
Abstract
Background
The SLC39A14, SLC30A10 and SLC39A8 are considered to be key genes involved in manganese (Mn) homeostasis in humans. Mn levels in plasma and urine are useful tools for early recognition of these disorders. We aimed to explore further biomarkers of Mn deposition in the central nervous system in two siblings presenting with acute dystonia and hypermanganesemia due to mutations in SLC39A14. These biomarkers may help clinicians to establish faster and accurate diagnosis and to monitor disease progression after chelation therapy is administered.
Results
A customized gene panel for movement disorders revealed a novel missense variant (c.311G > T; p.Ser104Ile) in SLC39A14 gene in two siblings presenting at the age of 10 months with acute dystonia and motor regression. Mn concentrations were analyzed using inductively coupled mass spectrometry in plasma and cerebrospinal fluid, disclosing elevated Mn levels in the index case compared to control patients. Surprisingly, Mn values were 3-fold higher in CSF than in plasma. We quantified the pallidal index, defined as the ratio between the signal intensity in the globus pallidus and the subcortical frontal white matter in axial T1-weighted MRI, and found significantly higher values in the SLC39A14 patient than in controls. These values increased over a period of 10 years, suggesting the relentless pallidal accumulation of Mn. Following genetic confirmation, a trial with the Mn chelator Na2CaEDTA led to a reduction in plasma Mn, zinc and selenium levels. However, parents reported worsening of cervical dystonia, irritability and sleep difficulties and chelation therapy was discontinued.
Conclusions
Our study expands the very few descriptions of patients with SLC39A14 mutations. We report for the first time the elevation of Mn in CSF of SLC39A14 mutated patients, supporting the hypothesis that brain is an important organ of Mn deposition in SLC39A14-related disease. The pallidal index is an indirect and non-invasive method that can be used to rate disease progression on follow-up MRIs. Finally, we propose that patients with inherited defects of manganese transport should be initially treated with low doses of Na2CaEDTA followed by gradual dose escalation, together with a close monitoring of blood trace elements in order to avoid side effects.
Similar content being viewed by others

Background
Manganese (Mn) is a trace metal with a key role as a cofactor of multiple enzymes, including hydrolases, lyases, glycosyltransferases, arginase, glutamine synthase and superoxide dismutase (SOD), in the synthesis of hormones and neurotransmitters [1] and during inflammatory events of the central nervous system [2].
Mn deposition in the brain can occur due to acquired causes (such as environmental exposure), as well as inherited defects in Mn transport and metabolism. Mn intoxication has been described in miners, welders, individuals working or living near ferro-alloy factories or in those drinking contaminated water, as well as in patients receiving total parenteral nutrition or those with acquired hepatocerebral degeneration [3]. Patients with severe Mn exposure may develop an extrapyramidal syndrome termed manganism, with rigidity, bradykinesia and dystonia [3]. Mn dyshomoeostasis may also result from inherited genetic defects in one of the transporters implicated in Mn homeostasis, namely SLC39A8, SLC30A10 and SLC39A14. Recently, several in vitro and in vivo models have elucidated their role in the transport of Mn and other divalent metals [2, 4,5,6,7,8,9,10]. SLC39A8 (MIM608732) encodes ZIP8, a Mn and Zn transporter that localizes to the hepatocyte canalicular membrane and reclaims Mn from bile [7]. Knock out ZIP8 mice showed markedly decreased Mn levels in multiple organs and whole blood, increased bile levels, and decreased activity of Mn-dependent enzymes, such as arginase and β-1, 4-galactosyltransferase [7]. Biallelic mutations in this gene lead to severe Mn depletion and a secondary congenital disorder of glycosylation (CDG) syndrome. Patients manifest developmental delay and intellectual disability, dwarfism, craniosynostosis, cerebellar atrophy, seizures and Leigh-like syndrome [11, 12].
