
Abstract
Background
Magnolia is a woody ornamental plant, which is widely used in urban landscaping. However, its lengthy juvenile period and recalcitrance to regeneration impedes functional characterization of its genes.
Results
We developed an efficient protoplast isolation and transient expression system for Magnolia denudata × Magnolia acuminata ‘Yellow River’. The highest yield of protoplasts was obtained from young leaves digested in 3% Cellulase R10, 0.8% Macerozyme R10, 0.04% pectinase and 0.4 M mannitol enzymolysis solution for 6 h. For transfection of protoplasts, 20% PEG4000 for 5 min was optimal. To verify the protoplast system and begin to understand heat tolerance in Magnolia, a heat shock transcription factor MdeHSF1 was cloned from ‘Yellow River’, which belongs to the HSF subfamily A and has significant homology with AtHSFA1A. Subcellular localization analysis indicated that MdeHSF1 was expressed in the cell nucleus. Furthermore, qPCR analysis of the MdeHSF1 transcript level in response to high temperature stress suggested that MdeHSF1 might be involved in regulating heat stress tolerance in ‘Yellow River’.
Conclusion
The described protocol provides a simple and straightforward method for isolating protoplast and exploring gene subcellular localization of MdeHSF1 in Magnolia. This expands the new research of protoplast isolation and transfection in Magnolia.
Similar content being viewed by others
Background
Magnolia is among the best known ornamental plant used in landscaping. The majority of Magnolia species are distributed in America and East Asia, including relict plants, some of which have survived for hundreds of millions of years [1]. Many species of Magnolia growing in the wild are endangered, and some face extinction. Several factors have contributed to this situation, including threats to their habitats, limited genetic diversity, and difficulties associated with breeding. Efforts are being made to protect, develop, and spread magnolias. Grafting and seeding are the main methods for propagating Magnolia. However, Magnolia has a long juvenile period (4–5 years or more) and is recalcitrant to regeneration. Currently, lack of any transformation system has become a limiting step for functional characterization of its genes including subcellular localization, protein–protein interaction, DNA–protein interaction, and transgenic studies [2,3,4]. Thus, transformation methods are urgently needed.
The plant protoplast can serve as a versatile cell-based experimental system to analyze gene expression during a transient time period [3, 5, 6]. Macromolecules, such as DNA, RNA and proteins, can be delivered into protoplasts using various methods, e.g., PEG-calcium fusion [7], electroporation [7] and microinjection [8]. Signal transduction and metabolic pathways as well as the transcription and translation machinery can be transiently manipulated to investigate cell-autonomous regulation and responses [9]. In addition, protoplasts can be used as a method for breeding. Plant protoplast isolation was first reported half a century ago using the root tip of tomato seedlings [10]. Subsequently, mesophyll cells [11,12,13], fruits [14, 15], stem [16], seed [17] and calluses [18] have been used as materials to isolate the protoplast. In recent years, protoplast isolation and transfection was well established for Arabidopsis [3, 4, 9, 19], Populus spp. [20, 21] and Bambusa vulgaris [22], etc. However, no published protocol for protoplast isolation and transfection is available for Magnolia.
As temperatures continue to rise globally, plants are increasingly exposed to heat stress. The major heat shock factors regulating heat stress response are heat shock transcription factors (HSFs) [23], which are important regulators for sensing and signaling heat stress [24]. HSFs have been characterized in Brassica rapa spp. pekinensis [23], Helianthus annuus [25], Arabidopsis [26], Zea mays [27], Glycine max [28] and Pyrus bretschneideri [24]. As most species of Magnolia appear to be particularly sensitive to heat stress, characterizing HSFs in Magnolia is of great significance to understanding and eventually improving their tolerance to heat stress.
In this study, we selected one classical species of Magnolia, Magnolia denudata × M. acuminata ‘Yellow River’ (‘Yellow River’), as the plant material to develop an efficient protoplast isolation and transfection protocol. ‘Yellow River’ is a hybrid of M. denudata × M. acuminata (Fig. 1). They have characteristic yellow flowers and are widely used in urban landscaping in China. As ‘Yellow River’ performs better than M. × soulangeana (Additional file 1: Table S1) in terms of heat stress tolerance, the molecular mechanism responsible for higher heat-stress resistance in ‘Yellow River’ is worth elucidating. The transient transformation system in protoplast is a cell-based functional genomics tool to explore this molecular mechanism. Therefore, we cloned its heat shock transcription factor MdHSF1 and characterized its subcellular localization using protoplast transfection in this paper, and the results confirm that the transient transformation system is viable. The method reported here for protoplast isolation and transfection of Magnolia ‘Yellow River’ lays a good foundation for the breeding of Magnolia, and expands the functional gene research in the field of Magnolia.

