
Abstract
Background
The status of vaginal microbiota in persistent high-risk human papilloma virus (HR-HPV) infection is unclear. The present work aimed to identify the vaginal microbiota of persistent HPV infection and explore the possible underlying microbiota factors.
Methods
A total of 100 women were recruited in this study, of which 28 presented HR-HPV persistent infection (P group), 30 showed clearance of any subtype of HR-HPV (C group), and 42 had no history of any HR-HPV infection (NC group). The vaginal microbiota and the community structure of the three groups were compared based on the 16S rRNA sequencing of the V3–V4 region. The microbiota diversity and differential analysis were carried out to detect the potential factors associated with HR-HPV infection.
Results
P and C groups showed an increase of Firmicutes and Actinobacteriota but a decrease in Proteobacteria compared to the NC group. The Chao1 index indicated that the microbial richness of the NC group was greater than C group (P < 0.05).The principal co-ordinate analysis(PCoA) revealed differences between the NC and P/C groups.The linear discriminant analysis effect size (LEfSe) method indicated that Proteobacteria phylum was significantly different in the mean relative abundance in the NC group,but the P and C groups did not show such indicative taxa. The Wilcox rank-sum test indicated that the Bifidobacterium (P = 0.002) and Lactobacillus (P = 0.005) of the C group were in a high mean relative abundance compared to the NC group.
Conclusions
The persistent HR-HPV infection is associated with dysbiosis of the vaginal microbiota. Microbiome regulation with Bifidobacteria and Lactobacillus may affect the clearance of HPV.
Similar content being viewed by others

Background
According to the World Health Organization (WHO) statistical data, cervical cancer is the fourth most common cancer in women. About 99% of the cases are linked to infection of high-risk human papillomavirus (HR-HPV) [1]. HPV infection is common in sexually active women and usually can be spontaneously eliminated from individuals within 6–24 months, and only a small proportion of infected women retain the HR-HPV [2]. However, epidemiological studies showed that the world prevalence of HPV infection is 11.7% [3]. Lacking a definitive treatment for the infection and the high cost of cervical cancer treatment makes HPV a major public health concern. Vaginal douching or sexual intercourse and biological factors, such as bacterial vaginosis or sexually transmitted infections (STIs) affect the vaginal microenvironment and act as cofactors in the persistent HPV infection [4,5,6,7]. Several studies explored the correlation between vaginal microbiota and HPV infection, precancerous lesions of cervix or cervical cancer. According to vaginal microbiota species composition, there were five community state types (CSTs). CST I, II, III, and V were dominated by Lactobacillus crispatus (L. crispatus), Lactobacillus gasseri, (L. gasseri), Lactobacillus iners (L. iners), and Lactobacillus jessenii (L. jessenii) respectively. CST IV was characterized by a high proportion of strictly anaerobic bacteria [8]. CST IV was correlated with persistent HR-HPV infection [9]. In addition, Brotman et al. [10] found that the CSTs were associated with changes in HPV status, and a small Lactobacillus community with high proportions of the genus Atopobium had the slowest rate of HPV clearance. The presence of Anaerococcus vaginae, Garderella vaginalis, and L. iners in the absence of L. crispatus were identified as the most high-risk combinations for the development of cervical cancer. Kyeong et al. [11] found that there was a marked decrease of L. crispatus in the CIN/cancer groups compared with that in the normal group. Atopobium vaginae, Dialister invisus, Finegoldia magna, Gardnerella vaginalis, Prevotella buccalis and Prevotella timonensis were significantly associated with the risk of CIN 2/3 or cervical cancer. So far, the majority of the studies had focused on the correlation between CSTs and HPV infection status or correlation between vaginal microbiota and the cervical lesions, while only a few studies aimed to identify the possible microbial biomarker that affects the susceptibility to HR-HPV before any cervical precancerous lesion occurred. Finding the biomarker could help clearing HPV before cervical lesions occur. Herein, we compared the composition and structure of vaginal microbiota in persistent HR-HPV infection, HR-HPV clearance, and HR-HPV-negative women based on 16S rRNA high-throughput sequencing to characterize the bacterial taxa.
