
Abstract
Efferocytosis is a physiologic phagocytic clearance of apoptotic cells, which modulates inflammatory responses and the immune environment and subsequently facilitates immune escape of cancer cells, thus promoting tumor development and progression. Efferocytosis is an equilibrium formed by perfect coordination among “find-me”, “eat-me” and “don’t-eat-me” signals. These signaling pathways not only affect the proliferation, invasion, metastasis, and angiogenesis of tumor cells but also regulate adaptive responses and drug resistance to antitumor therapies. Therefore, efferocytosis-related molecules and pathways are potential targets for antitumor therapy. Besides, supplementing conventional chemotherapy, radiotherapy and other immunotherapies with efferocytosis-targeted therapy could enhance the therapeutic efficacy, reduce off-target toxicity, and promote patient outcome.
Video abstract
Similar content being viewed by others


Background
Apoptosis is a mechanism of programmed cell death by which tissues self-renew and maintain homeostasis [1]. During apoptosis, phagocytes rapidly recognize and engulf dying cells before intracellular components are released. Subsequently, the membrane integrity of apoptotic cells is maintained, hence avoiding the exposure of immunogenic materials and the subsequent inflammatory responses [2]. This physiologic process, referred to as efferocytosis, efficiently removes apoptotic cells without subsequent secondary necrosis and damages [3]. Efferocytosis is induced by several physiological or pathological conditions and plays a crucial role in tissue differentiation, repair, and the resolution of inflammation [4]. Because of the pleiotropic role of efferocytosis, disruption and dysregulation of this process are associated with many pathological states, which may trigger some diseases. Previous research has linked efferocytosis to inflammatory diseases [5], autoimmune diseases [6] and atherosclerosis [7, 8].
Apoptotic cell removal is accomplished by either professional or non-professional phagocytes [9]. Professional phagocytes include macrophages and immature dendritic cells, which are recruited following the onset of apoptosis with a relatively faster and better motile ability in engulfment; while non-professional phagocytes are tissue-resident cells neighboring dying cells (e.g., epithelial cells, endothelial cells, fibroblasts and some stromal cells) and are comparatively slower in ingesting dead cells [10,11,12].
Tumor-associated macrophages are M2-polarized macrophages and a type of phagocyte involved in efferocytosis [13, 14]. Recent studies have demonstrated the vital role of efferocytosis in the tumor microenvironment, progression, and metastasis of tumors [15, 16]. Following engulfment, tumor-associated macrophages increase the production of anti-inflammatory cytokines and Treg cells (regulatory T cells), while inhibiting the production of pro-inflammatory cytokines and effector T cells [17,18,19]. Therefore, the immunologically silent clearance of apoptotic cells promotes inflammation resolution and immune suppression, which provide cancer cells with an environment to escape from immunological surveillance hence promoting tumor progression [1, 20]. This review explores the function and underlying mechanisms of efferocytosis in tumor progression and summarizes the promising targets and novel strategies for cancer therapy.
Biological mechanisms of efferocytosis
Recognition
Efferocytosis includes complicated and coordinated molecular communication and involves several signaling pathways that culminate in phagocytosis and clearance of dying cells. The first step in efferocytosis is recognition of the targeted apoptotic cells by phagocytes through “find-me” signals such as lipids, proteins, peptides, and complex structures released by dying cells [21]. These signals act as chemo-attractants for scavenger cells recruited to the apoptotic site during the early stages of efferocytosis. The first chemotactic factor to be identified as a “find-me” signal was a covalent dimer of the ribosomal protein S19 [22]. Lysophosphatidylcholine is a lipid attraction signal produced by apoptotic cells through caspase-3 mediated activation of the calcium-independent phospholipase A2 and stimulates the migration of monocytes and primary macrophages [23, 24]. Also, sphingosine-1-phosphate (S1P) is another lipid chemotactic factor that acts as a “find-me” signal in efferocytosis. S1P triggers the chemotaxis of macrophages to engulf apoptotic cells [25]. As a member of the chemokine family, CX3CL1 is also a “find-me” signal released by lymphocytes after apoptosis and attracts macrophages to the apoptotic site [26]. Elliott et al. demonstrated the role of extracellular nucleotides in phagocytosis and apoptotic cell recognition, as the release of ATP and UTP from apoptotic cells elicit an attraction signal through their receptor P2Y2 on phagocytes [27].
Engulfment
Following recognition and migration of phagocytes, apoptotic cells are engulfed by phagocytes. Engulfment is mediated by a series of molecular events called “eat-me” signals such as phosphatidylserine (PS) and calreticulin. These signals are unique surface markers on apoptotic cells identified by arrived phagocytes and enable them to exert a subsequent phagocytic function. The most common and widely studied “eat-me” signal is PS, which is usually confined to the inner leaflet of the plasma membrane but migrates to the outer leaflet during apoptosis [28, 29]. Apoptotic cells typically have exposed PS, phagocytes recognize and combine with this signal before the onset of engulfment.