SLC30A10 (also known as ZnT10) (MIM611146) and SLC39A14 (also known as ZIP14) (MIM608736) are efflux and influx transporters, respectively, that cooperatively regulate Mn homeostasis in humans. Recently, Liu et al. 2017 generated knockout (KO) mice models lacking SLC30A10, SLC39A14 and both transporters (double knockouts) demonstrating high blood and brain Mn levels in the three mice models, but only high liver Mn levels in the single SLC30A10 KO model. These findings are in agreement with those observed in patients with recessive mutations in SLC30A10 and SLC39A14, showing cerebral Mn deposition as a consequence of increased systemic Mn load in both disorders [13], but only polycythemia and liver cirrhosis in SLC30A10 [14].
Animal models demonstrated that SLC30A10 and SLC39A14 localized to the canalicular and basolateral domains of hepatocytes, respectively, thereby mediating Mn biliary excretion synergistically [8]. Moreover, SLC39A14 KO mice showed reduce Mn transport into enterocytes at the basolateral membrane, thereby decreasing Mn excretion via the gastrointestinal tract [4].
Patients with SLC30A10 and SLC39A14 defects show a progressive dystonia-parkinsonism syndrome as a consequence of Mn toxicity to the basal ganglia. Chelation therapy in both disorders increases Mn urinary excretion and decreases plasma Mn concentrations with variable clinical improvement [14,15,16,17]. SLC39A14 KO mice suffer from Mn brain deposition and motor dysfunction, thus recapitulating the disease in humans [4, 10].
In this study, we present two siblings with homozygous SLC39A14 mutations causing hypermanganesemia, progressive dystonia, severe disability and early death. Our study reports a likely novel pathogenic variant in SLC39A14, thereby expanding the reported mutations in this gene. We examine the levels of Mn and other trace elements in plasma and cerebrospinal fluid (CSF), as potential biomarkers for diagnosis and treatment monitoring. We also describe serial magnetic resonance imaging (MRI) abnormalities over time, confirming progressive changes with pallidal Mn deposition.
Methods
The index case was born to healthy consanguineous Senegalese parents. Detailed delineation of the patient’s history, disease course and clinical examination was undertaken, as well as molecular genetics, radiological and biochemical studies. Family history revealed a similarly affected older brother for whom a stored DNA sample was available. Parental DNA was also obtained. The study was approved by the ethics committee at Sant Joan de Déu Hospital and parents gave written informed consent for study participation.
We also performed a literature review on genetic causes of Mn dysregulation, by searching MEDLINE (through PubMed) the following keywords: #1 SLC30A10, #2 SLC39A14, #3 SLC39A8, #4 hypermanganesemia and #5 manganese homeostasis. A total of 12 clinical studies were finally selected (Table 1) [11, 12, 15,16,17,18,19,20,21,22,23,24,25].
Biochemical studies
CSF and plasma samples were analyzed with an Agilent 7500ce inductively coupled plasma mass spectrometer (Agilent Technologies, Waldbronn, Germany). The instrument uses a collision/reaction cell with hydrogen for selenium (Se) determination, and helium for zinc (Zn) and Mn. Briefly, after ionization of plasma or CSF samples in the plasma torch and elimination of interference in the collision/reaction cells, element concentrations were determined by mass spectrometry, as previously reported [26, 27]. Hemolysed plasma samples were excluded to avoid blood contamination that can significantly increase Zn and Mn values.
Plasma samples preparation: 25 μL of plasma samples were diluted (1:40; V:V) in 25 μL of distilled water and 950 μL of a solution containing 0.7 mmol/L EDTA (Merck, Darmstadt, Germany), 0.07% Triton-X-100 (Merck), 2% butanol, 0.5% ammoniac (Merck) and germanium as internal standard (Merck).
CSF samples preparation: 50 μL of CSF samples were diluted (1:20; V: V) in a 2% nitric solution and 10 μg/L of germanium as internal standard (Merck).
MRI studies
The pallidal index (PI), defined as the ratio between the signal intensity in the globus pallidus (SIGP) and the subcortical frontal white matter (SIFW) in axial T1-weighted MRI [28] was calculated in our patient on MRIs undertaken at age 11 months, 8 and 10 years. Values were compared to nine age-matched control patients. The MRIs in control patients were obtained as part of a diagnostic protocol for patients with chronic headache, and were classified as normal by expert neuroradiologists.
