
Abstract
Background and objectives
Sepsis is a leading cause of mortality and morbidity in the intensive care unit. Regulatory mechanisms underlying the disease progression and prognosis are largely unknown. The study aimed to identify master regulators of mortality-related modules, providing potential therapeutic target for further translational experiments.
Methods
The dataset GSE65682 from the Gene Expression Omnibus (GEO) database was utilized for bioinformatic analysis. Consensus weighted gene co-expression netwoek analysis (WGCNA) was performed to identify modules of sepsis. The module most significantly associated with mortality were further analyzed for the identification of master regulators of transcription factors and miRNA.
Results
A total number of 682 subjects with various causes of sepsis were included for consensus WGCNA analysis, which identified 27 modules. The network was well preserved among different causes of sepsis. Two modules designated as black and light yellow module were found to be associated with mortality outcome. Key regulators of the black and light yellow modules were the transcription factor CEBPB (normalized enrichment score = 5.53) and ETV6 (NES = 6), respectively. The top 5 miRNA regulated the most number of genes were hsa-miR-335-5p (n = 59), hsa-miR-26b-5p (n = 57), hsa-miR-16-5p (n = 44), hsa-miR-17-5p (n = 42), and hsa-miR-124-3p (n = 38). Clustering analysis in 2-dimension space derived from manifold learning identified two subclasses of sepsis, which showed significant association with survival in Cox proportional hazard model (p = 0.018).
Conclusions
The present study showed that the black and light-yellow modules were significantly associated with mortality outcome. Master regulators of the module included transcription factor CEBPB and ETV6. miRNA-target interactions identified significantly enriched miRNA.
Similar content being viewed by others

Background
Sepsis is defined as organ dysfunction syndrome caused by uncontrolled inflammatory response to infection. Sepsis is a leading cause of mortality in hospitalized patients [1, 2], and accounts for 30% of case fatality in hospitalized patients [3]. Despite the high mortality and morbidity, few agents are proven to be effective for the treatment of sepsis. Thus, more regulatory factors need to be identified to provide potential targets for the design of effective therapeutic agents.
Several studies have used transcriptome analysis to investigate potential biological pathways regulating the pathogenesis of sepsis [4,5,6,7,8]. These studies were performed by differential gene expression analysis, followed by enrichment analyses to established functional pathways. In these analyses, genes were tested individually. The sensitivity to identify biologically meaningful genes can be low due to multiple testing adjustment. Sepsis is a heterogeneous syndrome and its pathogenesis involves hundreds of genes. In this situation, the individual contribution of a single gene is too small to be detected with univariate test. In most diseases, genes function via networks of co-expressed genes with similar biological functions. Thus, identification of co-expression pattern could provide further insights into sepsis-associated biological pathways. Weighted gene co-expression network analysis (WGCNA) is a systems biology approach used for finding gene clusters with highly correlated expression levels and for relating them to phenotypic traits [9]. Rather than relating thousands of genes to the clinical trait, WGCNA focuses on the relationship between a few modules and the trait [10, 11]. To the best of our knowledge, WGCNA has been used to explore coexpression pattern in mouse model of sepsis [12], HIV infection [13], in vitro inflammatory cells [14], and pediatric sepsis [15]. Regulatory factors including transcription factors and miRNA were not systematically explored in adult sepsis.
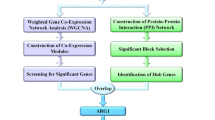
The present study aimed to identify gene co-expression modules in sepsis by using the consensus WGCNA (consensus from different causes of sepsis including pneumonia and abdominal sepsis). These consensus modules were related to clinical traits and enriched to functional biological pathways. Potential regulators of these modules were explored by using well curated databases.
Materials and methods
The GEO dataset and data preprocessing
The study used the publicly avaiable dataset GSE65682 from the Gene Expression Omnibus (GEO) database. The dataset contained 802 samples including healthy controls, non-sepsis critically ill patients and sepsis patients. Futhermore, the sepsis patients could be further categorized into pneumonia sepsis (n = 192), abdominal sepsis (n = 51) and others (n = 443) based on infection site. PAXgene blood RNA was isolated at intensive-care unit (ICU) admission and whole-blood leukocyte transcriptome was performed at the platform of Affymetrix Human Genome U219 Array. Key benefits of the gene chip include increased productivity and efficiency through parallel processing, excellent gene expression accuracy and reproducibility and complete coverage of the annotated genome. Further detials of the dataset can be found at other publications [16, 17]. Raw intensity expression data were preprocessed with the Robust Multi-array Average (RMA) method [18]. An advantage of this method is that normalization occurs at the probe level (rather than at the probeset level) across all of the selected hybridizations. The maximum expression intensity was used when multiple probe sets mapped an individual gene symbol. The quality of processed data were checked by using MA plot (Additional file 1: Figure S1).
