
Abstract
G protein-coupled receptors (GPCRs) are the largest family of membrane proteins in the human body and are responsible for accurately transmitting extracellular information to cells. Arrestin is an important member of the GPCR signaling pathway. The main function of arrestin is to assist receptor desensitization, endocytosis and signal transduction. In these processes, the recognition and binding of arrestin to phosphorylated GPCRs is fundamental. However, the mechanism by which arrestin recognizes phosphorylated GPCRs is not fully understood. The GPCR phosphorylation recognition “bar code model” and “flute” model describe the basic process of receptor phosphorylation recognition in terms of receptor phosphorylation sites, arrestin structural changes and downstream signaling. These two models suggest that GPCR phosphorylation recognition is a process involving multiple factors. This process can be described by a “QR code” model in which ligands, GPCRs, G protein-coupled receptor kinase, arrestin, and phosphorylation sites work together to determine the biological functions of phosphorylated receptors.
Graphical Abstract

Video Abstract
Similar content being viewed by others

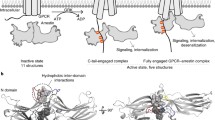
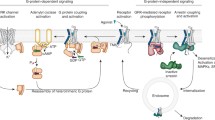
Background
G protein-coupled receptors (GPCRs) are the largest class of 7-transmembrane receptors on human cell membranes and can accurately transmit extracellular information, such as from hormones, neurotransmitters and odors, into the cell. GPCRs are an important information transmission hub [1,2,3]. There are two main mechanisms that mediate GPCR signaling: the G protein-dependent pathway and the G protein independent pathway [2, 4,5,6,7,8,9,10]. The G protein-dependent pathway is mainly regulated by guanine nucleotide-binding proteins (G proteins). G proteins are a class of highly conserved proteins, including Gs, Gi, Gq, and G12, whose main function is to transmit GPCR signals [11,12,13,14]. For example, in cardiomyocytes, catecholamine activates β1-adrenoceptor (β1-AR) and activates the Gs-adenylate cyclase (AC)-cyclic adenosine monophosphate (cAMP)-protein kinase A (PKA) pathway to exert positive chronotropic and inotropic effects. In heart failure, the release of a large amount of catecholamine and activation of the β2-adrenoceptor (β2-AR)-Gi pathway can inhibit the activity of AC and weaken β1-AR-Gs activation [15,16,17,18]. The G protein-independent pathway is mainly regulated by arrestin that recognizes and binds phosphorylated GPCRs catalyzed by G protein-coupled receptor kinase (GRK). Arrestin binding to a phosphorylated GPCR prevents G proteins from binding the GPCR and promotes receptor internalization [11, 19,20,21,22]. Arrestin binding to GPCRs can also transmit cellular signals. The ability of arrestin to recognize and bind phosphorylated GPCRs is the basis for achieving GPCR signaling. However, the specific mechanism by which this occurs is not completely clear.
Previous studies have found that rhodopsin phosphorylation can enhance arrestin binding [23]. It was further found that arrestin mainly recognizes the phosphorylated C-terminus of GPCRs [24]. The GPCR C-terminus phosphorylated by different protein kinases can transmit different downstream signals. To describe this, a “bar code” model [25, 26] has been proposed. The bar code model has given new insights into GPCR phosphorylation recognition, and the theory of the model has been continuously improved in several follow-up studies. Based on the bar code model, the “flute” model integrates structural changes of β-arrestin with GPCR phosphorylation recognition patterns. Different GPCR phosphorylation sites match different arrestin structures, and this matching determines the resulting signaling [27, 28]. Recently, molecular dynamics simulations and amino acid site mutations were used to verify the function of the vasopressin type 2 receptor (V2R) phosphorylation site in binding and activating β-arrestin. The different combinations of GPCR phosphorylation sites were found to directly affect the structural changes of β-arrestin and its binding to the receptor. This finding also indicated the diversity of the structural changes of β-arrestin [29, 30]. In a study of V2R, Qing-Tao He et al. analyzed the structure of the complex comprising the V2R C-terminus and β-arrestin1. The authors found that phosphorylation at different sites of V2R causes different structural changes in arrestin, which alters β-arrestin1 function [31]. A series of recent studies on bar code models and flute models have analyzed the phosphorylation recognition patterns of GPCRs and arrestin from the perspective of protein structure, bringing us closer to a comprehensive understanding of phosphorylation recognition patterns.
The purpose of this review was to discuss the mechanism by which arrestin recognizes GPCR phosphorylation, analyze the effects of the factors involved in this process (ligands, GPCR type, GRK, arrestin and GPCR phosphorylation sites), and briefly describe the historical development of phosphorylation recognition research.
Factors in GPCR phosphorylation recognition
The process of GPCR phosphorylation recognition is extremely complex and delicate. Ligands, GPCR types, GRK, arrestin, GPCR phosphorylation sites can all influence or even determine which function the receptor performs. Therefore, we first illustrate the effects of different factors on GPCR phosphorylation recognition.
Ligands
Ligands are the drivers of GPCR signaling. Light, odors, peptides, ions, hormones and antibodies are all become GPCR ligands [32, 33]. For example, light can activate rhodopsin in rod cells [34]. Angiotensin II type I receptor (AT1R) autoantibodies (AT1-AA) can continuously activate AT1R in vascular smooth muscle cells [35], and catecholamines can activate adrenaline receptors in cardiomyocytes [36]. GPCRs are also the target of many drugs. Some drugs, such as atropine, scopolamine (acetylcholine muscarinic receptor blocker), propranolol (β-adrenoceptor blocker), losartan (AT1R blocker) and phentolamine (alpha-adrenoceptor blocker), act by binding to GPCRs [37]. These are examples of drugs that are commonly used in the clinic [38,39,40,41].
Ligand classification is a complex matter. Based on the effect of GPCRs active state, ligands can be divided into agonists and antagonists (Fig. 1) [37]. According to their degree of GPCR activation, agonists can be divided into complete agonists and partial agonists. Complete agonists maximize the activation of GPCRs and result in the strongest signal transduction. PNU282987, a complete agonist of α7 nicotinic receptor (α7 nAChR), reverses depressive symptoms in Sprague Dawley (SD) rats induced by chronic mild stress [42]. Formoteru, a complete agonist of β2-AR, is used to treat asthma because of its bronchodilation effect [43]. Partial agonists partly activated a certain pathway when compared with the efficacy of a complete agonist. Δ9-Tetrahydrocannabinol (Δ9-THC) is a partial agonist of cannabinoid receptor 1 (CB1). It plays a role in the treatment of mental illnesses by activating CB1 [44, 45]. Antagonists are divided into inverse agonists and neutral antagonists. Inverse agonists inhibit basal activation of the receptor, reducing activation to lower than the basal level. Basal activation is inherent in some GPCRs and does not require ligand involvement. Neutral antagonists inhibit the effect of agonists but do not affect basal activation [37, 46].

Classification of GPCR ligands based on the effect of GPCRs active state. Complete agonists can maximize GPCR activation and signal transmission. Partial agonists can activate receptors but cannot maximize receptor activation. Inverse agonists inhibit the basal activation of the receptor. Neutral antagonists inhibit receptor activation but do not affect basal activation
However, the same ligand that plays a role of agonist in a response may become antagonist or inverse agonist in another response. This complicacy is the pluridimensional efficacy of a ligand [47]. For inverse agonist, the efficacy defined the ability to change receptor behavior become vectorial [48]. For example, carvedilol, as inverse agonist for β-adrenergic receptor, inhibited the production of cAMP medicated by Gs in congestive heart failure. However, carvedilol played the role of partial agonist for the activation of extracellular regulated protein kinases (ERK)1/2 mediated by β-arrestin [49, 50]. Similarly, [D-Trp12, Tyr34]-bPTH, the ligand of parathyroid hormone (PTH) receptor, was classified as antagonist of calcium signaling and an inverse agonist for cAMP production. However, it was also classified as partial agonist for ERK1/2 signaling [51, 52].
