
Abstract
The homeostasis of iron is vital to human health, and iron dyshomeostasis can lead to various disorders. Iron homeostasis is maintained by iron regulatory proteins (IRP1 and IRP2) and the iron-responsive element (IRE) signaling pathway. IRPs can bind to RNA stem-loops containing an IRE in the untranslated region (UTR) to manipulate translation of target mRNA. However, iron can bind to IRPs, leading to the dissociation of IRPs from the IRE and altered translation of target transcripts. Recently an IRE is found in the 5′-UTR of amyloid precursor protein (APP) and α-synuclein (α-Syn) transcripts. The levels of α-Syn, APP and amyloid β-peptide (Aβ) as well as protein aggregation can be down-regulated by IRPs but are up-regulated in the presence of iron accumulation. Therefore, inhibition of the IRE-modulated expression of APP and α-Syn or chelation of iron in patient’s brains has therapeutic significance to human neurodegenerative diseases. Currently, new pre-drug IRE inhibitors with therapeutic effects have been identified and are at different stages of clinical trials for human neurodegenerative diseases. Although some promising drug candidates of chemical IRE inhibitors and iron-chelating agents have been identified and are being validated in clinical trials for neurodegenerative diseases, future studies are expected to further establish the clinical efficacy and safety of IRE inhibitors and iron-chelating agents in patients with neurodegenerative diseases.
Similar content being viewed by others

Background
The roles of iron in hemoglobin formation and oxygen transport have been linked to human health and diseases [1]. Iron is important to the functioning of many prosthetic groups, including haem and iron-sulphur clusters, and iron depletion can contribute to anemia [2]. However, excess iron can promote the generation of deleterious reactive oxygen species (ROS) and is linked to both haemochromatosis and thalassaemia [3, 4]. The molecular mechanisms of iron metabolism in humans have been extensively studied. Ferrous iron that is absorbed from intestinal lumen into enterocytes can be exported into bloodstream via ferroportin (Fpn), inhibited by hepcidin or facilitated by hephaestin and ceruloplasmin (CP) with ferroxidase activities [5,6,7]. The iron in bloodstream can be captured by transferrin (Tf) in ferric state and transported to peripheral tissues [8]. In peripheral tissues the iron loaded Tf will be recognized by transferrin receptor (TfR) on cell membrane, followed by receptor-mediated endocytosis [8]. In acidic endosome the iron will be dissociated from Tf and released into cytoplasm via divalent metal transporter 1 (DMT1) after reduction to its ferrous state by a STEAP family reductase [8]. In the cytoplasm, free ferrous iron can be immediately used as a co-factor for enzyme such as tyrosine hydroxylase or taken up by mitochondria via mitoferrin for synthesis of Fe-S clusters and heme groups, which is indispensible for mitochondria functions [8, 9]. Considerable amount of iron in cells can be sequestered and stored in cytosol ferritin or mitochondrial ferritin (MtFt) in ferric state [9, 10]. Excess iron can be exported into bloodstream through Fpn to form ferric iron-Tf complex again for iron re-distribution [11].
A substantial amount of iron can be absorbed into brain, mainly in the substantia nigra pars compacta (SN) [12,13,14]. The iron in bloodstream can be transported across blood brain barrier (BBB) through brain capillary endothelial cells (BCECs) via Tf-TfR and DMT1-Fpn pathways [15]. The circulating iron-Tf complex can be captured by TfR on BCECs cell membrane, internalized via endocytosis, released to BCECs cytoplasm via DMT1 and exported into brain interstitial fluid via Fpn [15]. In brain iron plays multiple physiological roles including neurotransmitter synthesis, neuron myelination, mitochondrial functions and energy generation [15]. Iron homeostasis in the brain is precisely controlled and dysregulated brain iron homeostasis (iron overload or deficiency) can lead to brain disorders [15]. Brain iron deficiency (BID) can disturb brain development and functions [16]. BID can be associated with the pathogenesis of brain disorders including Attention Deficit Hyperactivity Disorder (ADHD) and Restless Legs Syndrome (RLS) [17,18,19,20,21]. The iron overload in brain can also be a pathological factor for brain disorders, including Alzheimer’s disease (AD) [22, 23], Parkinson’s disease (PD) [23, 24], and other human brain disorders [25,26,27,28,29,30].
Main text
Modulation of iron homeostasis by IRP-IRE signaling pathway
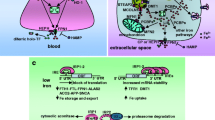
Iron homeostasis is elaborately regulated [8]. Although some control mechanisms exist at the transcriptional level, the absorption, transportation and storage of iron are meticulously modulated at the translational level by the iron regulatory protein (IRP) and iron-responsive element (IRE) signaling pathway [31,32,33]. IRP1 (90 KDa) and IRP2 (105 KDa) are two known RNA-binding proteins, and their inducible interactions with IRE function to control the translation of ferritin and Fpn mRNA and the stability of TfR mRNA. Briefly, the IRPs can control iron metabolism via binding to specific non-coding sequences, known as IREs, within the untranslated region (UTR) of mRNA transcripts [32]. The IREs are 30-nucleotide long RNA motifs containing the CAGUGN sequence (the classic IRE motif) and can form special stem-loop structures [32, 34]. IREs can be present in either the 3′-UTR or 5′-UTR of the target mRNA [32, 34]. Transcripts with IRE motifs in their 5′-UTR include the ferritin H and L subunits, Fpn and aminolevulinic acid synthetase [8, 35], whereas target mRNA with IRE motifs in the 3′-UTR include TfR (5 copies) and DMT-1 [35]. The detailed IRP-IRE signaling pathway translation modulation mechanisms are illustrated in Fig. 1.

Mechanism of translation modulation by the IRP-IRE signaling pathway. a The translation of transcripts containing an IRE in the 5′-UTR can occur in the absence of IRP binding to the IRE; (b) after the binding of IRP to the IRE in the 5′-UTR, translation of the transcripts is inhibited. c Iron can bind to IRP to induce a transformational change of IRP, leading to its dissociation from the IRE, which can promote the translation of transcripts. d The IRE can be present in the 3′-UTR of the transcript. Without IRP binding to the 3′-UTR, the transcript can be susceptible to endonuclease attack and degradation, leading to down-regulation of translation. e Binding of IRP to the IRE in the 3′-UTR can protect transcripts against endonuclease degradation and therefore promote the translation of transcripts. f Iron can bind to IRP, leading to the dissociation of IRP from the IRE in the 3′-UTR, endonuclease attack, and the degradation of transcripts, which down-regulates the translation of transcripts
IRPs can act as either a translational enhancer or a translational inhibitor [32, 34]. In iron-deficient cells, the interaction between IRP and the IRE motif in the 5′-UTR of target mRNA can abrogate translation via interruption of important interactions between the mRNA and ribosome for the initiation of translation [32]. However, in iron-replete cells, iron can bind with IRPs to induce a conformational change, which promotes the dissociation of IRPs from the target mRNA, leading to facilitation of the translation of the target mRNA [32]. In contrast, some mRNA transcripts contain one or more IRE motifs in their 3′-UTR. In iron-deficient cells, the binding of IRPs to an IRE at the 3′-UTR of transcripts can protect target mRNA against endonuclease cleavage [32]. Therefore, the interaction between IRPs and a 3′-UTR IRE can extend the half-life of transcripts and promote translation of target mRNA. However, in iron-replete cells, the dissociation of IRP from an IRE at the 3′-UTR renders target transcripts susceptible to endonuclease attack and degradation, leading to down-regulation of the translation of transcripts [32]. Both transcripts of ferritin and Fpn have IRE in their 5′-UTRs, so that under iron deficiency condition the translation of ferritin and Fpn can be inhibited by IRPs [8]. The decreased expression of ferritin and Fpn can reduce the unnecessary iron binding by ferritin and iron export by Fpn, leading to an increased level of free iron available for cell usage. However both TfR and DMT1 mRNA have 3′-UTR IREs, which can bind to IRPs under iron deficiency, leading to stabilization of transcripts and subsequent increased synthesis of TfR and DMT1 to promote iron absorption into cells [8]. In contrast, under a situation of iron accumulation, the increased iron level can disturb the IRP-IRE interaction to promote the translation of the ferritin and Fpn transcripts as well as to destabilize TfR and DMT1 mRNA [32]. Therefore under iron accumulation, the iron absorption will be inhibited, while iron storage and export can be enhanced. [8]. Pathological factors inducing a disturbance of the IRP-IRE signaling pathway will impair iron homeostasis, which can contribute to the onset and development of human disorders. However, one recent study reported a novel mechanism for the iron-induced modulation of translation of target mRNA [36]. They demonstrated that eukaryotic initiation factor 4F (eIF4F) can specifically bind to an IRE at the 5′-UTR of target mRNA with high affinity, which is vital for translation initiation [36]. However, iron can also directly bind to the IRE of target mRNA, leading to a conformational alteration of mRNA [36]. The conformational changes in mRNA induced by the binding of iron will facilitate the interaction between eIF4F and IRE-RNA, which can outcompete binding between IRP and the IRE [36]. Therefore, accumulated iron can contribute to up-regulated translation of target mRNA with a 5′-UTR IRE [36]. Recent evidence suggests that the IRP-IRE signaling pathway may play other physiological roles beyond iron homeostasis. Functional IREs are identified in both transcripts of CDC14A (cell division cycle 14 homologue A) and hypoxia-inducible factor 2a (HIF2a) [37,38,39]. The translation of CDC14A, a phosphatase involved in cell cycle, can be modulated by IRP-IRE signaling [37, 38]. The iron depletion can lead to growth inhibition at the G1–S transition [40]. These findings indicate other important physiological roles of the IRP-IRE signaling pathway beyond maintenance of iron homeostasis.