Previous studies have reported that phagocytes can directly bind to PS on apoptotic cells through numerous PS receptors. These receptors include T-cell immunoglobulin mucin (TIM) family (TIM-1, TIM-3, and TIM-4) [30, 31], brain-specific angiogenesis inhibitor 1 (BAI-1) [32], stabilin-2 family members [33], CD300 family members (CD300b and CD300f) [34, 35] and receptor for advanced glycation end products (RAGE) [36]. Alternatively, PS can indirectly bind to receptors on phagocytes via bridging molecules. For example, TAM receptors are a type of indirect PS receptors, and growth arrest-specific 6 (Gas6) and Protein S are bridging molecules that facilitate the binding of PS on apoptotic cells to TAM receptors (Tyro3, Axl and MerTK) on phagocytes [37]. Further, Gas 6 binds to all three receptors, while Protein S only binds to Tyro3 and MerTK [38]. Milk fat globule epidermal growth factor-8 (MFG-E8) bridges between PS and αvβ3/αvβ5 integrins, another type of indirect PS receptors [39]. Besides, the SCARF1 scavenger receptor on phagocytes uses C1q as a complement component to recognize PS on apoptotic cells [40]. Inversely, phagocytes can identify live and normal cells by detecting “don’t-eat-me” signals, which protects the cells from being engulfed. The most widely studied ligands of “don’t-eat-me” signals are CD47 [4] and CD31 [41].
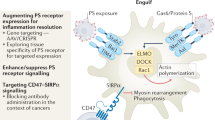
Following the “eat-me” signals recognition and tethering, cytoskeletal rearrangement occurs within phagocytes, leading to cell motility and phagosome formation to complete the process of engulfment. This process is mediated by ELMO/Dock180/Rac1 pathway [40, 41], which is proved to be downstream of PS receptors such as BAI-1 [30]. In addition, TAM receptors and αvβ5 integrin have been confirmed to have an effect on Rac1 activation and eventually causing phagocytosis [42]. However, mechanisms of some other receptors in post-receptor activation signaling are still unclear.
Immunomodulation
The clearance of apoptotic cells occurs via their interaction with phagocytes, which contributes to tissue homeostasis. This process is accompanied by the secretion of a series of anti-inflammatory cytokines, including transforming growth factor-beta (TGFβ), interleukin (IL)-10, prostaglandin E2 (PGE2) and platelet-activating factor (PAF). Meanwhile, the production of the pro-inflammatory cytokines IL-1β, tumor necrosis factor-alpha (TNF-α), and IL-12 is inhibited [42, 43]. This mechanism occurs via the reduction of M1 macrophage induced production of pro-inflammatory cytokines and suppression of the nuclear factor-kappa B (NF-ĸB) signaling [44]. In contrast, the anti-inflammatory response of M2 macrophages is enhanced [45]. Besides, TAM receptor activation dampens pro-inflammatory Toll-like receptor (TLR) signaling by upregulating the expression of suppressor of cytokine signaling (SOCS) 1 and SOCS3. Furthermore, the enhancement of efferocytosis is always in line with the decreased activation and function of CD8+ and CD4+ effector T cells as well as increased Treg cells-mediated immunosuppression [17, 46,47,48,49]. Indeed, CD8+ and CD4+ T cells play a critical role in tumor rejection response [50]. Particularly, CD8+ cytotoxic T cells are acknowledged to recognize tumor-specific antigens and target cancer cells, leading to the shrinkage of tumor [51].
Taken together, efferocytosis hinders the inflammatory response and modulates the immune environment, which facilitates immune escape and promotes tumorigenesis and progression (Fig. 1) [52]. Conversely, inhibition of efferocytosis in the context of cancer results in the release of intracellular components of dying cells. And the succeeding exposure of immunogenic materials triggers the robust innate and adaptive immune responses against cancer, due to the secretion of pro-inflammatory cytokines, the accumulation of inflammatory cells, as well as the enhanced recognition and presentation of tumor-specific antigens [1, 2, 20, 51].

Schematic representation of signaling pathways in the efferocytosis-induced immune suppression for tumor progression. The engulfment of apoptotic cells by tumor-associated macrophages triggers a series of signaling pathways, subsequently induces M2 polarization of macrophages while inhibiting M1 polarization, increases Treg cells while decreasing CD8+ T cells, and thereby resulting in the inflammation resolution and immune suppression, which may provide an environment for cancer to escape from immunological surveillance and promote tumor progression. NFκB = factor-κ-gene binding; JAK/STAT1 = Janus kinase/signal transducers and activators of transcription 1; PI3K/Akt = phosphatidylinositol 3 kinase/protein-serine-threonine kinase; PD-1/PD-L1 = programmed death-ligand 1/programmed cell death protein 1; TLR = Toll-like receptor; IL = interleukin; TGFβ = transforming growth factor-beta; PGE2 = prostaglandin E2; PAF = platelet-activating factor; TNF-α = tumor necrosis factor-alpha; Treg cells = regulatory t cell
The role of efferocytosis in the tumor microenvironment
The mechanisms of several efferocytosis-associated molecules and signaling pathways have been elucidated in the past few decades.
TAM receptors
TAM receptors promote the tumor microenvironment by promoting remodeling of the extracellular matrix and the release of factors conducive to cell proliferation, migration, and angiogenesis [53]. Under normal physiological conditions, TAM receptors are principally expressed on natural killer (NK) cells and antigen-presenting cells (APCs) such as macrophages and immature dendritic cells [54]. However, TAM receptors are overexpressed in various cancers, including lung cancer, leukemia, gastrointestinal cancer, and breast cancer, and the overexpression results in enhanced efferocytosis and a worse cancer outcome [55,56,57]. The underlying mechanisms of TAM receptors, particularly Axl and MerTK, have been widely researched.