Topographically, according the variant of binding sites, the GPCR ligands are divided into orthosteric ligands and allosteric ligands (also named allosteric modulators) [53]. The ligands that bind to the endogenous ligands (hormone or neurotransmitter) binding sites (orthosteric sites) are orthosteric ligands [54]. Allosteric ligands bind to recognition sites that are distinct from the orthosteric sites [55]. The allosteric ligands are divided into four categories, including negative allosteric modulators (NAMs), positive allosteric modulators (PAMs), allosteric agonists and silent allosteric modulators (SAMs). Ligands that bind to an allosteric site of the receptor resulting in inhibition of receptor function are considered NAMs. Because NAMs decrease the affinity of orthosteric agonist ligands by changing receptor conformation. PAMs have the opposite effect to NAMs. In the absence of orthosteric agonist, allosteric agonist can active GPCR by itself. SAMs bind to allosteric sites on the receptor but do no affect receptor function [56]. In the development of GPCR drugs, allosteric ligands have drawn more and more attention, especially for small molecules [57, 58]. For example, maraviroc was the allosteric modulator for chemokine receptor CCR5. The function of maraviroc was to treat Acquired Immune Deficiency Syndrome (AIDS) by allosteric inhibition of CCR5 chemokine signaling and the prevention of human immunodeficiency virus type 1 (HIV-1) entry [59].
The ligand is the initiating factor in GPCR signaling. The type and characteristics of a ligand will specifically change the structure of a GPCR, impacting the final biological results [60]. The complex and diverse array of ligands are the initial factors affecting the process of receptor phosphorylation.
GPCR type
Based on the homology of the GPCR sequence, receptors can be divided into class A (rhodopsin family), B (secretin family), C (glutamate family), the adhesin family, and class F (frizzled family) [61]. This large number of GPCRs constitutes a broad signal regulation network throughout the body that transforms external information into biological information and transmits it through signaling pathways [62].
GPCRs are widely distributed throughout the body, and their signal regulation, including phosphorylation, is extremely complex. The phosphorylation of GPCRs is the key to ensuring accurate receptor function. In the heart, catecholamines activate β1-AR to exert positive chronotropic and positive inotropic effects through the cAMP/PKA pathway. To avoid the overactivation of β1-AR, β1-AR is phosphorylated by GRK in a timely manner. β-Arrestin binds to phosphorylated β1-AR to assist receptor desensitization and endocytosis [16]. If GPCRs are not phosphorylated or if they are hyperphosphorylated, their signal can be disturbed, leading to the occurrence and development of disease. For example, GRK2 was a phosphatase in M2 macrophages. The downregulation of GRK2 expression overactivated the β2-AR/cAMP/PKA pathway in hepatocellular carcinoma, thereby promoting the growth of cancer cells [63]. GRK4-induced hyperphosphorylation of adiponectin receptors can lead to the development of hypertension [64]. These studies suggest that timely and appropriate phosphorylation ensures the accurate transmission of extracellular information by GPCRs and maintains homeostasis.
GRK
As an integral part of GPCR phosphorylation, the function of GRK is to phosphorylate the receptor. GRK catalyzes receptor phosphorylation, which enables the receptor to bind to arrestin and carry out its biological function. GRK is serine/threonine protease [65]. GRK was initially elucidated as an enzyme that promotes rhodopsin phosphorylation in a light-dependent manner. This enzyme was termed rhodopsin kinase (GRK1) [66]. Subsequently, β2-adrenoceptor kinase (GRK2) was found in a study of β2-AR [67]. To date, researchers have discovered seven GRKs (GRK1-GRK7) [66,67,68,69,70,71,72,73]. Among them, GRK2, 3, 5, and 6 are widely distributed throughout the body and regulate the phosphorylation process of most GPCRs [65, 74]. GRK1 and 7 are mainly distributed in the visual system and function in visual regulation. GRK1 is distributed in cones and rods, while GRK7 is mainly distributed in cones [75]. GRK4 is found in the testis [74].
The differences in GRK distribution means that different GRKs can only interact with tissue distribution-specific GPCRs. GRK1 mainly phosphorylates rhodopsin in rod cells [76]. GRK7 mainly compensates for the deficiency of GRK1 function in Oguchi patients [75]. Although GRK2, 3, 5, and 6 can interact with the vast majority of GPCRs, the effect of GPCR phosphorylation is not consistent. For example, GRK2 and GRK3 promote the desensitization of phosphorylated V2R, while GRK5 and GRK6 catalyze V2R phosphorylation and continue to transmit information to the ERK signaling [77]. Similarly, GRK2 and GRK3 catalyze AT1R phosphorylation to promote receptor desensitization and endocytosis, while GRK5 phosphorylates AT1R, leading to β-arrestin-dependent ERK activation [78]. The reason for this phenomenon may be related to the phosphorylation of different sites in GPCRs being catalyzed by different GRKs [27]. Thirteen serine (Ser)/threonine (Thr) residues (Ser246, Ser261, Ser262, Ser345, Ser346, Ser355, Ser356, Thr360, Ser364, Ser396, Ser401, Ser407, and Ser411) of β2-AR were identified by small interfering RNA (siRNA) and quantitative mass spectrometry. GRK2 mainly phosphorylated Thr360, Ser364, Ser396, Ser401, Ser407, and Ser411, while GRK6 phosphorylated only Ser355 and Ser356. This difference in phosphorylation sites eventually leads to the β2-AR-β-arrestin-ERK1/2 pathway being catalyzed by GRK6, while the phosphorylation of β2-AR catalyzed by GRK2 inhibits this signaling pathway [79]. These results suggest that different GRKs have different preferred phosphorylation sites and relatively fixed functions. GRK2-catalyzed receptor phosphorylation tends to promote endocytosis, while GRK6-catalyzed receptor phosphorylation tends to induce β-arrestin-dependent signal transduction.
The rich combination of seven GRKs, different tissue distributions and different preferences for phosphorylation sites allows a complex array of signaling that is regulated by GPCR phosphorylation. This complexity can accurately meet the signaling requirements of the body (Fig. 2). The diversity of phosphorylation site combinations also leads to different binding patterns of arrestin.

Ligand-GPCR-GRK-arrestin information transfer process. After the ligand activates the GPCR, GRK phosphorylates the activated receptor. Arrestin recognizes the phosphorylated receptor and binds to it, thereby causing receptor desensitization, endocytosis or the continuation of signal transmission. In this process, different GRKs phosphorylate different sites on the receptor. Arrestin recognizes these differences in phosphorylation sites and binds to the receptor in different structural conformations to perform different functions. The orange shapes represent different ligands, the gray dots represent sites that are not phosphorylated, and the red dots represent sites that are phosphorylated.
Arrestin
There are four types of arrestin: arrestin1, arrestin2, arrestin3, and arrestin4. Similar to GRK, the distribution of arrestin is tissue-specific. Arrestin1 and 4, also known as visual arrestins, are mainly distributed in the visual system; arrestin2 and 3 (β-arrestin1 and 2) are also widely distributed in the visual system [80]. Four types of arrestin have similar structure. Briefly, two crescent-shaped beta-sandwiches (N-and C-terminal domains, respectively) made up the structure of arrestin. The central crest, including finger loop, middle loop and C-loop, formed between the above domains. Figure loop was a key receptor-binding element. In the inactive state, the structure of finger loop was irregular. However, by interacting with the GPCR, α helix was formed in finger loop to assist arrestin to interact with phosphorylated GPCR easily [81]. The function of middle loop and C-loop was to stabilize the inactive basal-state arrestin. Besides, the basal structure of arrestin was also stabilized by the gate loop which consisted of C-terminal and polar core [81, 82].
Arrestins are multifunctional regulators of GPCR. When arrestin was discovered, its only function was identified as assisting in the desensitization of activated GPCRs [83, 84]. Further research has gradually discovered additional functions of arrestin. At present, it is believed that arrestin has three main functions: (1) assisting in the desensitization of activated receptors; (2) mediating receptor endocytosis; (3) signaling [85]. The basis for the above regulatory functions of arrestin lies in the changes in the structure of arrestin and the conformation with which it binds to the GPCR [86]. In the process of receptor desensitization, arrestin binds to GPCR transmembrane core, putting itself in a conformation named “core” conformation. G protein is excluded from the receptor by arrestin, thereby terminating the signal transduction of the G protein pathway [20]. In the process of receptor endocytosis, arrestin binds to phosphorylated GPCR C-terminal with “tail” conformation [21]. Arrestin recruits intracellular clathrin and β2-adaptin (AP2) in the form of cytoskeletal proteins to form endosomes and promote receptor internalization. Endocytosis receptors enter the lysosome and are either degraded, or they returned to the cell membrane to again carry out their function [87]. Arrestin can be considered as a transit point for GPCR signaling. During the signaling process, arrestin remains bound to the GPCR by “tail” conformation. Typically, activation of the SRC is seen in GPCR signaling via arrestin. Compared with the administration of β-arrestin1 alone, β-arrestin1 binding to phosphorylated β2-AR significantly increased the phosphorylation of SRC [9]. Biased activation of AT1R-β-arrestin1 promotes acute catecholamine secretion by recruiting transient receptor potential cation channel subfamily C 3 [88]. Besides, the mitogen-activated protein kinase (MAPK), tyrosine kinase Ser-Thr kinase Akt pathways are also transmitted by GPCR bound to arrestin and are used to regulate cell proliferation, migration, and apoptosis [89].
