
Abstract
Background
Heart failure with ejection fraction (HFpEF) is a syndrome resulting from several co-morbidities in which specific mediators are unknown. The platelet proteome responds to disease processes. We hypothesize that the platelet proteome will change composition in patients with HFpEF and may uncover mediators of the syndrome.
Methods and results
Proteomic changes were assessed in platelets from hospitalized subjects with symptoms of HFpEF (n = 9), the same subjects several weeks later without symptoms (n = 7) and control subjects (n = 8). Mass spectrometry identified 6102 proteins with five scans with peptide probabilities of ≥0.85. Of the 6102 proteins, 165 were present only in symptomatic subjects, 78 were only found in outpatient subjects and 157 proteins were unique to the control group. The S100A8 protein was identified consistently in HFpEF samples when compared with controls. We validated the fining that plasma S100A8 levels are increased in subjects with HFpEF (654 ± 391) compared to controls (352 ± 204) in an external cohort (p = 0.002). Recombinant S100A8 had direct effects on the electrophysiological and calcium handling profile in human induced pluripotent stem cell-derived cardiomyocytes.
Conclusions
Platelets may harbor proteins associated with HFpEF. S100A8 is present in the platelets of subjects with HFpEF and increased in the plasma of the same subjects. We further established a bedside-to-bench translational system that can be utilized as a secondary screen to ascertain whether the biomarkers may be an associated finding or causal to the disease process. S100A8 has been linked with other cardiovascular disease such as atherosclerosis and risk for myocardial infarction, stroke, or death. This is the first report on association of S100A8 with HFpEF.
Similar content being viewed by others

Background
The platelet proteome is an untapped resource for identifying proteins that may reflect a disease process. Platelets are easily accessible and free from major highly abundant proteins making them an attractive model for proteomic studies. Platelets change the composition of their proteins in diseases such as Alzheimer’s, cancer, diabetes, coronary artery disease and acute coronary syndrome [1–4]. Platelets are largely under-studied in heart failure, yet evidence indicates that both platelet function [5, 6] and platelet-derived proteins such as adhesion molecules and the natriuretic peptide receptor-C [7–10] are altered in heart failure. Therefore, changes in the platelet proteome may allow for the identification of proteins that influence the disease process in heart failure.
Heart failure with preserved ejection fraction (HFpEF) affects almost 50 % of patients with heart failure and is increasing in prevalence [11], yet the pathophysiological mechanisms are poorly understood. HFpEF is associated with diabetes, hypertension, renal dysfunction, atrial fibrillation and obesity. The systemic inflammatory state induced by these co-morbidities is predictive of HFpEF [12, 13]. Platelets are both contributors and responders of inflammatory processes [14]. Considering there are no targeted therapies for HFpEF and morbidity and mortality are high, it is paramount to identify biomarkers associated with HFpEF and clarify their mechanistic role in clinical heart failure in order to develop targeted treatments. Consequently, by examining the platelet proteome of subjects with HFpEF, there is the potential to identify proteins that may provide insight into the disease mechanisms.
We established a novel bed-to-bench translational system to identify potential mediators of HFpEF using both platelet proteome analysis and mechanistic studies in induced pluripotent stem cell-derived cardiomyocytes. The broad utility of this strategy is to incorporate bioactivity studies into guiding the selection of proteins from proteomic studies for further investigation. We sought to compare the platelet proteome among subjects with HFpEF in the uncompensated (hospitalized) state, compensated (outpatient) state, and controls combined with validation in plasma samples from an external cohort and bioactivity studies using human induced pluripotent stem cell (iPSC)-derived cardiomyocytes. We hypothesized that [1] platelet proteomic analysis would successfully identify a protein associated with HFpEF, and [2] human iPSC-derived cardiomyocytes treated with recombinant proteins could serve as further validation by demonstrating phenotypic changes in cardiomyocyte calcium handling, which is altered in HFpEF.
Methods
Study population
For the discovery phase, subjects ≥50 years old presenting with New York Heart Association class II–III heart failure symptoms, a left ventricular ejection fraction (LVEF) >50 %, echocardiographic evidence of diastolic dysfunction and increased LV filling pressure were evaluated at the Medical College of Wisconsin between June 2012 to December 2013 for participation in this study. Increased LV filling pressures were defined as E/e′ ≥ 15, or E/e′ ≥ 8 and ≤ 15 with either a BNP ≥ 200 pg/ml or a left atrial (LA) volume index > 40 ml/m2. Subjects were excluded if they had a clinical condition that potentially changed the platelet or plasma proteomic profile independent of HFpEF such as uncontrolled diabetes, an active infection or inflammatory disorder, chronic renal failure requiring dialysis, severe liver disease, malignancy, acute myocardial infarction, chronic obstructive pulmonary disease requiring steroids, or recent surgical or invasive cardiac procedures. Subjects were excluded if they had other cardiac causes for their symptoms such as severe valvular disease, amyloidosis, or hypertrophic cardiomyopathy. Blood was drawn from the nine subjects enrolled in the study (HFpEF hospitalized group). Five of these subjects (HFpEF outpatient group) returned ≥2 weeks after discharge for second blood draw. Subjects with an LVEF ≥50 % and without evidence of increased LV filling pressures served as the control group.