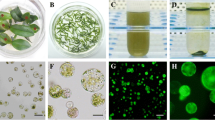
Different tissues for protoplast isolation in ‘Yellow River’. The shape of ‘Yellow River’ (a), mature leaves of ‘Yellow River’ (b), young leaves of ‘Yellow River’ (c), ‘cut-off’ leaves from ‘Yellow River’ (d, e), Protoplasts isolated from the young leaves (f, g), bar 50 μm
Methods
Plant material and growth conditions
M. denudata × M. acuminata ‘Yellow River’ was grown on the campus of Cornell University in Ithaca, New York, USA (Fig. 1a) was approximately 20 years old, with the diameter at breast height (DBH) of 11.0 cm. Mature leaves were sampled on July 7, 2014 (Fig. 1b). The annual branches with leaf buds were collected on February 16, 2015 (Fig. 1c). The annual branches were taken into the lab, and cultured in water at 23 °C. After one week, the sprouted leaves were used to isolate protoplasts. Meanwhile, the buds were also collected. All these materials were taken from the annual branches and they all faced in one direction. After being detached, mature leaves, young leaves and buds were washed twice with sterile water, and then sterilized with 70% ethanol for 5 s first and then with 0.1% HgCl2 for 5 min. Finally, the materials were used to isolate protoplast after being washed 4 times with sterile water (Fig. 1d–g).
Protoplast isolation
Protoplast isolation was performed following the methods of Wu [3], Zhai [29], Rezazadeh [11] and Guo [20], with some modifications. 2 g of leaves (Mature leaves or young leaves) were sliced to 1–2 mm width in a sterilized petri dish with a fresh razor blade (2 g of buds was also sliced to fragments with a razor blade), then rapidly transferred into 100 mL sterilized beaker which contained 10 mL enzymatic solution (The enzymatic solution was placed at 55 °C for 10 min, cooled to room temperature, and sterilized using a 0.45 µm filter prior to use).
Then three methods were used to treat samples for choosing the optimal way of protoplast isolation: Method 1: Four concentrations mannitol/sorbitol (0.2, 0.4, 0.6 or 0.8 M) were used to treat the samples for getting optimal enzymatic solution (replicated three times each experiment); Method 2: Six combinations with different concentrations of Cellulase R10 and Macerozyme R10 (a) 1% (w/v) Cellulase R10, 0.4% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (b) 1.5% (w/v) Cellulase R10, 0.5% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (c) 2% (w/v) Cellulase R10, 0.6% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (d) 2.5% (w/v) Cellulase R10, 0.7% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (e) 3% (w/v) Cellulase R10, 0.8% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (f) 3.5% (w/v) Cellulase R10, 0.9% (w/v) Macerozyme R10, and 0.04% (w/v) Pectinase). Each combination was prepared to the appropriate concentration using a solution containing 20 mM KCl; 20 mM MES-KOH (pH 5.7); 10 mM CaCl2; 10 mM β-mercaptoethanol and 0.1% (w/v) bovine serum albumin (replicated three times each experiment). The third method was based on the results from the methods 1 and 2, and involved vacuum-infiltrating leaf strips for 30 min and incubated in the enzymatic solution at 25 °C in darkness with rotation (60 rpm) for about 2, 4, 6, 8, 10 or 12 h (the beaker with the leaf strips was covered with parafilm and aluminum foil) (replicated three times each experiment). After digestion, an equal volume of W5 solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl, 2 mM MES-KOH (pH 5.7), sterilized using a 0.45-mm filter) was added. Next, the solution was filtered through a sterilized nylon membrane (200 µm mesh size) and transferred into a 50 mL sterilized centrifuge tube. Then the nylon membrane was washed twice with 10 mL W5 solution and all the filtrate was transferred to a 50 mL centrifuge tube.