Genetic analysis
A customized gene panel for movement disorders was design by Sure Design Tool (Agilent Technologies, Santa Clara, CA, USA). This panel included 78 genes causing basal ganglia disease which were classified in four groups: Aicardi-Goutières syndrome, thiamine metabolism, mitochondrial disorders and other neurometabolic disorders, including SLC30A10 and SLC39A14 related to Mn dysregulation. Library construction was performed according to manufacturer’s protocol using HaloPlex technology. Sequencing was carried out on MiSeq sequencer (Illumina, San Diego, CA, USA). Data processing, variant calling and variant annotation were done by DNAnexus platform and Variant Studio software. The average of mean-coverage in the sample gene panel was 95% for a read depth of 20X. Filtering was performed by minor allele frequency < 1% and possible pathogenicity based on mutation effects (frameshift, insertions deletions, missense, stop gain and splice site regions). Variant validation and segregation studies were done by PCR with Sanger sequencing using the Big Dye Terminator Cycle Sequencing System (Applied Biosystems). Primers for validation of the identified change in SLC39A14 were forward primer 5’-GAAGGCTGAGTAGGTTGCTG- 3′ and reverse primer 5’-CTCCTCGTTTTCCTGGTTCT-3′.
Results
Clinical presentation
The proband was born from consanguineous healthy Senegalese parents, after an uneventful pregnancy and delivery. He had a normal perinatal period and early neurodevelopmental milestones were on average. At 11 months he developed acute generalized dystonia and neurological regression following an intercurrent viral respiratory infection. On neurological examination there was evidence of skull deformity with normal head circumference, dystonic tetraparesis, oromandibular dystonia and opisthotonos. A plain skull radiogram revealed multiple-suture craniosynostosis. Metabolic investigations in blood, CSF and urine were normal at that moment, except for a mild decrease in 5-hydroxyindolacetic acid concentrations (104 nmol/L, reference values (RV): 170–490). The family moved to Senegal and they returned to our hospital at the age of 9 years. At this time, the patient had developed microcephaly (head circumference 51 cm, 6th percentile), severe dystonic tetraparesis, anarthria, dysphagia and malnutrition. He required enteral tube feeding and received baclofen, diazepam and gabapentin for symptomatic control of dystonia.
On reviewing the family history, it became apparent that there was a similarly affected older brother, born in 1997, who developed acute dystonic tetraparesis associated with rigidity, hypokinesia and pyramidal signs, following a viral illness at the age of 10 months. Brain MRI revealed bilateral pallidal T2-hypointensity and pallidal and cerebral and cerebellar white matter T1-hyperintensity. His clinical status remained unchanged until age 21 months, when he died in Senegal of unknown cause.
The clinical picture of these siblings was analyzed in the context of the existing literature on 46 patients previously identified with genetic defects leading to Mn dysregulation (Table 1).
Biochemical studies
Biochemical studies in the proband detected normal full blood count, liver function, Fe metabolism, Zn, Se and copper (Cu) concentrations in plasma. We also found elevated plasma Mn (10.5 μg/L, RV: 0.4–0.9 μg/L) and extremely elevated CSF Mn concentrations (34 μg/L, RV: 0.5–1.7 μg/L).
MRI analysis
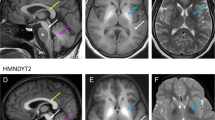
Brain MRI of the patient at 11 months showed a symmetrical high-T1 and low T2 signal in both the pallidum and dentate nuclei. White matter signal intensity of the cerebrum, cerebellum and brainstem was also very high in T1 (Fig. 1a). Follow-up MRI at 10 years showed persistent T1-hyperintensity of the globi pallidi, volume loss, gliosis and atrophy of the dentate nuclei and moderate atrophy of cerebellar folia. Moreover, quantitative assessments of the PI was significantly higher in the patient at age 11 months, 8 and 10 years as compared to nine age-matched controls (U = 0.000; p = 0.009, Mann-Whitney U test). We also observed slightly increased signal intensity over time (Fig. 1b).