For further biomarker validation, an additional set of 25 HFpEF subjects and 18 age and co-morbidity matched control subjects were recruited from Northwestern University. All subjects gave written informed consent to participate in the study. The Institutional Review Board at the Medical College of Wisconsin and Northwestern University approved the respective study protocols, which conformed to the principles of the Declaration of Helsinki.
Reagents
Supplies and other reagents were purchased from Sigma-Aldrich (St. Louis, MO) unless specified. Recombinant S100A8 was purchased from Creative BioMart (Shirley, NY).
Platelet preparation
Blood was separated into serum and platelet fractions. Platelets were extensively washed in buffer (45 mM sodium citrate, 25 mM citric acid, 80 mM d-glucose). During all steps, care was taken to avoid activation of platelets. Flow cytometry with anti-CD41 (Life Technologies, Grand Island, NY) and anti-P-selectin (BioLegend, San Diego, CA) was performed to assess for platelet activation (Additional file: 1. Figure S1). Microscopy confirmation verified that the purified platelets had leukocyte and red blood cell contamination that was less than 0.02 and 1 %, respectively (Additional file: 2. Figure S2).
Global proteomic studies
Platelets from individual samples were resuspended in lysis buffer (125 mM Tris pH 6.8, 4 % SDS, 10 % glycerol, 5 % β-mercaptoethanol, Roche Complete Protease Inhibitor, Thermo HALT Phosphatase Inhibitor Cocktail). After determining protein concentration, the protein sample was separated by 1-dimensional SDS-PAGE gel (Bis-Tris 4–12 %) with internal DNA markers as described in our earlier publication [15]. The gel was stained with indoine blue and divided into three pieces. The proteins were reduced with 100 mM dithiotreitol (DTT) in 25 mM NH4HCO3 for 30 min at 56 °C and alkylated with 55 mM iodoacetamide (IAA) in 25 mM NH4HCO3 for 30 min at room temperature followed by trypsin digestion overnight. Peptides were extracted with 0.1 % trifluoroacetic acid (TFA) and 70 % acetonitrile/5 % TFA in water, respectively. Extracts were dried in a Speedvac and subsequently acidified to 0.1 % TFA. The samples were desalted using a ZipTip (C18).
For biomarker discovery, all samples were subject to tandem mass spectrometry. Three injection replicates of each fraction (three fractions per sample) were run on an LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific). For each injection replicate, 1.5 µl sample was separated via C18 column over the course of a 150 min gradient from buffer A (2 % acetonitrile, 98 % H2O, 0.1 % formic acid) to buffer B (98 % acetonitrile, 2 % H2O, 0.1 % formic acid). The gradient program began with 2 min at 98 % A, followed by a 3 min ramp to 95 % A, a 115 min ramp to 60 % A, a 15 min ramp to 2 % A, 3 min at 2 % A, 2 min ramp to 98 % A, then a 10 min equilibration in 98 % A. MS1 scans were detected in the FTMS section of the Orbitrap Velos in profile mode at a resolution of 30,000 (full width of peak at half-maximum at 400 m/z). The ten most abundant parent ions from each MS1 scan were selected for fragmentation via collision induced dissociation. Results of SEQUEST searches against UniProt human database (version April 2013) and all nine runs of each sample were combined using Visualize software. Visualize software was also used to generate comparison data [16]. The protein lists include proteins identified with at least five scans that were observed with peptide probability >0.85.
S100A8 expression
S100A8 levels were determined using a S100A8 enzyme-linked immunoassay kit from MBL International (Des Plaines, IL).
Induced pluripotent stem cell induced-cardiomyocyte differentiation
The induced pluripotent stem cell (iPSC) line used in this study was a generous gift from Dr. Stephan Duncan. This iPSC line was generated from human foreskin fibroblasts and previously characterized [17]. The iPSC line was maintained on Matrigel (BD Biosciences, San Jose, CA) in mTeSR-1 media (Stem Cell Technologies, BC, Canada) and differentiated into cardiomyocytes according to published protocols [18, 19]. Differentiated cells were maintained in cardiomyocyte maintenance media (RPMI/B27; Life Technologies, Grand Island, NY). For all experiments, 35 ± 5 day old contracting cardiomyocytes were used.
Electrophysiology
Action potentials were recorded from the human iPSC-derived cardiomyocytes using the current clamp configuration of the patch clamp technique, as previously described [20, 21]. Briefly, patch pipettes were pulled from borosilicate glass capillaries (King Precision Glass, Claremont, CA) with a micropipette puller (PC-10; Harishige, Tokyo, Japan) and heat polished using a microforge (MF-830; Narishige). The pipette resistances ranged from 3–5 MΩ when filled with the intracellular recording solution. This pipette solution contained 60 mM K-glutamate, 50 mM KCL, 10 mM HEPES, 1 mM MgCl2, 11 mM EGTA, 1 mM CaCl2, and 5 mM K2-ATP (pH adjusted to 7.4 with KOH). The extracellular bath solution contained 132 mM NaCl, 4.8 mM KCl, 1.2 mM MgCl2, 1.0 mM CaCl2, 5 mM dextrose, and 10 mM HEPES (pH adjusted 7.4 with NaOH). Action potentials were recorded using a Multiclamp 700B amplifier and Digidata 1440A interface (Molecular Devices, Sunnyvale, CA). pClamp 10 software (Molecular Devices) was used for data acquisition and analysis. Spontaneously beating nodal-, atrial-, and ventricular-like cells were characterized based on the maximum rate of depolarization (dV/dt), action potential duration (APD) at 50 and 90 % repolarization, and maximum diastolic potential. Recordings were conducted at physiological temperature (37 °C). The temperature of the recording chamber was controlled via a temperature control unit (TC 344B; Warner Instruments, Hamden, CT).