Consensus weighted gene co-expression network analysis
The first step in constructing a consensus WGCNA was to choose the soft threshoulding power to which co-expression similarity was raised to calculte adjaency. We chose from a set of values from 4 to 20 based on the criterion of approximate scale-free topology [10]. Since the topological overlap matrices (TOM) of different sepsis may have different statistical property, we performed quantile normalization over the three types of sepsis (e.g. pneumonia, abdomianl and other sepsis). The consensus TOM was calculated by taking the component-wise (“parallel”) minimum of the TOMs in individual dataset, which was then input to hierarchical clustering. Finally, modules were identified in the resulting dendrogram using the Dynamic Tree Cut algorithm [19]. This algorithm has several advantages such as capability of identifying nested clusters and flexibility. Modules with similar expression profiles were merged at the threshold of 0.25.
Gene significance was defined as the Student t-test statistic for testing differential expression between sepsis and healthy controls. The significance level was adjusted for multiple testing with Bonferroni correction. The dataset also contained critically ill patients such as those with major abdominal surgery without infection. There were common pathways between critical illness and severe infection, the comparison between sepsis versus non-infectious critical illness would omit some important genes. Thus, the differential expression was tested between sepsis versus healthy controls.
Relating consensus module to pneumonia-specific sepsis module
Modules specific to pnenomia sepsis were identified by the method as described above. Pneumonia sepsis modules were then related to consensus modules. We calculated the overlaps of each pair of pneumonia-consensus modules, and used the hypergeometric test to assign a p-value to each of the pairwise overlaps. This is also known as the cross-tabulation based comparison of modules. This method is justified by the idea that if a module is well preserved and reproducible in all types of sepsis (pneumonia, abdominal and other sepsis), this module could represent the common pathways involving the pathogenesis of sepsis [20].
Module preservation across all three datasets were explored by pairwise comparing eigengene networks in penumonia, abdominal and other sepsis. Mortality was added as an additional “eigengene”. Network preservation is simply the difference between adjacencies in the two compared sets [20]. A small difference of the adjacency matrix between two sets indicate the modules are well preserved between the two comparing sets.
Relating consensus modules to clinical traits
Module eigengene was calculated for each module as the first principal component of gene expressions for that module. Correlation analysis was performed to relate module eigengene to external traits including age, gender, mortality and survival time (e.g. survivors were censored at 28 days). We combined three datasets into one and performed correlation analysis.
Gene significance for mortality was correlated to the module membership to investigate whether genes significantly associated with mortality outcome was also associated with module membership. Module membership (eigengene-based connectivity) for each gene was calculated by correlating its gene expression profile with the module eigengene of a given module. For a given module, a module membership value of 0 indicates that a gene is not part of the module; whereas a module memberhsip of − 1 or 1 is highly connected to the module.
Enrichment analysis for biological function and transcription factors
Modules associated with important clinical trait such as mortality were further analyzed for their enrichment in Gene Ontology (GO) pathways [21]. Specifically, the gene set from a given module were enriched to GO terms to find whether some of functional GO terms are over-represented using annotations for that gene set. Upset plot was employed to display overlapped genes among different GO terms. Dotplot shows the gene ratio and adjusted p values for each enriched GO terms. Enriched terms were organized into a network with edges connecting overlapping gene sets. In this way, mutually overlapping gene sets are tend to cluster together, making it easy to identify functional modules. The category netplot depicts the linkages of genes and GO terms as a network, which is helpful to see which genes are involved in enriched pathways and genes that may belong to multiple annotation categories.
Modules (gene lists) significantly correlated with the mortality trait were tested for its over-representation in transcription factor (TF) binding motifs by using RcisTarget [22]. Two types of databases (i.e. Gene-motif rankings and the annotation of motifs to transcription factors) were employed in the analysis: Gene-motif rankings which provides the rankings of all the genes for each motif and the annotation of motifs to transcription factors. Parameter settings for the score of each pair of gene-motif were: species = Homo sapiens, Scoring/search space = 500 bp uptream the transcription start site (TSS), Number of orthologous species = 10. The annotation of motifs to transcription factors was performed using the motifAnnotations_hgnc ('mc9nr', 24,453 motifs).
Identification of miRNA-target interactions
The multiMiR package was employed for the retrieval of miRNA-target interactions from 14 external databases in R. These databases are comprehensive collections of predicted and validated miRNA-target interactions and their associations with diseases and drugs [23]. The module of interest was those associated with mortlaity outcome. It was interesting to check whether some, or all, of these genes within a module were targeted by the same miRNA(s). We restrited our search to the “mirtarbase” table because this table included only experimentally validated miRNA-target interactions.
Survival analysis
The association of each module with the survival outcome was determined by using Cox proportional model. The eigengene value of each module was added into the Cox regression model for univariate analysis. Genes matched to the module with the strongest association with survival were used to cluster patients into two groups using reversed graph embedding (DDRTree), which projects data into a reduced dimensional space while constructs a principal tree which passes through the middle of the data simultaneously [24]. Samples were clustered into two groups using k-means clustering. Survival probablity of the two groups were comapred using the log-rank test.