Radiological findings from the patient. a: Brain MRI of the patient at 11 months (first line) shows a high-T1 and low T2 signal in the pallidi and dentate nuclei. MRI at 10 years (second line) shows persistent T1-hyperintensity of the globi pallidi, volume loss, gliosis and atrophy of the dentate nuclei and moderate atrophy of cerebellar folia. b: Distribution of individual PI scores in patient and controls
Genetic analysis
We identified a homozygous missense variant (c.311G > T; p.Ser104Ile) in exon 3 of SLC39A14 (NM_001128431), which was confirmed by Sanger sequencing in both the patient and his affected brother. Both parents were heterozygous carriers of this variant (Fig. 2). Unfortunately, DNA samples from three unaffected siblings were not available for the analysis. This novel variant was not found in HGMD, dbSNP, 1000 Genome project, ExAC database or CIBERER Spanish Variant Server. This variant affected a highly conserved amino acid residue, located in the N-terminus extracellular loop, studied by UCSC browser and Clustal Omega software and it was categorized as pathogenic by SIFT (0.002), PROVEAN (−3.03) and Mutation Taster (142), and as possibly damaging by PolyPhen-2 (0.664).

The figure shows segregation of the novel variant by Sanger sequencing in the index case and family members. Footnote: The proband is marked with an arrow. The consanguinity is represented by a double line. Filled and unfilled symbols indicate affected and unaffected individuals, respectively. The parents are represented as a carrier status
Treatment
Following genetic confirmation, we instigated compassionate treatment with the Mn chelator, Na2CaEDTA, 1 g/m2/day in two divided oral doses for a five-day course. This protocol was initially proposed for lead intoxication and has more recently been used in patients with SLC30A10 and SLC39A14 deficiency [14, 15]. No clinical side effects were recorded during the administration. On the fifth day, we observed a reduction of plasma Mn (from 10.5 to 4.5 μg/L, 57% reduction, RV: 0.4–0.9 μg/L), Zn (from 1260 to 381 ng/L, 69.8% reduction, RV: 628–1200 ng/L) and Se (from 84 to 58 ng/L, 31.5% reduction, RV: 67–104 ng/L) (Fig. 3), and therefore he was supplemented with Zn acetate (10 mg/day) and Se (50 mg every 2 days). There was also a mild decrease in plasma Cu and Fe but values remained within the normal range. Two weeks after treatment, the family referred worsening of cervical dystonia, irritability and sleep difficulties, which improved with incremental doses of diazepam. The family decided to discontinue chelation therapy. Neurological examination 3 weeks later was comparable to baseline.

Biochemical analysis before and after treatment. Footnote: Graph shows a reduction of plasma Mn, Zn and Se five days after Na2CaEDTA therapy
Discussion
We present two siblings with a homozygous missense variant in SLC39A14, manifesting a rapidly progressive generalized dystonia in infancy and hypermanganesemia, similar to a cohort of eight SLC39A14 mutated children reported [15]. Our patient did not show polycythemia or liver disease, as observed in SLC30A10-deficient patients (Table 1). We measured plasma and CSF Mn, both showing very high values comparing to our control patients and literature reference ranges [29]. Importantly, Mn values were 3-fold higher in CSF than in plasma; in keeping with the finding that brain Mn levels are four to 20-fold higher in slc39a14 mutant zebra fish [15] and knockout mice [4, 6] compared to wild-type. Our findings support the hypothesis that brain is likely to be the main organ of Mn deposition in SLC39A14 deficiency.
Previously, Chang et al., 2009 reported a correlation between pallidal index (PI) and plasma Mn concentrations of 43 manganese-exposed welders and concluded that high PI might be attributed to Mn brain deposition [30]. We used PI to quantify pallidal Mn accumulation in our patient, and observed significantly higher values than in controls. Furthermore, our patient had higher PI values compared to occupationally Mn exposed workers [31]. These values slightly increased over time, suggesting the relentless pallidal accumulation of Mn in SLC39A14 deficiency.