Ratiometric Ca2+ microfluorometry
Briefly, human iPSC-derived cardiomyocytes plated on coverslips were exposed to Fura-2-AM (5 µM) for 30 min at room temperature, washed three times with extracellular bath solution, and given 30 min for de-esterification. For Ca2+ microfluorometry, the fluorophore was excited alternately with 340 and 380 nm wavelength illumination and images were acquired at 510 nm through a 20× objective. Recordings from each cell were obtained at a rate of 3 Hz. After background subtraction, the fluorescence ratio R for individual cell was determined as the intensity of emission during 340 nm excitation (I340) divided by I380, on a pixel-by-pixel basis. Activation-induced transients were generated by depolarization produced by microperfusion application of 50 mM KCl [22].
Statistical analysis
Data is presented as either mean ± SD or as total percentage. Continuous variables were compared using the Student t test, assuming equal variance and dichotomous variables were compared using the Fisher exact test. Mass spectrometry measurements between groups were compared for either the presence (assigned a number value of 1) or absence (assigned a number of value of 0) of the protein identified in the sample using non-parametric Wilcoxon rank-sum tests without adjusting for multiple testing. Mass spectrometry data analysis was performed by the biostatical consulting service at the Medical College of Wisconsin.
Results
Clinical and echocardiographic characteristics of the discovery cohort
As described in Table 1 the median age of the HFpEF subjects is slightly greater than the control subjects (p = 0.04). The HFpEF group had a higher incidence of atrial fibrillation and cerebral vascular accident/transient ischemia in comparison to control subjects. Although not statistically significant, HFpEF subjects were more likely to have diabetes, coronary heart disease, hyperlipidemia and a distant smoking history. A significant number of HFpEF subjects were taking beta blockers compared to the control group. Echocardiogram studies confirmed the presence of diastolic dysfunction and increased LV pressure in the HFpEF group (Table 2). Left atrial volume indices were significantly elevated along with an increase in LV wall thickness in the HFpEF group compared to control.
Overall description of proteomic findings
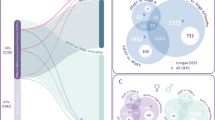
Global proteomic experiments were performed using 21 separate platelet preparations. Combining these experiments, a total of 6102 proteins were identified with at least five scans with a protein probability of >0.85. The HFpEF hospitalized group had a total of 5546 proteins, the HFpEF outpatient group had a total of 4854 proteins and the control group had a total of 5498 proteins identified. A total of 4172 proteins were found to be shared among all three groups. When comparing two groups, 321 proteins were identified as being shared amongst the outpatient and control group. A total of 361 proteins were found in both the hospitalized and outpatient groups and a total of 848 proteins were found in both the control and hospitalized groups. The number of unique proteins in each group consisted of 165 proteins in the HFpEF hospitalized group, 78 proteins in the HFpEF outpatient group, and 157 unique proteins in the control group (Fig. 1). To assess for possible contamination from other blood cells, the data set was scanned for the presence of CD45 and MHC II chains; proteins that are expressed in leukocytes. These proteins were not found in the data set; therefore, the contamination from leukocytes was likely to be minimal. However, complement C5 and β-2-glycoprotein were identified in the data sets denoting some serum contamination was present.

Global proteomic analysis of platelets identifies 6102 proteins. The Venn diagram displays he results of the analysis of platelet proteins from the individual subjects by in-depth LC–MS/MS. In total, 6102 proteins were identified with 4172 common among all data sets. There were 165, 78, and 157 proteins identified that were unique to the HFpEF Symptomatic, HFpEF outpatient and Control groups
Unique proteins in each study group
The platelet proteome from nine subjects were analyzed in the HFpEF hospitalized group, five subjects in the HFpEF outpatient and seven subjects in the control group. The unique proteins identified with a scan count of >9 are listed in Table 3. In addition after applying the non-parametric Wilcoxon rank-sum test, 37 proteins were found to be more prevalent amongst the combined HFpEF groups than with the control and 77 proteins were identified that were found to be more prevalent amongst the control with a p value <0.05. These proteins are listed Table 4.