Methods
Data and sample collection
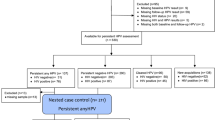
A total of 100 women were recruited at the Gynecology Outpatient Department of West China Second Hospital in Chengdu between January 2015 and March 2021. All the women underwent the screening test for both HPV DNA detection and cytology. We use polymerase chain reaction(PCR) reverse dot blot hybridization to qualitatively detect 17 types of HR-HPV DNA, namely, 16, 18, 31, 33, 35, 39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 73 and 82. The including criteria were as follows: (1) The cytological examination of the uterine cervix indicated negative intraepithelial lesions or malignancies (NILMs); (2) If HPV16 or HPV18 was positive, women were referred for colposcopy, and no cervical intraepithelial neoplasia (CIN) or cervical cancer should be detected; (3) Women with persistent infection of specific HR-HPV subtypes for ≥ 12 months comprised the P group;(4) Women with clearance of some HR-HPV subtypes after observation for 12 months comprised the C group; (5) Women without HR-HPV infection records for the recent 2 years constituted the NC group. According to the HPV subtypes, the samples from P and C groups were further divided into PC (persistent infection with subtype 16 or 18), PO (persistent infection with subtypes other than subtype 16 and 18), CC (clearance of subtype 16 or 18), and CO (clearance of subtypes other than 16 and18) subgroups. The excluding criteria were as follows: (1) Women had sexual activity in the 48 h before the visit; (2) Women used douches, vaginal medications, or had reported vaginal discharge in the past 48 h; (3) Pregnant women; (4) Women had taken any antibiotics or antimycotics, hormone replacement treatment, or oral contraceptive medication in the past 30 days; (5) Women received any antivirus treatment, such as topical interferon. After obtaining informed consent, mid-vaginal secretion samples were obtained from these women using sterile cotton swabs. For each sample, two swabs were collected in sterilized tubes on ice until genomic DNA extractions within 3 h.
Whole genomic DNA extraction
The TIANamp Swab DNA kit (TIANGEN BIOTECH Beijing Co. Ltd) was used to extract the whole genomic DNA of vaginal bacteria species, according to the manufacturer's instructions. The swab suspensions were mixed with 20 μL proteinase K. The mixture was incubated at 56 °C for 60 min. Then, the samples were subjected to mechanical lysis by bead beating in a Fast Prep 24 machine (MPBio, USA) at 6 m/s for 40 s. The lysates were subjected to centrifugation at 12,000 rpm for 60 s to pellet the beads and filtered using the CR2 columns. DNA was purified, and the quality of total genomic DNA was evaluated by agarose gel electrophoresis (1% E-gel, Invitrogen, USA).
DNA amplification and sequencing of 16S rRNA gene fragments
The V3-V4 region of the bacterial 16S ribosomal RNA gene was amplified by PCR (95 °C for 2 min, followed by 25 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s and a final extension at 72 °C for 5 min) using primers 341F (CCTAYGGGRBGCASCAG) and 806R(GGACTACNNGGGTWTCTAAT). The 20 μL PCR reaction consisted of 10 ng template DNA, 0.8 μL of each primer (5 μM), 2μL of 2.5 mM dNTPs, 4 μL of 5X FastPfu Buffer, and 0.4μL of FastPfu polymerase. The amplicons were excised from 2% agarose gels and purified using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) according to the manufacturer’s instructions. Purified PCR products were quantified using Qubit®3.0 (Life Invitrogen). The pooled DNA product was used to construct Illumina pair-end library following Illumina’s genomic DNA library preparation procedure. Then, the amplicon library was paired-end sequenced (2 × 250) on an Illumina MiSeq platform (Shanghai BIOZERON Co., Ltd), according to the standard protocols.
Data analysis
The raw reads were deposited into the Genome Sequence Archive(GSA)database.(Accession Number: CRA005466) After merging the paired reads and chimera filtering, sequences were clustered into operational taxonomic units (OTUs) at 97% similarity using the Deblur denoising algorithm. The phylogenetic affiliation of each 16S rRNA gene sequence was analyzed by uclust algorithm (http://www.drive5.com/usearch/manual/uclust_algo.html) against the silva (SSU138.1) 16S rRNA database using the confidence threshold of 80%.