The oncogenic and cancer-promoting mechanisms of MerTK are associated with the classic cell proliferation pathways phosphatidylinositol 3 kinase (PI3K)/protein-serine-threonine kinase (Akt) and mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) [55]. Through the PI3K/Akt signaling axis, MerTK induces polarization of M2 macrophages while inhibiting polarization of M1 macrophages, and thereby resulting in the resolution of inflammation [58]. M2 macrophages play a crucial role in the cancer microenvironment by facilitating tumor progression and invasion due to their immunosuppressive characteristics [59]. In addition to innate immunity, MerTK is involved in the modulation in adaptive immunity as genetic ablation of MerTK parallels increased intratumoral CD8+ T lymphocytes and promoted lymphocyte proliferation [48]. Moreover, MerTK-driven efferocytosis also induces the expression of the programmed death-ligand 1 (PD-L1), a molecule that promotes tumor escape, and therefore enhances the immune suppression microenvironment for cancer cells [57]. Notably, MerTK is essential for TIM-4 induced efferocytosis [60]. Since TIM-4 lacks an extensive intracellular domain, it works in tandem with MerTK to facilitate the phagocytosis of apoptotic cells [60]. Furthermore, it has been shown in many malignant tumors that MerTK is involved in the resistance of multiple anticancer therapies. In non-small cell lung cancer, for instance, MerTK participates in drug-resistance of epidermal growth factor receptor (EGFR) inhibitors [61].
Axl is typically activated by the Gas6 ligand, and the affinity of Gas6 for Axl is higher than to Tyro3 and MerTK receptors [62]. The Gas6/Axl signaling pathway influences cancer development and progression through its effect on tumor cell proliferation, invasion, metastasis, epithelial-mesenchymal transition (EMT), and angiogenesis [38, 63, 64]. A previous study demonstrated that Axl is required for EMT, which promotes metastasis of HER-2 positive breast cancer [65]. Several signaling pathways, including Janus kinase (JAK)/signal transducers and activators of transcription 1 (STAT1), PI3K/Akt, and NF-ĸB, MAPK/ERK are downstream of the Gas6/Axl pathway [66]. Therefore, macrophage polarization induced by the PI3K/Akt signaling axis is also involved in the cancer-promoting mechanisms of Axl. Moreover, Axl could mediate resistance to chemotherapy [67], radiotherapy, and targeted molecular therapy in many cancers [68]. For instance, overexpression of Axl promotes resistance to EGFR-tyrosine kinase inhibitor (TKI) through the PI3K/Akt and MAPK/ERK signaling pathways [69]. In small-cell lung cancer, Axl promotes primary and acquired resistance to WEE1 (WEE1 G2 checkpoint kinase) inhibition. This resistance occurs via activation of another G2-checkpoint protein, the checkpoint kinase 1 (CHK1), which is a parallel pathway for the repair of DNA damage [70].
Furthermore, TAM receptors are characterized as ubiquitylation substrates for the E3 ligase casitas B-lineage lymphoma-b (Cbl-b), which suppresses antitumor activities of NK cells, CD8+ T cells and CD4+ T cells while enhances Treg immunosuppressive activity [46, 47, 71, 72]. Consequently, the inhibition of Cbl-b by blocking TAM receptors has the potential to boost immunity against cancer.
Direct PS receptors
Among the TIM family, TIM-4 is expressed on the surface of phagocytes and is tightly involved in the efferocytosis [73, 74]. Although the overall function of TIM-4 remains obscure, several pieces of evidence suggest that TIM-4 is involved in tumor progression. Overexpression of TIM-4 has been shown to promote the proliferation of non-small cell lung carcinoma [75]. Also, a previous study demonstrated that TIM-4 attenuates the effect of chemotherapy and increases immune tolerance to cancer via interaction with AMPKα1 [76]. A recent study explored the role of TIM-4 in colorectal cancer and reported that TIM-4 promotes angiogenesis by upregulating vascular endothelial growth factor (VEGF). TIM-4 also recruits tumor-associated macrophages through the PI3K/Akt signaling pathway, thereby promoting cancer progression [77]. The roles of other direct PS receptors (BAI-1, stabilin-2 family members, and CD300 family members) remain elusive.
Bridging molecules
Gas6 bridges between PS and TAM receptors, thereby promoting cancer cell proliferation and migration [78]. Notably, leukocytes infiltrating through cancerous tissues upregulate Gas6, which contributes to tumor growth and invasion [79]. The role of the Gas6/Axl signaling pathway in cancers has been described in the “TAM receptors” section.
MFG-E8 bridges between PS and αvβ3/αvβ5 integrins [39] and its expression is upregulated in tumors [80]. MFG-E8 attenuates inflammation and increases Treg cells, efferocytosis, angiogenesis, allograft tolerance, tumorigenicity, and cancer metastasis [81, 82]. Moreover, overexpression of MFG-E8 is negatively associated with prognosis in various cancers including breast, colorectal and esophageal cancers [83,84,85]. According to Yamada et al., MFG-E8 promotes angiogenesis by upregulating the expression of VEGF and endothelin (ET)-1 in bone marrow-derived mesenchymal stromal cells to trigger tumor progression in melanomas [86]. Besides, MFG-E8 enhances M2 polarization of macrophages [86, 87], and blockade of MFG-E8 enhances antitumor effector T cells but inhibits Treg cells, leading to tumor destruction [17].
The implication of efferocytosis in cancer therapy
Given the vital role of efferocytosis in the tumor microenvironment, progression, and metastasis, efferocytosis-targeted approaches could offer a novel therapeutic strategy in tumorigenesis and cancer management [1, 20]. We have summarized some representative agents of efferocytosis-targeted therapy in Table. 1. Also, chemotherapy and radiotherapy induce apoptosis of cancer cells and increase the subsequent efferocytosis, which suppresses inflammatory responses. Therefore, combining these traditional therapies with efferocytosis-targeted therapy or other types of immunotherapy could enhance their efficacy and improve patient outcomes [73].