Results
Demographic data
A total of 802 subjects were initially identified from the GEO database. 116 subjects were excluded because they were either healthy or non-infectious controls, and 4 subjects were excluded because they were outliers (Additional file 1: Figure S2). As a result, a number of 682 subjects were included in our analysis. These subjects were classified by the infection site into pneumonia sepsis, abdominal sepsis and unselected sepsis (other sepsis). Demographic data were comparable between the three groups (Table 1). The median age was 63 years (IQR: 53 to 72), and 58% (394/682) patients were male. The overall 28 day mortality rate was 17% (113/682).
Consensus Network construction and module detection
A soft-thresholding power of 6 was used to obtain approximate scale-free topology for the network (Additional file 1: Figure S3). Consensus WGCNA identified 27 modules (Fig. 1). Gene differential expression analysis between healthy controls and sepsis showed that genes such as ARG1, CD177, MMP8 and C19orf59 were upregulated. Up-or down-regulation of modules were determined by the median of gene significance T value and fold changes. For instance, the black model was significantly upregulated with median T value > 3 and log2 fold change > 0.5 (Fig. 2).

Consensus weighted gene coexporession network analysis. Modules were identified in the resulting dendrogram using the Dynamic Tree Cut algorithm. A total of 27 modules were identified. Modules were distinguished from each other by assigning different colors

Gene differential expression analysis (sepsis against healthy control) across modules. a Boxplot of the distribution of significance T values. The black and darkorange modules showed the most significant difference between sepsis versus controls. The red dashed lines indicate statistical significance level of two-tailed T test that have been adjusted by Bonferroni method; b fold changes of the differential expression across modules. The darkorange and black modules were among the most up-regulated modules; and c volcano plot showing differentially expressed genes between sepsis and healthy controls. Genes such as ARG1, CD177 and MMP8 were most differentially expressed
Consensus eigengene networks and their differential analysis are shown in Fig. 3. The results showed that the eigengene networks in the three datasets were well preserved. To explore whether modules identified in the pneumonia sepsis could also be identified in consensus modules, the correspondence of pneumonia set specific and consensus modules was explored (Fig. 4). The result indicates that most peumonia set-specific modules have a consensus counterpart (Fig. 4). The black color module in the consensus analysis corresponds to the red module in the pneumonia specific set analysis (275 genes overlap with significant p value).

Summary plot of consensus eigengene networks and their differential analysis. The top 3 panels show the dendrograms (clustering trees) of the consensus module eigengenes in the 3 datasets of pneumonia, abdominal and other sepsis. Below, the eigengene networks in the three sets are shown as heatmaps labeled penumonia sepsis, obdominal sepsis and other sepsis. In the heatmaps, red denotes high adjacency (positive correlation) and blue denotes low adjacency. The Preservation heatmap shows the preservation network, defined as one minus the absolute difference of the eigengene networks in the two datasets. The barplot shows the mean preservation of adjacency for each of the eigengenes to all other eigengenes; in other words, the barplot depicts the column means of the preservation heatmap

Correspondence of pneumonia set-specific modules and the consensus modules. Each row of the table corresponds to one pneumonia set-specific module (labeled by color as well as text), and each column corresponds to one consensus module. Numbers in the table indicate gene counts in the intersection of the corresponding modules. Coloring of the table encodes − log(p), with p being the Fisher’s exact test p value for the overlap of the two modules. The stronger the red color, the more significant the overlap is. The table indicates that most peumonia set-specific modules have a consensus counterpart
Relating Consensus modules to external clinical traits
The correlation between module eigengene and clinical traits were explored by Pearson’s correlation analysis (Fig. 5). The black module was significantly associated with mortality with higher module express correlated to lower mortality rate (coefficient = -0.16; p < 0.001). However, the light-yellow module was positively associated with mortality (coefficient = 0.14; p < 0.001). The black module was positively associated with survival time (coefficient = 0.16; p < 0.001). The correlations between gene significance for mortality and module membership were explored and the results showed that there was a highly significant correlation between module membership and gene significance for mortality in three modules inclusing the light-yellow, ligh cyan and pink modules (Additional file 1: Figure S4).

Consensus module-trait relationship. Each module was represented by its eigengene value and correaltion analyses between each of the eigengene values and clinical traits were performed. The first number in the matrix cell represents the corelation coefficient and the number in the parenthesis is the p value for the corelation. The black module was negatively correlated to the mortality and the light-yellow module was positively related to the mortality
Module biological function
The most enriched pathways of the black module included myeloid leukocyte mediated immunity, neutrophil mediated immunity, leukocyte degranulation and myeloid cell activation involved in immune response (Fig. 6). The light-yellow module was enriched in biological pathways such as translation, nucleobase-containing compound catabolic process, heterocycle catabolic process and cellular nitrogen compound catabolic process (Fig. 7).