Finally, the filtrate was centrifuged at 100g for 10 min, and the supernatant was carefully removed using a pipettor. The protoplasts were washed twice with W5 solution and re-suspended to a final concentration of 1 × 106 protoplasts/mL for transfection.
The protoplast yield of different tissue types (mature leaves, young leaves and bud) was also compared. The viability of the isolated protoplasts was also determined with FDA (fluorescein diacetate) dye [30]. The FDA stock solution was added into the isolated protoplasts at a final concentration of 0.01%. After being incubated in darkness for 5 min and washed three times with W5 solution, protoplast viability was checked under a fluorescence microscope.
Protoplast transfection
The isolated protoplasts were placed on ice, and an equal volume of PEG solution (20% w/v polyethylene glycol, PEG4000, 0.3 M mannitol and 100 mM CaCl2) (with 10% PEG4000 as a control, replicated three times) was added. In order to select the optimal concentration of PEG4000, concentrations of 10, 15, 20, 25 and 30% PEG4000, w/v were tested. Next, 20 µg of the pEZS-NL plasmid, which contained a 35S promoter and the coding sequence fused to EGFP and OCS 3′ terminator which can used for transient expression [31] (Isolated by PureLink® Quick Plasmid Miniprep Kit, Invitrogen, cat. no. K210010), 100 µL of isolated protoplasts and 120 µL of the PEG solution were combined and mixed gently in a 2 mL microfuge tube. The mixture was incubated at room temperature for 5, 10, 15 and 30 min in triplicate (to determine the optimum transfection time) before 220 µL of W5 solution was added to terminate the reaction. The mixture was centrifuged at 120g for 5 min, and the supernatant was carefully removed with a pipettor. The protoplasts were re-suspended overnight at room temperature in 2 mL of incubation buffer (0.4 mM mannitol, 4 mM MES-KOH (pH 5.7), 20 mM KCl). Finally, the protoplasts were centrifuged at 120g for 2 min and re-suspended with an appropriate amount of WI solution (0.5 M mannitol, 4 mM MES-KOH (pH 5.7), 20 mM KCl). A Zeiss LSM 710 fluorescence microscope was used to visualize the EGFP signal. The transformation efficiency was determined by (%) = (fluorescent protoplast number in view/total protoplast number in view) × 100%.
Note
All data were analyzed by SPSS.19. The data of transfection rate was analyzed through the square root of the arcsine difference.
RNA isolation and cloning of MdeHSF1 and construction of its expression vector
RNA was extracted from leaves of ‘Yellow River’ according to a modified CTAB method and digested with DNase I. RNA was detected by electrophoresis. Total RNA was used to synthesize first-strand cDNA using the SuperScript reverse transcriptase with Oligo-dT primer. The cDNA was then used as templates for PCR amplification.
Our preliminary data obtained on ‘Yellow River’ at Hangzhou, Zhejiang Province of China (Additional file 1: Table S1) showed that ‘Yellow River’ has stronger tolerance to heat stress than M. × soulangana. We also determined the transcript profile of ‘Yellow River’ leaves using RNA-seq in response to heat treatment. Based on the transcriptome data of M. sinostellata, the primers were designed, F: 5′-ATGGACGGTGCTCATGGCAGCA-3′, R: 5′-CAGCCTTTGTTTTCTGATGTAAGAA-3′, for homology-based cloning of MdeHSF1 via PCR. The PCR products were cloned into the pMD19-T simple vector using T4 DNA Ligase, Then the vector was transferred into E. coli. DH5a competent cells and grown on the LB medium with Ampicillin. Positive bacterial colonies were detected by PCR using the M13 primer shown in the pMD19-T vector user manual; then sequenced at Cornell’s Biotechnology Resources Center. Finally, MdeHSF1 was analyzed from the website of http://www.ncbi.nlm.nih.gov/Structure/cdd/wrpsb.cgi, the protein structure was analyzed using the smart website http://smart.embl-heidelberg.de/, the protein prediction was from the website https://www.predictprotein.org/, and MdeHSF1 was compared with HSF of Arabidopsis thaliana.