Linking IRP-IRE signaling pathway to human neurodegenerative disorders
However, recent findings indicate that the iron accumulation-induced dysfunction of the IRP-IRE signaling pathway may contribute significantly to protein aggregation and neuron loss in AD and PD, which sheds new light on the pathogenesis and therapy of the neurodegeneration observed in AD and PD.
AD is the most common form of dementia in the elderly [41, 42]. The pathology of AD is characterized by synaptic defects, neuron loss and the formation of amyloid β-peptide (Aβ) plaques in the hippocampus and cortical areas [41, 42]. Aβ plaques contain Aβ aggregates and neurofibrillary tangles formed by aggregated forms of the microtubule-associated protein tau [41, 42]. Aβ is generated during protease cleavage of amyloid precursor protein (APP) and is critical in AD pathogenesis [43]. APP is a single transmembrane metalloprotein that can be cleaved via two main proteolytic procedures: amyloidogenic processing and non-amyloidogenic processing [43]. The amyloidogenic processing of APP can generate Aβ peptides 40–42 amino acids in length. Among the Aβ peptides produced, Aβ42 is more apt to form fibrillar protein aggregates [43]. Up-regulated APP expression and Aβ levels are associated with neuronal loss in AD [43]. Furthermore, increased levels of APP can also be linked to amyloidosis in Down syndrome (DS), since the APP gene is located on chromosome 21, which is triplicated in DS [44, 45]. The increased level of APP can be a pathological factor and a therapeutic target linked to Aβ amyloidosis and neuron loss in AD and DS.
However, recent findings suggest that the increased APP level, aggravated Aβ deposition and neuron loss in AD can be induced by iron accumulation and disturbance of the IRP-IRE signaling pathway [46, 47]. A novel functional IRE is found in the 5′-UTR of the APP mRNA transcript (+51 to +94 from the 5′-cap site) [48,49,50]. The IRE in APP mRNA is located immediately upstream of an interleukin-1 responsive acute box domain (+101 to +146) [48,49,50]. The IRE in APP mRNA can bind specifically to IRP, which can be abrogated by a mutation in the core IRE motif [50]. In the presence of iron chelators, the translation of APP mRNA can be selectively down-regulated [48, 51]. In contrast, iron influx or an increased iron level can enhance APP mRNA translation and Aβ generation [48, 51]. Therefore, these findings demonstrate that iron accumulation can be a pathological factor triggering the up-regulated expression of the APP holoprotein and subsequent Aβ deposition. However, therapeutic strategies targeting the IRE in APP mRNA should alleviate Aβ deposition and neuron loss in AD brains. The strategy of targeting the APP 5′-UTR to reduce APP expression and Aβ amyloid formation has been validated by the usage of novel IRE chemical inhibitors to alleviate APP and amyloid levels as well as cognitive decline in the TgCRND8 AD mouse model [52]. Identified potent IRE inhibitors, such as Posiphen, have been reported to be bona fide APP 5′-UTR-directed translation blockers that can reduce Aβ generation in the cerebrospinal fluid (CSF) of humans and have entered clinical trials for AD therapy [53]. In contrast, iron species can be a factor promoting ROS generation and inducing Aβ aggregation [54]. The increased iron level in AD patient brains is found to be relevant to the formation of neurofibrillary tangles [15]. The therapeutic strategies using iron chelators plus IRE inhibitors may act collaboratively to alleviate Aβ aggregation and rescue neuron loss in AD patient brains.
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, and the estimated incidence of PD is approximately 1% of the population over the age of 55 [55, 56]. PD is characterized by progressive DA neuron loss in the SN as well as Lewy body formation in affected brain areas [55, 56]. The pathogenesis of the progressive DA neuron degeneration in PD is still unclear as of yet, and no effective therapy has been developed to alleviate the progressive DA neuron degeneration in PD. However, recent new findings on the roles of the IRP-IRE signaling pathway in PD add to the pathogenesis and therapy of iron-relevant DA neuron degeneration in PD. The involvement of IRPs in DA neuron degeneration can be supported by 2 in vivo studies on IRP2 knockout mice models [57, 58]. In middle to late age (18 to 24 months), mice lacking IRP2 will develop abnormal motoric PD-like behaviors including tremors at rest, abnormal gait, and bradykinesia [57, 58]. Furthermore, significant iron accumulation in the brains of IRP2 knockout mice precede the onset of DA neuron degeneration and PD-like symptoms by many months, accompanied by the deposition of ubiquitin-positive protein aggregates and inclusions in the mouse brain [58]. The DA level in the dorsal striatum of IRP2 knockout mice decreased (approximately 20%) significantly compared with that of control mice, suggesting that IRP2 knockout induced DA neuron degeneration in the mouse brain [57]. Previous findings demonstrated that iron retention and iron dependent toxicity to DA neurons can be induced by nitric oxide (NO) via facilitation of interactions between IRP and IRE to inhibit translation of ferritin, Fpn and other iron metabolism relevant proteins [59, 60]. Furthermore the NO can inhibit APP translation to aggravate the iron retention in DA neurons, as APP stabilizes Fpn to promote iron export [61]. NO is supposed to be a causative factor upstream of nigral iron accumulation and DA neuron degeneration in PD pathogenesis [61]. These findings suggest a close association of iron and the IRP-IRE signaling pathway with DA neurodegeneration in PD.
Furthermore previous evidence suggests that iron accumulation and dysfunction of the IRP-IRE signaling pathway can be linked to α-Syn-induced toxicity of DA neurons [62, 63]. α-Syn is the central protein involved in the pathogenesis and therapy of PD [64, 65]. Lewy bodies consisting of aggregated α-Syn can be found in almost all PD patient brains [66, 67]. α-Syn mutations are known to be linked to the onset of the familial form of PD (FPD) [68,69,70]. Furthermore, the increased level of the α-Syn protein in the brain due to gene duplication or triplication is also linked to the onset of FPD [71, 72]. However, human post-mortem brain studies have demonstrated that while the mRNA levels of α-Syn in PD brains are either unchanged or decreased, higher levels of the insoluble α-Syn protein occur during PD progression [62]. In contrast, disrupted iron metabolism in the SN is a common feature of PD [46]. Studies have shown that the iron levels can be up-regulated to almost 2-fold in a single SN DA neuron in PD patient brains [24]. The elevated iron levels in PD patient brains are thought to be linked to the accelerated deleterious formation of α-Syn protein aggregates [63]. Furthermore, novel evidence has shown that elevated iron levels in the brain can cause enhanced α-Syn protein translation in the brain [49]. A unique RNA stem-loop with an IRE motif (CAGUGN) is found in the 5′-UTR of α-Syn mRNA [49, 62, 73]. The IRE stem-loop of the α-Syn transcript spans two exons [73]. IRPs can bind with the IRE of α-Syn mRNA and inhibit translation of the α-Syn transcript [49]. However, iron can reverse the translation inhibition of α-Syn by IRPs and therefore facilitate α-Syn expression and aggregation [49]. The translation of α-Syn mRNA can be inhibited by new IRE chemical inhibitors [73, 74]. These findings suggest a pathological relevance of iron and the IRP-IRE signaling pathway in the α-Syn-dependent toxicity of DA neurons in PD. Iron chelators and new IRE inhibitors should alleviate iron-dependent α-Syn toxicity and DA neuron degeneration in PD [73, 75, 76]. The detailed IRE sequences and stem-loop structures as well as maps of the IRE stem-loop in α-Syn and APP transcripts are illustrated in Fig. 2.