Blockade of “eat-me” signaling
Notably, “find-me” signals are not tumor-specific. More research has, therefore, focused on therapies targeted to the “eat-me” signaling pathway, among which the previously described PS signaling is the most common and the most widely studied.
PS targeting
Several PS targeting agents, such as annexin proteins and PS targeting antibodies, have been widely studied [1]. Annexin proteins, the naturally occurring ligands for PS, saturate and block the externalized PS, thus inhibiting the “eat-me” signaling pathway [102]. This blockage triggers a pro-inflammatory response, increases the immunogenicity of apoptotic tumor cells, and shifts the immunosuppressive environment towards an antitumor response [20, 103, 104]. PS targeting antibodies specifically bind to PS with high affinity. As PS is also expressed in vascular endothelial cells, these antibodies not only target PS-expressing tumors but also target tumor blood vessels [105,106,107]. The interaction between PS targeting antibodies and exposed PS increases the expression of inflammatory cytokines and reduces the expression of immunosuppressive myeloid-derived suppressor cells [108]. Besides, PS targeting antibodies induce the polarization of M1 macrophages and recruitment of mature dendritic cells, leading to an increase of tumor-specific cytotoxic T cells [108]. When used in combination with either chemotherapy, radiotherapy, or immune checkpoint antibodies (anti-CTLA-4 and anti-PD-1), PS targeting agents have been shown to facilitate the curative effect of these therapies [20, 103]. As such, pre-clinical agents associated with PS targeting antibodies such as Annexin A5 of annexin proteins and 3G4, 2aG4 and chimeric 1 N11 have been developed [20]. Multiple clinical trials of bavituximab, a PS targeting antibody, have also been carried out [88,89,90].
However, subsequent phase II study and phase III trial did not provide evidence on the substantial improvement of efficacy following the addition of bavituximab compared to the chemotherapy alone group [54, 109]. Besides efferocytosis, PS targeting therapy also interferes with the function of antigen-presenting cell (APCs) and induces non-selective inhibition of all PS-dependent phagocytic activity. Thus, PS inhibition may cause other harmful side effects on the body [54]. Notably, PS receptor-blocking approaches also inhibit PS signaling pathway.
TAM targeting
TAM receptors play a pleiotropic role in tumor pathophysiology and drug resistance. Previous studies have reported that all three TAM receptors are overexpressed in various cancers. This overexpression promotes oncogenic signaling and efferocytosis, resulting in a worse cancer outcome [55,56,57].
The Axl inhibitors potentiate the apoptosis of live cancer cells, reduce migration and invasion of tumor cells, and suppress efferocytosis [110]. Previous studies have also reported that Axl targeting synergizes with chemotherapy and other targeted therapies such as VEGF, EGFR, PI3K, PARP (poly ADP-ribose polymerase), and HER2 inhibitors to promote therapeutic efficacy [111,112,113]. There are currently five types of Axl targeting agents under development. These agents include small-molecule TKIs, nucleotide aptamers, monoclonal antibodies (mAbs), soluble receptors, and several natural compounds [38]. Research on reputable Axl inhibitors, especially the TKIs, has now progressed into clinical trial phases [38]. Nonetheless, Axl TKIs have increased clinical off-target toxicity and drug resistance [114, 115]. Nucleotide aptamers and mAbs are emerging therapy with higher affinity and specificity, and lower toxicity and drug resistance [92, 116,117,118], although their application is currently at preclinical stages [38]. Other than the transmembrane form, Axl can also exist in a soluble form once it is cleaved in the extracellular domain. Soluble Axl binds to Gas6 or Axl itself, thereby inhibiting the transmembrane Axl and Gas6 signaling pathway [93]. Besides, it has been shown that natural compounds such as celastrol [94] and dihydroartemisinin [95] show therapeutic potential through Axl inhibition.
MerTK can induce intrinsic and adaptive resistance of Axl-targeted agents, which advocates a dual targeting of Axl and MerTK for the hindrance of downstream signaling and tumor growth [91]. However, the ablation of MerTK may cause visual impairment [119, 120]. Hence, the safety of MerTK-targeted therapies should be explored further. Several preclinical studies have reported similar adaptive responses caused by single TKI therapy, and co-targeting of the receptor tyrosine kinase family could, therefore, be a novel strategy for overcoming drug resistance and increasing efficacy [61, 121,122,123,124,125]. Notably, cells of the innate immune system are involved in the initiation and regulation of adaptive immune response. Co-targeting innate immune checkpoints such as TAM receptors may thus enhance the recruitment and activation of adaptive immune cells and increase the therapeutic efficacy compared to the single adaptive immune checkpoint-targeted therapy [54].
MFG-E8 targeting
Although the administration of MFG-E8 nucleotide aptamer alone may not be effective enough for averting tumor progression and boosting immunity [96]. Previous studies have demonstrated that the down-regulation of MFG-E8 increases the sensitivity of tumor cells to TKIs and cytotoxic agents in vitro [126, 127]. Besides, the combination of chemotherapy and MFG-E8 RNA interference contributes to sustained inhibition of tumor survival and growth [96]. These synergistic actions could be attributed to several mechanisms since the down-regulation of MFG-E8 signaling results in several different effects, including (1) decrease in chemotherapy resistance of tumor cells; (2) inhibition of MFG-E8-mediated efferocytosis; (3) blockage of VEGF-induced angiogenesis; (4) enhanced cross-presentation between the dying tumor cells and dendritic cells; (5) reduced Treg cells and increased activation and function of CD4+ and CD8+ effector T cells [17, 96, 128, 129]. Indeed, MFG-E8 bridges between PS exposed on apoptotic cells and αvβ3/αvβ5 integrins expressed on macrophages [39, 130]. Therefore, integrin-targeted molecules such as extracellular matrix ligands, high-mobility group box 1, and inhibitory antibodies also suppress efferocytosis [97, 98].