GO enrichment analysis for the black module. a The upset plot showing overlap of genes across enriched biological pathways. Biological pathways such as myeloid leukocyte mediated immunity, leukocyte degranulation and neutrophil mediated immunity shared large number of genes. b Dot plot showing the ordered enrichment biological pathways; the most enriched pathways included myeloid leukocyte mediated immunity and neutrophil mediated immunity. c Enriched terms were organized into a network with edges connecting overlapping gene sets. In this way, mutually overlapping gene sets are tend to cluster together, making it easy to identify functional modules; and d The category netplot depicts the linkages of genes and GO terms as a network, which is helpful to see which genes are involved in enriched pathways and genes that may belong to multiple annotation categories. The most important biological pathways in the module involves the immune response, including the immune response including myeloid leukocyte mediated immunity, neutrophil mediated immunity, leukocyte degranulation and myeloid cell activation

GO enrichment analysis for the light-yellow module. a The upset plot showing overlap of genes across enriched biological pathways; b dot plot showing the ordered enrichment biological pathways; c Enriched terms were organized into a network with edges connecting overlapping gene sets. In this way, mutually overlapping gene sets are tend to cluster together, making it easy to identify functional modules; and d The category netplot depicts the linkages of genes and GO terms as a network, which is helpful to see which genes are involved in enriched pathways and genes that may belong to multiple annotation categories. The most important biological pathways in the module involves the translation and catabolic processes, including nucleobase-containing compound catabolic process, heterocycle catabolic process and cellular nitrogen compound catabolic process
Enrichment analysis for transcription factors
Modules were made up of co-expressed genes, indicating that they were regulated by common mechanisms such as the transcription factors. Thus, we performed enrichment analysis for transcription factors. The enrichment analysis involved three steps: (1) motif enrichment analysis with cumulative recovery curves (Additional file 1: Figures S5, S6 and S7), (2) motif-TF annotation, and (3) selection of significant genes. The results showed that the transcription factor CEBPB was the master regulator for the black module, which was annotated to the motif dbcorrdb__CEBPB__ENCSR000BQI_1__m1. A total of 93 genes in the black module was enriched in this motif. The normalized enrichment score (NES) was 5.53. This motif was directly annotated to the transcription factor CEBPB which is an important transcription factor regulating the expression of genes involved in immune and inflammatory responses [25,26,27]. All enriched motifs and corresponding transcription factors for the black module were shown in Additional file 2: Supplemental Digital Content (SDC) 2. The network of the top 3 enriched motifs and corresponding genes are shown in Additional file 1: Figure S8.
The transcription factor ETV6 was the master regulator for the light-yellow module, which was annotated to the motif taipale__ETV6_full_CCGGAASCGGAAGTN_repr and cisbp__M5425. A total of 12 genes in the light-yellow module were enriched in the motifs. The NES was 6 and 5.98 for these two motifs [Additional file 3: Supplemental Digital Content (SDC) 3].
Identification of miRNA-target interactions
From the “mirtarbase” table with validated miRNA-target interactions, we identified 1,981 miRNA that were potential regulators of the black module [Additional file 4: Supplemental Digital Content (SDC) 4]. The top 5 miRNA regulated the most number of genes were hsa-miR-335-5p (n = 59), hsa-miR-26b-5p (n = 57), hsa-miR-16-5p (n = 44), hsa-miR-17-5p (n = 42), and hsa-miR-124-3p (n = 38). A network connecting miRNA and target genes are shown in Additional file 1: Figure S9.
For the light-yellow module, we identified 893 miRNA that were potential regulators of the module [Additional file 5: Supplemental Digital Content (SDC) 5]. The top 5 miRNA regulated the most number of genes were hsa-miR-16-5p (n = 14), hsa-miR-92a-3p (n = 12), hsa-miR-26b-5p (n = 9), hsa-miR-615-3p (n = 9), and hsa-let-7b-5p (n = 8).
Survival analysis
To further validate that the black module was related to the mortality outcome, the high-dimension space was reduced to lower dimension space by using reversed graph embedding (DDRTree). The method provided non-linear dimension reduction that retains the intrinsic structure of the sample data. The silhouette method showed that 2-cluster would be the optimal number (Fig. 8a). In the 2-dimension space (Fig. 8b), the samples were classified into two clusters. Heatmap of all module genes showed that the two clusters could be well separated (Fig. 8c), which supported that the intrinsic structure was well preserved with the dimension reduction. The two clusters were significantly associated with survival outcome (p = 0.018 in the Cox proportional hazard model, Fig. 8d). Cluster 2, as compared with Cluster 1, was characrized by activated functions including myeloid cell activation, cell activation, leukocyte activation and myeloid leukocyte activation (Fig. 9).