Comparison of the 5′-UTR IRE stem-loop of the α-Syn, APP, H-ferritin and L-ferritin transcripts. a Stem-loop structures of α-Syn, APP, H-ferritin and L-ferritin. b Relative alignment of the sequences encoding the 5′-UTR-specific IRE stem-loops in α-Syn, APP, H-ferritin and L-ferritin. The key IRE motifs of AGU or AGA are highlighted in purple. B. Homological analysis of the IRE stem-loop sequences of the α-Syn, APP, H-ferritin and L-ferritin transcripts. Key IRE motifs are highlighted in yellow, while homological nucleotides in the flank areas of the key IRE motifs are highlighted in cyan. c Maps of the 5′-UTR IRE stem-loops in the α-Syn, APP, H-ferritin and L-ferritin transcripts
The iron and IRP-IRE signaling pathway also seem to be implicated in the pathogenesis of ALS [77, 78]. ALS is a progressive neurodegenerative disorder induced by multiple pathogenic factors and is characterized by the progressive and selective loss of motor neurons in the cerebral cortex, brainstem, and spinal cord [77]. α-Syn-positive inclusion bodies can be identified in ALS [79, 80]. Increasing evidence indicates a pathological role of iron dysregulation in neuronal cell death in ALS [77, 81,82,83]. The role of iron in the pathogenesis of ALS is supported by the therapeutic effects of an iron chelation strategy in ALS mouse models [84,85,86]. Furthermore, a potential IRE has been computer-predicted to exist in the 5′-UTR of mRNA encoding C9orf72, a key ALS-linked gene [87]. This RNA structure is proposed to post-transcriptionally regulate the expression of the C9orf72 protein in response to iron dyshomeostasis [87]. Such a hypothesis can be supported by previous findings on the increased levels of iron and ferritin in patients with ALS [87]. Thus, a potential mechanistic link may be present between iron and the iron-modulated translation of C9orf72, which is associated with ALS pathogenesis [87].
Linking iron and the IRP-IRE signaling pathway to other human disorders
Although none of the point mutations in IRP1 or IRP2 have been confirmed to cause human disease, some preliminary studies have implicated the potential association of SNPs or mutations of IRPs with human diseases. A genome-wide association study (GWAS) identified three IRP2 SNPs associated with patients with chronic obstructive pulmonary disease (COPD) [88]. The levels of IRP2 protein and mRNA have been found to be higher in lung tissue samples from COPD patients than from controls [88]. These findings have indicated that IRP2 is a COPD-susceptible gene. Further studies are needed to establish the potential pathogenic role for IRP2 in COPD.
Mutations in the IRE motif have been identified in other human disorders beyond human neurodegenerative diseases [89,90,91,92,93,94,95]. Multiple point mutations in the IRE motif of the L-ferritin transcript have been linked to hyperferritinemia-cataract syndrome (HHCS) with hyperferritinemia and autosomal dominant congenital cataract [89,90,91]. The disease is characterized by early-onset, bilateral nuclear cataracts and up-regulated levels of serum ferritin [93]. However, HHCS patients demonstrate no hematological or biochemical abnormalities in iron metabolism [89, 90]. The serum iron level and transferrin saturation of HHCS patients are normal or low [89, 90]. The level of body iron is also not increased, therefore excluding iron overload as the pathogenic cause of HHCS [89, 90]. Studies have demonstrated that the point mutation in the IRE motif of L-ferritin mRNA disturbs the IRP interactions of the highly conserved IRE stem-loop [89, 90]. Furthermore, the disease-relevant point mutations in the IRE motif of L-ferritin lead to the abolishment of the binding of IRPs to the L-ferritin transcript, resulting in significantly increased synthesis of L-ferritin in cells from HHCS patients, which is in agreement with the up-regulated serum ferritin levels observed in these patients [93]. The point mutation in the IRE of L-ferritin mRNA induces hyperferritinemia and may lead to the accumulation and aggregation of L-ferritin in the lens and the early onset of cataract. Furthermore, mutations in the IRE motif of the H-ferritin gene may also be relevant to human disease [92]. A point mutation (A49U) in the 5′-UTR IRE motif of the human H-ferritin gene has been identified in members of a Japanese family with dominantly inherited iron overload [92]. This point mutation in the IRE motif of H-ferritin increases its affinity for IRP binding, leading to suppression of H-ferritin synthesis, an increase in iron uptake, autosomal dominant iron overload and tissue iron deposition [92]. In addition to mutation-induced alterations in H- and L-ferritin, mutation-induced alterations in the expression of other IRE-modulated protein can also be disease-relevant. Mutations in the 5′-UTR downstream of the IRE motif of Fpn mRNA have been detected in a patient affected by hemochromatosis with iron overload [94]. Fpn is the major and sole iron exporter that transports ion out of cells [95]. The mutation in the 5′-UTR of the Fpn transcript may alter the IRE-IRP interaction, inhibit translation of Fpn, impair the exportation of iron from cells, and induce iron overload.
On the other hand, mutations in the coding areas of IRE-modulated genes have also been found to be relevant to human disease. The mutations of ferritin that lead to the impaired capacity of ferritin to retain iron within its iron core have been linked to PD as well as neuroferritinopathy, a severe dominantly inherited movement disorder characterized by the deposition of iron and ferritin in the brain, normal or low serum ferritin levels, and highly variable clinical features [96,97,98]. Mutations at the C-terminus of L-ferritin impair its stability and decrease its capacity to interact with iron, validated by the transgenic mouse model expressing mutant L-ferritin [99,100,101]. Other disease-relevant coding region mutations have been reported in other IRE-encoded genes, including erythroid 5-aminolevulinate synthase (eALAS, linked to sideroblastic anemia) [102,103,104], Fpn (hereditary hemochromatosis) [105, 106] and DMT-1 [107,108,109].
Potential novel therapies using IRE inhibitors for human neurodegenerative diseases
The IRP-IRE signaling pathway has been implicated in the modulation of APP and α-Syn translation, which is important to neurodegeneration in PD and AD. Therefore, the identification of small molecular IRE chemical inhibitors to reduce APP and α-Syn levels and alleviate protein aggregation can have therapeutic significance to human neurodegenerative diseases [73, 110]. In principle, identified chemical IRE inhibitors can decrease ferritin and Tf expression to alleviate the excess iron accumulation in AD or PD brains. Furthermore, therapeutic IRE inhibitors that down-regulate APP and α-Syn protein translation and inhibit protein aggregation can promote neuronal survival. Therefore, potent non-toxic pre-drug IRE inhibitors with excellent BBB penetrating capacity should have high therapeutic significance in neurodegenerative diseases. So far, some promising drug candidates of IRE inhibitors have been identified and are being tested in different human clinical trials for AD and PD.
In one recent study, thirteen potent APP translation blockers that act selectively towards the uniquely configured IRE RNA stem-loop in the 5′-UTR of APP mRNA were identified from 110,000 compounds of a chemical library at Harvard [110]. Some of these chemicals were able to selectively reduce neural APP production in SH-SY5Y cells without affecting cell viability or the levels of α-Syn and ferritin [110]. In this study, the identified APP blocker-9 (JTR-009), a benzimidazole, was found to be superior to the other APP blockers in its ability to reduce the production of toxic Aβ in SH-SY5Y neuronal cells [110]. JTR-009 is thoughts to directly interact with the IRE in the 5′-UTR of APP mRNA and constitutively repress APP translation [110]. Furthermore, pifithrin-α (PFTα), an anti-stroke agent and a p53 inhibitor, was also found to have selective APP translation inhibition capacity [110]. However, JTR-009 was able to selectively inhibit APP expression, while PFTα could cause a dose-dependent down-regulation of APP, α-Syn and actin proteins in cells [110]. Furthermore, the potency of JTR-009 to inhibit the APP 5′-UTR-conferred translation was greater than that of posiphen ((+)-phenserine), a well-recognized and tolerated IRE inhibitor with both APP and α-Syn translation inhibitory capacity [110].