In addition to the blockade of the above “eat-me” signaling pathways, targeting the TIM receptor family and Gas6, which are the direct PS receptors and bridging molecule for efferocytosis respectively, could also improve the current immunotherapies [1, 47]. However, the blockade of “eat-me” signaling pathways may produce excessive pro-inflammatory mediators and fail to produce sufficient factors for tissue repair [131, 132], which compromises on the rationality of this therapeutic strategy.
Blockade of “don’t-eat-me” signaling
Efferocytosis is an equilibrium formed following proper coordination among “find-me”, “eat-me” and “don’t-eat-me” signals [73]. The “don’t-eat-me” signaling are primarily emitted by CD47, whose receptor is signal regulatory protein-α (SIRP-α), a protein expressed on the surface of phagocytes [73, 133]. Previous studies have revealed that CD47 is overexpressed (approximately three-fold compared to healthy cells) on the plasma membrane of all human cancers and enables cancer cells to evade phagocytosis [49, 134]. Notably, the expression of CD47 mRNA is negatively correlated with patient survival rates [134]. Dysregulation of CD47, therefore, affects tumor-associated efferocytosis and represents a promising therapeutic strategy.
Anti-CD47 mAbs facilitate phagocytosis of cancer cells [134]. In addition, anti-CD47 mAbs decrease the ability of Treg cells to overcome immune evasion by cancer cells and increase the capacity of CD8+ T cells to exhibit an effective antitumor cytotoxic function [49]. Furthermore, anti-CD47 mAbs prevent or eliminate metastatic lesions and circulating tumor cells [135, 136]. Consequently, the blockade of CD47 signals inhibits the growth and metastasis of human tumors [134]. Since CD47 is also expressed on normal cells at varying levels, looming safety concerns on targeting CD47 should be addressed [137]. Previous studies have revealed that anti-CD47 mAbs could produce potent antitumor responses without causing any severe side effects, even at doses that exceed the minimum effective dose. This observation could be attributed to the lack of secondary prophagocytic “eat-me” signals on the surface of healthy cells [134, 138]. Under cellular stress, however, the calreticulin of healthy cells may translocate to the cell surface and result in phagocytosis of healthy cells by nearby macrophages. Thus, targeting CD47 should be applied with caution in the context of recent ongoing inflammatory or cytotoxic treatments [139, 140]. The findings of a study by Willingham et al. point out that a better efficacy of anti-CD47 mAbs therapy is correlated with smaller tumor size at the onset of treatment. The study further proposes that the optimal time to effect an anti-CD47 therapy is after maximal cytoreductive surgery [134].
Combination of anti-CD47 mAbs and other antitumor antibodies such as trastuzumab (anti-HER2 antibody), rituximab (anti-CD20 antibody), alemtuzumab (anti-CD52 antibody), and cetuximab (anti-epidermal growth factor receptor antibody) elicits a synergistic effect. This effect may be attributed to a magnifying effect of Fc receptor-dependent phagocytosis by the second antitumor antibody [49, 134, 141]. Such a combined therapy does not only result in increased therapeutic efficacy but also possesses several advantages. These advantages include (1) decreased off-target toxicity compared to chemotherapy; (2) reduced potential antibody toxicity compared to monotherapies; (3) enhanced cancer cell elimination, even when the epitope of a single drug target mutates [49]. Several anti-CD47 mAbs have so far been developed, including B6H12.2 and BRIC126 [49, 99, 100], and efficacy studies are ongoing. However, the research is based on xenotransplantation models, and more experimental studies and clinical trials should, therefore, be done to validate the reported efficacies.
Intercellular cell adhesion molecule-1
Intercellular cell adhesion molecule-1 (ICAM-1) is a transmembrane glycoprotein of the immunoglobulin supergene family, which is expressed in all leukocytes and is a ligand for β2 integrins [142, 143]. ICAM-1 is also expressed on the cell surface of many cancer types and facilitates tumor progression and metastasis [101]. ICAM-1 inhibits the efferocytosis of apoptotic tumor cells through the suppression of the PI3K/Akt signaling pathway, and the downregulation of efferocytosis leads to the decrease of M2 macrophage polarization, consequently inhibiting tumor progression and tumor metastasis [101, 144]. Therefore, the role of ICAM-1 in efferocytosis makes it a promising target for cancer treatment. Other agents that alter the polarization of tumor-associated macrophages are also of therapeutic potential.
Combined inhibition of apoptosis and secondary necrosis
A recent study [52] described two distinct mechanisms for cell death: apoptosis and secondary necrosis, both of which affect the tumor microenvironment in different ways. In the study, inhibition of efferocytosis did not suppress the production of tumor myeloid-derived suppressor cells, Treg cells, and some immunosuppressive mediators. Further research uncovered that this phenomenon could be because decreased efferocytosis induced secondary necrosis of apoptotic cells, which stimulated the expression of inflammation-resolving factor indoleamine-2,3-dioxygenase (IDO) 1, leading to the restoration of the immunosuppressive environment for cancer cells. The results indicate that apoptotic and necrotic cancer cells promote tumor progression through efferocytosis and IDO1 respectively. Combined inhibition of these two processes showed a better result in tumor regression and inhibition of metastasis [52].