The community structures of P, C, and NC groups were analyzed. Venn diagrams were drawn using the online tool “Draw Venn Diagram” (http://bioinformatics.psb.ugent.be/webtools/Venn) to analyze overlapped and unique OTUs during the processes. The rarefaction analysis based on Mothur v.1.21.1 was conducted to reveal the alpha diversity by Chao index. The beta diversity was analyzed using bray_curtis [12] to compare the results of the principal co-ordinates component analysis (PCA) by R-forge (Vegan 2.0 package). The biomarkers for HR-HPV infection or clearance were identified by linear discriminant analysis effect size (LEfSe) analysis [13]. Kruskal–Wallis sum-rank test was performed to examine the changes and dissimilarities among classes, followed by linear discriminant analysis (LDA) to determine the size effect of each distinctively abundant taxa. To explore the correlation between vaginal microbiota and the infection status of certain HPV subtypes, Wilcox rank-sum test was performed on the PC, PO, CC, CO, and NC groups to identify the putative biomarker for persistent HR-HPV infection or elimination of HR-HPVs.
The statistical analysis was performed using the SPSS Statistics software 17.0 (IBM, USA). Student's t-test, χ2 or Fisher's exact test, and one-way analysis of variance (ANOVA) were conducted to determine the statistical significance as appropriate. P ≤ 0.05 was considered statistically significant.
Results
Baseline of the P, C, and NC groups and quality assurance
The age of the cohort was 21–64 (median, 36) years old. Of these, 19 patients were in menopausal transition or postmenopause, one patient of group C had levonorgestrel intrauterine system. P group consisted of 28 cases, the C group consisted of 30 cases, and 42 cases comprised the NC group. The clinical characteristics are shown in Table 1, and the baseline was balanced.
The sequences of each sample varied from 10,021 to 58,982, and the average length of the valid tags was 421.54bps. A total of 1459 OTUs were identified using a cutoff of 97% sequence similarity. Subsequently, 19 phyla, 215 genera, and 240 species were identified, and the average goods coverage index was 0.998, indicating that 99.8% of the bacteria present in our samples were likely to have been identified. An additional figure file shows this in more detail (see Additional file 1).
Comparison of the community structure of the P , C, and NC groups
The microbial community analysis indicated that Firmicutes, Actinobacteriota, and Proteobacteria were the dominant three phyla in all the study groups, with relative abundances of 61.18, 10.68, and 18.47%, respectively, in the NC group, and 70.92, 13.89, and 9.08%, respectively, in the P group, and 74.26, 18.28, and 1.85%, respectively, in the C group. Samples of P and C groups were associated with an increase in Firmicutes and Actinobacteriota and decrease in Proteobacteria compared to the samples of NC group (Fig. 1A). At the genus level, the C group was associated with a bloom of Lactobacillus compared to the NC and P group samples. Samples of NC group were associated with an increase in Tepidimonas and Escherichia-shigella and decrease in Gardnerella compared to the samples of P and C groups (Fig. 1B).The Venn diagram revealed that there were 373 common OTUs within the samples of all the three groups. NC, P and C groups had 233, 375 and 84 unique OTUs respectively, indicating that the microbiota community of each group was varied. (Fig. 1C).

Microbial community structures of the P, C, and NC groups. A The microbial community bar plot of the three groups at the phyla level. B The microbial community bar plot of the three groups at the genus level. C OTU Venn diagram of the three groups. Different colors indicate various groups. The number in the overlapped area means the number of common OTUs of the groups
Comparison of microbiota diversity in P, C, and NC groups
The Chao1 index indicated that the microbial richness of the NC group was greater than the C group with statistical difference (P < 0.05) and P group without significant statistical difference (P = 0.23). No statistical difference of microbial richness in P and C groups had been revealed. (Fig. 2).The principal co-ordinates analysis (PCoA) of bray_curtis distances was used to compare the beta diversity among the three groups. The results revealed a distinction between NC and P/C groups. The three principal component scores accounted for 38.37%, 32.41%, and 12.52% (Fig. 3).The analysis of similarity (anosim) of bray_curtis indicated a significant beta diversity difference within the three groups(P = 0.001).