Posiphen is a small molecule drug derived from Calabar beans and is a phenyl carbamoyl analogue of (+)-physostigmine [73]. Identified from a nature product (NP) chemical library, posiphen is found to inhibit both APP and α-Syn protein translation [73]. Furthermore, the inhibitory effects of posiphen are potent and not toxic [73]. However, phenserine, its enantiomer with cholinesterase inhibitory capacity, has less inhibitory potency than posiphen as well as minor toxicity [73]. Another study demonstrated that posiphen and its metabolites, as well as phenserine, possess neuroprotective / neurotrophic capacities at concentrations of clinical relevance [111]. All compounds are found to potently inhibit the protein translation of APP and α-Syn in neuronal cells [111]. Therefore, posiphen and its metabolites, as well as phenserine, may be drug candidates for AD and PD. The posiphen-induced inhibition of IRE has been validated by various in vivo and in vitro studies [74, 112,113,114]. Recent phase 1 human clinical trials and a proof-of-concept study in subjects with mild cognitive impairment (MCI) have demonstrated the brain entry and capacity of posiphen to lower APP production in subjects, as assessed in CSF, by as much as 50% [115]. Currently phenserine is under phase 3 clinical trials for AD, while posiphen is in a phase 2 human clinical trial for AD and PD [116].
Other potential chemical IRE inhibitors have also been reported. In a pilot study, paroxetine (a serotonin reuptake blocker), N-acetyl cysteine (anti-oxidant and Fe2+ chelator, NAC), and erythromycin (macrolide antibiotic) were employed to assess their anti-amyloid capacity in the transgenic TgCRND8 AD mouse model [52]. The levels of Aβ peptide were found to be reduced after the exposure of the mice to paroxetine, NAC, and erythromycin [52]. Other studies have verified that paroxetine can modulate APP expression via its actions on the 5′-UTR of the APP transcript [116, 117]. Therefore, paroxetine may be a chemical IRE inhibitor that can modulate APP expression. Another high-throughput screening study to search for novel, nontoxic, and selective small-molecule inhibitors of α-Syn expression has been performed [118]. From 303,224 screened compounds, one compound (ML150) displayed very potent inhibition of α-Syn expression [118]. ML150 specifically reduced α-Syn translation and likely acts via an interaction with and inhibition of the IRE of the α-Syn transcript [118]. These findings suggest that ML150 is a novel IRE inhibitor that specifically modulates α-Syn expression. The detailed molecular structures of the identified IRE chemical inhibitors as well as their potential relevance to therapies against human neuron degenerative diseases are illustrated and summarized in Fig. 3.

Identified small molecular IRE inhibitors that modulate the translation of α-Syn and APP. a Identified IRE inhibitors with a capacity to inhibit α-Syn translation. b Identified IRE inhibitors with a capacity to inhibit APP translation. c Identified IRE inhibitors with a capacity to inhibit both α-Syn and APP translation. d Table 1. Summary of IRE inhibitors potential for AD and PD therapies
The pathological and therapeutic targets in the iron-IRP-IRE signaling pathway for AD and PD
There has been an accumulation of evidence regarding the pathological iron accumulation in AD and PD brains [22,23,24, 29, 30]. Excess iron deposits in the brain can induce oxidative stress, leading to protein aggregation and neuron vulnerability [119, 120]. Furthermore, an increased iron level can lead to up-regulation of translation of APP and α-Syn, 2 key proteins vital to neuron degeneration in AD and PD [48, 51, 63]. Therefore, BBB-penetrating iron chelators should have therapeutic significance for AD and PD patients [50, 116, 121]. The administration of desferrioxamine (DFO), an iron chelator, can slow dementia in AD patients [120]. However, DFO is unstable with poor BBB permeability [122]. Clioquinol (CQ), an iron/Cu/Zn chelator with BBB-penetrating capacity, can clinically alleviate cognitive loss and decrease plasma Aβ levels in AD patients [123]. However, CQ has been identified to be associated with myelinopathies [124]. A second-generation 8-hydroxyquinoline analog metal chaperone, PBT2, was found to be superior to CQ with little adverse events [125, 126]. PBT2 can improve cognition in aged APP C57Bl/6 and Tg2576 transgenic mice, with the effects associated with a decreased interstitial Aβ level [127, 128]. Furthermore, a phase 2 trial with PBT2 in 78 AD patients showed a reduction in Aβ levels in CSF and enhanced performance metrics [125, 126]. Other iron chelators, including VK28 (Fe3+ chelator), HLA20 and M30 (Fe3+ chelator with N-propargylamine-like properties), can both suppress APP expression and lower the Aβ level in the brain [122]. These results suggest that iron chelators induce down-regulation of APP and Aβ levels via IRP inhibition of APP translation. Iron-chelating agents have also been validated to be effective in PD therapy [129,130,131,132,133]. A natural iron chelator, phytic acid (IP6), has been found to protect DA neurons in an in vitro PD model [129]. Furthermore, iron chelators, including deferasirox, deferiprone, deferrioxamine, VAR10303 and D-607, can all significantly attenuate the loss of DA neurons in various in vivo PD models [130,131,132,133]. Detailed future studies are expected to further establish clinical efficacy and safety of iron-chelating strategies in AD and PD therapies.
The intactness of the IRE motif is vital to the functions of the IRP-IRE signaling pathway. Conceivably, point mutations in key nucleotides of the IRE motif of target mRNA can disturb the IRE stem-loop structure and abrogate the interaction between IRPs and the IRE, which can be a pathogenic factor contributing to neuron degeneration in AD and PD. The point mutations in the IRE motif of the H-ferritin gene can lead to inherited human diseases associated with perturbed iron metabolism (iron overload) [92, 134]. Studying whether mutations in the IREs of H-ferritin and other iron metabolism-relevant genes are susceptibility factors for PD and AD will be intriguing. Furthermore, point mutations in the IRE motif of APP and α-Syn genes can be a pathogenic factor for AD and PD. A well-described single nucleotide polymorphism (SNP) associated with AD risk exists in the 5′-UTR of the APP gene [135]. The pathogenic SNP in the 5′-UTR of APP is hypothesized to disturb the IRE motif, leading to increased AD risk [135]. Therefore, more attention should be paid to potential disease-relevant mutations or SNPs in the IRE motifs of the APP and α-Syn genes.
In contrast, point mutations in the coding area of IRP genes, especially the IRP2 gene, may lead to a conformational change of IRP proteins, which can disturb the interaction of IRP proteins with the IRE motifs of APP and α-Syn transcripts. The impaired functional interaction between IRP and the IRE of APP and α-Syn transcripts may up-regulate translation of APP and α-Syn, leading to susceptibility to AD or PD. So far, no mutations in IRPs have been confirmed to be relevant to human disease. However, some SNP polymorphisms in the promoter region of the IRP2 gene have been associated with AD susceptibility [136]. A potentially functional single SNP in the IRP2 promoter region located in the cis-element that interacts with transcription factors may explain the altered IRP2 level in AD patients [136]. Future intriguing findings on these aspects are expected, which should be able to add to our knowledge of the pathogenesis and therapy of AD and PD.
Conclusions
In summary, recent evidence suggests a pathological and therapeutic link between the iron-IRP-IRE signaling pathway and neuron degeneration in human disorders, especially neurodegenerative diseases. Interesting and significant findings relevant to the pathophysiological roles of the IRP-IRE signaling pathway in human disorders are expected, which can add to our knowledge of the pathogenesis and therapy of comprehensive human disorders, especially incurable human neurodegenerative diseases.