Na2CaEDTA is a chelating agent that combines with metal ions to form stable and soluble complexes that are excreted in the urine. It ameliorated dystonia and parkinsonism in SLC30A10 mutated patients [18, 22], and its efficacy proved to be persistent over time is some cases with long-term follow-up [22, 32]. More recently, two SLC39A14 patients received Na2CaEDTA, one, given chelation early in the disease course showed clinical improvement, whereas the other (older) patient continued to deteriorate despite treatment [15]. Interestingly, two SLC39A14 KO mice models recently demonstrated a positive effect with two different chelators: a zinc-supplemented diet significantly decreased brain Mn uptake [4], and the metal chelator Na2CaEDTA reduced serum Mn levels and rescued motor deficits [10].
The administration of Na2CaEDTA in our patient led to the reduction of not only plasma Mn values, but also Se and Zn, which are cofactors of important enzymes such as SOD and selenoproteins, and hence, they were supplemented in our patient. Even though Se is not a cation, the decrease might be an effect of EDTA on renal tubules during chelation therapy [33]. Unfortunately, worsening of dystonia resulted in discontinuation of Na2CaEDTA and the long-term efficacy of this treatment could not be tested in this patient.
Conclusions
In this study we present two infants presenting with dystonia and hypermanganesemia caused by a homozygous missense variant in SLC39A14, a recently recognized gene involved in Mn homeostasis in humans, thus expanding the very few descriptions of this disorder. We also report for the first time the elevation of Mn in CSF of SLC39A14 mutated patients, supporting the hypothesis that brain is an important organ of Mn deposition in SLC39A14-related disease. The measurement of PI values on MRI is a non-invasive method that may help monitor Mn pallidal deposition over time. Finally, we propose that patients with inherited defects of manganese transport should be initially treated with low doses of Na2CaEDTA followed by gradual dose escalation, together with a close monitoring of blood trace elements in order to avoid side effects.
Abbreviations
- CDG:
-
Congenital disorder of glycosylation
- CSF:
-
Cerebrospinal fluid
- Cu:
-
Copper
- Mn:
-
Manganese
- MRI:
-
Magnetic resonance imaging
- PI:
-
Pallidal index
- RV:
-
Reference values
- Se:
-
Selenium
- SIFW:
-
Subcortical frontal white matter
- SIGP:
-
Signal intensity in the globus pallidus
- SOD:
-
Superoxide dismutase
- Zn:
-
Zinc
References
Santos D, Batoreu MC, Almeida I, Ramos R, Sidoryk-Wegrzynowicz M, Aschner M, et al. Manganese alters rat brain amino acids levels. Biol Trace Elem Res. 2012;150(1–3):337–41.
Fujishiro H, Yoshida M, Nakano Y, Himeno S. Interleukin-6 enhances manganese accumulation in SH-SY5Y cells: implications of the up-regulation of ZIP14 and the down-regulation of ZnT10. Metallomics. 2014;6(4):944–9.
Chen P, Parmalee N, Aschner M. Genetic factors and manganese-induced neurotoxicity. Front Genet. 2014;5:265.
Aydemir TB, Kim MH, Kim J, Colon-Perez LM, Banan G, Mareci TH, et al. Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces Neurotoxic signatures and diminished motor activity. J Neurosci. 2017;37(25):5996–6006.
Girijashanker K, He L, Soleimani M, Reed JM, Li H, Liu Z, et al. Slc39a14 gene encodes ZIP14, a metal/bicarbonate symporter: similarities to the ZIP8 transporter. Mol Pharmacol. 2008;73(5):1413–23.
Hutchens S, Liu C, Jursa T, Shawlot W, Chaffee BK, Yin W, et al. Deficiency in the manganese efflux transporter SLC30A10 induces severe hypothyroidism in mice. J Biol Chem. 2017;292(23):9760–73.
Lin W, Vann DR, Doulias PT, Wang T, Landesberg G, Li X, et al. Hepatic metal ion transporter ZIP8 regulates manganese homeostasis and manganese-dependent enzyme activity. J Clin Invest. 2017;127(6):2407–17.