Box plot of the Chao1 index for the three groups. Each box plot represents one of the groups. The dark horizontal bar represents the median value of each group, while the boxes represent the 25th and 75th percentile values

PCoA analysis of the P, C, and NC groups. NC samples are indicated in blue, P samples are in orange, and C samples are in red. The distance within the groups at the Pc1, Pc2, and Pc3 axes indicates the beta-diversity of the three groups
The feature taxa of P, C, and NC groups
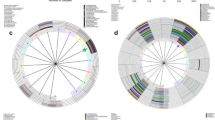
To identify the feature bacterial taxa, the samples of the three groups were compared by LEfSe method. Proteobacteria phylum differed significantly in the mean relative abundance in the NC group, while the P and C groups had no such indicative taxa (Fig. 4).To identify whether there is any biomarker for HPV16 and HPV18 infection or elimination, the subgroups PC (persistent infection with subtype 16 or 18), PO (persistent infection with subtypes other than 16 and 18), CC (clearance of subtype16 or 18), and CO (clearance of subtypes other than 16 and 18) were analyzed further. The Wilcox rank-sum test indicated that genera Vulcaniibacterium and Tepidimonas of the NC group had a statistically higher mean relative abundance compared to the PO, PC, CO, and CC subgroups (P < 0.05). Escherichia-Shigella of the NC group had a higher mean relative abundance compared to the CC, CO, and PO subgroups (P < 0.05). Thermus of NC group had a higher mean relative abundance compared to CC and CO groups (P < 0.05). Compared to the CO and NC groups, the Bifidobacterium was highly abundant in the CC group (P < 0.05). The Lactobacillus in the CO group was in a high mean relative abundance (P < 0.05) compared to the NC, CC, PC and PO groups. Compared to the CC group, Prevotella had a higher mean relative abundance in the CO group (P < 0.05) (Fig. 5). Some other genera differed significantly, but the mean relative abundance was very low (< 0.01%), so these genera were ignored.

LEfSe analysis of the three groups. A A cladogram of the indicative taxa in the vaginal microbiota of the NC group compared to the other two groups. The point in the center represents the root of the tree (bacteria). The rings represent the taxonomic levels in descending order, from phylum to genus. The diameters of the circles represent the relative taxonomic abundances. B Histogram of the LDA scores of the taxa enriched in the NC group. The LDA scores indicate the effect sizes and rankings of the taxa with different relative abundance between the NC and P/C groups. (LDA, linear discriminant analysis)

The Wilcox rank-sum test of the NC, PO, PC, CO, and CC subgroups. The horizontal axis indicates different genus, and the vertical axis indicates the mean relative abundance of the genus. The bars in different colors represent different groups. (PC: persistent infection with subtype HPV16 or 18. PO: persistent infection with subtypes other than 16 and18. CC: clearance of subtype16 or18. CO: clearance of subtypes other than 16 and18. *: P < 0.05; **: P < 0.01)
Discussion
Accumulating evidence showed that the human microbiota mirrors the host physiology and plays a major role in human health [14]. Some cross-sectional studies have addressed the association between the microbiota in the female reproductive tract and HPV infection and related diseases. Firstly, we attempted to investigate the putative pathogen that might relate to persistent HR-HPV infection without any cytological change through high phylogenetic resolution sequencing and we found that the infection was associated with disturbed microbiota profiles. Although Lactobacillus is the most abundant genus in all the three groups, the microbial richness of the NC group was greater than in the C group. The beta diversity analysis revealed a significant difference between the NC and P/C groups. It has been shown that increased diversity of vaginal microbiota has relation with HPV acquisition and persistence [15] However, Gajer et al. [16] found that neither variation in community composition, nor high levels of diversity are necessarily indicative of dysbiosis. Our results indicated that P and C groups had the lower vaginal microbiota diversity. It revealed that the alpha and beta diversities of the P and C groups were similar and could be interpreted as part of the samples in C group with infection of more than one subtype HR-HPVs not achieving complete elimination of all the infected HR-HPVs; thus, there was some degree of similarity in the vaginal microbiota of the C and P groups.