Conclusion and future perspectives
Efferocytosis inhibits inflammatory responses, modulates the immune environment, and facilitates immune escape of cancer cells. Subsequently, efferocytosis promotes tumor development and progression. Efferocytosis is an equilibrium formed by perfect coordination among “find-me”, “eat-me” and “don’t-eat-me” signals. Among the three signals, the “find-me” signals do not exhibit specificity for antitumor targeting, and many studies have, therefore, focused on the “eat-me” and “don’t-eat-me” signals. These signaling pathways influence cancer development and progression by affecting the proliferation, invasion, and metastasis of tumor cells, epithelial-mesenchymal transition, and angiogenesis. Thus, the signals represent potential therapeutic targets for cancer treatment.
On the other hand, chemotherapy and radiotherapy induce apoptosis of cancer cells and increases the subsequent efferocytosis. Combining these traditional therapies with efferocytosis-targeted therapy could, therefore, enhance the efficacy and promote patient outcomes. Single TKI therapy also elicits adaptive responses and drug resistance, and co-targeting receptors of the tyrosine kinase family are promising for overcoming TKI-related drug resistance and increasing treatment efficacy. Also, the combined therapies could result in reduced side effects. However, the mechanisms underlying efferocytosis are obscure; for instance, the Tyro3 pathway has not been extensively explored. Besides, no compelling evidence on the efficacy of several efferocytosis inhibitors such as bavituximab has been established, and the potential clinical off-target toxicity may limit the clinical application. Furthermore, the safety of some efferocytosis-associated including MerTK inhibitors and anti-CD47 mAbs needs to be further studied.
Availability of data and materials
Not applicable.
Abbreviations
- S1P:
-
Sphingosine-1-phosphate
- PS:
-
Phosphatidylserine
- TIM:
-
T-cell immunoglobulin mucin
- BAI-1:
-
Brain-specific angiogenesis inhibitor 1
- TAM:
-
Tyro3, Axl and MerTK
- Gas:
-
Growth arrest-specific protein 6
- TGFβ:
-
Transforming growth factor-beta
- PGE2:
-
Prostaglandin E2
- PAF:
-
Platelet-activating factor
- IL:
-
Interleukin
- NFκB:
-
Factor-κ-gene binding
- TNF-α:
-
Tumor necrosis factor-alpha
- TLR:
-
Toll-like receptor
- SOCS:
-
Suppressor of cytokine signaling
- APCs:
-
Antigen-presenting cells
- JAK/STAT1:
-
Janus kinase/signal transducers and activators of transcription 1
- PI3K/Akt:
-
Phosphatidylinositol 3 kinase/protein-serine-threonine kinase
- PD-1:
-
Programmed death-ligand 1
- PD-L1:
-
Programmed cell death protein 1
- Treg cells:
-
Regulatory T cell
- TKI:
-
Tyrosine kinase inhibitor
- MFG-E8:
-
Milk fat globule epidermal growth factor-8
- CD:
-
Cluster of differentiation
- ICAM-1:
-
Intercellular cell adhesion molecule-1
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-mesenchymal transition
- CHK1:
-
Checkpoint kinase 1
- NK cell:
-
Natural killer cell
- VEGF:
-
Vascular endothelial growth factor
- ET:
-
Endothelin
- CTLA-4:
-
Cytotoxic T-lymphocyte-associated protein 4
- PARP:
-
Poly ADP-ribose polymerase
- IDO:
-
Indoleamine-2,3-dioxegenase
References
Kumar S, Calianese D, Birge RB. Efferocytosis of dying cells differentially modulate immunological outcomes in tumor microenvironment. Immunol Rev. 2017;280:149–64. https://doi.org/10.1111/imr.12587.
Werfel TA, Cook RS. Efferocytosis in the tumor microenvironment. Semin Immunopathol. 2018;40:545–54. https://doi.org/10.1007/s00281-018-0698-5.
Sachet M, Liang YY, Oehler R. The immune response to secondary necrotic cells. Apoptosis. 2017;22:1189–204. https://doi.org/10.1007/s10495-017-1413-z.
Gardai SJ, et al. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell. 2005;123:321–34. https://doi.org/10.1016/j.cell.2005.08.032.
Vandivier RW, Henson PM, Douglas IS. Burying the dead: the impact of failed apoptotic cell removal (efferocytosis) on chronic inflammatory lung disease. Chest. 2006;129:1673–82. https://doi.org/10.1378/chest.129.6.1673.
Tian L, et al. Enhanced efferocytosis by dendritic cells underlies memory T-cell expansion and susceptibility to autoimmune disease in CD300f-deficient mice. Cell Death Differ. 2016;23:1086–96. https://doi.org/10.1038/cdd.2015.161.
Kojima Y, Weissman IL, Leeper NJ. The role of Efferocytosis in atherosclerosis. Circulation. 2017;135:476–89. https://doi.org/10.1161/circulationaha.116.025684.
Kojima Y, et al. CD47-blocking antibodies restore phagocytosis and prevent atherosclerosis. Nature. 2016;536:86–90. https://doi.org/10.1038/nature18935.
Albert ML, Kim JI, Birge RB. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol. 2000;2:899–905. https://doi.org/10.1038/35046549.
Monks J, Smith-Steinhart C, Kruk ER, Fadok VA, Henson PM. Epithelial cells remove apoptotic epithelial cells during post-lactation involution of the mouse mammary gland. Biol Reprod. 2008;78:586–94. https://doi.org/10.1095/biolreprod.107.065045.