Liu C, Hutchens S, Jursa T, Shawlot W, Polishchuk EV, Polishchuk RS, et al. Hypothyroidism induced by loss of the manganese efflux transporter SLC30A10 may be explained by reduced thyroxine production. J Biol Chem in press https://doi.org/10.1074/jbc.M117.804989.
Pinilla-Tenas JJ, Sparkman BK, Shawki A, Illing AC, Mitchell CJ, Zhao N, et al. Zip14 is a complex broad-scope metal-ion transporter whose functional properties support roles in the cellular uptake of zinc and nontransferrin-bound iron. Am J Physiol Cell Physiol. 2011;301(4):C862–71.
Xin Y, Gao H, Wang J, Qiang Y, Imam MU, Li Y, et al. Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov. 2017;3:17025.
Riley LG, Cowley MJ, Gayevskiy V, Roscioli T, Thorburn DR, Prelog K, et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. 2017;40(2):261–9.
Park JH, Hogrebe M, Grüneberg M, DuChesne I, von der Heiden AL, Reunert J, et al. SLC39A8 deficiency: a disorder of manganese transport and Glycosylation. Am J Hum Genet. 2015;97(6):894–903.
Aschner M, Erikson KM, Herrero Hernández E, Tjalkens R. Manganese and its role in Parkinson's disease: from transport to neuropathology. NeuroMolecular Med. 2009;11(4):252–66.
Tuschl K, Mills PB, Parsons H, Malone M, Fowler D, Bitner-Glindzicz M, et al. Hepatic cirrhosis, dystonia, polycythaemia and hypermanganesaemia - a new metabolic disorder. J Inherit Metab Dis. 2008;31(2):151–63.
Tuschl K, Meyer E, Valdivia LE, Zhao N, Dadswell C, Abdul-Sada A, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7:11601.
Tuschl K, Clayton PT, Gospe SM Jr, Gulab S, Ibrahim S, Singhi P, et al. Syndrome of hepatic cirrhosis, dystonia, polycythemia, and hypermanganesemia caused by mutations in SLC30A10, a manganese transporter in man. Am J Hum Genet. 2012;90(3):457–66.
Zaki MS, Issa MY, Elbendary HM, El-Karaksy H, Hosny H, Ghobrial C, et al. Hypermanganesemia with dystonia, polycythemia and cirrhosis in 10 patients: six novel SLC30A10 mutations and further phenotype delineation. Clin Genet. 2017; https://doi.org/10.1111/cge.13184.
Quadri M, Federico A, Zhao T, Breedveld GJ, Battisti C, Delnooz C, et al. Mutations in SLC30A10 cause parkinsonism and dystonia with hypermanganesemia, polycythemia, and chronic liver disease. Am J Hum Genet. 2012;90(3):467–77.
Quadri M, Kamate M, Sharma S, Olgiati S, Graafland J, Breedveld GJ, et al. Manganese transport disorder: novel SLC30A10 mutations and early phenotypes. Mov Disord. 2015;30(7):996–1001.
Gospe SM Jr, Caruso RD, Clegg MS, Keen CL, Pimstone NR, Ducore JM, et al. Paraparesis, hypermanganesaemia, and polycythaemia: a novel presentation of cirrhosis. Arch Dis Child. 2000;83(5):439–42.
Brna P, Gordon K, Dooley JM, Price V. Manganese toxicity in a child with iron deficiency and polycythemia. J Child Neurol. 2011;26(7):891–4.
Stamelou M, Tuschl K, Chong WK, Burroughs AK, Mills PB, Bhatia KP, et al. Dystonia with brain manganese accumulation resulting from SLC30A10 mutations: a new treatable disorder. Mov Disord. 2012;27(10):1317–22.
Lechpammer M, Clegg MS, Muzar Z, Huebner PA, Jin LW, Gospe SM Jr. Pathology of inherited manganese transporter deficiency. Ann Neurol. 2014;75(4):608–12.
Mukhtiar K, Ibrahim S, Tuschl K, Mills P. Hypermanganesemia with Dystonia, Polycythemia and cirrhosis (HMDPC) due to mutation in the SLC30A10 gene. Brain and Development. 2016;38(9):862–5.