The survival analysis with the black module. a Optimal number of classes derived from DDRTree reduced dimension space with the silhouette width; the two-class model was the best fit model with the highest silhouette width. b Discriminative dimension reduction (DDR) graph of the black module gene list. K-means clustering was used to separate samples into two groups (blue and red). c Heatmap of gene expression sorted by DDR score. The two clusters identified by the DDR score were consistent with that identified by black module genes. Genes showing on the right were those from the black module. d Kaplan–Meier curves for groups defined by DDR k-means clustering of black module genes. Cox’s proportional hazard modelling of module groups showed that the cluster membership was significant association with survival (p = 0.018)

The differences in biological functions between cluster 2 and cluster 1 as identified by the black module. a Volcano plot showing differentially expressed genes in the black module between cluster 2 and cluster 1. b enriched GO biological pathways. c Gene set enrichment analysis with GO term; as compared to cluster 1, cluster 2 was characterized by activated functions such as leukocyte activation, cell activation and myeloid leukocyte activation. d Enrichment Map for enrichment result of over-representation test
The light-yellow module was also related to the mortality outcome. The silhouette method showed that 2-cluster would be the optimal number (Fig. 10a). The study population could be classified into two clusters in a 2-dimension space. Heatmap (Fig. 10b, c) showed that the study population could be well separated into two clusters. Cluster 2 showed significantly lower survival probability than cluster 1 (p = 0.01, Fig. 10d).

The survival analysis with the light-yellow module. a Optimal number of classes derived from DDRTree reduced dimension space with the silhouette width; the two-class model was the best fit model with the highest silhouette width. b Discriminative dimension reduction (DDR) graph of the light-yellow module gene list. K-means clustering was used to separate samples into two groups (blue and red). c Heatmap of gene expression sorted by DDR score. The two clusters identified by the DDR score were consistent with that identified by light-yellow module genes. Genes showing on the right were those from the light-yellow module. d Kaplan–Meier curves for groups defined by DDR k-means clustering of light-yellow module genes. Cox’s proportional hazard modelling of module groups showed that the cluster membership was significant association with survival (p = 0.01)
Discussion
The study employed consensus network analysis to identify gene co-expression modules. These modules were involved in distinct biological functions and were associated with clinical traits. Regulatory mechanisms of some important modules involving transcription regulators and miRNA were explored by validated databases. Consistent with previously published studies employing high-throughput dataset to examine sepsis, these modules were enriched for pathways related to immune response [28, 29]. The black module was significantly associated with survival outcome and the transcription factor CEBPB was the master regulator of this module. Several miRNAs including hsa-miR-335-5p, hsa-miR-26b-5p, hsa-miR-16-5p, hsa-miR-17-5p and hsa-miR-124-3p were identified to be important regulators of the gene expressions in the module.
Our analysis has several implications for research and clinical practice. First, sepsis is shown to have a dysregulated immune response highlighted by the upregulation of the black module involving biological functions such as myeloid leukocyte mediated immunity, neutrophil mediated immunity, leukocyte degranulation and myeloid cell activation involved in immune response. Immune dysfunction has long been recognized as an important mediator of sepsis [4, 30, 31]. However, previous studies mostly analyzed high-throughput data at individual gene level involving differential expression analysis followed by functional pathway enrichment [4, 15, 31, 32]. In neonate sepsis, Meng and colleagues identified 7 hub genes in key pathways. However, the study did not relate these gene expression profiling to clinical outcomes and the results cannot be extrapolated to adult sepsis [33]. The present study employed WGCNA to identify modules associated with mortality by treating co-expressed genes as a module. The idea is that genes are working together to take their functions in disease processes. Second, by focusing on modules most significantly associated with mortality, we identified several important regulators including transcription factors and miRNAs. The results support the hypothesis that co-expressed genes are very likely to be regulated by common factors. These transcription factors (such as CEBPB and ETV6) and miRNAs are potential targets for the treatment of sepsis. Since the upregulation of the black module is significantly associated with mortality, drugs targeting these sites may improve the survival outcome. Third, although sepsis is a heterogeneous syndrome [34,35,36,37,38], different types of sepsis share some common important immune regulatory pathways. Our study employed consensus WGCNA and examined whether the module identified in one cause of sepsis can also be found in other causes of sepsis. The results show that most modules are well preserved despite the various causes of sepsis, indicating common pathways associated with the sepsis. Furthermore, the module network constructed by TOM is also well preserved across various causes of sepsis. Collectively, these findings support the notion that sepsis can be considered as a clinical symdrome because various causes of sepsis lead to common pathways. Drugs designed to target these common pathways can be helpful in improving clinical outcomes. For example, our study showed that cluster 2 identified by the black module was associated with lower survival probability, and this subtypes of sepsis was characterized by leukocyte activation. Such over-activation of inflammatory response may indicate that immunoregulatory agents such as arachidonic acid and eicosapentaenoic acid can help to improve the survival outcome [39].