Regardless of which function arrestin performs, the specific combination of phosphorylated GPCRs is a prerequisite. The four classes of arrestin and their distribution characteristics form the basis of its finely tuned regulation of the GPCR signaling pathway. The four types of arrestin have similar structures and amino acid sequences [87, 90], but their differences in distribution and function suggest that there may be differences in the process of phosphorylation recognition. In the visual system, the homology between arrestin1 and arrestin4 is approximately 58%. Arrestin1 is mainly distributed in rod cells and binds to light-activated phosphorylated rhodopsin, while arrestin4 is only found in cones and has the highest affinity for human green cone opsin [91, 92]. Arrestin1 can inactivate both rhodopsin and cone pigment, while arrestin4 can inactivate only cone pigment [93,94,95]. Although arrestin2 and arrestin3 have high homology (76% identical) [92], they have different affinities for different combinations of GPCR phosphorylation.
According to their differences in affinity for arrestins, GPCRs can be divided into two types: type A and type B. Type A GPCRs do not interact with visual arrestin, and their affinity for β-arrestin2 is higher than that for β-arrestin1. Examples of type A GPCRs include β2-AR, μ opioid receptors, endothelin receptor A, dopamine D1A receptor, and α1 adrenoceptor. Type B GPCRs interact with visual arrestin, and there is no difference in affinity for β-arrestin1/2; examples of type B GPCRs include AT1R, neurotensin receptor 1, V2R, and thyrotropin releasing hormone receptor [96]. Although arrestins have similar structure and function in a broad sense, the difference between them may be the crux to affect the different functions for each type of arrestin. However, at present, the understanding of GPCR-arrestin phosphorylation coding is mainly based on arrestin2 (β-arrestin1) [27, 29, 97]. Therefore, our understanding of the phosphorylation recognition pattern needs to be further clarified.
Recently, Latorraca et al. and Dwivedi-Agnihotri et al. found that when binding different phosphorylation sites at the C-terminus of GPCRs, arrestin undergoes different structural changes, and the structural changes of one region are not necessarily accompanied by structural changes in other regions [29, 30]. For example, the phosphorylation of Ser350 in V2R caused structural changes in the β-arrestin1 gate loop and finger loop but had little effect on the C-terminal structure of β-arrestin. However, during the phosphorylation of Ser360 in V2R, the change in the C-terminal structure of β-arrestin was significantly greater than that of the gate loop and finger loop [29]. This discovery contrasted with the original view that the structural changes of arrestin switch between an activated state and an inactive state [26, 29]. The diversity of structural changes may be one of the reasons why only four types of arrestin are needed to regulate the function of more than 800 GPCRs.
In summary, although there are only four types of arrestin, they can sufficiently and accurately transmit complex information to cells. This is mainly due to the diversity in the distribution, function, and structural changes of arrestins (Fig. 2). This diversity is fundamental to the process of arrestin’s recognition of phosphorylation.
GPCR phosphorylation sites
The phosphorylation sites of GPCRs are mainly located in the third intracellular loop and at the C-terminus. The arrestin binding sites are primarily found in the C-terminus of GPCRs. At these sites, serine (Ser, S) and threonine (Thr, T) form the S/T amino acid cluster. The function of the S/T cluster is to stabilize the binding between GPCRs and arrestin [98, 99] and to promote GPCRs to complete arrestin-clathrin-dependent endocytosis [100]. However, the number of phosphorylation sites differs among GPCRs. For example, neurotensin receptor subtype 1 (NTS1) has six phosphorylation sites in the C-terminus; AT1R, CXCR2 and CXCR5 all have seven phosphorylation sites; and CXCR3 and CCR7 have eight phosphorylation sites [98].
In the process of GPCR–arrestin interaction, different phosphorylation sites have different functions (Fig. 2). There are eight phosphorylation sites at the C-terminus of V2R. Using molecular dynamics simulation, researchers found that the Ser350 phosphorylation site promotes the activation of β-arrestin1, while the Thr360 site contributes to the binding of β-arrestin1 to V2R. However, when Ser350 and Ser362 were jointly phosphorylated, the binding ability of β-arrestin1 decreased significantly. Similarly, when Ser357 and T360 were jointly phosphorylated, the activation ability of β-arrestin1 also decreased significantly [29]. This suggests that the quality of a phosphorylation site might be more important to the interaction between a GPCR and arrestin than the number of phosphorylation sites. This conclusion was supported by experiments that investigated the phosphorylation sites of V2R by amino acid mutation [30]. Thr347 and Ser350 were not necessary for the recruitment of β-arrestin. Ser357 affected the recruitment and migration of β-arrestin but had no effect on the activation of ERK. Both Ser362 and Ser363 affected the recruitment of β-arrestin, and the common influence of these two sites was significantly stronger than that of each single site alone. When S362 and S363 were mutated to alanine (Ala, A), V2R lost its ability to recruit β-arrestin and become a G protein-biased receptor. Thr360 played a key role in recruiting V2R and β-arrestin, determining β-arrestin migration, and activating ERK, but the role of Thr359 in these functions was much smaller.
The phosphorylation of different sites is mainly affected by GRK. GRK2 and GRK6 can cause V2R to form different phosphorylation combination patterns. Β-Arrestin1 recognizes difference in phosphorylation patterns and determines downstream signaling [27].
Exploration of GPCR phosphorylation recognition by arrestin
Early exploration
The C-terminal phosphorylation of GPCRs is the basis of arrestin binding [101, 102]. However, this was not clear in the early stages of elucidating the function of arrestin. In 1984, researchers found that the binding of arrestin to rhodopsin is strictly based on the light-dependent mode, that is, rhodopsin is phosphorylated and binds arrestin as a result of light activation. However, in the absence of light activation, the two proteins do not bind. This suggested that the phosphorylation of rhodopsin enhances arrestin binding. This was also the initial understanding of GPCR phosphorylation and arrestin binding [23]. Using limited proteolysis and other methods, Krzysztof Palczewski and colleagues found that the 163–182 amino acid residues of arrestin constitute a region that recognizes phosphorylated rhodopsin [103]. This was basically consistent with the findings of Vsevolod V. Gurevich and Jeffrey L. Benovicarrestin who used arrestin complementary deoxyribo nucleic acid (cDNA) truncation to modify arrestin expression and found that the region in arrestin that recognizes the C-terminus of phosphorylated rhodopsin is composed of amino acid residues 158–185 [24].
In 1987, with the discovery of visual arrestin analogs, the understanding of arrestin gradually expanded from rhodopsin to all GPCRs and made it clear that arrestin binding requires GPCR phosphorylation [104]. Using amino acid mutation techniques, Sergey A. Vishnivetskiy and others found that the polar core in arrestin plays an important role in the recognition of phosphorylated rhodopsin. The polar core is composed of Arg175, Asp30, Asp296, and Asp303, in which the function of Arg175 is a phosphorylation-sensitive trigger. The role of the polar core is to stabilize arrestin in the inactive state. The phosphorylated rhodopsin can bind to Arg175 and break the electrostatic stability of the polar core, which promotes the transformation of arrestin from an inactive state to an activated state [105].
These studies suggest that GPCR phosphorylation is a prerequisite for arrestin binding and the transduction of signal through the G protein-independent pathway. In this context, the role of GPCR phosphorylation recognition patterns in signal transduction must be clarified. With a further in-depth understanding of GPCR phosphorylation and arrestin binding, researchers have proposed and refined the GPCR phosphorylation recognition bar code model and flute model.
GPCR phosphorylated bar code model
Previous studies have found that GPCRs phosphorylated by different protein kinases can have different biological functions. For example, β2-AR phosphorylation catalyzed by GRK promotes β-arrestin binding to the receptor, while phosphorylation by PKA does not result in β-arrestin recruitment. However, both kinases cause β2-AR desensitization [106,107,108,109,110]. Protein kinase CK2 activates the ERK and Jun kinase pathways after the phosphorylation of M3 muscarinic receptors [111]. To explain these phenomena, Andrew B. Tobin et al. proposed a phosphorylated bar code model (Fig. 3) to elaborate the relationship between GPCR phosphorylation and downstream signaling. The bar code model proposes that different protein kinases catalyze the phosphorylation of GPCRs at different sites, resulting in the activation of different signaling pathways [25, 26]. When the bar code model was initially proposed, receptor phosphorylation was not limited to the activity of GRK, and the signal transduction process did not only occur through arrestin. In later research, the phosphorylation recognition model tended to state that GRK catalyzed receptor phosphorylation, and signal transduction was mediated by arrestin.