Hermetet F, et al. Efferocytosis of apoptotic human papillomavirus-positive cervical cancer cells by human primary fibroblasts. Biol Cell. 2016;108:189–204. https://doi.org/10.1111/boc.201500090.
Parnaik R, Raff MC, Scholes J. Differences between the clearance of apoptotic cells by professional and non-professional phagocytes. Curr Biol. 2000;10:857–60. https://doi.org/10.1016/s0960-9822(00)00598-4.
Bingle L, Brown NJ, Lewis CE. The role of tumour-associated macrophages in tumour progression: implications for new anticancer therapies. J Pathol. 2002;196:254–65. https://doi.org/10.1002/path.1027.
Kurahara H, et al. Significance of M2-polarized tumor-associated macrophage in pancreatic cancer. J Surg Res. 2011;167:e211–9. https://doi.org/10.1016/j.jss.2009.05.026.
Roy S, et al. Macrophage-derived Neuropilin-2 exhibits novel tumor-promoting functions. Cancer Res. 2018;78:5600–17. https://doi.org/10.1158/0008-5472.can-18-0562.
Lewis CE, Pollard JW. Distinct role of macrophages in different tumor microenvironments. Cancer Res. 2006;66:605–12. https://doi.org/10.1158/0008-5472.can-05-4005.
Jinushi M, et al. Milk fat globule epidermal growth factor-8 blockade triggers tumor destruction through coordinated cell-autonomous and immune-mediated mechanisms. J Exp Med. 2009;206:1317–26. https://doi.org/10.1084/jem.20082614.
Voll RE, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–1. https://doi.org/10.1038/37022.
Joseph M, Enting D. Immune responses in bladder Cancer-role of immune cell populations, prognostic factors and therapeutic implications. Front Oncol. 2019;9:1270. https://doi.org/10.3389/fonc.2019.01270.
Birge RB, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016;23:962–78. https://doi.org/10.1038/cdd.2016.11.
Ravichandran KS. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–55. https://doi.org/10.1016/j.immuni.2011.09.004.
Nishimura T, et al. Apoptotic cells of an epithelial cell line, AsPC-1, release monocyte chemotactic S19 ribosomal protein dimer. J Biochem. 2001;129:445–54. https://doi.org/10.1093/oxfordjournals.jbchem.a002876.
Lauber K, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. 2003;113:717–30. https://doi.org/10.1016/s0092-8674(03)00422-7.
Lauber K, Blumenthal SG, Waibel M, Wesselborg S. Clearance of apoptotic cells: getting rid of the corpses. Mol Cell. 2004;14:277–87. https://doi.org/10.1016/s1097-2765(04)00237-0.
Gude DR, et al. Apoptosis induces expression of sphingosine kinase 1 to release sphingosine-1-phosphate as a “come-and-get-me” signal. FASEB J. 2008;22:2629–38. https://doi.org/10.1096/fj.08-107169.
Truman LA, et al. CX3CL1/fractalkine is released from apoptotic lymphocytes to stimulate macrophage chemotaxis. Blood. 2008;112:5026–36. https://doi.org/10.1182/blood-2008-06-162404.
Elliott MR, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature. 2009;461:282–6. https://doi.org/10.1038/nature08296.
Martin SJ, et al. Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med. 1995;182:1545–56. https://doi.org/10.1084/jem.182.5.1545.
Fadok VA, et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol (Baltimore, Md : 1950). 1992;148:2207–16.
Miyanishi M, et al. Identification of Tim4 as a phosphatidylserine receptor. Nature. 2007;450:435–9. https://doi.org/10.1038/nature06307.
Kobayashi N, et al. TIM-1 and TIM-4 glycoproteins bind phosphatidylserine and mediate uptake of apoptotic cells. Immunity. 2007;27:927–40. https://doi.org/10.1016/j.immuni.2007.11.011.
Park D, et al. BAI1 is an engulfment receptor for apoptotic cells upstream of the ELMO/Dock180/Rac module. Nature. 2007;450:430–4. https://doi.org/10.1038/nature06329.
Park SY, et al. Rapid cell corpse clearance by stabilin-2, a membrane phosphatidylserine receptor. Cell Death Differ. 2008;15:192–201. https://doi.org/10.1038/sj.cdd.4402242.
Murakami Y, et al. CD300b regulates the phagocytosis of apoptotic cells via phosphatidylserine recognition. Cell Death Differ. 2014;21:1746–57. https://doi.org/10.1038/cdd.2014.86.
Tian L, et al. p85alpha recruitment by the CD300f phosphatidylserine receptor mediates apoptotic cell clearance required for autoimmunity suppression. Nat Commun. 2014;5:3146. https://doi.org/10.1038/ncomms4146.
He M, et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011;12:358–64. https://doi.org/10.1038/embor.2011.28.
van der Meer JH, van der Poll T, van ‘t Veer C. TAM receptors, Gas6, and protein S: roles in inflammation and hemostasis. Blood. 2014;123:2460–9. https://doi.org/10.1182/blood-2013-09-528752.
Zhu C, Wei Y, Wei X. AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer. 2019;18:153. https://doi.org/10.1186/s12943-019-1090-3.
Hanayama R, et al. Autoimmune disease and impaired uptake of apoptotic cells in MFG-E8-deficient mice. Science (New York, NY). 2004;304:1147–50. https://doi.org/10.1126/science.1094359.
Ramirez-Ortiz ZG, et al. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat Immunol. 2013;14:917–26. https://doi.org/10.1038/ni.2670.