Mortality is an important clinical trait and thus we tried to identify modules most significantly assocated with mortlaity. Two methods were employed to validate that the black module was associated with mortality. Firstly, we simply correlate the module eigengene to the mortality. Module eigengene is computed as the first component in principal component analysis (PCA), which however is a linear transformation of the high-dimensional space. It is well recognized that linear transformation cannnot fully recover the intrinsic structure of a high-dimensional space [40,41,42]. Thus, we also emplyed manifold learning to better capture the black module [24]. The result is consistent with the above observation that the black module can well separate survivors from non-survivors. Most clinical trials of sepsis are focusing on how to reduce mortality. The identified black module in our study can help to design drugs that potentially useful for reducing mortality rate. For example, our analysis shows that hsa-miR-335-5p is the top ranked miRNA in the regulation of the black module. Since the black module consists mostly genes involved in inflammatory response, the upregulation of hsa-miR-335-5p is proposed to have inflammatory suppression effects [43,44,45]. More recently, hsa-miR-335-5p is shown to reduce inflammation via negative regulation of the TPX2-mediated AKT/GSK3β signaling pathway in a chronic rhinosinusitis mouse model [46]. Collectively, these observations strongly support our results that hsa-miR-335-5p can be a candidate therapeutic target.
The CEBPB was annotated to the most significantly enriched motifs for genes in the black module. The eigengene of black module is the most significant associated with mortality, and thus the master regulator CEBPB is also an important risk factor for mortality via immunomodulation [47, 48]. Our finding is also consistent with a recent study comparing sepsis with and without shock, in which CEBPB is significantly enriched in septic shock versus non-shock patients [49]. Since the presence of shock is a significant risk factor for mortality, the result supports the notion that CEBPB can be a potential target for improving survival outcome.
The strength of the study is the large sample size. To the best of our knowledge, the MARS consortium is the largest sepsis cohort with genome-wide blood transcriptional profiling [16]. Furthermore, the included sepsis subjects were classified into different causes of sepsis, allowing for the consensus network analysis. The consensus analysis identified common functional modules involved in sepsis. However, there are limitations in the study. First, the severity of illness is not available in the cohort, prohibiting correlation analysis for modules and severity scores. However, since most severity scores are developed with the mortality as the end point, the mortality can be used as a surrogate for the severity of illness. Second, the transcription regulators were predicted by bioinformatic analysis, which might have high false positive rate. The in vivo function of these transcription factors and miRNAs should be validated in experimental studies. In this regard, results from the current analysis can be considered as hypothesis-generating. Third, there are more causes of sepsis that have not been categorized in the study. For example, urinary tract infection is also an important cause of sepsis in clinical practice, however, the dataset did not contain this subclass of sepsis.
Conclusion
In conclusion, the present study identified the black and light-yellow modules to be the most significantly associated with mortality. Master regulators of the black module included transcription factor CEBPB. miRNA-target interactions identified significantly enriched miRNA. These regulators can be potential therapeutic targets for the treatment of sepsis.
Availability of data and materials
Data were available on request.
Abbreviations
- GEO:
-
Gene Expression Omnibus
- WGCNA:
-
Consensus weighted gene co-expression netwoek analysis
- ICU:
-
Intensive care unit
- TOM:
-
Topological overlap matrices
- GO:
-
Gene Ontology
- TF:
-
Transcription factor
- TSS:
-
Transcription start site
References
Abe T, Yamakawa K, Ogura H, Kushimoto S, Saitoh D, Fujishima S, et al. Epidemiology of sepsis and septic shock in intensive care units between sepsis-2 and sepsis-3 populations: sepsis prognostication in intensive care unit and emergency room (SPICE-ICU). J Intensive Care. 2020;8:44–9.
Markwart R, Saito H, Harder T, Tomczyk S, Cassini A, Fleischmann-Struzek C, et al. Epidemiology and burden of sepsis acquired in hospitals and intensive care units: a systematic review and meta-analysis. Intensive Care Med. 2020;315:801–16.
Rhee C, Jones TM, Hamad Y, Pande A, Varon J, O'Brien C, et al. Prevalence, underlying causes, and preventability of sepsis-associated mortality in US acute care hospitals. JAMA Netw Open. 2019;2:e187571.
Zhai J, Qi A, Zhang Y, Jiao L, Liu Y, Shou S. Bioinformatics analysis for multiple gene expression profiles in sepsis. Med Sci Monit. 2020;26:e920818.
Scicluna BP, van Vught LA, Zwinderman AH, Wiewel MA, Davenport EE, Burnham KL, et al. Classification of patients with sepsis according to blood genomic endotype: a prospective cohort study. Lancet Respir Med. 2017;5:816–26.