Bar code model diagram. This picture is from the review by Tobin et al. [25]
In 2017, X. Edward Zhou et al. reported an activated rhodopsin-arrestin complex. Two phosphorylation coding modes for the combination of GPCR and arrestin, namely, “PXPXXP/E/D” and “PXXPXXP/E/D”, were identified. P represents phosphorylated serine or threonine, X represents any amino acid residue, except that proline cannot be present at either of the second XX sites. When the GPCR C-terminus exhibits this pattern of phosphorylated amino acids, it is recognized by arrestin, which then binds to it. In an analysis of 825 GPCRs, one or two phosphorylation coding modes were found in 436 GPCR C-termini. These two phosphorylation coding patterns may therefore be universal arrestin recognition codes of phosphorylated GPCRs [97]. This finding defined the environment of GPCR phosphorylation sites recognized by arrestin.
Recently, researchers have used molecular dynamics simulations and site-directed spectroscopy to analyze phosphorylated V2R binding to activated β-arrestin1 [29]. The results revealed several aspects of GPCR–arrestin binding. (1) The binding of arrestin to GPCRs depended on the spatial arrangement of phosphorylated amino acids rather than on the number of phosphorylated amino acids. Simulating the phosphorylation of all 8 phosphorylation sites at the V2R C-terminus (Thr347, Ser350, Ser357, Thr359, Thr360, Ser362, Ser363, Ser364) promoted the binding and activation of β-arrestin1. The same effect could be achieved by phosphorylating the second site (pSer350, p2) alone. However, phosphorylation of the third site (pSer357, p3) alone or the simultaneous phosphorylation of the third and fifth sites (pSer357 + pThr360, p3, 5) did not induce β-arrestin binding and activation. (2) The phosphorylation patterns of GPCRs that favor arrestin binding were different from the phosphorylation patterns of GPCRs that favor arrestin activation. Among the eight phosphorylation sites of V2R, pThr360 (p5) simultaneously favored β-arrestin1 binding and activation. pSer350 (p2) and pThr359 (p4) mainly favored β-arrestin1 activation. pSer363 (p7) mainly favored β-arrestin1 binding. (3) Arrestin has a complicated spatial structure, and the structural changes of each part were not synchronized upon GPCR binding and activation. When the gate loop of β-arrestin1 was in an inactive state, the finger loop was able to be in an activated state that bound to V2R. (4) Different patterns of phosphorylation led to a variety of arrestin structures. The study found that arrestin has multiple forms after binding to GPCRs, and the form depends on the spatial arrangement of the phosphorylation sites and charge attraction. These findings enrich our understanding of the active and inactive states of arrestin. This research extended the study of the bar code model from two-dimensional space to three-dimensional space and clarified the structural basis of the bar code model. However, the way in which complex phosphorylation coding and arrestin spatial structures correspond to downstream signaling requires further study.
GPCR phosphorylation flute model
The bar code model explains the diversity of GPCR phosphorylation signaling. However, it is not clear how only a few arrestins and GRKs can transmit thousands of GPCR phosphorylation signals. In 2015, Fan Yang et al. proposed a flute model of GPCR phosphorylation signaling (Fig. 4) [27]. Using unnatural amino acid incorporation and fluorine-19 nuclear magnetic resonance (19F-NMR) spectroscopy, the researchers described the diverse phosphorylation signals of GPCRs from the perspective of molecular structure. Different GRKs catalyze phosphorylation at distinct sites, and β-arrestin recognizes phosphorylated sites in different structures, finally achieving specific signaling pathways. Seven phosphorylation sites (p1–p7) were detected in the V2R/β-arrestin1 crystal complex. GRK2 mainly catalyzes p1, p4, p6 and p7 phosphorylation. Β-Arrestin1 could identify these sites and recruit clathrin. GRK6 catalyzes p1 and p5 phosphorylation, enabling β-arrestin1 to attract SRC. The main role of clathrin is to mediate receptor endocytosis [112]. As a protein kinase, SRC plays an important role in cell morphology, proliferation, movement and survival [113].

Flute model diagram. This picture is from the review by Yang et al. [28]
Recently, Qing-Tao He et al. designed four V2R C-termini with different phosphorylation sites and analyzed the binding ability, structural changes and functional differences in relation to β-arrestin1 [31]. V2Rpp-1, V2Rpp-3, V2Rpp-4, V2Rpp-6, and 7 short peptides were designed according to the 7 phosphorylation sites (Thr347, Ser350, Ser357, Thr360, Ser362, Ser363, Ser364) at the C-terminus of V2R. The suffixed number represents the position of the C-terminal phosphorylation site. V2Rpp-1 not only activated β-arrestin but also recruited c-Raf-1 kinase to the V2Rpp-1-β-arrestin1 complex. The V2Rpp-3-β-arrestin1 complex interacted with MEK1. Both c-Raf-1 and MEK1 are effectors of V2R. V2Rpp-6,7 was mainly responsible for regulating the nuclear localization sequence (NLS) structure in the N-terminal domain of β-arrestin1. An analysis of the structure of V2Rpp-4 and β-arrestin1 showed that this complex causes the rearrangement of C-terminal pS357 and pT359 and causes β-arrestin1 and pT359 to form phospho interactions. In the study of V2R C-terminal phosphorylation sites, T359 was not included as one of the seven phosphorylation sites studied by the flute model [27, 28, 31]. However, this site was included in the bar code model studies [29, 30]. This is a primary difference between the two models in the study of V2R.
The flute model is similar to the bar code model; however, the flute model not only enriches the bar code model theory but also explains the GPCR phosphorylation recognition theory from the perspective of structural changes.
GPCR phosphorylation QR code model
There are more than 800 known GPCRs in the human body, but research on receptor phosphorylation recognition patterns has mainly focused on rhodopsin and V2R [27, 29, 30, 97]. Ligands, GPCR type, GRK, arrestin, the phosphorylation sites may all affect phosphorylation recognition. These factors play a combined role to ultimately determine the biological impact of phosphorylation (Table 1). For instance, angiotensin II (Ang II) is the agonist for AT1R. Under the stimulation of Ang II, the activation of AT1R is maintains operational for only a short period of time. β-arrestin recruited to the AT1R phosphorylated by GRK2/3 and promoted AT1R endocytosis; however, ERK activation mediated by β-arrestin was enhanced when AT1R was phosphorylated by GRK5/6 [78]. AT1-AA was another agonist for AT1R. Under the stimulation of AT1-AA, AT1R was sustained activation, making the recruitment of β-arrestin restricted and the endocytosis of AT1R thus limited [35].
A similar phenomenon exists in the β1-AR. The β-adrenergic receptor agonist isoproterenol (ISO) activates receptors and signals via the Gs pathway [17]. Phosphorylated β1-AR recruits β-arrestin and drives receptor desensitization and endocytosis [114]. Meanwhile, the presence of β1-adrenergic receptor autoantibody (β1-AA) in patients with cardiovascular disease, although also an agonist of β1-AR, allows for sustained activation of β1-AR. Although it increases β1-AR phosphorylation, it inhibits the recruitment of the receptor to β-arrestin [115]. The reason for this needs further investigation. We speculate that it may be related to a mismatch of phosphorylation sites. That is, the β1-AR phosphorylation site is not efficiently recognized by β-arrestin under the action of β1-AA. Carvedilol as a representative of β-blocker could biased active β1-AR-Gi-β-arrestin pathway. Activation of this pathway could then exert cardioprotective effects by transactivating the epidermal growth factor receptor (EGFR)- extracellular signal-regulated kinase (ERK) pathway [49, 116]. The above evidence illustrates the phosphorylation of GPCR and its complex and variable functions. Therefore, only the precise regulation of the multiple factors involved in this process can ensure the accuracy of signaling.
This modulation is similar to a “QR” code (Fig. 5), which produces the final information after integrating various components. Ligands, the GPCR type, the GRK, arrestin and GPCR phosphorylation sites can all be thought of as information that is integrated into a QR code. Receptor desensitization, endocytosis, signaling and other functions represent information that is generated by the QR code.