Abbreviations
- AD:
-
Alzheimer’s disease
- ADHD:
-
Attention Deficit Hyperactivity Disorder
- ALS:
-
amyotrophic lateral sclerosis
- APP:
-
amyloid precursor protein
- Aβ:
-
amyloid β-peptide
- BBB:
-
blood-brain barrier
- BCECs:
-
brain capillary endothelial cells
- BID:
-
brain iron deficiency
- CDC14A:
-
cell division cycle 14 homologue A
- COPD:
-
chronic obstructive pulmonary disease
- CP:
-
ceruloplasmin
- CQ:
-
clioquinol
- CSF:
-
cerebrospinal fluid
- DA:
-
dopamine
- DFO:
-
desferrioxamine
- DMT-1:
-
divalent metal transporter 1
- DS:
-
Down syndrome
- eALAS:
-
erythroid 5-aminolevulinate synthase
- eIF4F:
-
eukaryotic initiation factor 4F
- FPD:
-
familial form of PD
- Fpn:
-
ferroportin
- GWAS:
-
genome-wide association study
- HHCS:
-
hyperferritinemia-cataract syndrome
- HIF:
-
hypoxia-inducible factor
- IP6:
-
phytic acid
- IRE:
-
iron-responsive element
- IRP:
-
iron regulatory protein
- JTR-009:
-
APP blocker-9
- MCI:
-
mild cognitive impairment
- MtFt:
-
mitochondrial ferritin
- NAC:
-
N-acetyl-cysteine
- NBIA 1:
-
neurodegeneration with brain iron accumulation Type 1
- NO:
-
nitric oxide
- NP:
-
nature product
- PD:
-
Parkinson’s disease
- PFTα:
-
pifithrin-α
- RLS:
-
Restless Legs Syndrome
- ROS:
-
reactive oxygen species
- SN:
-
substantia nigra
- Tf:
-
transferring
- TfR:
-
transferrin receptor
- UTR:
-
untranslated region
- α-Syn:
-
α-synuclein
References
The mineral nutrition of livestock, 3rd ed. Br J Nutr. 2000; 84: 393.
Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol. 2001;33:940–59.
Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol. 2005;202:199–211.
Fontecave M, Pierre JL. Iron: metabolism, toxicity and therapy. Biochimie. 1993;75:767–73.
Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434:365–81.
Fleming MD. The regulation of hepcidin and its effects on systemic and cellular iron metabolism. Hematology Am Soc Hematol Educ Program. 2008;1:151–8.
Anderson GJ, Frazer DM, McKie AT, Vulpe CD. The ceruloplasmin homolog hephaestin and the control of intestinal iron absorption. Blood Cells Mol Dis. 2002;29:367–75.
Hentze MW, Muckenthaler MU, Galy B, Camaschella C. Two to tango: regulation of mammalian iron metabolism. Cell. 2010;142:24–38.
Horowitz MP, Greenamyre JT. Mitochondrial iron metabolism and its role in neurodegeneration. J Alzheimers Dis. 2010;20(Suppl 2):S551–68.
Philpott CC, Ryu MS. Special delivery: distributing iron in the cytosol of mammalian cells. Front Pharmacol. 2014;5:173.
Ganz T. Cellular iron: ferroportin is the only way out. Cell Metab. 2005;1:155–7.
Koeppen AH. The history of iron in the brain. J Neurol Sci. 1995;134(Suppl):1–9.
Koeppen AHA. Brief history of brain iron research. J Neurol Sci. 2003;207:95–7.
Gotz ME, Double K, Gerlach M, Youdim MB, Riederer P. The relevance of iron in the pathogenesis of Parkinson's disease. Ann N Y Acad Sci. 2004;1012:193–208.
Hare D, Ayton S, Bush A, Lei PA. Delicate balance: iron metabolism and diseases of the brain. Front Aging Neurosci. 2013;5:34.
Hurtado EK, Claussen AH, Scott KG. Early childhood anemia and mild or moderate mental retardation. Am J Clin Nutr. 1999;69:115–9.
Cortese S, Azoulay R, Castellanos FX, Chalard F, Lecendreux M, Chechin D, Delorme R, Sebag G, Sbarbati A, Mouren MC, et al. Brain iron levels in attention-deficit/hyperactivity disorder: a pilot MRI study. World J Biol Psychiatry. 2012;13:223–31.
Konofal E, Cortese S, Lecendreux M, Arnulf I, Mouren MC. Effectiveness of iron supplementation in a young child with attention-deficit/hyperactivity disorder. Pediatrics. 2005;116:e732–4.
Ghanizad A. Regarding the effects of iron supplementation on attention deficit hyperactivity disorder in children. Pediatr Neurol. 2008;39:73. author reply 73-4
Youdim MB, Ben-Shachar D, Yehuda S. Putative biological mechanisms of the effect of iron deficiency on brain biochemistry and behavior. Am J Clin Nutr. 1989;50:607–15. discussion 615-7
Connor JR, Boyer PJ, Menzies SL, Dellinger B, Allen RP, Ondo WG, Earley CJ. Neuropathological examination suggests impaired brain iron acquisition in restless legs syndrome. Neurology. 2003;61:304–9.
Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat Rev Neurosci. 2004;5:863–73.
Ward RJ, Zucca FA, Duyn JH, Crichton RR, Zecca L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014;13:1045–60.
Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA, Elstner M, Morris CM. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology. 2007;68:1820–5.
Li K, Besse EK, Ha D, Kovtunovych G, Rouault TA. Iron-dependent regulation of frataxin expression: implications for treatment of Friedreich ataxia. Hum Mol Genet. 2008;17:2265–73.
Wilson RB. Iron dysregulation in Friedreich ataxia. Semin Pediatr Neurol. 2006;13:166–75.
Kurian MA, McNeill A, Lin JP, Maher ER. Childhood disorders of neurodegeneration with brain iron accumulation (NBIA). Dev Med Child Neurol. 2011;53:394–404.
Mehnaaz L. Neurodegeneration with brain iron accumulation (NBIA) formerly Hallervorden- Spatz disease. J Assoc Physicians India. 2016;64:132.
Fernaeus S, Reis K, Bedecs K, Land T. Increased susceptibility to oxidative stress in scrapie-infected neuroblastoma cells is associated with intracellular iron status. Neurosci Lett. 2005;389:133–6.
Olsen MK, Roberds SL, Ellerbrock BR, Fleck TJ, McKinley DK, Gurney ME. Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord. Ann Neurol. 2001;50:730–40.
Gray NK, Hentze MW. Iron regulatory protein prevents binding of the 43S translation pre-initiation complex to ferritin and eALAS mRNAs. EMBO J. 1994;13:3882–91.
Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci. 2004;1012:1–13.
Zhang DL, Ghosh MC, Rouault TA. The physiological functions of iron regulatory proteins in iron homeostasis - an update. Front Pharmacol. 2014;5:124.
Piccinelli P, Samuelsson T. Evolution of the iron-responsive element. RNA. 2007;13:952–66.
Kato J, Kobune M, Ohkubo S, Fujikawa K, Tanaka M, Takimoto R, Takada K, Takahari D, Kawano Y, Kohgo Y, et al. Iron/IRP-1-dependent regulation of mRNA expression for transferrin receptor, DMT1 and ferritin during human erythroid differentiation. Exp Hematol. 2007;35:879–87.
Ma J, Haldar S, Khan MA, Sharma SD, Merrick WC, Theil EC, Goss DJ. Fe2+ binds iron responsive element-RNA, selectively changing protein-binding affinities and regulating mRNA repression and activation. Proc Natl Acad Sci U S A. 2011;109:8417–22.
Fu D, Richardson DR. Iron chelation and regulation of the cell cycle: 2 mechanisms of posttranscriptional regulation of the universal cyclin-dependent kinase inhibitor p21CIP1/WAF1 by iron depletion. Blood. 2007;110:752–61.
Yu Y, Kovacevic Z, Richardson DR. Tuning cell cycle regulation with an iron key. Cell Cycle. 2007;6:1982–94.
Sanchez M, Galy B, Muckenthaler MU, Hentze MW. Iron-regulatory proteins limit hypoxia-inducible factor-2alpha expression in iron deficiency. Nat Struct Mol Biol. 2007;14:420–6.
Nurtjahja-Tjendraputra E, Fu D, Phang JM, Richardson DR. Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood. 2007;109:4045–54.
Kayed R, Jackson GR, Estes DM, Barrett AD. Alzheimers disease: review of emerging treatment role for intravenous immunoglobulins. J Cent Nerv Syst Dis. 2011;3:67–73.
Abbasowa L, Heegaard NHA. Systematic review of amyloid-beta peptides as putative mediators of the association between affective disorders and Alzheimers disease. J Affect Disord. 2014;168:167–83.
Sadigh-Eteghad S, Sabermarouf B, Majdi A, Talebi M, Farhoudi M, Mahmoudi J. Amyloid-beta: a crucial factor in Alzheimer's disease. Med Princ Pract. 2014;24:1–10.