The microbial community analysis showed that P and C group samples were associated with an increase in Firmicutes and Actinobacteriota while a decline in Proteobacteria compared to the samples of the NC group. Also, LEfSe analysis found that the most abundant discriminant taxon was Proteobacteria in the NC group. Studies revealed that cervicovaginal dysbiotic states reduce cervicovaginal barrier function [17]and alter metabolic profiles [18], and these may, in turn, facilitate HPV acquisition and CIN/cancer development, respectively. So, the increased abundance of Proteobacteria in vagina may facilitate keeping microbiota stability.
It has been shown that the reduction of genus Lactobacillus also has relation with HPV acquisition and persistence. Wei et al. [19] found the abundance of Lactobacillales was significantly decreased in the HPV-positive group as compared to the HPV-negative group. Chao et al [20] also found that persistent HR-HPV infection group had a lower relative abundance of Lactobacillus than the incident infection and uninfected groups but without statistical difference. In our study, comparing to the NC group, the relative abundance of Lactobacillus decreased a little in the P group without statistical difference but increased in the C group significantly. In the subgroup analysis, it revealed that samples of clearing HPV16 or 18 had a higher mean relative abundance of Bifidobacterium, and samples of clearing the other HR-HPVs had a higher mean relative abundance of Lactobacillus compared to the NC groups. In addition, Lactobacillus and Bifidobacterium were significantly higher in the CO and CC groups, respectively, indicating that Bifidobacterium might exert a protective effect against HPV16 and 18, while Lactobacillus might be protective against the other HR-HPVs.
Bifidobacterium and Lactobacillus have been studied extensively in the virus infection. In an animal experiment, the mice infected with influenza were fed probiotics of Lactobacillus mucosae 1025, Bifidobacterium breve CCFM1026, and their mixture MIX for 19 days. B. breve CCFM1026 significantly increased the lymphocyte count and the expressions of TLR7, MyD88, TRAF6, and TNF-α to restore the immune balance and decrease viral loading [21]. Another study revealed that protein-based metabolic products of L. casei, L. fermentun, B. adolescentis, and B. bifidum showed significant anti-rotavirus activity [22]. B. adolescentis was also reported as a microorganism with a potential antiviral activity against herpes simplex virus type 1, hepatitis B virus, and coxsackie virus [23,24,25]. A recent study [26] analyzed the expression of CASP3 and HPV18 E6 and E7 genes in HeLa cells before and after treatment with L. crispatus and L. rhamnosus culture supernatants. The results indicated that the expression of HPV18 E6 in HeLa cells was significantly decreased after treatment with Lactobacilli culture supernatants. These studies suggested that Lactobacillus and Bifidobacterium were promising anti-HPV infection probiotics but further investigation into the prophylactic properties and mechanisms was needed.
To date, there is no definitive treatment for HPV, however, probiotics may be an effective treatment. Several probiotics comprised of living bacteria (such as Bifidobacteria species, Lactobacilli, and Streptococci), have been proven to improve the immune system and inflammatory state clinically and also reduce the risk of diabetes, allergic disorders, and certain cancers [27]. Palma et al. found that antibiotic treatment plus vaginal Lactobacillus implementation was effective in viral clearance in women affected by bacterial vaginosis with concomitant HPV infections [28]. Another clinical trial demonstrated that oral administration of HPV16 E7-expressing L. casei resulted in the regression of HPV16-related CIN3. Therapeutic vaccine immunization with E7-bound L. casei showed the induction of E7-specific mucosal IFNc-producing cells [29]. Therefore, considering these findings and our results, probiotics containing Bifidobacteria and/or Lactobacillus may be helpful in HR-HPV clearance in HPV-positive women.
Conclusions
In conclusion, persistent HR-HPV infection might be associated with dysbiosis of the vaginal microbiota. Microbiome regulation with Bifidobacteria and Lactobacillus may affect the clearance of HPV, but further research is needed to provide an in-depth insight into the prophylactic properties and the antivirus mechanism.
Availability of data and materials
The datasets used and analyzed during the current study were deposited into the Genome Sequence Archive (GSA) database. Before released, they are available from the corresponding author on reasonable request.