Boycott KM, Beaulieu CL, Kernohan KD, Gebril OH, Mhanni A, Chudley AE, et al. Autosomal-recessive intellectual disability with Cerebellar atrophy syndrome caused by mutation of the manganese and zinc transporter gene SLC39A8. Am J Hum Genet. 2015;97(6):886–93.
Heitland P, Köster HD. Biomonitoring of 37 trace elements in blood samples from inhabitants of northern Germany by ICP-MS. J Trace Elem Med Biol. 2006;20(4):253–62.
Wahlen R, Evans L, Turner J, Hearn R. The use of collision/reaction cell ICP-MS for the simultaneous determination of 18 elements in blood and serum samples. Santa Clara: Agilent Technologies. 2005. http://www.agilent.com/chem. Accesed 28 Ago 2009.
Krieger D, Krieger S, Jansen O, Gass P, Theilmann L, Lichtnecker H. Manganese and chronic hepatic encephalopathy. Lancet. 1995;346(8970):270–4.
Harrington JM, Young DJ, Essader AS, Sumner SJ, Levine KE. Analysis of human serum and whole blood for mineral content by ICP-MS and ICP-OES: development of a mineralomics method. Biol Trace Elem Res. 2014;160(1):132–42.
Chang Y, Kim Y, Woo ST, Song HJ, Kim SH, Lee H, et al. High signal intensity on magnetic resonance imaging is a better predictor of neurobehavioral performances than blood manganese in asymptomatic welders. Neurotoxicology. 2009;30(4):555–63.
Li SJ, Jiang L, Fu X, Huang S, Huang YN, Li XR, et al. Pallidal index as biomarker of manganese brain accumulation and associated with manganese levels in blood: a meta-analysis. PLoS One. 2014;9(4):1–7.
Di Toro Mammarella L, Mignarri A, Battisti C, Monti L, Bonifati V, Rasi F, et al. Two-year follow-up after chelating therapy in a patient with adult-onset parkinsonism and hypermanganesaemia due to SLC30A10 mutations. Neurol. 2014;261(1):227–8.
Elmer M. Cranton MD. A Textbook on EDTA Chelation Therapy. 2nd ed. Hampton Roads Publishing Company, Inc. 2001.
Acknowledgements
Not applicable.
Fundings
This work is funded by the Plan Nacional de I + D + I and Instituto de Salud Carlos III- Subdirección General de Evaluación y Fomento de la Investigación Sanitaria, project PI15/00287, the European Regional Development Fund (FEDER) and the Fundació La Marató TV3 (20,143,130 to BPD). LMS has a grant from Fundació Sant Joan de Déu. JD Ortigoza-Escobar has a grant from Rio Hortega, Instituto de Salud Carlos III (CM16/00084). MAK holds a Wellcome Intermediate Clinical Fellowship.
Available of data and materials
The biochemical, radiological and genetic data analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Contributions
LMS participated in the study design, acquisition, analysis and interpretation of data, and drafting of the manuscript. LMS, MML, MB and RA participated in the analysis and interpretation of biochemical and molecular genetic studies. JDOE, MIV, HB, AD, LA, JM and AM performed the analysis and interpretation of clinical and radiological studies. MAK, JDOE, RA and AM participated in the analysis and interpretation of data, and in the drafting and revision of the manuscript. BPD conceived the idea for the study, designed study, supervised study, interpreted data, drafted and revised manuscript content. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The study was approved by the ethics committee at Sant Joan de Déu Hospital (PIC-158-15) and parents gave written informed consent for study participation.
Consent for publication
Written informed consent was obtained from the patient’s parents for publication of this case report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Competing interests
Authors disclose no financial or personal relationships with other people or organisations that could inappropriately influence the present work.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Marti-Sanchez, L., Ortigoza-Escobar, J.D., Darling, A. et al. Hypermanganesemia due to mutations in SLC39A14: further insights into Mn deposition in the central nervous system. Orphanet J Rare Dis 13, 28 (2018). https://doi.org/10.1186/s13023-018-0758-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-018-0758-x