Burnham KL, Davenport EE, Radhakrishnan J, Humburg P, Gordon AC, Hutton P, et al. Shared and distinct aspects of the sepsis transcriptomic response to fecal peritonitis and pneumonia. Am J Respir Crit Care Med. 2017;196:328–39.
Antcliffe DB, Burnham KL, Al-Beidh F, Santhakumaran S, Brett SJ, Hinds CJ, et al. Transcriptomic signatures in sepsis and a differential response to steroids from the VANISH randomized trial. Am J Respir Crit Care Med. 2019;199:980–6.
Weiss SL, Cvijanovich NZ, Allen GL, Thomas NJ, Freishtat RJ, Anas N, et al. Differential expression of the nuclear-encoded mitochondrial transcriptome in pediatric septic shock. Crit Care. 2014;18:623.
Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559–613.
Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17.
Horvath S, Zhang B, Carlson M, Lu KV, Zhu S, Felciano RM, et al. Analysis of oncogenic signaling networks in glioblastoma identifies ASPM as a molecular target. Proc Natl Acad Sci U S A. 2006;103:17402–7.
Huang J, Sun R, Sun B. Identification and evaluation of hub mRNAs and long non-coding RNAs in neutrophils during sepsis. Inflamm Res. 2020;69:321–30.
Ding J, Ma L, Zhao J, Xie Y, Zhou J, Li X, et al. An integrative genomic analysis of transcriptional profiles identifies characteristic genes and patterns in HIV-infected long-term non-progressors and elite controllers. J Transl Med. 2019;17:35.
Godini R, Fallahi H, Ebrahimie E. Network analysis of inflammatory responses to sepsis by neutrophils and peripheral blood mononuclear cells. Mishra A, editor. PLoS ONE. 2018;13:e0201674.
Li Y, Li Y, Bai Z, Pan J, Wang J, Fang F. Identification of potential transcriptomic markers in developing pediatric sepsis: a weighted gene co-expression network analysis and a case-control validation study. J Transl Med. 2017;15:254–311.
Scicluna BP, Klein Klouwenberg PMC, van Vught LA, Wiewel MA, Ong DSY, Zwinderman AH, et al. A molecular biomarker to diagnose community-acquired pneumonia on intensive care unit admission. Am J Respir Crit Care Med. 2015;192:826–35.
Scicluna BP, Wiewel MA, van Vught LA, Hoogendijk AJ, Klarenbeek AM, Franitza M, et al. Molecular biomarker to assist in diagnosing abdominal sepsis upon ICU admission. Am J Respir Crit Care Med. 2018;197:1070–3.
Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64.
Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54–17.
Langfelder P, Luo R, Oldham MC, Horvath S. Is my network module preserved and reproducible? Bourne PE, editor. PLoS Comput Biol. 2011;7:e1001057.
Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–7.
Aibar S, González-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods. 2017;14:1083–6.
Ru Y, Kechris KJ, Tabakoff B, Hoffman P, Radcliffe RA, Bowler R, et al. The multiMiR R package and database: integration of microRNA-target interactions along with their disease and drug associations. Nucleic Acids Res. 2014;42:e133–e143143.
Mao Q, Wang L, Goodison S, Sun Y. Dimensionality reduction via graph structure learning. New York: Association for Computing Machinery; 2015. p. 765–774.
Kinoshita S, Akira S, Kishimoto T. A member of the C/EBP family, NF-IL6 beta, forms a heterodimer and transcriptionally synergizes with NF-IL6. Proc Natl Acad Sci U S A. 1992;89:1473–6.
Chinery R, Brockman JA, Dransfield DT, Coffey RJ. Antioxidant-induced nuclear translocation of CCAAT/enhancer-binding protein beta. A critical role for protein kinase A-mediated phosphorylation of Ser299. J Biol Chem. 1997;272:30356–61.
Roy SK, Hu J, Meng Q, Xia Y, Shapiro PS, Reddy SPM, et al. MEKK1 plays a critical role in activating the transcription factor C/EBP-beta-dependent gene expression in response to IFN-gamma. Proc Natl Acad Sci U S A. 2002;99:7945–50.
Cheng L, Nan C, Kang L, Zhang N, Liu S, Chen H, et al. Whole blood transcriptomic investigation identifies long non-coding RNAs as regulators in sepsis. J Transl Med. 2020;18:217–313.
Zador Z, Landry A, Balas M, Marshall JC, Cusimano MD. Data driven analysis reveals shared transcriptome response, immune cell composition, and distinct mortality rates across differing etiologies of critical illness. Crit Care Med. 2020;48:338–43.
Delano MJ, Ward PA. Sepsis-induced immune dysfunction: can immune therapies reduce mortality? J Clin Invest. 2016;126:23–31.
Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, et al. Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med. 2016;4:259–71.
Maslove DM, Wong HR. Gene expression profiling in sepsis: timing, tissue, and translational considerations. Trends Mol Med. 2014;20:204–13.