The correct clone of MdeHSF1 was cloned into the transient expression vector pEZS-NL through double enzyme digestion (BamHI and XhoI), then the vector was transferred into the DH5a competent cells. The correct clone was detected by PCR using the 35S promoter primer F and HSF primer R. The clone was cultured on LB liquid medium (Ampicillin) at 37 °C with rotation (120 rpm) for 12 h. The plasmid was isolated using PureLink® Quick Plasmid Miniprep Kit.
RNA extraction and quantitative real-time PCR (qRT-PCR)
‘Yellow River’ and M. × soulangeana leaf samples were finely grounded in liquid nitrogen, and RNA was extracted using the CTAB method as described by Gasic et al. [32]. After treatment with RQ1 DNase, RNA was quantified using a NanoDrop spectrophotometer, and the RNA integrity was confirmed by agarose gel electrophoresis. One milligram of total RNA was reverse-transcribed into cDNA using the iScript cDNA Synthesis kit. Actin gene served as the internal control. Every qRT-PCR was performed in 3 replicates on an Icycler iQ5 (BioRad) using the SYBR Green Supermix kit (Bio-Rad) according to the instruction manual. Data were analyzed using iQ5 2.0 software (Bio-Rad) with the ddCT method.
Note
The sources of all relative reagents were showed in Additional file 1: Table S2.
Results
An efficient method of protoplast isolation in ‘Yellow River’
The yield of protoplast in the treatment of mannitol was significantly higher than that under sorbitol treatment (p < 0.01). The optimal concentration was 0.4 M mannitol, which yielded 1.89 × 105 protoplasts/g FW (fresh weight), the highest yield of all the treatments (p < 0.01) (Fig. 2a).

The protoplast yield under different treatments. In response to a range (0.2, 0.4, 0.6 and 0.8 M) of mannitol and sorbitol concentrations in digestion solution with 3% Cellulase R10, 0.8% Macerozyme R10, and 0.4% Pectinase (a), protoplast yield as affected by different concentrations of digestion enzymes (b): (a) 1% (w/v) Cellulase R10, 0.4% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (b) 1.5% (w/v) Cellulase R10, 0.5% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (c) 2% (w/v) Cellulase R10, 0.6% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (d) 2.5% (w/v) Cellulase R10, 0.7% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (e) 3% (w/v) Cellulase R10, 0.8% (w/v) Macerozyme R10, 0.04% (w/v) Pectinase; (f) 3.5% (w/v) Cellulase R10, 0.9% (w/v) Macerozyme R10, and 0.04% (w/v) Pectinase, protoplast yield as affected by digestion time (2, 4, 6, 8, 10 or 12 h) (c), protoplast yield as affected by tissue type (buds, young leaves and mature leaves) (d)
Furthermore, we tested the effects of different concentration combinations of cell wall-degrading enzymes on protoplast yield. Via the addition of various concentrations of the enzymes, the protoplast yield tended to increase until the concentration reached 3% (w/v) Cellulase (R10), 0.8% (w/v) Macerozyme (R10) and 0.04% (w/v) Pectinase. The protoplast yield in this combination was significantly higher than in other combinations (p < 0.01) (Fig. 2b). Effects of enzymolysis time on protoplast yield were also tested. As digestion time increased from 2 to 6 h, protoplast yield increased, but with further increases in digestion time, protoplast yield decreased significantly (Fig. 2c). These results indicated that the optimal digestion time was 6 h.
Finally, we examined the effect of three tissue types (buds, young leaves and mature leaves) on protoplast yield and found that protoplast yield obtained from young leaves was significantly higher than from the other two types (p < 0.01) (Fig. 2d). In summary, the optimal method for protoplast isolation in ‘Yellow River’ is to digest strips of young leaves in a 3% (w/v) Cellulase (R10), 0.8% Macerozyme (R10), 0.04% (w/v) Pectinase, and 0.4 M mannitol enzymolysis solution for 6 h.
Establishment of a ‘Yellow River’ protoplast transient expression system
We established a PEG-mediated transient expression system for ‘Yellow River’ by testing a range of PEG4000 concentrations and transfection time. The transient expression vector pEZS-NL harboring an EGFP-tag [31] was used to study the transformation efficiency in response to PEG concentrations and transfection time (Fig. 3). The results showed that 20% w/v of PEG4000 was the optimal to get the high transformation efficiency (p < 0.01) (Fig. 3a). The transformation efficiency was significantly higher after 10 min and 15 min than after 30 min (p < 0.01), and did not differ significantly from efficiency after 5 min (Fig. 3b). Therefore, 5 min was taken as the optimal transfection time.