Discovery and validation cohort ELISA confirmation
One particularly interesting finding was the identification of S100A8. The m/z ratio graph representing S100A8 is shown in Fig. 2. Even though the p value was 0.08, it was identified in six out of the nine HFpEF subjects. S100A8 has not been previously associated with HFpEF but has been linked to advanced heart failure [23]. Additionally, S100A8 has been found to correlate with traditional cardiovascular risk factors and the manifestation of cardiovascular disease [24, 25]. For these reasons, we decided to look more closely at S100A8 to verify its association with HFpEF. S100A8 is found in platelets [26, 27] and the plasma [25, 28]; because we used the platelet lysates for the mass spect analysis, we used the plasma samples for quantitative ELISA analysis. Figure 3 shows that plasma S100A8 levels are increased symptomatic HFpEF when compared to control (MCW cohort). We then validated these findings by studying a larger cohort of subjects recruited from the Northwestern University HFpEF Program. In this larger cohort, we saw a similar increase in plasma S100A8 levels in the HFpEF group (Fig. 3; Northwestern cohort).

Representative MS/MS scan for S100A8 peptide sequence ALNSIIDVYHK. Raw m/z spectral images with peak assignments and b and y ion lists along with a representation of peptide sequencing by tandem mass spectrometry

Plasma levels of S100A8 in control vs. HFpEF groups. a S100A8 is found in increased levels in the plasma of subjects with HFpEF vs. control subjects as detected by ELISA. The MCW columns include the control (n = 7) and HFpEF (n = 9) from the discovery cohort and the NWU colums include the control (n = 18) and HFpEF (n = 25) samples from the validation cohort. *p < 0.006 vs MCW Control. # p < 0. 002 vs NWU Control
Exogenously applied rS100A8 affects cardiomyocyte function in vitro
To ascertain whether S100A8 may play a causal role in the HFpEF disease process; we developed a bedside-to-bench translational system (Fig. 4) to screen for biological effects of identified proteins on cardiomyocyte function in vitro. We added recombinant S100A8 (800 ng/ml) to iPSC-derived cardiomyocytes in vitro and measured action potentials and intracellular Ca2+ concentrations separately. This specific concentration of rS100A8 was selected as it was the average plasma concentration observed in the HFpEF group (Fig. 3).

Overview of primary and secondary screening methods to identify potential mediators of HFpEF. a Platelet proteomes were subject to mass spectral analysis and novel proteins were identified. b Human cardiomyocytes derived from induced pluripotent stem cells were used to determine whether proteins that were identified in a had direct effects on cardiomyocytes function in vitro. Purified recombinant protein S100A8 was tested in this assay
Action potentials (APs) were recorded in the current clamp mode using the patch clamp technique. The recordings were acquired from spontaneously beating cells. External application of rS100A8 slowed the spontaneous pacing within 25 s which suggests the interaction with a membrane receptor. In the example shown in Fig. 5a, the spontaneous generation of APs with atrial-like properties was slowed in the presence of rS100A8. The peak-to-peak AP interval increased from 1.5 to 2.4 s. This effect was reversible upon washout of rS100A8 (results not shown). In a different beating cell cluster, the recorded atrial-like APs showed arrhythmogenic tendencies characterized by infrequent incidents of failed triggering of APs, as shown in Fig. 5b. The rS100A8 exacerbated this trend by increasing the frequency of these failed events. Thus, the electrophysiological profile of these iPSC-derived cardiomyocytes is profoundly impacted by rS100A8.

S100A8-mediated effects on human iPSC-derived cardiomyocytes. a Shows example action potentials recorded from rS100A8 treated iPSC derived human cardiomyocytes. The addition of rS100A8 to the buffer extended the period between action potentials. This period is phase 4; the diastolic membrane potential between action potentials. b rS100A8 exacerbates the arrhythmic tendencies of human cardiomyocytes. c Spontaneous Ca2+ transients recorded from human cardiomyocytes treated with rS100A8 as indicated by the blue line. rS100A8 significantly delayed the recovery of depolarization. Wash out of rS100A8 reversed these effects
Intracellular Ca2+ concentrations ([Ca2+]i) were measured using the ratiometric Ca2+ microfluorometry technique with Fura-2-AM fluorescent dye. The [Ca2+]i were monitored in spontaneously beating cells. The sample trace (Fig. 5c) shows a spontaneous Ca2+ transient recording that was interrupted by activity-induced depolarization (50 mM K+; duration of application as noted) at certain time points (indicated by the red arrows) using a microperfusion system. Of particular note is the recovery of the spontaneous Ca2+ transient following each depolarizing pulse. In the absence of rS100A8, the recovery was relatively fast. In contrast, the recovery was considerably slower in the presence of rS100A8. Following a third depolarizing pulse, recovery was not evident until the washout of rS100A8; this observation also suggests that rS100A8 effects are mediated through a membrane receptor. In summary, rS100A8 adversely affected the calcium handling of iPSC-derived cardiomyocytes.
Conclusions
The key finding of this study was that it was possible to derive platelet protein data sets specific for HFpEF patients. These proof-of-concept findings suggest that the platelet proteome might provide a useful tool for screening for HFpEF-associated biomarkers. Although several platelet proteins were identified in HFpEF subjects; their exact connection to HFpEF has yet to be determined. Though our data is limited by the small size, our discovery cohort has similar characteristics of larger HFpEF cohorts reported in the literature [29–31]. By combining proteomics with bioactivity assays, we have demonstrated that the platelet proteome is an untapped resource for determining disease mediators in HFpEF.
