Apolipoprotein E and Alzheimer disease: pathobiology and targeting strategies. Nat Rev Neurol. 2019;15:501–18.
Li Z, Shue F, Zhao N, Shinohara M, Bu G. APOE2: protective mechanism and therapeutic implications for Alzheimer's disease. Mol Neurodegener. 2020;15:63.
Zhao N, Attrebi ON, Ren Y, Qiao W, Sonustun B, Martens YA, et al. APOE4 exacerbates alpha-synuclein pathology and related toxicity independent of amyloid. Sci Transl Med. 2020;12(529):eaay1809.
Shi Y, Yamada K, Liddelow SA, Smith ST, Zhao L, Luo W, et al. ApoE4 markedly exacerbates tau-mediated neurodegeneration in a mouse model of tauopathy. Nature. 2017;549:523–7.
Zhao N, Liu CC, Van Ingelgom AJ, Linares C, Kurti A, Knight JA, et al. APOE epsilon2 is associated with increased tau pathology in primary tauopathy. Nat Commun. 2018;9:4388.
Wu M, He Y, Zhang J, Yang J, Qi J. Co-injection of Abeta1-40 and ApoE4 impaired spatial memory and hippocampal long-term potentiation in rats. Neurosci Lett. 2017;648:47–52.
Twohig D. Nielsen HM: alpha-synuclein in the pathophysiology of Alzheimer's disease. Mol Neurodegener. 2019;14:23.
Wennberg AM, Tosakulwong N, Lesnick TG, Murray ME, Whitwell JL, Liesinger AM, et al. Association of Apolipoprotein E epsilon4 With Transactive Response DNA-Binding Protein 43. JAMA Neurol. 2018;75:1347–54.
Vossel KA, Bien-Ly N, Bernardo A, Rascovsky K, Karydas A, Rabinovici GD, et al. ApoE and TDP-43 neuropathology in two siblings with familial FTLD-motor neuron disease. Neurocase. 2013;19:295–301.
Koike Y, Sugai A, Hara N, Ito J, Yokoseki A, Ishihara T, et al. Age-related demethylation of the TDP-43 autoregulatory region in the human motor cortex. Commun Biol. 2021;4:1107.
Nag S, Yu L, Capuano AW, Wilson RS, Leurgans SE, Bennett DA, et al. Hippocampal sclerosis and TDP-43 pathology in aging and Alzheimer disease. Ann Neurol. 2015;77:942–52.
Nelson PT, Dickson DW, Trojanowski JQ, Jack CR, Boyle PA, Arfanakis K, et al. Limbic-predominant age-related TDP-43 encephalopathy (LATE): consensus working group report. Brain. 2019;142:1503–27.
Nelson PT, Schmitt FA, Lin Y, Abner EL, Jicha GA, Patel E, et al. Hippocampal sclerosis in advanced age: clinical and pathological features. Brain. 2011;134:1506–18.
Uchino A, Takao M, Hatsuta H, Sumikura H, Nakano Y, Nogami A, et al. Incidence and extent of TDP-43 accumulation in aging human brain. Acta Neuropathol Commun. 2015;3:35.
Nag S, Yu L, Boyle PA, Leurgans SE, Bennett DA, Schneider JA. TDP-43 pathology in anterior temporal pole cortex in aging and Alzheimer's disease. Acta Neuropathol Commun. 2018;6:33.
Tsuiji H, Inoue I, Takeuchi M, Furuya A, Yamakage Y, Watanabe S, et al. TDP-43 accelerates age-dependent degeneration of interneurons. Sci Rep. 2017;7:14972.
Dickson DW, Davies P, Bevona C, Van Hoeven KH, Factor SM, Grober E, et al. Hippocampal sclerosis: a common pathological feature of dementia in very old (> or = 80 years of age) humans. Acta Neuropathol. 1994;88:212–21.
Amador-Ortiz C, Dickson DW. Neuropathology of hippocampal sclerosis. Handb Clin Neurol. 2008;89:569–72.
Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum Mol Genet. 2008;17:3631–42.
Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–9.
Dickson DW, Baker M, Rademakers R. Common variant in GRN is a genetic risk factor for hippocampal sclerosis in the elderly. Neurodegener Dis. 2010;7:170–4.
Pao WC, Dickson DW, Crook JE, Finch NA, Rademakers R, Graff-Radford NR. Hippocampal sclerosis in the elderly: genetic and pathologic findings, some mimicking Alzheimer disease clinically. Alzheimer Dis Assoc Disord. 2011;25:364–8.
Aoki N, Murray ME, Ogaki K, Fujioka S, Rutherford NJ, Rademakers R, et al. Hippocampal sclerosis in Lewy body disease is a TDP-43 proteinopathy similar to FTLD-TDP Type A. Acta Neuropathologica. 2015;129:53–64.