QR code model for GPCR phosphorylation recognition. Schematic diagram of the QR code model: ligands, GPCR type, GRK, arrestin, GPCR phosphorylation sites and other factors work together to determine the fate of phosphorylated receptor desensitization, endocytosis, the continuation of signal transmission, or other functions
Conclusion
Accurately understanding the process of GPCR phosphorylation recognition will help to fully describe the way in which arrestin binds to receptors and its functional mechanism. It will also help us to better understand the physiological state of the body and provide potential targets and ideas for disease prevention and treatment. At present, research on the mode of GPCR phosphorylation recognition mainly focuses on the bar code model and the flute model [28,29,30, 62, 79]. These two models show the process of receptor phosphorylation recognition from the perspective of GPCRs, GRK, and arrestin, laying the foundation for GPCR phosphorylation recognition.
Analysis of the barcode model and the flute model revealed that the detection of the phosphorylation recognition process by these two models was mainly between GPCR and GRK or GPCR and arrestin [28, 29], with less detection of the mutual recognition between the three [79]. Our knowledge of the two recognition models is mainly focused on V2R and rhodopsin, with V2R being the most studied receptor carried out [27, 29, 30, 97]. But this is less than one fourth of one percent compared to the more than 800 GPCRs in the human body. This indicates that our recognition of GPCR phosphorylation patterns is still lacking. During receptor phosphorylation, factors such as ligand, receptor, GRK, arrestin, and phosphorylation site all affect the final function of the receptor. Therefore, we are proposing the QR code model to theoretically explain the effect of multiple factors acting together on receptor function.
The difference between the QR code model and the barcode model or the flute model is that the QR code model integrates the factors influencing the whole process from GPCR activation to arrestin function, such as ligands, GRK, etc. This model is developed on the existing theory and is based on the two existing recognition models. Based on our re-thinking on the recognition model of GPCR phosphorylation, we find both of the earlier models hold their own strengths, but limited. The barcode model and the flute model can determine the interactions between GPCR and GRK or arrestin with experimental precision, while the QR code model considers the receptor phosphorylation process macroscopically and is more theoretically oriented.
We are also conscious of the fact that the QR code model still has limitations and does not fully explain some specific phenomena of the GPCR phosphorylation recognition process. (1) Phosphorylation of some receptors is not involved in receptor endocytosis. Human lutropin receptor belongs to the class A GPCRs [117]. Although its agonist, human chorionic gonadotropin, can cause β-arrestin2-mediated receptor endocytosis, it is the receptor activity rather than the phosphorylation of the receptor that determines receptor endocytosis [118]. It is showed that mutations in the extracellular second loop of the receptor, F515A and T521A, enhance agonist-induced receptor endocytosis, while mutations in S512A and V519A impair agonist-induced receptor endocytosis. However, agonists have no effect on phosphorylation of either mutant receptor [119]. A comparative study of mutant receptors with impaired activity and impaired phosphorylation revealed that it is the activity of the receptor, but not the impaired phosphorylation, that that lengthens the time of receptor endocytosis [120]. (2) The presence of basal activation state of some receptors. The basal activation state of β2-AR in cardiomyocytes maintains cardiomyocyte beating without the involvement of ligands [17, 121]. (3) Intracellular activation of GPCR. β1-AR is distributed in both cell and organelle membranes and both can signal through the cAMP-PKA pathway [122, 123]. However, how the activated GPCR on the organelle membrane is desensitized and endocytosed remains to be further investigated. The above phenomena again illustrate the complexity of the GPCR signaling process. The existing theories or hypotheses are only summaries of known experimental results and speculations on the unknown. Our knowledge and understanding of GPCR phosphorylation recognition patterns still need to be further improved.
GPCR signal recognition, including phosphorylation, is the basis for the development of biased drugs. The presentation of allosteric microprocessors theory elaborated the mechanism of GPCR biased signaling. In this theory, biased ligands, GPCRs, transducers such as GRKs, G proteins and arrestins interacted allosterically. Biased ligands transmitted distinct GPCRs structural information that was processed into distinct biological outputs [124]. A biased agonist is a ligand that preferentially activates one receptor signaling pathway over another [37]. Oliceridine, a biased agonist of opioid receptors, specifically activates the μ-opioid receptor-G protein pathway and inhibits the binding of β-arrestin to the receptor, thereby achieving an analgesic effect. Unlike morphine, another agonist of μ-opioid receptors, the biased activation of oliceridine achieves an analgesic effect while avoiding adverse reactions such as respiratory depression caused by β-arrestin activation [125, 126]. Recent studies have also found that carvedilol, which has biased characteristics to activate the β2-AR-β-arrestin pathway, enhanced skeletal muscle contractility in mice without causing skeletal muscle cell hypertrophy. This feature might have therapeutic value for patients with sarcopenia and frailty. Although clenbuterol, another agonist of β2-AR, also enhanced the contractility of skeletal muscle cells, it caused cell hypertrophy and caused side effects such as arrhythmia [127]. Biased activation of the desired signaling pathway can minimize side effects while achieving the therapeutic effect, which is a characteristic of biased drugs. GPCR signaling pathways have been intensely studied for drug research and development owing to their characteristics of phosphorylation, and select pathways can be accurately activated or blocked to treat diseases.
Availability of data and materials
Not applicable.
Abbreviations
- GPCRs:
-
G protein-coupled receptors
- GRK:
-
G protein-coupled receptor kinase
- G proteins:
-
Guanine nucleotide-binding proteins
- AC:
-
Adenylate cyclase
- cAMP:
-
Cyclic adenosine monophosphate
- PKA:
-
Protein kinase A
- β1-AR:
-
β1-Adrenoceptor
- β2-AR:
-
β2-Adrenoceptor
- V2R:
-
Vasopressin type 2 receptor
- AT1R:
-
Angiotensin II type I receptor
- AT1-AA:
-
Autoantibodies
- α7 nAChR:
-
α7 Nicotinic receptor
- SD:
-
Sprague Dawley rats
- Δ9-THC:
-
Δ9-Tetrahydrocannabinol
- CB1:
-
Cannabinoid receptor 1
- ERK:
-
Extracellular regulated protein kinases
- PTH:
-
Parathyroid hormone
- NAMs:
-
Negative allosteric modulators
- PAMs:
-
Positive allosteric modulators
- SAMs:
-
Silent allosteric modulators
- AIDS:
-
Acquired Immune Deficiency Syndrome
- HIV-1:
-
Human immunodeficiency virus type 1
- siRNA:
-
Small interfering RNA
- MAPK:
-
Mitogen-activated protein kinase
- NTS1:
-
Neurotensin receptor subtype 1
- cDNA:
-
Complementary deoxyribo nucleic acid
- 19F-NMR:
-
Fluorine-19 nuclear magnetic resonance
- NLS:
-
Nuclear localization sequence
- β1-AA:
-
β1-Adrenergic receptor autoantibody
- EGFR:
-
Epidermal growth factor receptor
- Ang II:
-
Angiotensin II
References
Pierce KL, Premont RT, Lefkowitz RJ. Seven-transmembrane receptors. Nat Rev Mol Cell Biol. 2002;3(9):639–50.
Kahsai AW, Pani B, Lefkowitz RJ. GPCR signaling: conformational activation of arrestins. Cell Res. 2018;28(8):783–4.
Yang F, Mao C, Guo L, Lin J, Ming Q, Xiao P, et al. Structural basis of GPBAR activation and bile acid recognition. Nature. 2020;587(7834):499–504.
Seyedabadi M, Ghahremani MH, Albert PR. Biased signaling of G protein coupled receptors (GPCRs): molecular determinants of GPCR/transducer selectivity and therapeutic potential. Pharmacol Ther. 2019;200:148–78.
Du Y, Duc NM, Rasmussen S, Hilger D, Kubiak X, Wang L, et al. Assembly of a GPCR-G protein complex. Cell. 2019;177(5):1232–42.
Gurevich VV, Gurevich EV. Arrestins and G proteins in cellular signaling: the coin has two sides. Sci Signal. 2018;11(549):eaav1646.
Reiter E, Ayoub MA, Pellissier LP, Landomiel F, Musnier A, Trefier A, et al. beta-arrestin signalling and bias in hormone-responsive GPCRs. Mol Cell Endocrinol. 2017;449:28–41.
Xiao P, Yan W, Gou L, Zhong YN, Kong L, Wu C, et al. Ligand recognition and allosteric regulation of DRD1-Gs signaling complexes. Cell. 2021;184(4):943–56.
Yang F, Xiao P, Qu CX, Liu Q, Wang LY, Liu ZX, et al. Allosteric mechanisms underlie GPCR signaling to SH3-domain proteins through arrestin. Nat Chem Biol. 2018;14(9):876–86.