The platelet proteome in healthy individuals is remarkably stable with only minor differences in protein expression patterns [32]. Veitinger et al. suggests the difference in platelet proteins between individuals is a results of the uptake of plasma proteins by the platelet [33]. Inflammation is closely linked with HFpEF [34] and considering that platelets are involved in the inflammatory process, it is not surprising that our proteomics screen led to the identification of several proteins also involved in inflammation. These include serum amyloid A (SAA), Lipopolysaccharide binding protein, apolipoprotein A1 and S100A8. Two proteins, serum amyloid-A (SAA) protein 1 and apolipoprotein A1 were increased in the sera of non-human primates after drug-induced cardiac injury [35]. In addition, increased levels of SAA in serum have been associated with coronary heart disease [36], as well as systolic heart failure [37] and has been shown to be a predictor of cardiovascular outcomes in women [38].
S100A8 is a member of the S100 calcium-binding family of proteins, which exhibit increased levels in a number of inflammatory states. S100A8 is commonly mentioned with its binding partner, S100A9. Even though S100A8 is found in the plasma [23], it is known that platelets and megakaryocytes might serve as an additional source of S100A8 and might contribute to the plasma pool of S100A8/A9 in inflammatory diseases and cardiovascular events [26, 27, 39].
S100A8 and S100A9 are not normally expressed in cardiomyocytes [40] although its cardiac expression can be induced by endotoxins or angiotensin II [40, 41]. Release of S100A8/A9 from cells allows it to act in a paracrine or autocrine fashion. These extracellular functions are mediated by the toll-like receptor 4 (TLR4) [42, 43] or the receptor for advanced glycation end products (RAGE) [40, 44, 45]. More recently, CD36 has been identified as a receptor [26]. In the mouse, S100A8/A9 signals through RAGE to promote inflammation and fibrosis after angiotensin II or hypoxic-induced cardiac injury [41, 45].
Increased platelet S100A8 mRNA and plasma protein levels were present in patients with acute myocardial infarction [39]. Plasma levels of S100A8/A9 predicted risk of future myocardial infarction, stroke or death in post-menopausal healthy women [25]. Elevated S100A8 levels have also been found in other inflammatory disorders which are associated with abnormalities of vascular and cardiac function, particularly diastolic dysfunction, such as diabetes [46–48], end-stage renal disease [49, 50], and inflammatory bowel disease [51, 52]. This is the first association of S100A8 with HFpEF, yet its role in the disease process still needs be elucidated. S100A8 has immediate effects on the electrophysiological and Ca2+ handling profiles of human induced cardiomyocytes suggesting that S100A8 is acting through a membrane receptor. S100A8 interaction with RAGE affects calcium flux in neonatal rat ventricular cardiomyocytes and HL-1 cardiomyocytes [40, 53]. The adverse effects on the electrophysiological and Ca2+ handling profiles resulting from S100A8 treatment of human induced cardiomyocytes; validates our bedside-to-bench translational screen as an approach to identify bioactive proteins that may contribute to the disease mechanisms in HFpEF.
We also considered the possibility that subjects progress to HFpEF through loss of cardioprotective proteins. Therefore, we searched amongst our control group and were able to identify four proteins that could potentially have protective qualities against the development of heart failure. Cyclic nucleotide phosphodiesterase 3A1 (PDE3A) regulates β-adrenergic signaling to effect physiological cardiac performance. Furthermore, PDE3A protects the heart against angiotensin II-induced cardiac remodeling in mice [54]. Copper Chaperone for Superoxide Dismutase (CCS) plays a role in copper delivery to tissues; disturbances in copper homeostasis mediates cardiomyopathy [55]. Zinc finger protein 451 a negative regulator of TGF-beta signaling [56]. The transient receptor potential cation channel subfamily M member 2 (TRPM2) protein limits oxidative stress injury and dampens the inflammatory response [57].
The present study must be interpreted within the context of its limitations. First of all, this was a discovery effort and not designed as a quantitative proteomic analysis. Therefore, we cannot determine if specific proteins are up- or down-regulated. In addition, it is unlikely that one protein is responsible for a complex disease as HFpEF, but our findings offer new perspectives regarding HFpEF and further confirmation of the platelet proteins identified in this study will need to be validated in a larger cohort. In addition, combining proteomics with functional bioactivity assessments may be a strategy to complement and strengthen the search for biomarkers by combining protein identified with biological activity in a relevant in vitro model system.
In conclusion, from the discovery set in HFpEF patients, we derived a panel of platelet proteins that may be specific for HFpEF. Furthermore, this set distinguished a set of platelet proteins which are consistent in HFpEF subjects whether they are decompensated and hospitalized or compensated after discharge. We further established a bedside-to-bench translational system that can be utilized as a secondary screen to ascertain whether the biomarkers may be an associated finding or causal to the disease process.
References
Banfi C, Brioschi M, Marenzi G, De Metrio M, Camera M, Mussoni L, et al. Proteome of platelets in patients with coronary artery disease. Exp Hematol. 2010;38(5):341–50.