Abbreviations
- ANOVA:
-
One-way analysis of variance
- Anosim:
-
Analysis of similarity
- CIN:
-
Cervical intraepithelial neoplasia
- CSTs:
-
Community state types
- HR-HPV:
-
High-risk human papilloma virus
- LDA:
-
Linear discriminant analysis
- LefSe:
-
Linear discriminant analysis effect size
- NILMs:
-
Negative intraepithelial lesions or malignancies
- PcoA:
-
Principal co-ordinate analysis
- qRT-PCR:
-
Real-time quantitative polymerase chain reaction
- STIs:
-
Sexually transmitted infections
- WHO:
-
World Health Organization
References
Borgogna JC, Shardell MD, Santori EK, Nelson TM, Rath JM, Glover ED, et al. The vaginal metabolome and microbiota of cervical HPV-positive and HPV-negative women: a cross-sectional analysis. BJOG. 2020;127(2):182–92.
Tolstov Y, Hadaschik B, Pahernik S, Hohenfellner M, Duensing S. Human papillomaviruses in urological malignancies: a critical assessment. Urol Oncol. 2014;32(1):46.e19-46.e27.
Gillet E, Meys JF, Verstraelen H, Bosire C, De Sutter P, Temmerman M, et al. Bacterial vaginosis is associated with uterine cervical human papillomavirus infection: a meta-analysis. BMC Infect Dis. 2011;1110:1.
Guo YL, You K, Qiao J, Zhao YM, Geng L. Bacterial vaginosis is conducive to the persistence of HPV infection. Int J STD AIDS. 2012;23(8):581–4.
Vriend HJ, Bogaards JA, van Bergen JE, Brink AA, van den Broek IV, Hoebe CJ, et al. Incidence and persistence of carcinogenic genital human papillomavirus infections in young women with or without Chlamydia trachomatis co-infection. Cancer Med. 2015;4(10):1589–98.
Clarke MA, Rodriguez AC, Gage JC, Herrero R, Hildesheim A, Wacholder S, et al. A large, population-based study of age-related associations between vaginal pH and human papillomavirus infection. BMC Infect Dis. 2012;12:33.
Ravel J, Gajer P, Abdo Z, Schneider GM, Koenig SS, McCulle SL, et al. Vaginal microbiome of reproductive-age women. Proc Natl Acad Sci USA. 2011;108(Suppl 1):4680–7.
Di Paola M, Sani C, Clemente AM, Iossa A, Perissi E, Castronovo G, et al. Characterization of cervico-vaginal microbiota in women developing persistent high-risk Human Papillomavirus infection. Sci Rep. 2017;7(1):10200.
Brotman RM, Shardell MD, Gajer P, Tracy JK, Zenilman JM, Ravel J, et al. Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. J Infect Dis. 2014;210(11):1723–33.
So KA, Yang EJ, Kim NR, Hong SR, Lee JH, Hwang CS, et al. Changes of vaginal microbiota during cervical carcinogenesis in women with human papillomavirus infection. PLoS ONE. 2020;15;(9): e0238705.
Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R. UniFrac: an effective distance metric for microbial community comparison. ISME J. 2011;5(2):169–72.
Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, et al. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60.
Schwarzer M. Gut microbiota: puppeteer of the host juvenile growth. Curr Opin Clin Nutr Metab Care. 2018;21(3):179–83.
Mitra A, Macintyre DA, Marchesi JR, Lee YS, Bennett PR, Kyrgiou M. The vaginal microbiota, human papillomavirus infection and cervical intraepithelial neoplasia: what do we know and where are we going next? Microbiome. 2016;4:58.
Gajer P, Brotman RM, Bai G, Sakamoto J, Schutte UME, Zhong X, et al. Temporal dynamics of the human vaginal microbiota. Sci Transl Med. 2012;4:132.
Borgdorff H, Gautam R, Armstrong SD, Xia D, Ndayisaba GF, van Teijlingen NH, et al. Cervicovaginal microbiome dysbiosis is associated with proteome changes related to alterations of the cervicovaginal mucosal barrier. Mucosal Immunol. 2016;9:621–33.
Ilhan ZE, Laniewski P, Thomas N, Roe DJ, Chase DM, Herbst-Kralovetz MM. Deciphering the complex interplay between microbiota, HPV, inflammation and cancer through cervicovaginal metabolic profiling. EBioMedicine. 2019;44:675–90.