Meng Y, Cai XH, Wang L. Potential genes and pathways of neonatal sepsis based on functional gene set enrichment analyses. Comput Math Methods Med. 2018;2018:6708520–610.
Leligdowicz A, Matthay MA. Heterogeneity in sepsis: new biological evidence with clinical applications. Crit Care. 2019;23:80–8.
Bhavani SV, Carey KA, Gilbert ER, Afshar M, Verhoef PA, Churpek MM. Identifying novel sepsis subphenotypes using temperature trajectories. Am J Respir Crit Care Med. 2019;200:327–35.
Kim WY, Jung J-W, Choi JC, Shin JW, Kim JY. Subphenotypes in patients with septic shock receiving vitamin c, hydrocortisone, and thiamine: a retrospective cohort analysis. Nutrients. 2019;11:2976.
Wiersema R, Jukarainen S, Vaara ST, Poukkanen M, Lakkisto P, Wong H, et al. Two subphenotypes of septic acute kidney injury are associated with different 90-day mortality and renal recovery. Crit Care. 2020;24:150–21010.
Zhang Z, Zhang G, Goyal H, Mo L, Hong Y. Identification of subclasses of sepsis that showed different clinical outcomes and responses to amount of fluid resuscitation: a latent profile analysis. Crit Care. 2018;22:347.
Das UN. Combination of aspirin with essential fatty acids is superior to aspirin alone to prevent or ameliorate sepsis or ARDS. Lipids Health Dis. 2016;15:206–8.
Tenenbaum JB, de Silva V, Langford JC. A global geometric framework for nonlinear dimensionality reduction. Science. 2000;290:2319–23.
Zhu B, Liu JZ, Cauley SF, Rosen BR, Rosen MS. Image reconstruction by domain-transform manifold learning. Nature. 2018;555:487–92.
Tang C, Bian M, Liu X, Li M, Zhou H, Wang P, et al. Unsupervised feature selection via latent representation learning and manifold regularization. Neural Netw. 2019;117:163–78.
Shang Z, Sun W, Zhang M, Xu L, Jia X, Zhang R, et al. Identification of key genes associated with multiple sclerosis based on gene expression data from peripheral blood mononuclear cells. PeerJ. 2020;8:e8357.
Yue J, Wang P, Hong Q, Liao Q, Yan L, Xu W, et al. MicroRNA-335-5p plays dual roles in periapical lesions by complex regulation pathways. J Endod. 2017;43:1323–8.
Zhu L, Chen L, Shi C-M, Xu G-F, Xu L-L, Zhu L-L, et al. MiR-335, an adipogenesis-related microRNA, is involved in adipose tissue inflammation. Cell Biochem Biophys. 2014;68:283–90.
Gu X, Yao X, Liu D. Up-regulation of microRNA-335-5p reduces inflammation via negative regulation of the TPX2-mediated AKT/GSK3β signaling pathway in a chronic rhinosinusitis mouse model. Cell Signal. 2020;70:109596.
Hu Y, Zhong W, Chen M, Zhang Q. Identifying crucial genes for prognosis in septic patients: Gene integration study based on PRISMA guidelines. Medicine (Baltimore). 2019;98:e16807.
Dai J, Kumbhare A, Youssef D, Yao ZQ, McCall CE, Gazzar EM. Expression of C/EBPβ in myeloid progenitors during sepsis promotes immunosuppression. Mol Immunol. 2017;91:165–72.
Braga D, Barcella M, Herpain A, Aletti F, Kistler EB, Bollen Pinto B, et al. A longitudinal study highlights shared aspects of the transcriptomic response to cardiogenic and septic shock. Crit Care. 2019;23:414–24.
Acknowledgements
None
Funding
Z.Z. received funding from the National Natural Science Foundation of China (Grant No. 81901929). Ping Xu received funding from Research project of Health and Family Planning Commission Of Sichuan Province (17PJ136),Research project of Zigong City Science & Technology and Intellectual Property Right Bureau (2017SF04) and Research project of Zigong City Science & Technology and Intellectual Property Right Bureau (2018SF04).
Author information
Authors and Affiliations
Contributions
ZZ and LC conceived the idea, performed analysis and drafted the manuscript; PX, PC and YH performed data acquisition and figure preparations, LX, PC and PX performed manifold learning algorithm. PX and LC interpreted the results and help to revise the manuscript. The authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study was an analysis of the third-party anonymized databases with pre-existing IRB approval.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1.
Supplemental digital content 1.
Additional file 2.
Supplemental digital content 2.
Additional file 3.
Supplemental digital content 3.
Additional file 4.
Supplemental digital content 4.
Additional file 5.
Supplemental digital content 5.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhang, Z., Chen, L., Xu, P. et al. Gene correlation network analysis to identify regulatory factors in sepsis. J Transl Med 18, 381 (2020). https://doi.org/10.1186/s12967-020-02561-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-020-02561-z