Parguiña AF, Grigorian-Shamajian L, Agra RM, Teijeira-Fernández E, Rosa I, Alonso J, et al. Proteins involved in platelet signaling are differentially regulated in acute coronary syndrome: a proteomic study. PLoS One. 2010;5(10):e13404.
Springer DL, Miller JH, Spinelli SL, Pasa-Tolic L, Purvine SO, Daly DS, et al. Platelet proteome changes associated with diabetes and during platelet storage for transfusion. J Proteome Res. 2009;8(5):2261–72.
Veitinger M, Oehler R, Umlauf E, Baumgartner R, Schmidt G, Gerner C, et al. A platelet protein biochip rapidly detects an Alzheimer’s disease-specific phenotype. Acta Neuropathol. 2014;128(5):665–77.
Chung I, Choudhury A, Lip GY. Platelet activation in acute, decompensated congestive heart failure. Thromb Res. 2007;120(5):709–13.
Schafer A, Eigenthaler M, Bauersachs J. Platelet activation in heart failure. Clin Lab. 2004;50(10):559–66.
Andreassi MG, Del Ry S, Palmieri C, Clerico A, Biagini A, Giannessi D. Up-regulation of ‘clearance’ receptors in patients with chronic heart failure: a possible explanation for the resistance to biological effects of cardiac natriuretic hormones. Eur J Heart Fail. 2001;3(4):407–14.
Chung I, Choudhury A, Patel J, Lip GY. Soluble, platelet-bound, and total P-selectin as indices of platelet activation in congestive heart failure. Ann Med. 2009;41(1):45–51.
Stumpf C, Lehner C, Eskafi S, Raaz D, Yilmaz A, Ropers S, et al. Enhanced levels of CD154 (CD40 ligand) on platelets in patients with chronic heart failure. Eur J Heart Fail. 2003;5(5):629–37.
Gurbel PA, Gattis WA, Fuzaylov SF, Gaulden L, Hasselblad V, Serebruany VL, et al. Evaluation of platelets in heart failure: is platelet activity related to etiology, functional class, or clinical outcomes? Am Heart J. 2002;143(6):1068–75.
UlHaq MA, Wong C, Hare DL. Heart failure with preserved ejection fraction: an insight into its prevalence, predictors, and implications of early detection. Rev Cardiovasc Med. 2015;16(1):20–7.
Putko BN, Wang Z, Lo J, Anderson T, Becher H, Dyck JR, et al. Circulating levels of tumor necrosis factor-alpha receptor 2 are increased in heart failure with preserved ejection fraction relative to heart failure with reduced ejection fraction: evidence for a divergence in pathophysiology. PLoS One. 2014;9(6):e99495.
Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, et al. Inflammatory markers and incident heart failure risk in older adults: the health ABC (health, aging, and body composition) study. J Am Coll Cardiol. 2010;55(19):2129–37.
Smyth SS, McEver RP, Weyrich AS, Morrell CN, Hoffman MR, Arepally GM, et al. Platelet functions beyond hemostasis. J Thromb Haemost. 2009;7(11):1759–66.
Heroux MS, Chesnik MA, Halligan BD, Al-Gizawiy M, Connelly JM, Mueller WM, et al. Comprehensive characterization of glioblastoma tumor tissues for biomarker identification using mass spectrometry-based label-free quantitative proteomics. Physiol Genomics. 2014;46(13):467–81.
Halligan BD, Greene AS. Visualize: a free and open source multifunction tool for proteomics data analysis. Proteomics. 2011;11(6):1058–63.
Si-Tayeb K, Noto FK, Sepac A, Sedlic F, Bosnjak ZJ, Lough JW, et al. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev Biol. 2010;10:81.
Lian X, Zhang J, Azarin SM, Zhu K, Hazeltine LB, Bao X, et al. Directed cardiomyocyte differentiation from human pluripotent stem cells by modulating Wnt/beta-catenin signaling under fully defined conditions. Nat Protoc. 2013;8(1):162–75.
Lian X, Hsiao C, Wilson G, Zhu K, Hazeltine LB, Azarin SM, et al. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc Natl Acad Sci. 2012;109(27):E1848–57.
Bhattacharya S, Burridge PW, Kropp EM, Chuppa SL, Kwok WM, Wu JC, et al. High efficiency differentiation of human pluripotent stem cells to cardiomyocytes and characterization by flow cytometry. J Vis Exp. 2014;91:52010.
Suzuki A, Aizawa K, Gassmayr S, Bosnjak ZJ, Kwok WM. Biphasic effects of isoflurane on the cardiac action potential: an ionic basis for anesthetic-induced changes in cardiac electrophysiology. Anesthesiology. 2002;97(5):1209–17.
Stoehr A, Neuber C, Baldauf C, Vollert I, Friedrich FW, Flenner F, et al. Automated analysis of contractile force and Ca2 + transients in engineered heart tissue. Am J Physiol Heart Circ Physiol. 2014;306(9):H1353–63.
Ma LP, Haugen E, Ikemoto M, Fujita M, Terasaki F, Fu M. S100A8/A9 complex as a new biomarker in prediction of mortality in elderly patients with severe heart failure. Int J Cardiol. 2012;155(1):26–32.