Wei ZT, Chen HL, Wang CF, Yang GL, Han SM, Zhang SL. Depiction of vaginal microbiota in women with high-risk human papillomavirus infection. Front Public Health. 2021;8:587298.
Chao X, Sun T, Wang S, Tan X, Fan Q, Shi H, et al. Research of the potential biomarkers in vaginal microbiome for persistent high-risk human papillomavirus infection. Ann Transl Med. 2020;8(4):100.
Lu W, Fang Z, Liu X, Li L, Zhang P, Zhao J, et al. The potential role of probiotics in protection against influenza virus infection in mice. Foods. 2021;10(4):1–16.
Fernandez-Duarte KP, Olaya-Galán NN, Salas-Cárdenas SP, Lopez-Rozo J, Gutierrez-Fernandez MF. Bifidobacterium adolescentis (DSM 20083) and Lactobacillus casei (Lafti L26-DSL): Probiotics Able to Block the In Vitro Adherence of Rotavirus in MA104 Cells. Probiotics Antimicrob Proteins. 2018;10(1):56–63.
An HM, Lee DK, Kim JR, Lee SW, Cha MK, Lee KO, et al. Antiviral activity of Bifidobacterium adolescentis SPM 0214 against herpes simplex virus type 1. Arch Pharm Res. 2012;35(9):1665–71.
Lee DK, Kang JY, Shin HS, Park IH, Ha NJ. Antiviral activity of Bifidobacterium adolescentis SPM0212 against hepatitis B virus. Arch Pharm Res. 2013;36(12):1525–32.
Kim MJ, Lee DK, Park JE, Park IH, Seo JG, Ha NJ. Antiviral activity of Bifidobacterium adolescentis SPM1605 against Coxsackievirus B3. Biotechnol Biotechnol Equip. 2014;28(4):681–8.
Motevaseli E, Azam R, Akrami SM, Mazlomy M, Saffari M, Modarressi MH, et al. The effect of Lactobacillus crispatus and Lactobacillus rhamnosus culture supernatants on expression of autophagy genes and HPV E6 and E7 oncogenes in the HeLa cell line. Cell J. 2016;17(4):601–7.
Li Y, Yu T, Yan H, Li D, Yu T, Yuan T, et al. Vaginal microbiota and HPV infection: novel mechanistic insights and therapeutic strategies. Infect Drug Resist. 2020;13:1213–20.
Palma E, Recine N, Domenici L, Giorgini M, Pierangeli A, Panici PB. Long-term Lactobacillus rhamnosus BMX 54 application to restore a balanced vaginal ecosystem: a promising solution against HPV-infection. BMC Infect Dis. 2018;18(1):13–9.
Komatsu A, Igimi S, Kawana K. Optimization of human papillomavirus (HPV) type 16 E7-expressing lactobacillus-based vaccine for induction of mucosal E7-specific IFNγ-producing cells. Vaccine. 2018;36(24):3423–6.
Acknowledgements
Not applicable.
Funding
This study was supported by the National Key Research and Development Program of China (2021YFC2009100, 2021YFC2009105) and the foundation of Science & Technology Department of Sichuan Province (2019YJ0044).
Author information
Authors and Affiliations
Contributions
LM and TW were responsible for data acquisition, analysis and interpretation, drafting the article and final approval of the version to be published. YC, DW, TC, YZ and JM were responsible for clinical samples and data collection. XZ, YL and LD were responsible for DNA extraction and data acquisition. NX: responsible for the initial concept, design of study and final review of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was conducted according to the Chinese National Code for Health Research Ethics. Ethical approved to conduct this study was obtained from the Ethics Committee in the Sichuan University (NO. 044). Informed consent was obtained from all participants prior to enrolment in the study.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Additional file 1.
Rarefaction curves. The horizontal axis indicates the sampling depth, and the vertical axis indicates the index. All the curves become flat as the sampling depth increases, indicating that the high sampling coverage was achieved in all samples.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Mei, L., Wang, T., Chen, Y. et al. Dysbiosis of vaginal microbiota associated with persistent high-risk human papilloma virus infection. J Transl Med 20, 12 (2022). https://doi.org/10.1186/s12967-021-03201-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-021-03201-w