Wang MW, Yang Z, Chen X, Zhou SH, Huang GL, Sun JN, et al. Activation of PTH1R alleviates epididymitis and orchitis through Gq and beta-arrestin-1 pathways. Proc Natl Acad Sci U S A. 2021;118(45).
Hilger D, Masureel M, Kobilka BK. Structure and dynamics of GPCR signaling complexes. Nat Struct Mol Biol. 2018;25(1):4–12.
Syrovatkina V, Alegre KO, Dey R, Huang XY. Regulation, signaling, and physiological functions of G-proteins. J Mol Biol. 2016;428(19):3850–68.
Milligan G, Kostenis E. Heterotrimeric G-proteins: a short history. Br J Pharmacol. 2006;147(Suppl 1):S46-55.
Northup JK, Sternweis PC, Smigel MD, Schleifer LS, Ross EM, Gilman AG. Purification of the regulatory component of adenylate cyclase. Proc Natl Acad Sci U S A. 1980;77(11):6516–20.
Johnson JA, Liggett SB. Cardiovascular pharmacogenomics of adrenergic receptor signaling: clinical implications and future directions. Clin Pharmacol Ther. 2011;89(3):366–78.
Woo AY, Song Y, Xiao RP, Zhu W. Biased beta2-adrenoceptor signalling in heart failure: pathophysiology and drug discovery. Br J Pharmacol. 2015;172(23):5444–56.
Devic E, Xiang Y, Gould D, Kobilka B. Beta-adrenergic receptor subtype-specific signaling in cardiac myocytes from beta(1) and beta(2) adrenoceptor knockout mice. Mol Pharmacol. 2001;60(3):577–83.
Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, et al. Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res. 1999;84(1):43–52.
Santos-Otte P, Leysen H, van Gastel J, Hendrickx JO, Martin B, Maudsley S. G protein-coupled receptor systems and their role in cellular senescence. Comput Struct Biotechnol J. 2019;17:1265–77.
Shukla AK, Westfield GH, Xiao K, Reis RI, Huang LY, Tripathi-Shukla P, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512(7513):218–22.
Cahill TR, Thomsen AR, Tarrasch JT, Plouffe B, Nguyen AH, Yang F, et al. Distinct conformations of GPCR-beta-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci U S A. 2017;114(10):2562–7.
Lymperopoulos A. Arrestins in the cardiovascular system: an update. Prog Mol Biol Transl Sci. 2018;159:27–57.
Kuhn H, Hall SW, Wilden U. Light-induced binding of 48-kDa protein to photoreceptor membranes is highly enhanced by phosphorylation of rhodopsin. FEBS Lett. 1984;176(2):473–8.
Gurevich VV, Benovic JL. Visual arrestin interaction with rhodopsin. Sequential multisite binding ensures strict selectivity toward light-activated phosphorylated rhodopsin. J Biol Chem. 1993;268(16):11628–38.
Tobin AB, Butcher AJ, Kong KC. Location, location, location…site-specific GPCR phosphorylation offers a mechanism for cell-type-specific signalling. Trends Pharmacol Sci. 2008;29(8):413–20.
Tobin AB. G-protein-coupled receptor phosphorylation: where, when and by whom. Br J Pharmacol. 2008;153(Suppl 1):S167–76.
Yang F, Yu X, Liu C, Qu CX, Gong Z, Liu HD, et al. Phospho-selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat Commun. 2015;6:8202.
Yang Z, Yang F, Zhang D, Liu Z, Lin A, Liu C, et al. Phosphorylation of G protein-coupled receptors: from the barcode hypothesis to the flute model. Mol pharmacol. 2017;92(3):201–10.
Latorraca NR, Masureel M, Hollingsworth SA, Heydenreich FM, Suomivuori CM, Brinton C, et al. How GPCR phosphorylation patterns orchestrate arrestin-mediated signaling. Cell. 2020;183(7):1813–25.
Dwivedi-Agnihotri H, Chaturvedi M, Baidya M, Stepniewski TM, Pandey S, Maharana J, et al. Distinct phosphorylation sites in a prototypical GPCR differently orchestrate beta-arrestin interaction, trafficking, and signaling. Sci Adv. 2020;6(37):eabb8368.
He QT, Xiao P, Huang SM, Jia YL, Zhu ZL, Lin JY, et al. Structural studies of phosphorylation-dependent interactions between the V2R receptor and arrestin-2. Nat Commun. 2021;12(1):2396.
Candia M, Kratzer B, Pickl WF. On peptides and altered peptide ligands: from origin, mode of action and design to clinical application (immunotherapy). Int Arch Allergy Immunol. 2016;170(4):211–33.
Allen JA, Roth BL. Strategies to discover unexpected targets for drugs active at G protein-coupled receptors. Annu Rev Pharmacol Toxicol. 2011;51:117–44.
Varma N, Mutt E, Muhle J, Panneels V, Terakita A, Deupi X, et al. Crystal structure of jumping spider rhodopsin-1 as a light sensitive GPCR. Proc Natl Acad Sci U S A. 2019;116(29):14547–56.
Bian J, Lei J, Yin X, Wang P, Wu Y, Yang X, et al. Limited AT1 receptor internalization is a novel mechanism underlying sustained vasoconstriction induced by AT1 receptor autoantibody from preeclampsia. J Am Heart Assoc. 2019;8(6):e11179.
Molinoff PB. Alpha- and beta-adrenergic receptor subtypes properties, distribution and regulation. Drugs. 1984;28(Suppl 2):1–15.
Wacker D, Stevens RC, Roth BL. How ligands illuminate GPCR molecular pharmacology. Cell. 2017;170(3):414–27.
Lakstygal AM, Kolesnikova TO, Khatsko SL, Zabegalov KN, Volgin AD, Demin KA, et al. DARK classics in chemical neuroscience: atropine, scopolamine, and other anticholinergic deliriant hallucinogens. ACS Chem Neurosci. 2019;10(5):2144–59.
Angiotensin II receptor antagonists. 2012.
Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. 2002;359(9311):995–1003.
Laczo J, Markova H, Lobellova V, Gazova I, Parizkova M, Cerman J, et al. Scopolamine disrupts place navigation in rats and humans: a translational validation of the Hidden Goal Task in the Morris water maze and a real maze for humans. Psychopharmacology. 2017;234(4):535–47.
Neves GA, Grace AA. alpha7 nicotinic receptor full agonist reverse basolateral amygdala hyperactivity and attenuation of dopaminergic neuron activity in rats exposed to chronic mild stress. Eur Neuropsychopharmacol. 2019;29(12):1343–53.
O’Byrne PM, FitzGerald JM, Bateman ED, Barnes PJ, Zhong N, Keen C, et al. Inhaled combined budesonide-formoterol as needed in mild asthma. N Engl J Med. 2018;378(20):1865–76.
Hua T, Vemuri K, Nikas SP, Laprairie RB, Wu Y, Qu L, et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature. 2017;547(7664):468–71.
Mechoulam R, Hanus LO, Pertwee R, Howlett AC. Early phytocannabinoid chemistry to endocannabinoids and beyond. Nat Rev Neurosci. 2014;15(11):757–64.
Roth BL. DREADDs for neuroscientists. Neuron. 2016;89(4):683–94.
Kenakin T. Biased receptor signaling in drug discovery. Pharmacol Rev. 2019;71(2):267–315.
Kenakin T. New concepts in pharmacological efficacy at 7TM receptors: IUPHAR review 2. Br J Pharmacol. 2013;168(3):554–75.
Kim IM, Tilley DG, Chen J, Salazar NC, Whalen EJ, Violin JD, et al. Beta-blockers alprenolol and carvedilol stimulate beta-arrestin-mediated EGFR transactivation. Proc Natl Acad Sci U S A. 2008;105(38):14555–60.
Wisler JW, DeWire SM, Whalen EJ, Violin JD, Drake MT, Ahn S, et al. A unique mechanism of beta-blocker action: carvedilol stimulates beta-arrestin signaling. Proc Natl Acad Sci U S A. 2007;104(42):16657–62.
Kenakin TP. Pharmacological onomastics: what’s in a name? Br J Pharmacol. 2008;153(3):432–8.
Gesty-Palmer D, Chen M, Reiter E, Ahn S, Nelson CD, Wang S, et al. Distinct beta-arrestin- and G protein-dependent pathways for parathyroid hormone receptor-stimulated ERK1/2 activation. J Biol Chem. 2006;281(16):10856–64.
Valant C, Sexton PM, Christopoulos A. Orthosteric/allosteric bitopic ligands: going hybrid at GPCRs. Mol Interv. 2009;9(3):125–35.