Rovelet-Lecrux A, Frebourg T, Tuominen H, Majamaa K, Campion D, Remes AMAPP. Locus duplication in a Finnish family with dementia and intracerebral haemorrhage. J Neurol Neurosurg Psychiatry. 2007;78:1158–9.
Guyant-Marechal L, Rovelet-Lecrux A, Goumidi L, Cousin E, Hannequin D, Raux G, Penet C, Ricard S, Mace S, Amouyel P, et al. Variations in the APP gene promoter region and risk of Alzheimer disease. Neurology. 2007;68:684–7.
Altamura S, Muckenthaler MU. Iron toxicity in diseases of aging: Alzheimer's disease, Parkinson's disease and atherosclerosis. J Alzheimers Dis. 2009;16:879–95.
Rogers JT, Bush AI, Cho HH, Smith DH, Thomson AM, Friedlich AL, Lahiri DK, Leedman PJ, Huang X, Cahill CM. Iron and the translation of the amyloid precursor protein (APP) and ferritin mRNAs: riboregulation against neural oxidative damage in Alzheimer's disease. Biochem Soc Trans. 2008;36:1282–7.
Bandyopadhyay S, Huang X, Cho H, Greig NH, Youdim MB, Rogers JT. Metal specificity of an iron-responsive element in Alzheimer's APP mRNA 5'untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J Neural Transm Suppl. 2006;71:237–47.
Cahill CM, Lahiri DK, Huang X, Rogers JT. Amyloid precursor protein and alpha synuclein translation, implications for iron and inflammation in neurodegenerative diseases. Biochim Biophys Acta. 2009;1790:615–28.
Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer's amyloid precursor protein transcript. J Biol Chem. 2002;277:45518–28.
Cho HH, Cahill CM, Vanderburg CR, Scherzer CR, Wang B, Huang X, Rogers JT. Selective translational control of the Alzheimer amyloid precursor protein transcript by iron regulatory protein-1. J Biol Chem. 2010;285:31217–32.
Tucker S, Ahl M, Cho HH, Bandyopadhyay S, Cuny GD, Bush AI, Goldstein LE, Westaway D, Huang X, Rogers JTRNA. Therapeutics directed to the non coding regions of APP mRNA, in vivo anti-amyloid efficacy of paroxetine, erythromycin, and N-acetyl cysteine. Curr Alzheimer Res. 2006;3:221–7.
Maccecchini ML, Chang MY, Pan C, John V, Zetterberg H, Greig NH. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-beta peptide and tau levels: target engagement, tolerability and pharmacokinetics in humans. J Neurol Neurosurg Psychiatry. 2013;83:894–902.
Tahmasebinia F, Emadi S. Effect of metal chelators on the aggregation of beta-amyloid peptides in the presence of copper and iron. Biometals. 2017;30:285–93.
Khan MA, Quadri SA, Tohid HA. Comprehensive overview of the neuropsychiatry of Parkinson's disease: a review. Bull Menn Clin. 2017;81:53–105.
Venuto CS, Potter NB, Ray Dorsey E, Kieburtz KA. Review of disease progression models of Parkinson's disease and applications in clinical trials. Mov Disord. 2016;31:947–56.
Salvatore MF, Fisher B, Surgener SP, Gerhardt GA, Rouault T. Neurochemical investigations of dopamine neuronal systems in iron-regulatory protein 2 (IRP-2) knockout mice. Brain Res Mol Brain Res. 2005;139:341–7.
LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat Genet. 2001;27:209–14.
Weiss G, Goossen B, Doppler W, Fuchs D, Pantopoulos K, Werner-Felmayer G, Wachter H, Hentze MW. Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J. 1993;12:3651–7.
Jaffrey SR, Cohen NA, Rouault TA, Klausner RD, Snyder SH. The iron-responsive element binding protein: a target for synaptic actions of nitric oxide. Proc Natl Acad Sci U S A. 1994;91:12994–8.
Ayton S, Lei P, Hare DJ, Duce JA, George JL, Adlard PA, McLean C, Rogers JT, Cherny RA, Finkelstein DI, et al. Parkinson's disease iron deposition caused by nitric oxide-induced loss of beta-amyloid precursor protein. J Neurosci. 2015;35:3591–7.
Febbraro F, Giorgi M, Caldarola S, Loreni F, Romero-Ramos M. Alpha-Synuclein expression is modulated at the translational level by iron. Neuroreport. 2012;23:576–80.
Li WJ, Jiang H, Song N, Xie JX. Dose- and time-dependent alpha-synuclein aggregation induced by ferric iron in SK-N-SH cells. Neurosci Bull. 2010;26:205–10.
Rocha EM, De Miranda B, Sanders LH. Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson's disease. Neurobiol Dis. 2017;17:30080–3.
Wrasidlo W, Tsigelny IF, Price DL, Dutta G, Rockenstein E, Schwarz TC, Ledolter K, Bonhaus D, Paulino A, Eleuteri S, et al. A de novo compound targeting alpha-synuclein improves deficits in models of Parkinson's disease. Brain. 2016;139:3217–36.
Lindstrom V, Ihse E, Fagerqvist T, Bergstrom J, Nordstrom E, Moller C, Lannfelt L, Ingelsson M. Immunotherapy targeting alpha-synuclein, with relevance for future treatment of Parkinson's disease and other Lewy body disorders. Immunotherapy. 2014;6:141–53.
Wakabayashi K, Tanji K, Mori F, Takahashi H. The Lewy body in Parkinson's disease: molecules implicated in the formation and degradation of alpha-synuclein aggregates. Neuropathology. 2007;27:494–506.
Kruger R, Kuhn W, Muller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schols L, Riess O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet. 1998;18:106–8.
Vaughan J, Durr A, Tassin J, Bereznai B, Gasser T, Bonifati V, De Michele G, Fabrizio E, Volpe G, Bandmann O, et al. The alpha-synuclein Ala53Thr mutation is not a common cause of familial Parkinson's disease: a study of 230 European cases. European consortium on genetic susceptibility in Parkinson's disease. Ann Neurol. 1998;44:270–3.
Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, Sherman H, Yu I, Shah B, Weir D, Thompson C, Szu-Tu C, Trinh J, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson's disease. Mov Disord. 2013;28:811–3.
Nishioka K, Hayashi S, Farrer MJ, Singleton AB, Yoshino H, Imai H, Kitami T, Sato K, Kuroda R, Tomiyama H, et al. Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson's disease. Ann Neurol. 2006;59:298–309.
Bradbury J. Alpha-synuclein gene triplication discovered in Parkinson's disease. Lancet Neurol. 2003;2:715.
Rogers JT, Mikkilineni S, Cantuti-Castelvetri I, Smith DH, Huang X, Bandyopadhyay S, Cahill CM, Maccecchini ML, Lahiri DK, Greig NH. The alpha-synuclein 5'untranslated region targeted translation blockers: anti-alpha synuclein efficacy of cardiac glycosides and Posiphen. J Neural Transm (Vienna). 2011;118:493–507.
QS Y, Reale M, Kamal MA, Holloway HW, Luo W, Sambamurti K, Ray B, Lahiri DK, Rogers JT, Greig NH. Synthesis of the Alzheimer drug Posiphen into its primary metabolic products (+)-N1-norPosiphen, (+)-N8-norPosiphen and (+)-N1, N8-bisnorPosiphen, their inhibition of amyloid precursor protein, alpha-Synuclein synthesis, interleukin-1beta release, and cholinergic action. Antiinflamm Antiallergy Agents Med Chem. 2013;12:117–28.
Febbraro F, Andersen KJ, Sanchez-Guajardo V, Tentillier N, Romero-Ramos M. Chronic intranasal deferoxamine ameliorates motor defects and pathology in the alpha-synuclein rAAV Parkinson's model. Exp Neurol. 2013;247:45–58.
Finkelstein DI, Hare DJ, Billings JL, Sedjahtera A, Nurjono M, Arthofer E, George S, Culvenor JG, Bush AI, Adlard PA. Clioquinol improves cognitive, motor function, and microanatomy of the alpha-Synuclein hA53T transgenic mice. ACS Chem Neurosci. 2016;7:119–29.