The protoplast transfection ratio and transient expression. The ratio of transfection at 10, 15, 20, 25 and 30% (w/v) PEG4000 concentration (a), the ratio of transfection after 5, 10, 15 and 20 min (b), images of GFP and chlorophyll autofluorescence (Chl) as well as bright field images of protoplasts captured 12 h after transfection (c, d). Bar in c, 50 μm. Bar in d, 10 μm
Cloning a heat shock transcription factor MdeHSF1 and its expression pattern under heat stress in ‘Yellow River’
The complete sequence of MdeHSF1 gene was cloned from ‘Yellow River’. The length of ORF was 1521 bp, coding 506 AA, with PI value of 4.97. The gene contained a conserved high HSF-DNA-binding domain at N terminal, belonging to the heat shock transcription factor family (Fig. 4). The phylogenetic tree showed that MdeHSF1 had the highest homology to AtHSFA1A (Fig. 5a). Furthermore, the subcellular localization of MdeHSF1 was also predicted to be in the cell nucleus, a typical characteristic of transcription factors (Fig. 5b).

The protein of MdeHSF1 and three AtHSF proteins were compared using DNAman6.0 software. AtHSFA1A is At4g17750; AtHSFB1 is At4g36990; AtHSFC1 is At3g24520

The HSF DNA binding domain is underlined (a). Phylogenetic relationship between MdeHSF1 and AtHSF proteins drawn using MEGA6.0 (b), the conserved domain analysis of the MdeHSF1 and AtHSF proteins using the MEME website (c)
Using expression changes of MdeHSF1 as a proxy, we estimated the difference in the heat-stress resistance of two species in Magnolia, ‘Yellow River’ and M. × soulangeana. MdeHSF1 shared similar expression patterns in two species, exemplified by the increase in expression levels increased with the longer duration of heat stress. The highest expression of MdeHSF1 in both species occurred after 5 h of heat stress. However, the expression of MdeHSF1 was more induced by heat stress in ‘Yellow River’ than in M. × soulangeana. Higher expression of MdeHSF1 was consistent with the phenomenon that ‘Yellow River’ exhibited higher heat-stress resistance in nature, than that in M. × soulangeana (Fig. 6).

qPCR analysis of the MdeHSF1 expression level under high temperature (42 °C) from 0 to 5 h in ‘Yellow River’ and M. × soulangeana leaves
Subcellular localization analysis of MdeHSF1 in ‘Yellow River’ protoplast
To determine the subcellular localization of MdeHSF1, an MdeHSF1-EGFP fusion protein vector, pESZ-NL-MdeHSF1, was constructed. Protoplast isolation and transformation was performed as previously described, and the MdeHSF1 transient expression vector was successfully expressed in the ‘Yellow River’ protoplast (Fig. 7a). These results confirmed that MdeHSF1 was expressed in the cell nucleus (Fig. 7b), and demonstrated the utility of the protoplast isolation and transient expression system we developed.

Plasmids can be transferred to protoplasts for transient expression of MdeHSF1. The protected picture of the subcellular location of MdeHSF1 (a), the subcellular location of MdeHSF1 in ‘Yellow River’ protoplast (b) Bar 10 μm
Discussion
Protoplast isolation and transient expression is widely used in plant molecular research to examine gene function, subcellular position and protein–protein interactions. In Arabidopsis, the protoplast transient expression system is very mature, and some rapid and simple methods have been developed. For example, the Tape Sandwich system, allows for rapid isolation of high-quality protoplasts from Arabidopsis leaves [3]. There are also efficient protoplast isolation and transient expression systems developed for other model plants and economically important plants, such as Nicotiana tabacum [33], Oryza sativa [16, 34], Malus × domestica [35] and Fragaria × ananassa [36]. These protoplast transient expression systems have been used in a variety of experimental studies and have facilitated the advancement of gene function research [16, 34] In contrast, there are a limited number of reports on protoplast isolation and transient expression in ornamental plants. In this study, we developed an efficient protoplast isolation and transient expression system for ‘Yellow River’ after testing different tissue types, enzymatic combinations, enzymolysis solution osmotic potentials, and enzymolysis time. Similar to previous reports on other plants, the effective concentration of PEG solution for the transient expression system is approximately 20%. Under optimal condition, the protoplast yield of ‘Yellow River’ young leaves can reach approximately 1.89 × 105 protoplasts/g FW. This value was higher than the protoplast yield of Arabidopsis, Oryza sativa and Populus [3, 20, 29, 34]. Compared with mature leaves, the young leaves grown in vitro or in a lab, are more suitable to isolate protoplasts.