Schiopu A, Cotoi OS. S100A8 and S100A9: DAMPs at the crossroads between innate immunity, traditional risk factors, and cardiovascular disease. Mediators Inflamm. 2013;2013:828354.
Cotoi OS, Duner P, Ko N, Hedblad B, Nilsson J, Bjorkbacka H, et al. Plasma S100A8/A9 correlates with blood neutrophil counts, traditional risk factors, and cardiovascular disease in middle-aged healthy individuals. Arterioscler Thromb Vasc Biol. 2014;34(1):202–10.
Wang Y, Fang C, Gao H, Bilodeau ML, Zhang Z, Croce K, et al. Platelet-derived S100 family member myeloid-related protein-14 regulates thrombosis. J Clin Invest. 2014;124(5):2160–71.
Morrow DA, Wang Y, Croce K, Sakuma M, Sabatine MS, Gao H, et al. Myeloid-related protein 8/14 and the risk of cardiovascular death or myocardial infarction after an acute coronary syndrome in the pravastatin or atorvastatin evaluation and infection therapy: thrombolysis in myocardial infarction (PROVE IT-TIMI 22) trial. Am Heart J. 2008;155(1):49–55.
Hirata A, Kishida K, Nakatsuji H, Hiuge-Shimizu A, Funahashi T, Shimomura I. High serum S100A8/A9 levels and high cardiovascular complication rate in type 2 diabetics with ultra sonographic low carotid plaque density. Diabetes Res Clin Pract. 2012;97(1):82–90.
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355(3):251–9.
Bhatia RS, Tu JV, Lee DS, Austin PC, Fang J, Haouzi A, et al. Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl JMed. 2006;355(3):260–9.
Ho JE, Lyass A, Lee DS, Vasan RS, Kannel WB, Larson MG, et al. Predictors of new-onset heart failure: differences in preserved versus reduced ejection fraction. Circ Heart Fail. 2013;6(2):279–86.
Burkhart JM, Gambaryan S, Watson SP, Jurk K, Walter U, Sickmann A, et al. What can proteomics tell us about platelets? Circ Res. 2014;114(7):1204–19.
Veitinger M, Umlauf E, Baumgartner R, Badrnya S, Porter J, Lamont J, et al. A combined proteomic and genetic analysis of the highly variable platelet proteome: from plasmatic proteins and SNPs. J Proteomics. 2012;75(18):5848–60.
Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. 2013;62(4):263–71.
Song B, Liu Y, Parman T, Liu S, Miller JK, Liu X, et al. Quantitative proteomics for cardiac biomarker discovery using isoproterenol-treated nonhuman primates. J Proteome Res. 2014;13(12):5909–17.
Wang DX, Liu H, Yan LR, Zhang YP, Guan XY, Xu ZM, et al. The relationship between serum amyloid A and apolipoprotein A-I in high-density lipoprotein isolated from patients with coronary heart disease. Chin Med J (Engl). 2013;126(19):3656–61.
Kim JB, Hama S, Hough G, Navab M, Fogelman AM, Maclellan WR, et al. Heart failure is associated with impaired anti-inflammatory and antioxidant properties of high-density lipoproteins. Am J Cardiol. 2013;112(11):1770–7.
Johnson BD, Kip KE, Marroquin OC, Ridker PM, Kelsey SF, Shaw LJ, et al. Serum amyloid A as a predictor of coronary artery disease and cardiovascular outcome in women: the National Heart, Lung, and Blood Institute-Sponsored Women’s Ischemia Syndrome Evaluation (WISE). Circulation. 2004;109(6):726–32.
Healy AM, Pickard MD, Pradhan AD, Wang Y, Chen Z, Croce K, et al. Platelet expression profiling and clinical validation of myeloid-related protein-14 as a novel determinant of cardiovascular events. Circulation. 2006;113(19):2278–84.
Boyd JH, Kan B, Roberts H, Wang Y, Walley KR. S100A8 and S100A9 mediate endotoxin-induced cardiomyocyte dysfunction via the receptor for advanced glycation end products. Circ Res. 2008;102(10):1239–46.
Wu Y, Li Y, Zhang C, A X, Wang Y, Cui W, et al. S100a8/a9 released by CD11b + Gr1 + neutrophils activates cardiac fibroblasts to initiate angiotensin II-Induced cardiac inflammation and injury. Hypertension. 2014;63(6):1241–50.
Kuipers MT, Vogl T, Aslami H, Jongsma G, van den Berg E, Vlaar AP, et al. High levels of S100A8/A9 proteins aggravate ventilator-induced lung injury via TLR4 signaling. PLoS ONE. 2013;8(7):e68694.
Schelbergen RF, Blom AB, van den Bosch MH, Sloetjes A, Abdollahi-Roodsaz S, Schreurs BW, et al. Alarmins S100A8 and S100A9 elicit a catabolic effect in human osteoarthritic chondrocytes that is dependent on toll-like receptor 4. Arthritis Rheum. 2012;64(5):1477–87.
Ghavami S, Rashedi I, Dattilo BM, Eshraghi M, Chazin WJ, Hashemi M, et al. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J Leukoc Biol. 2008;83(6):1484–92.