Digby GJ, Conn PJ, Lindsley CW. Orthosteric- and allosteric-induced ligand-directed trafficking at GPCRs. Curr Opin Drug Discov Dev. 2010;13(5):587–94.
De Amici M, Dallanoce C, Holzgrabe U, Trankle C, Mohr K. Allosteric ligands for G protein-coupled receptors: a novel strategy with attractive therapeutic opportunities. Med Res Rev. 2010;30(3):463–549.
Burford NT, Watson J, Bertekap R, Alt A. Strategies for the identification of allosteric modulators of G-protein-coupled receptors. Biochem Pharmacol. 2011;81(6):691–702.
O’Brien JB, Wilkinson JC, Roman DL. Regulator of G-protein signaling (RGS) proteins as drug targets: progress and future potentials. J Biol Chem. 2019;294(49):18571–85.
Chen H, Nwe PK, Yang Y, Rosen CE, Bielecka AA, Kuchroo M, et al. A forward chemical genetic screen reveals gut microbiota metabolites that modulate host physiology. Cell. 2019;177(5):1217–31.
Tan Q, Zhu Y, Li J, Chen Z, Han GW, Kufareva I, et al. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science. 2013;341(6152):1387–90.
Imai S, Yokomizo T, Kofuku Y, Shiraishi Y, Ueda T, Shimada I. Structural equilibrium underlying ligand-dependent activation of beta2-adrenoreceptor. Nat Chem Biol. 2020;16(4):430–9.
Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E, et al. The concise guide to pharmacology 2015/16: overview. Br J Pharmacol. 2015;172(24):5729–43.
Bahouth SW, Nooh MM. Barcoding of GPCR trafficking and signaling through the various trafficking roadmaps by compartmentalized signaling networks. Cell Signal. 2017;36:42–55.
Wu JJ, Yang Y, Peng WT, Sun JC, Sun WY, Wei W. G protein-coupled receptor kinase 2 regulating beta2-adrenergic receptor signaling in M2-polarized macrophages contributes to hepatocellular carcinoma progression. Onco Targets Ther. 2019;12:5499–513.
Zhang Y, Wang S, Huang H, Zeng A, Han Y, Zeng C, et al. GRK4-mediated adiponectin receptor-1 phosphorylative desensitization as a novel mechanism of reduced renal sodium excretion in hypertension. Clin Sci (Lond). 2020;134(18):2453–67.
Komolov KE, Benovic JL. G protein-coupled receptor kinases: past, present and future. Cell Signal. 2018;41:17–24.
Kuhn H, Dreyer WJ. Light dependent phosphorylation of rhodopsin by ATP. FEBS Lett. 1972;20(1):1–6.
Benovic JL, Strasser RH, Caron MG, Lefkowitz RJ. Beta-adrenergic receptor kinase: identification of a novel protein kinase that phosphorylates the agonist-occupied form of the receptor. Proc Natl Acad Sci U S A. 1986;83(9):2797–801.
Benovic JL, Onorato JJ, Arriza JL, Stone WC, Lohse M, Jenkins NA, et al. Cloning, expression, and chromosomal localization of beta-adrenergic receptor kinase 2. A new member of the receptor kinase family. J Biol Chem. 1991;266(23):14939–46.
Lorenz W, Inglese J, Palczewski K, Onorato JJ, Caron MG, Lefkowitz RJ. The receptor kinase family: primary structure of rhodopsin kinase reveals similarities to the beta-adrenergic receptor kinase. Proc Natl Acad Sci U S A. 1991;88(19):8715–9.
Ambrose C, James M, Barnes G, Lin C, Bates G, Altherr M, et al. A novel G protein-coupled receptor kinase gene cloned from 4p16.3. Hum Mol Genet. 1992;1(9):697–703.
Kunapuli P, Benovic JL. Cloning and expression of GRK5: a member of the G protein-coupled receptor kinase family. Proc Natl Acad Sci U S A. 1993;90(12):5588–92.
Benovic JL, Gomez J. Molecular cloning and expression of GRK6. A new member of the G protein-coupled receptor kinase family. J Biol Chem. 1993;268(26):19521–7.
Hisatomi O, Matsuda S, Satoh T, Kotaka S, Imanishi Y, Tokunaga F. A novel subtype of G-protein-coupled receptor kinase, GRK7, in teleost cone photoreceptors. FEBS Lett. 1998;424(3):159–64.
Premont RT, Macrae AD, Stoffel RH, Chung N, Pitcher JA, Ambrose C, et al. Characterization of the G protein-coupled receptor kinase GRK4. Identification of four splice variants. J Biol Chem. 1996;271(11):6403–10.
Chen CK, Zhang K, Church-Kopish J, Huang W, Zhang H, Chen YJ, et al. Characterization of human GRK7 as a potential cone opsin kinase. Mol Vis. 2001;7:305–13.
Kuhn H, Wilden U. Assay of phosphorylation of rhodopsin in vitro and in vivo. Methods Enzymol. 1982;81:489–96.
Ferguson SS, Downey WR, Colapietro AM, Barak LS, Menard L, Caron MG. Role of beta-arrestin in mediating agonist-promoted G protein-coupled receptor internalization. Science. 1996;271(5247):363–6.
Kim J, Ahn S, Ren XR, Whalen EJ, Reiter E, Wei H, et al. Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc Natl Acad Sci U S A. 2005;102(5):1442–7.
Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, et al. Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci Signal. 2011;4(185):a51.
Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53(1):1–24.
Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523(7562):561–7.
Scheerer P, Sommer ME. Structural mechanism of arrestin activation. Curr Opin Struct Biol. 2017;45:160–9.
Pfister C, Chabre M, Plouet J, Tuyen VV, De Kozak Y, Faure JP, et al. Retinal S antigen identified as the 48K protein regulating light-dependent phosphodiesterase in rods. Science. 1985;228(4701):891–3.
Wilden U, Wust E, Weyand I, Kuhn H. Rapid affinity purification of retinal arrestin (48 kDa protein) via its light-dependent binding to phosphorylated rhodopsin. FEBS Lett. 1986;207(2):292–5.
Jean-Charles PY, Kaur S, Shenoy SK. G protein-coupled receptor signaling through beta-arrestin-dependent mechanisms. J Cardiovasc Pharmacol. 2017;70(3):142–58.
Liu Q, He QT, Lyu X, Yang F, Zhu ZL, Xiao P, et al. DeSiphering receptor core-induced and ligand-dependent conformational changes in arrestin via genetic encoded trimethylsilyl (1)H-NMR probe. Nat Commun. 2020;11(1):4857.
Kendall RT, Luttrell LM. Diversity in arrestin function. Cell Mol Life Sci. 2009;66(18):2953–73.
Liu CH, Gong Z, Liang ZL, Liu ZX, Yang F, Sun YJ, et al. Arrestin-biased AT1R agonism induces acute catecholamine secretion through TRPC3 coupling. Nat Commun. 2017;8:14335.
Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by beta-arrestins. Science. 2005;308(5721):512–7.
Shilton BH, McDowell JH, Smith WC, Hargrave PA. The solution structure and activation of visual arrestin studied by small-angle X-ray scattering. Eur J Biochem. 2002;269(15):3801–9.
Chen J. The physiological roles of arrestin-1 in rod photoreceptor cells. Handb Exp Pharmacol. 2014;219:85–99.
Craft CM, Deming JD. Cone arrestin: deciphering the structure and functions of arrestin 4 in vision. Handb Exp Pharmacol. 2014;219:117–31.
Chan S, Rubin WW, Mendez A, Liu X, Song X, Hanson SM, et al. Functional comparisons of visual arrestins in rod photoreceptors of transgenic mice. Invest Ophthalmol Vis Sci. 2007;48(5):1968–75.
Nikonov SS, Brown BM, Davis JA, Zuniga FI, Bragin A, Pugh EJ, et al. Mouse cones require an arrestin for normal inactivation of phototransduction. Neuron. 2008;59(3):462–74.
Shi G, Yau KW, Chen J, Kefalov VJ. Signaling properties of a short-wave cone visual pigment and its role in phototransduction. J Neurosci. 2007;27(38):10084–93.
Oakley RH, Laporte SA, Holt JA, Caron MG, Barak LS. Differential affinities of visual arrestin, beta arrestin1, and beta arrestin2 for G protein-coupled receptors delineate two major classes of receptors. J Biol Chem. 2000;275(22):17201–10.