With continued rise in temperatures globally, plants are increasingly exposed to high temperatures. This is more of a problem for Magnolia as it is often used in urban landscaping. Compared with other Magnolia species, such as M. × soulangeana, ‘Yellow River’ has higher tolerance to heat stress, but the molecular mechanism has remained unclear. Several studies showed that heat-shock transcription factors (HSF) are central regulators of heat-stress responsive genes. The plant HSF family shows a striking multiplicity, showing a strong diversification of expression pattern and function within the family [37]. In plants, there are three classes in the HSF protein family (classes A, B, and C), which are differentiated by peculiarities of their flexible linkers and HR-A/B regions [38]. And many researchers showed that AtHSFA1A was overexpressed in the heat shock response [39,40,41]. HSFs trigger the expression of Heat-shock Protein (HSP) in response to various stresses in living organisms, and makes HSP complex [40]. In the present paper, one HSF transcription factor, MdeHSF1 was cloned, and we found that the expression level of MdeHSF1 was much higher in ‘Yellow River’ than that of M. × soulangeana under high temperature stress. This finding indicates that MdeHSF1 might be an important gene regulator controlling heat tolerance in ‘Yellow River’. Furthermore, the protein–protein interaction prediction results suggest that MdeHSF1 exhibits homology with AtHSFA1A and plays an important role in the cell signal network.
Conclusion
These results not only verify the efficiency of the protoplast transient expression system but also provide important clues to understanding heat tolerance in ‘Yellow River’. In future studies, responses of proteins to the expression level of MdeHSF1 should be explored in more detail and the use of the protoplast system for detecting protein–protein interactions via bimolecular fluorescence complementation assays, could be investigated.
References
Law Y, Xia N, Yang H. The origin, evolution and phytogeography of Magnoliaceae. J Trop Subtrop Bot. 1995;3(4):1–12.
Faraco M, Di Sansebastiano GP, Spelt K, Koes RE, Quattrocchio FM. One protoplast is not the other! Plant Physiol. 2011;156(2):474–8.
Wu F-H, Shen S-C, Lee L-Y, Lee S-H, Chan M-T, Lin C-S. Tape-Arabidopsis Sandwich—a simpler Arabidopsis protoplast isolation method. Plant Methods. 2009;5(1):16.
Yoo S-D, Cho Y-H, Sheen J. Arabidopsis mesophyll protoplasts: a versatile cell system for transient gene expression analysis. Nat Protoc. 2007;2(7):1565–72.
Eeckhaut T, Lakshmanan PS, Deryckere D, Van Bockstaele E, Van Huylenbroeck J. Progress in plant protoplast research. Planta. 2013;238(6):991–1003.
Lung S-C, Yanagisawa M, Chuong SD. Protoplast isolation and transient gene expression in the single-cell C4 species, Bienertia sinuspersici. Plant Cell Rep. 2011;30(4):473–84.
Hassanein A, Hamama L, Loridon K, Dorion N. Direct gene transfer study and transgenic plant regeneration after electroporation into mesophyll protoplasts of Pelargonium × hortorum, ‘Panaché Sud’. Plant Cell Rep. 2009;28(10):1521–30.
Masani MYA, Noll GA, Parveez GKA, Sambanthamurthi R, Prüfer D. Efficient transformation of oil palm protoplasts by PEG-mediated transfection and DNA microinjection. PLoS ONE. 2014;9(5):e96831.
Sheen J. Signal transduction in maize and Arabidopsis mesophyll protoplasts. Plant Physiol. 2001;127(4):1466–75.
Cocking E. A method for the isolation of plant protoplasts and vacuoles. Nature. 1960;187(4741):962–3.
Rezazadeh R, Harrison DK, Williams RR. Intraspecific somatic hybridization of mango (Mangifera indica L.) through protoplast fusion. J Appl Hortic. 2011;13(2):101–7.