Gajowiak A, Stys A, Starzynski RR, Staron R, Lipinski P. Misregulation of iron homeostasis in amyotrophic lateral sclerosis. Postepy Hig Med Dosw (Online). 2016;70:709–21.
Jeong SY, Rathore KI, Schulz K, Ponka P, Arosio P, David S. Dysregulation of iron homeostasis in the CNS contributes to disease progression in a mouse model of amyotrophic lateral sclerosis. J Neurosci. 2009;29:610–9.
Takei Y, Oguchi K, Koshihara H, Hineno A, Nakamura A, Ohara S. Alpha-Synuclein coaggregation in familial amyotrophic lateral sclerosis with SOD1 gene mutation. Hum Pathol. 2013;44:1171–6.
Koch Y, Helferich AM, Steinacker P, Oeckl P, Walther P, Weishaupt JH, Danzer KM, Otto M. Aggregated alpha-Synuclein increases SOD1 oligomerization in a mouse model of amyotrophic lateral sclerosis. Am J Pathol. 2016;186:2152–61.
Yu J, Qi F, Wang N, Gao P, Dai S, Lu Y, Su Q, Du Y, Che F. Increased iron level in motor cortex of amyotrophic lateral sclerosis patients: an in vivo MR study. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:357–61.
Ignjatovic A, Stevic Z, Lavrnic S, Dakovic M, Bacic G. Brain iron MRI: a biomarker for amyotrophic lateral sclerosis. J Magn Reson Imaging. 2013;38:1472–9.
Deschauer M, Gaul C, Behrmann C, Prokisch H, Zierz S, Haack TB. C19orf12 mutations in neurodegeneration with brain iron accumulation mimicking juvenile amyotrophic lateral sclerosis. J Neurol. 2012;259:2434–9.
Golko-Perez S, Mandel S, Amit T, Kupershmidt L, Youdim MB, Weinreb O. Additive neuroprotective effects of the multifunctional iron chelator M30 with enriched diet in a mouse model of amyotrophic lateral sclerosis. Neurotox Res. 2016;29:208–17.
Kupershmidt L, Weinreb O, Amit T, Mandel S, Carri MT, Youdim MB. Neuroprotective and neuritogenic activities of novel multimodal iron-chelating drugs in motor-neuron-like NSC-34 cells and transgenic mouse model of amyotrophic lateral sclerosis. FASEB J. 2009;23:3766–79.
Wang Q, Zhang X, Chen S, Zhang X, Zhang S, Youdium M, Le W. Prevention of motor neuron degeneration by novel iron chelators in SOD1(G93A) transgenic mice of amyotrophic lateral sclerosis. Neurodegener Dis. 2011;8:310–21.
Qureshi M, Brown RH Jr, Rogers JT, Cudkowicz ME. Serum ferritin and metal levels as risk factors for amyotrophic lateral sclerosis. Open Neurol J. 2008;2:51–4.
DeMeo DL, Mariani T, Bhattacharya S, Srisuma S, Lange C, Litonjua A, Bueno R, Pillai SG, Lomas DA, Sparrow D, et al. Integration of genomic and genetic approaches implicates IREB2 as a COPD susceptibility gene. Am J Hum Genet. 2009;85:493–502.
Miyajima H. Genetic disorders affecting proteins of iron and copper metabolism: clinical implications. Intern Med. 2002;41:762–9.
Aguilar-Martinez P, Schved JF, Brissot P. The evaluation of hyperferritinemia: an updated strategy based on advances in detecting genetic abnormalities. Am J Gastroenterol. 2005;100:1185–94.
Cao W, McMahon M, Wang B, O'Connor R, Clarkson MA. Ccase report of spontaneous mutation (C33>U) in the iron-responsive element of L-ferritin causing hyperferritinemia-cataract syndrome. Blood Cells Mol Dis. 2010;44:22–7.
Kato J, Fujikawa K, Kanda M, Fukuda N, Sasaki K, Takayama T, Kobune M, Takada K, Takimoto R, Hamada H, et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am J Hum Genet. 2001;69:191–7.
Cazzola M, Bergamaschi G, Tonon L, Arbustini E, Grasso M, Vercesi E, Barosi G, Bianchi PE, Cairo G, Arosio P. Hereditary hyperferritinemia-cataract syndrome: relationship between phenotypes and specific mutations in the iron-responsive element of ferritin light-chain mRNA. Blood. 1997;90:814–21.
Liu W, Shimomura S, Imanishi H, Iwamoto Y, Ikeda N, Saito M, Ohno M, Hara N, Yamamoto T, Nakamura H, et al. Hemochromatosis with mutation of the ferroportin 1 (IREG1) gene. Intern Med. 2005;44:285–9.
Mok H, Jelinek J, Pai S, Cattanach BM, Prchal JT, Youssoufian H, Schumacher A. Disruption of ferroportin 1 regulation causes dynamic alterations in iron homeostasis and erythropoiesis in polycythaemia mice. Development. 2004;131:1859–68.
Levi S, Cozzi A, Arosio P. Neuroferritinopathy: a neurodegenerative disorder associated with L-ferritin mutation. Best Pract Res Clin Haematol. 2005;18:265–76.
Garringer HJ, Irimia JM, Li W, Goodwin CB, Richine B, Acton A, Chan RJ, Peacock M, Muhoberac BB, Ghetti B, et al. Effect of systemic iron overload and a chelation therapy in a mouse model of the neurodegenerative disease hereditary Ferritinopathy. PLoS One. 2016;11:e0161341.
Friedman A, Arosio P, Finazzi D, Koziorowski D, Galazka-Friedman J. Ferritin as an important player in neurodegeneration. Parkinsonism Relat Disord. 2011;17:423–30.
Luscieti S, Santambrogio P, Langlois d'Estaintot B, Granier T, Cozzi A, Poli M, Gallois B, Finazzi D, Cattaneo A, Levi S, et al. mutant ferritin L-chains that cause neurodegeneration act in a dominant-negative manner to reduce ferritin iron incorporation. J Biol Chem. 2010;285:11948–57.
Muhoberac BB, Baraibar MA, Vidal R. Iron loading-induced aggregation and reduction of iron incorporation in heteropolymeric ferritin containing a mutant light chain that causes neurodegeneration. Biochim Biophys Acta. 1812;2011:544–8.
Vidal R, Miravalle L, Gao X, Barbeito AG, Baraibar MA, Hekmatyar SK, Widel M, Bansal N, Delisle MB, Ghetti B. Expression of a mutant form of the ferritin light chain gene induces neurodegeneration and iron overload in transgenic mice. J Neurosci. 2008;28:60–7.
Cotter PD, May A, Fitzsimons EJ, Houston T, Woodcock BE, Al-Sabah AI, Wong L, Bishop DF. Late-onset X-linked sideroblastic anemia. Missense mutations in the erythroid delta-aminolevulinate synthase (ALAS2) gene in two pyridoxine-responsive patients initially diagnosed with acquired refractory anemia and ringed sideroblasts. J Clin Invest. 1995;96:2090–6.
Cox TC, Bottomley SS, Wiley JS, Bawden MJ, Matthews CS, May BKX. Linked pyridoxine-responsive sideroblastic anemia due to a Thr388-to-Ser substitution in erythroid 5-aminolevulinate synthase. N Engl J Med. 1994;330:675–9.
Whatley SD, Ducamp S, Gouya L, Grandchamp B, Beaumont C, Badminton MN, Elder GH, Holme SA, Anstey AV, Parker M, et al. C-terminal deletions in the ALAS2 gene lead to gain of function and cause X-linked dominant protoporphyria without anemia or iron overload. Am J Hum Genet. 2008;83:408–14.
Mayr R, Griffiths WJ, Hermann M, McFarlane I, Halsall DJ, Finkenstedt A, Douds A, Davies SE, Janecke AR, Vogel W, et al. Identification of mutations in SLC40A1 that affect ferroportin function and phenotype of human ferroportin iron overload. Gastroenterology 2011; 140: 2056–2063, 2063 e1.
De Domenico I, Ward DM, Musci G, Kaplan J. Iron overload due to mutations in ferroportin. Haematologica. 2006;91:92–5.
Mims MP, Guan Y, Pospisilova D, Priwitzerova M, Indrak K, Ponka P, Divoky V, Prchal JT. Identification of a human mutation of DMT1 in a patient with microcytic anemia and iron overload. Blood. 2005;105:1337–42.