Zhou XE, He Y, de Waal PW, Gao X, Kang Y, Van Eps N, et al. Identification of phosphorylation codes for arrestin recruitment by G protein-coupled receptors. Cell. 2017;170(3):457–69.
Marchese A, Paing MM, Temple BR, Trejo J. G protein-coupled receptor sorting to endosomes and lysosomes. Annu Rev Pharmacol Toxicol. 2008;48:601–29.
Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Molecular determinants underlying the formation of stable intracellular G protein-coupled receptor-beta-arrestin complexes after receptor endocytosis*. J Biol Chem. 2001;276(22):19452–60.
Okamoto Y, Shikano S. Differential phosphorylation signals control endocytosis of GPR15. Mol Biol Cell. 2017;28(17):2267–81.
Gurevich VV, Gurevich EV. The structural basis of the arrestin binding to GPCRs. Mol Cell Endocrinol. 2019;484:34–41.
Lohse MJ, Hoffmann C. Arrestin interactions with G protein-coupled receptors. Handb Exp Pharmacol. 2014;219:15–56.
Palczewski K, Buczylko J, Imami NR, McDowell JH, Hargrave PA. Role of the carboxyl-terminal region of arrestin in binding to phosphorylated rhodopsin. J Biol Chem. 1991;266(23):15334–9.
Benovic JL, Kuhn H, Weyand I, Codina J, Caron MG, Lefkowitz RJ. Functional desensitization of the isolated beta-adrenergic receptor by the beta-adrenergic receptor kinase: potential role of an analog of the retinal protein arrestin (48-kDa protein). Proc Natl Acad Sci U S A. 1987;84(24):8879–82.
Vishnivetskiy SA, Paz CL, Schubert C, Hirsch JA, Sigler PB, Gurevich VV. How does arrestin respond to the phosphorylated state of rhodopsin? J Biol Chem. 1999;274(17):11451–4.
John B, Weissman DB. Cytogenetic components of reproductive isolation in Trimerotropis thalassica and T. occidentalis. Chromosoma. 1977;60(2):187–203.
Tran TM, Friedman J, Qunaibi E, Baameur F, Moore RH, Clark RB. Characterization of agonist stimulation of cAMP-dependent protein kinase and G protein-coupled receptor kinase phosphorylation of the beta2-adrenergic receptor using phosphoserine-specific antibodies. Mol Pharmacol. 2004;65(1):196–206.
Roth NS, Campbell PT, Caron MG, Lefkowitz RJ, Lohse MJ. Comparative rates of desensitization of beta-adrenergic receptors by the beta-adrenergic receptor kinase and the cyclic AMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1991;88(14):6201–4.
Lefkowitz RJ. G protein-coupled receptors. III. New roles for receptor kinases and beta-arrestins in receptor signaling and desensitization. J Biol Chem. 1998;273(30):18677–80.
Pitcher JA, Freedman NJ, Lefkowitz RJ. G protein-coupled receptor kinases. Annu Rev Biochem. 1998;67:653–92.
Torrecilla I, Spragg EJ, Poulin B, McWilliams PJ, Mistry SC, Blaukat A, et al. Phosphorylation and regulation of a G protein-coupled receptor by protein kinase CK2. J Cell Biol. 2007;177(1):127–37.
Mettlen M, Chen PH, Srinivasan S, Danuser G, Schmid SL. Regulation of clathrin-mediated endocytosis. Annu Rev Biochem. 2018;87:871–96.
Roskoski RJ. Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Commun. 2004;324(4):1155–64.
Ranjan R, Gupta P, Shukla AK. GPCR signaling: beta-arrestins kiss and remember. Curr Biol. 2016;26(7):R285–8.
Chen H, Cao N, Wang L, Wu Y, Wei H, Li Y, et al. Biased activation of beta2-AR/Gi/GRK2 signal pathway attenuated beta1-AR sustained activation induced by beta1-adrenergic receptor autoantibody. Cell Death Discov. 2021;7(1):340.
Wang J, Hanada K, Staus DP, Makara MA, Dahal GR, Chen Q, et al. Galphai is required for carvedilol-induced beta1 adrenergic receptor beta-arrestin biased signaling. Nat Commun. 2017;8(1):1706.
Shinozaki H, Butnev V, Tao YX, Ang KL, Conti M, Segaloff DL. Desensitization of Gs-coupled receptor signaling by constitutively active mutants of the human lutropin/choriogonadotropin receptor. J Clin Endocrinol Metab. 2003;88(3):1194–204.
Min L, Galet C, Ascoli M. The association of arrestin-3 with the human lutropin/choriogonadotropin receptor depends mostly on receptor activation rather than on receptor phosphorylation. J Biol Chem. 2002;277(1):702–10.
Li S, Liu X, Min L, Ascoli M. Mutations of the second extracellular loop of the human lutropin receptor emphasize the importance of receptor activation and de-emphasize the importance of receptor phosphorylation in agonist-induced internalization. J Biol Chem. 2001;276(11):7968–73.
Min L, Ascoli M. Effect of activating and inactivating mutations on the phosphorylation and trafficking of the human lutropin/choriogonadotropin receptor. Mol Endocrinol. 2000;14(11):1797–810.
Xiang Y, Devic E, Kobilka B. The PDZ binding motif of the beta 1 adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J Biol Chem. 2002;277(37):33783–90.
Wang Y, Shi Q, Li M, Zhao M, Reddy GR, Teoh JP, et al. Intracellular beta1-adrenergic receptors and organic cation transporter 3 mediate phospholamban phosphorylation to enhance cardiac contractility. Circ Res. 2021;128(2):246–61.
Irannejad R, Pessino V, Mika D, Huang B, Wedegaertner PB, Conti M, et al. Functional selectivity of GPCR-directed drug action through location bias. Nat Chem Biol. 2017;13(7):799–806.
Smith JS, Lefkowitz RJ, Rajagopal S. Biased signalling: from simple switches to allosteric microprocessors. Nat Rev Drug Discov. 2018;17(4):243–60.
DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, et al. A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J Pharmacol Exp Ther. 2013;344(3):708–17.
Boom M, Niesters M, Sarton E, Aarts L, Smith TW, Dahan A. Non-analgesic effects of opioids: opioid-induced respiratory depression. Curr Pharm Des. 2012;18(37):5994–6004.
Kim J, Grotegut CA, Wisler JW, Mao L, Rosenberg PB, Rockman HA, et al. The beta-arrestin-biased beta-adrenergic receptor blocker carvedilol enhances skeletal muscle contractility. Proc Natl Acad Sci U S A. 2020;117(22):12435–43.
Namkung Y, Dipace C, Javitch JA, Sibley DR. G protein-coupled receptor kinase-mediated phosphorylation regulates post-endocytic trafficking of the D2 dopamine receptor. J Biol Chem. 2009;284(22):15038–51.
Pals-Rylaarsdam R, Hosey MM. Two homologous phosphorylation domains differentially contribute to desensitization and internalization of the m2 muscarinic acetylcholine receptor. J Biol Chem. 1997;272(22):14152–8.
Pals-Rylaarsdam R, Xu Y, Witt-Enderby P, Benovic JL, Hosey MM. Desensitization and internalization of the m2 muscarinic acetylcholine receptor are directed by independent mechanisms. J Biol Chem. 1995;270(48):29004–11.
Jones KT, Echeverry M, Mosser VA, Gates A, Jackson DA. Agonist mediated internalization of M2 mAChR is beta-arrestin-dependent. J Mol Signal. 2006;1:7.
Wu G, Bogatkevich GS, Mukhin YV, Benovic JL, Hildebrandt JD, Lanier SM. Identification of Gbetagamma binding sites in the third intracellular loop of the M(3)-muscarinic receptor and their role in receptor regulation. J Biol Chem. 2000;275(12):9026–34.
Poulin B, Butcher A, McWilliams P, Bourgognon JM, Pawlak R, Kong KC, et al. The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/arrestin-dependent manner. Proc Natl Acad Sci U S A. 2010;107(20):9440–5.
Acknowledgements
We thank Ruiqi Hou for her help in creating the table in this review.
Funding
This work was supported by funding from the National Natural Science Foundation of China (81970334, 81770393, 31771267).
Author information
Authors and Affiliations
Contributions
HC researched data for the article, discussed its contents and wrote the manuscript. XZ and SZ researched data for the article and discussed its contents. HL designed, reviewed and edited the manuscript before submission. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Chen, H., Zhang, S., Zhang, X. et al. QR code model: a new possibility for GPCR phosphorylation recognition. Cell Commun Signal 20, 23 (2022). https://doi.org/10.1186/s12964-022-00832-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-022-00832-4