Wallin A, Johansson L. Plant regeneration from leaf mesophyll protoplasts of in vitro cultured shoots of a columnar apple. J Plant Physiol. 1990;135(5):565–70.
Wallin A, Welander M. Improved yield of apple leaf protoplasts from in vitro cultured shoots by using very young leaves and adding l-methionine to the shoot medium. Plant Cell Tissue Organ. 1985;5(1):69–72.
Fieuw S, Willenbrink J. Isolation of protoplasts from tomato fruit (Lycopersicon esculentum): first uptake studies. Plant Sci. 1991;76(1):9–17.
Maddumage R, Fung RM, Weir I, Ding H, Simons JL, Allan AC. Efficient transient transformation of suspension culture-derived apple protoplasts. Plant Cell Tissue Organ. 2002;70(1):77–82.
Chen S, Tao L, Zeng L, Vega-Sanchez ME, Umemura K, Wang GL. A highly efficient transient protoplast system for analyzing defence gene expression and protein–protein interactions in rice. Mol Plant Pathol. 2006;7(5):417–27.
Lu D, Cooper-Bland S, Pental D, Cocking E, Davey M. Isolation and sustained division of protoplasts from cotyledons of seedlings and immature seeds of Glycine max L. Zeitschrift für Pflanzenphysiologie. 1983;111(5):389–94.
Clement WKF, Lai KS, Wong MY, Maziah M. Heat and hydrolytic enzymes treatment improved the Agrobacterium-mediated transformation of recalcitrant indica rice (Oryza sativa L.). Plant Cell Tissue Organ. 2016;125(1):183–90.
Asai T, Stone JM, Heard JE, Kovtun Y, Yorgey P, Sheen J, Ausubel FM. Fumonisin B1-induced cell death in Arabidopsis protoplasts requires jasmonate-, ethylene-, and salicylate-dependent signaling pathways. Plant Cell. 2000;12(10):1823–35.
Guo J, Morrell-Falvey JL, Labbé JL, Muchero W, Kalluri UC, Tuskan GA, Chen J-G. Highly efficient isolation of Populus mesophyll protoplasts and its application in transient expression assays. PLoS ONE. 2012;7(9):e44908.
Tan B, Xu M, Chen Y, Huang M. Transient expression for functional gene analysis using Populus protoplasts. Plant Cell Tissue Organ. 2013;114(1):11–8.
Hisamoto Y, Kobayashi M. Protoplast isolation from bamboo leaves. Plant Biotechnol. 2010;27(4):353–8.
Ma J, Xu Z-S, Wang F, Tan G-F, Li M-Y, Xiong A-S. Genome-wide analysis of HSF family transcription factors and their responses to abiotic stresses in two Chinese cabbage varieties. Acta Physiol Plant. 2014;36(2):513–23.
Qiao X, Li M, Li L, Yin H, Wu J, Zhang S. Genome-wide identification and comparative analysis of the heat shock transcription factor family in Chinese white pear (Pyrus bretschneideri) and five other Rosaceae species. BMC Plant Biol. 2015;15(1):12.
Almoguera C, Rojas A, Dı́az-Martı́n J, Prieto-Dapena P, Carranco R, Jordano J. A seed-specific heat-shock transcription factor involved in developmental regulation during embryogenesis in sunflower. J Bio Chem. 2002;277(46):43866–72.
Schöffl F, Hübel A, Lee JH. Derepression of the heat shock response and genetic engineering of thermotolerance in Arabidopsis using recombinant heat shock factor. In: Grillo S, Leone A, editors. Physical stresses in plants. Berlin, Heidelberg: Springer; 1996. p. 39–46.
Lin Y-X, Jiang H-Y, Chu Z-X, Tang X-L, Zhu S-W, Cheng B-J. Genome-wide identification, classification and analysis of heat shock transcription factor family in maize. BMC Genom. 2011;12(1):76.
Li P-S, Yu T-F, He G-H, Chen M, Zhou Y-B, Chai S-C, Xu Z-S, Ma Y-Z. Genome-wide analysis of the Hsf family in soybean and functional identification of GmHsf-34 involvement in drought and heat stresses. BMC Genom. 2014;15(1):1009.