Josephs KA, Mackenzie I, Frosch MP, Bigio EH, Neumann M, Arai T, et al. LATE to the PART-y. Brain. 2019;142:e47.
Katsumata Y, Abner EL, Karanth S, Teylan MA, Mock CN, Cykowski MD, et al. Distinct clinicopathologic clusters of persons with TDP-43 proteinopathy. Acta Neuropathol. 2020;140:659–74.
Robinson JL, Porta S, Garrett FG, Zhang PP, Xie SX, Suh E, et al. Limbic-predominant age-related TDP-43 encephalopathy differs from frontotemporal lobar degeneration. Brain. 2020;143:2844–57.
Koga S, Sanchez-Contreras M, Josephs KA, Uitti RJ, Graff-Radford N, van Gerpen JA, et al. Distribution and characteristics of transactive response DNA binding protein 43 kDa pathology in progressive supranuclear palsy. Mov Disord. 2017;32:246–55.
Fujishiro H, Uchikado H, Arai T, Hasegawa M, Akiyama H, Yokota O, et al. Accumulation of phosphorylated TDP-43 in brains of patients with argyrophilic grain disease. Acta Neuropathol. 2009;117:151–8.
Uryu K, Nakashima-Yasuda H, Forman MS, Kwong LK, Clark CM, Grossman M, et al. Concomitant TAR-DNA-binding protein 43 pathology is present in Alzheimer disease and corticobasal degeneration but not in other tauopathies. J Neuropathol Exp Neurol. 2008;67:555–64.
Schwab C, Arai T, Hasegawa M, Yu S, McGeer PL. Colocalization of transactivation-responsive DNA-binding protein 43 and huntingtin in inclusions of Huntington disease. J Neuropathol Exp Neurol. 2008;67:1159–65.
Koga S, Kouri N, Walton RL, Ebbert MTW, Josephs KA, Litvan I, et al. Corticobasal degeneration with TDP-43 pathology presenting with progressive supranuclear palsy syndrome: a distinct clinicopathologic subtype. Acta Neuropathol. 2018;136:389–404.
Koga S, Lin WL, Walton RL, Ross OA, Dickson DW. TDP-43 pathology in multiple system atrophy: colocalization of TDP-43 and alpha-synuclein in glial cytoplasmic inclusions. Neuropathol Appl Neurobiol. 2018;44:707–21.
Outeiro TF, Koss DJ, Erskine D, Walker L, Kurzawa-Akanbi M, Burn D, et al. Dementia with Lewy bodies: an update and outlook. Mol Neurodegener. 2019;14:5.
Kumar S, Phaneuf D, Cordeau P Jr, Boutej H, Kriz J, Julien JP. Induction of autophagy mitigates TDP-43 pathology and translational repression of neurofilament mRNAs in mouse models of ALS/FTD. Mol Neurodegener. 2021;16:1.
Chen X, Li Y, Wang C, Tang Y, Mok SA, Tsai RM, et al. Promoting tau secretion and propagation by hyperactive p300/CBP via autophagy-lysosomal pathway in tauopathy. Mol Neurodegener. 2020;15:2.
Lynch-Day MA, Mao K, Wang K, Zhao M, Klionsky DJ. The role of autophagy in Parkinson's disease. Cold Spring Harb Perspect Med. 2012;2:a009357.
Rahman MA, Rhim H. Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep. 2017;50:345–54.
Liscic RM, Alberici A, Cairns NJ, Romano M, Buratti E. From basic research to the clinic: innovative therapies for ALS and FTD in the pipeline. Mol Neurodegener. 2020;15:31.
Acknowledgements
We thank Dr. Hongmei Li for proofreading of the manuscript.
Funding
This work was supported by the Coins for Alzheimer’s Research Trust (CART) Foundation (to N.Z.), Mayo Alzheimer’s Disease Research Center developmental grant (to N.Z.), CurePSP (to S.K.), and NIH grants U19 AG069701 (to G.B., D.W.D., and N.Z.), P30 AG026276 (to D.W.D. and G.B.), and U54 NS110435 (to G.B., D.W.D., and N.Z.).
Author information
Authors and Affiliations
Contributions
A.M. led the writing of the manuscript with significant input from S.K. and J.O.. A.M., S.K., and N.Z. prepared the figures and tables. D.W.D., G.B., and N.Z. co-edited the manuscript and supervised the writing. All authors have read and agreed on the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors agreed to publish.
Competing interests
All the authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Meneses, A., Koga, S., O’Leary, J. et al. TDP-43 Pathology in Alzheimer’s Disease. Mol Neurodegeneration 16, 84 (2021). https://doi.org/10.1186/s13024-021-00503-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-021-00503-x