Volz HC, Laohachewin D, Seidel C, Lasitschka F, Keilbach K, Wienbrandt AR, et al. S100A8/A9 aggravates post-ischemic heart failure through activation of RAGE-dependent NF-kappaB signaling. Basic Res Cardiol. 2012;107(2):250.
Ortega FJ, Sabater M, Moreno-Navarrete JM, Pueyo N, Botas P, Delgado E, et al. Serum and urinary concentrations of calprotectin as markers of insulin resistance and type 2 diabetes. Eur J Endocrinol. 2012;167(4):569–78.
Tribouilloy C, Rusinaru D, Mahjoub H, Souliere V, Levy F, Peltier M, et al. Prognosis of heart failure with preserved ejection fraction: a 5 year prospective population-based study. Eur Heart J. 2008;29(3):339–47.
Lindman BR, Davila-Roman VG, Mann DL, McNulty S, Semigran MJ, Lewis GD, et al. Cardiovascular phenotype in HFpEF patients with or without diabetes: a RELAX trial ancillary study. J Am Coll Cardiol. 2014;64(6):541–9.
Malíčková K, Brodská H, Lachmanová J, Dusilová Sulková S, Janatková I, Marečková H, et al. Plasma calprotectin in chronically dialyzed end-stage renal disease patients. Inflamm Res. 2010;59(4):299–305.
Han JH, Han JS, Kim EJ, Doh FM, Koo HM, Kim CH, et al. Diastolic dysfunction is an independent predictor of cardiovascular events in incident dialysis patients with preserved systolic function. PLoS ONE. 2015;10(3):e0118694.
Cayatte C, Joyce-Shaikh B, Vega F, Boniface K, Grein J, Murphy E, et al. Biomarkers of therapeutic response in the IL-23 pathway in inflammatory bowel disease. Clin Trans Gastroenterol. 2012;3:e10.
Caliskan Z, Gokturk HS, Caliskan M, Gullu H, Ciftci O, Ozgur GT, et al. Impaired coronary microvascular and left ventricular diastolic function in patients with inflammatory bowel disease. Microvasc Res. 2015;97:25–30.
Yan D, Luo X, Li Y, Liu W, Deng J, Zheng N, et al. Effects of advanced glycation end products on calcium handling in cardiomyocytes. Cardiology. 2014;129(2):75–83.
Iwaya S, Oikawa M, Chen Y, Takeishi Y. Phosphodiesterase 3A1 protects the heart against angiotensin II–induced cardiac remodeling through regulation of transforming growth factor-β expression. Int Heart J. 2014;55(2):165–8.
Hordyjewska A, Popiolek L, Kocot J. The many “faces” of copper in medicine and treatment. Biometals. 2014;27(4):611–21.
Feng Y, Wu H, Xu Y, Zhang Z, Liu T, Lin X, et al. Zinc finger protein 451 is a novel Smad corepressor in transforming growth factor-β signaling. J Biol Chem. 2014;289(4):2072–83.
Knowles H, Li Y, Perraud A-L. The TRPM2 ion channel, an oxidative stress and metabolic sensor regulating innate immunity and inflammation. Immunol Res. 2013;55(1–3):241–8.
Authors’ contributions
JLS conceived and designed the research. RR, DP and SJS contributed clinical samples. SPM designed the proteomics experiments and MAC assisted in performing mass spectral analysis. RR, DP, JLS, WMK and HEW performed research and analyzed the data. RR DP and JLS drafted the paper and all coauthors edited the paper. All authors read and approved final manuscript.
Acknowledgements
This work was supported by funds awarded to J.L.S. from the National Institutions of Health K08 Grant Number HL111148, Steve Cullen Healthy Heart Walk/Run Event and also by grant 1UL1RR031973 from the Clinical and Translational Science Award (CTSI) program of the National Center for Research Resources, National Institutes of Health. We appreciate study subject referrals from Dr. Joshua Meskin and the biostatistics consulting services provided by Drs. Tao Wang and Shi Zhao from the Division of Biostatistics at the Medical College of Wisconsin.
Competing interests
The authors declare that they have no competing interests.
Author information
Authors and Affiliations
Corresponding author
Additional information
Roseanne Raphael and Diana Purushotham contributed equally to this work
Additional files
12967_2016_774_MOESM1_ESM.tif
Additional file 1: Figure S1. Flow cytometry to assess platelet activation. Purified platelet samples were incubated with fluorescently labeled antibodies against CD62P and CD41 and subject to flow cytometry. Platelets positive for CD41 + but negative for CD62P are non-activated. CD62P positive platelets are activated.
12967_2016_774_MOESM2_ESM.tif
Additional file 2: Figure S2. Microscopy for Platelet Purity. Isolated platelets were observed under the microscope. Visible red blood cells and leukocytes were counted and calculated as a percentage of platelets in each field. Microscopy confirmation verified that the purified platelet had a leukocyte contamination < 0.02 % and a red blood cell contamination of < 1 %.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Raphael, R., Purushotham, D., Gastonguay, C. et al. Combining patient proteomics and in vitro cardiomyocyte phenotype testing to identify potential mediators of heart failure with preserved ejection fraction. J Transl Med 14, 18 (2016). https://doi.org/10.1186/s12967-016-0774-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12967-016-0774-3