Zhai Z, Jung H-I, Vatamaniuk OK. Isolation of protoplasts from tissues of 14-day-old seedlings of Arabidopsis thaliana. Jove-J Vis Exp. 2009;30:e1149-e.
Larkin P. Purification and viability determinations of plant protoplasts. Planta. 1976;128(3):213–6.
Meng D, Gu Z, Li W, Wang A, Yuan H, Yang Q, Li T. Apple MdABCF assists in the transportation of S-RNase into pollen tubes. Plant J. 2014;78(6):990–1002.
Gasic K, Hernandez A, Korban SS. RNA extraction from different apple tissues rich in polyphenols and polysaccharides for cDNA library construction. Plant Mol Biol Rep. 2004;22(4):437–8.
Locatelli F, Vannini C, Magnani E, Coraggio I, Bracale M. Efficiency of transient transformation in tobacco protoplasts is independent of plasmid amount. Plant Cell Rep. 2003;21(9):865–71.
Zhang Y, Su J, Duan S, Ao Y, Dai J, Liu J, Wang P, Li Y, Liu B, Feng D. A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Methods. 2011;7(1):30.
Doughty S, Power J. Callus formation from leaf mesophyll protoplasts of Malus X domestica Borkh. cv. Greensleeves. Plant Cell Rep. 1988;7(3):200–1.
Nyman M, Wallin A. Transient gene expression in strawberry (Fragaria x ananassa Duch.) protoplasts and the recovery of transgenic plants. Plant Cell Rep. 1992;11(2):105–8.
von Koskull-Döring P, Scharf K-D, Nover L. The diversity of plant heat stress transcription factors. Trends Plant Sci. 2007;12(10):452–7.
Nover L, Bharti K, Döring P, Mishra SK, Ganguli A, Scharf K-D. Arabidopsis and the heat stress transcription factor world: how many heat stress transcription factors do we need? Cell Stress Chaperon. 2001;6(3):177–89.
H-C Liu, Y-Y Charng. Common and distinct functions of Arabidopsis class A1 and A2 heat shock factors in diverse abiotic stress responses and development. Plant Physiol. 2013;163(1):276–90.
Liu Y, Zhang C, Chen J, Guo L, Li X, Li W, Yu Z, Deng J, Zhang P, Zhang K. Arabidopsis heat shock factor HsfA1a directly senses heat stress, pH changes, and hydrogen peroxide via the engagement of redox state. Plant Physiol Bioch. 2013;64:92–8.
Qian J, Chen J, Liu Y, Yang L, Li W, Zhang L. Overexpression of Arabidopsis HsfA1a enhances diverse stress tolerance by promoting stress-induced Hsp expression. Genet Mol Res. 2014;13(1):1233–43.
Authors’ contributions
Conceived and designed the experiments: YS, DM, and LC. Performed the experiments: YS and DM. Data analyses: YS, DM and JZ. Contributed to the writing of the manuscript: YS, DM, LC, JZ and KM. All authors read and approved the final manuscript.
Acknowledgements
We thank the Olena Vatamaniuk Lab for sharing a Zeiss LSM 710 fluorescence microscope and Jiapei Yan and Yutian Dong for assistance with the microscope. Tianzhong Li lab provided the pEZS-NL plasmid.
Competing interests
The authors declare that they have no competing interests.
Availability of data and material
All data generated in this study are included in this article and additional files. Material is available from the corresponding author on reasonable request.
Funding
This work was supported in part by the National Natural Science Foundation of China (No. 31400599), the 13th Five-Year-Plan Projects for Floriculture Special Breeding of Zhejiang Province, China (No. 2016C02056-12), the Public Welfare Forestry Industry Project of State Forestry Administration, China (No. 201504322), the Science Foundation of Zhejiang A & F University (KF201323).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Author information
Authors and Affiliations
Corresponding author
Additional file
13007_2017_193_MOESM1_ESM.docx
Additional file 1: Table S1. Injury degree of landscape plants in the parks and roads in Hang Zhou, China under nature high-temperature. Table S2. The sources of all relative reagents
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shen, Y., Meng, D., McGrouther, K. et al. Efficient isolation of Magnolia protoplasts and the application to subcellular localization of MdeHSF1 . Plant Methods 13, 44 (2017). https://doi.org/10.1186/s13007-017-0193-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13007-017-0193-3