Lam-Yuk-Tseung S, Camaschella C, Iolascon A, Gros P. A novel R416C mutation in human DMT1 (SLC11A2) displays pleiotropic effects on function and causes microcytic anemia and hepatic iron overload. Blood Cells Mol Dis. 2006;36:347–54.
Iolascon A, d'Apolito M, Servedio V, Cimmino F, Piga A, Camaschella C. Microcytic anemia and hepatic iron overload in a child with compound heterozygous mutations in DMT1 (SCL11A2). Blood. 2006;107:349–54.
Bandyopadhyay S, Cahill C, Balleidier A, Huang C, Lahiri DK, Huang X, Rogers JT. Novel 5′ untranslated region directed blockers of iron-regulatory protein-1 dependent amyloid precursor protein translation: implications for down syndrome and Alzheimer's disease. PLoS One. 2013;8:e65978.
Lilja AM, Luo Y, QS Y, Rojdner J, Li Y, Marini AM, Marutle A, Nordberg A, Greig NH. Neurotrophic and neuroprotective actions of (−)- and (+)-phenserine, candidate drugs for Alzheimer's disease. PLoS One. 2013;8:e54887.
Lahiri DK, Chen D, Maloney B, Holloway HW, QS Y, Utsuki T, Giordano T, Sambamurti K, Greig NH. The experimental Alzheimer's disease drug posiphen [(+)-phenserine] lowers amyloid-beta peptide levels in cell culture and mice. J Pharmacol Exp Ther. 2007;320:386–96.
Shaw KT, Utsuki T, Rogers J, QS Y, Sambamurti K, Brossi A, Ge YW, Lahiri DK, Greig NH. Phenserine regulates translation of beta -amyloid precursor protein mRNA by a putative interleukin-1 responsive element, a target for drug development. Proc Natl Acad Sci U S A. 2001;98:7605–10.
Morse LJ, Payton SM, Cuny GD, Rogers JT. FDA-preapproved drugs targeted to the translational regulation and processing of the amyloid precursor protein. J Mol Neurosci. 2004;24:129–36.
Maccecchini ML, Chang MY, Pan C, John V, Zetterberg H, Greig NH. Posiphen as a candidate drug to lower CSF amyloid precursor protein, amyloid-beta peptide and tau levels: target engagement, tolerability and pharmacokinetics in humans. J Neurol Neurosurg Psychiatry. 2012;83:894–902.
Bandyopadhyay S, Rogers JT. Alzheimer's disease therapeutics targeted to the control of amyloid precursor protein translation: maintenance of brain iron homeostasis. Biochem Pharmacol. 2014;88:486–94.
Payton S, Cahill CM, Randall JD, Gullans SR, Rogers JT. Drug discovery targeted to the Alzheimer's APP mRNA 5′-untranslated region: the action of paroxetine and dimercaptopropanol. J Mol Neurosci. 2003;20:267–75.
Ross NT, Metkar SR, Le H, Burbank J, Cahill C, Germain A, MacPherson L, Bittker J, Palmer M, Rogers J, et al. Identification of a small molecule that selectively inhibits alpha-synuclein translational expression. Probe reports from the NIH molecular libraries. Program. 2011;
Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci U S A. 1991;88:10540–3.
Rogers JT, Lahiri DK. Metal and inflammatory targets for Alzheimer's disease. Curr Drug Targets. 2004;5:535–51.
Jiang H, Luan Z, Wang J, Xie J. Neuroprotective effects of iron chelator Desferal on dopaminergic neurons in the substantia nigra of rats with iron-overload. Neurochem Int. 2006;49:605–9.
Bandyopadhyay S, Huang X, Lahiri DK, Rogers JT. Novel drug targets based on metallobiology of Alzheimer's disease. Expert Opin Ther Targets. 2010;14:1177–97.
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A, et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: a pilot phase 2 clinical trial. Arch Neurol. 2003;60:1685–91.
Zhang YH, Raymick J, Sarkar S, Lahiri DK, Ray B, Holtzman D, Dumas M, Schmued LC. Efficacy and toxicity of clioquinol treatment and A-beta42 inoculation in the APP/PSI mouse model of Alzheimer's disease. Curr Alzheimer Res. 2013;10:494–506.
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H, et al. PBT2 rapidly improves cognition in Alzheimer's disease: additional phase II analyses. J Alzheimers Dis. 2010;20:509–16.
Lannfelt L, Blennow K, Zetterberg H, Batsman S, Ames D, Harrison J, Masters CL, Targum S, Bush AI, Murdoch R, et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer's disease: a phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008;7:779–86.
Adlard PA, Sedjahtera A, Gunawan L, Bray L, Hare D, Lear J, Doble P, Bush AI, Finkelstein DI, Cherny RA. A novel approach to rapidly prevent age-related cognitive decline. Aging Cell. 2013;13:351–9.
Adlard PA, Bica L, White AR, Nurjono M, Filiz G, Crouch PJ, Donnelly PS, Cappai R, Finkelstein DI, Bush AI. Metal ionophore treatment restores dendritic spine density and synaptic protein levels in a mouse model of Alzheimer's disease. PLoS One. 2011;6:e17669.
Xu Q, Kanthasamy AG, Reddy MB. Neuroprotective effect of the natural iron chelator, phytic acid in a cell culture model of Parkinson's disease. Toxicology. 2008;245:101–8.
Ghosh B, Antonio T, Reith ME, Dutta AK. Discovery of 4-(4-(2-((5-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)ethyl)piperaz in-1-yl)quinolin-8-ol and its analogues as highly potent dopamine D2/D3 agonists and as iron chelator: in vivo activity indicates potential application in symptomatic and neuroprotective therapy for Parkinson's disease. J Med Chem. 2010;53:2114–25.
Dexter DT, Statton SA, Whitmore C, Freinbichler W, Weinberger P, Tipton KF, Della Corte L, Ward RJ, Crichton RR. Clinically available iron chelators induce neuroprotection in the 6-OHDA model of Parkinson's disease after peripheral administration. J Neural Transm (Vienna). 2011;118:223–31.
Bar-Am O, Amit T, Kupershmidt L, Aluf Y, Mechlovich D, Kabha H, Danovitch L, Zurawski VR, Youdim MB, Weinreb O. Neuroprotective and neurorestorative activities of a novel iron chelator-brain selective monoamine oxidase-a/monoamine oxidase-B inhibitor in animal models of Parkinson's disease and aging. Neurobiol Aging. 2015;36:1529–42.
Das B, Kandegedara A, Xu L, Antonio T, Stemmler T, Reith MEA, Dutta AKA. Novel iron(II) preferring dopamine agonist chelator as potential symptomatic and neuroprotective therapeutic agent for Parkinson's disease. ACS Chem Neurosci. 2017;8:723–30.
Beaumont C, Leneuve P, Devaux I, Scoazec JY, Berthier M, Loiseau MN, Grandchamp B, Bonneau D. Mutation in the iron responsive element of the L ferritin mRNA in a family with dominant hyperferritinaemia and cataract. Nat Genet. 1995;11:444–6.
Athan ES, Lee JH, Arriaga A, Mayeux RP, Tycko B. Polymorphisms in the promoter of the human APP gene: functional evaluation and allele frequencies in Alzheimer disease. Arch Neurol. 2002;59:1793–9.
Coon KD, Siegel AM, Yee SJ, Dunckley TL, Mueller C, Nagra RM, Tourtellotte WW, Reiman EM, Papassotiropoulos A, Petersen FF, et al. Preliminary demonstration of an allelic association of the IREB2 gene with Alzheimer's disease. J Alzheimers Dis. 2006;9:225–33.
Acknowledgments
We thank Singapore National Medical Research Council (STaR and Transition awards and the clinical translational research programme in Parkinson’s disease) for their support.
Funding
This study was supported by Singapore National Medical Research Council (NMRC) grants including STaR and Transition awards and a clinical translational research programme in Parkinson’s disease.
Availability of data and materials
All tables and figures are original for this review.
Author information
Authors and Affiliations
Contributions
ZZD reviewed the literature, drafted and critically revised the manuscript. TEK drafted and critically revised the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, Z.D., Tan, EK. Iron regulatory protein (IRP)-iron responsive element (IRE) signaling pathway in human neurodegenerative diseases. Mol Neurodegeneration 12, 75 (2017). https://doi.org/10.1186/s13024-017-0218-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13024-017-0218-4