Brown S, et al. Apoptosis disables CD31-mediated cell detachment from phagocytes promoting binding and engulfment. Nature. 2002;418:200–3. https://doi.org/10.1038/nature00811.
Fadok VA, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–8. https://doi.org/10.1172/jci1112.
Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. https://doi.org/10.1172/jci11638.
Tibrewal N, et al. Autophosphorylation docking site Tyr-867 in Mer receptor tyrosine kinase allows for dissociation of multiple signaling pathways for phagocytosis of apoptotic cells and down-modulation of lipopolysaccharide-inducible NF-kappaB transcriptional activation. J Biol Chem. 2008;283:3618–27. https://doi.org/10.1074/jbc.M706906200.
Camenisch TD, Koller BH, Earp HS, Matsushima GK. A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol (Baltimore, Md : 1950). 1999;162:3498–503.
Loeser S, et al. Spontaneous tumor rejection by cbl-b-deficient CD8+ T cells. J Exp Med. 2007;204:879–91. https://doi.org/10.1084/jem.20061699.
Paolino M, et al. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature. 2014;507:508–12. https://doi.org/10.1038/nature12998.
Cook RS, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. 2013;123:3231–42. https://doi.org/10.1172/jci67655.
Chao MP, et al. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713. https://doi.org/10.1016/j.cell.2010.07.044.
Knocke S, et al. Tailored tumor immunogenicity reveals regulation of CD4 and CD8 T cell responses against Cancer. Cell Rep. 2016;17:2234–46. https://doi.org/10.1016/j.celrep.2016.10.086.
Desrichard A, Snyder A, Chan TA. Cancer Neoantigens and applications for immunotherapy. Clin Cancer Res. 2016;22:807–12. https://doi.org/10.1158/1078-0432.Ccr-14-3175.
Werfel TA, et al. Treatment-induced tumor cell apoptosis and secondary necrosis drive tumor progression in the residual tumor microenvironment through MerTK and IDO1. Cancer Res. 2019;79:171–82. https://doi.org/10.1158/0008-5472.can-18-1106.
Pollard JW. Tumour-educated macrophages promote tumour progression and metastasis. Nat Rev Cancer. 2004;4:71–8. https://doi.org/10.1038/nrc1256.
Akalu YT, Rothlin CV, Ghosh S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol Rev. 2017;276:165–77. https://doi.org/10.1111/imr.12522.
Linger RM, Keating AK, Earp HS, Graham DK. TAM receptor tyrosine kinases: biologic functions, signaling, and potential therapeutic targeting in human cancer. Adv Cancer Res. 2008;100:35–83. https://doi.org/10.1016/s0065-230x(08)00002-x.
Rochlitz C, et al. Axl expression is associated with adverse prognosis and with expression of Bcl-2 and CD34 in de novo acute myeloid leukemia (AML): results from a multicenter trial of the Swiss Group for Clinical Cancer Research (SAKK). Leukemia. 1999;13:1352–8. https://doi.org/10.1038/sj.leu.2401484.
Nguyen KQ, et al. Overexpression of MERTK receptor tyrosine kinase in epithelial cancer cells drives efferocytosis in a gain-of-function capacity. J Biol Chem. 2014;289:25737–49. https://doi.org/10.1074/jbc.M114.570838.
Vergadi E, Ieronymaki E, Lyroni K, Vaporidi K, Tsatsanis C. Akt signaling pathway in macrophage activation and M1/M2 polarization. J Immunol (Baltimore, Md : 1950). 2017;198:1006–14. https://doi.org/10.4049/jimmunol.1601515.
Myers KV, Amend SR, Pienta KJ. Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer. 2019;18:94. https://doi.org/10.1186/s12943-019-1022-2.
Nishi C, Toda S, Segawa K, Nagata S. Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol Cell Biol. 2014;34:1512–20. https://doi.org/10.1128/mcb.01394-13.
Yan D, et al. MERTK promotes resistance to irreversible EGFR tyrosine kinase inhibitors in non-small cell lung cancers expressing wild-type EGFR family members. Clin Cancer Res. 2018;24:6523–35. https://doi.org/10.1158/1078-0432.ccr-18-0040.
Lew ED, et al. Differential TAM receptor-ligand-phospholipid interactions delimit differential TAM bioactivities. eLife. 2014;3. https://doi.org/10.7554/eLife.03385.
Vouri M, et al. Axl-EGFR receptor tyrosine kinase hetero-interaction provides EGFR with access to pro-invasive signalling in cancer cells. Oncogenesis. 2016;5:e266. https://doi.org/10.1038/oncsis.2016.66.
Korshunov VA, Mohan AM, Georger MA, Berk BC. Axl, a receptor tyrosine kinase, mediates flow-induced vascular remodeling. Circ Res. 2006;98:1446–52. https://doi.org/10.1161/01.RES.0000223322.16149.9a.
Goyette MA, et al. The receptor tyrosine kinase AXL is required at multiple steps of the metastatic Cascade during HER2-positive breast Cancer progression. Cell Rep. 2018;23:1476–90. https://doi.org/10.1016/j.celrep.2018.04.019.
Dent P. Crosstalk between ERK, AKT, and cell survival. Cancer Biol Ther. 2014;15:245–6. https://doi.org/10.4161/cbt.27541.
Lin JZ, et al. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget. 2017;8:41064–77. https://doi.org/10.18632/oncotarget.17026.
Giles KM, et al. Axl mediates acquired resistance of head and neck cancer cells to the epidermal growth factor receptor inhibitor erlotinib. Mol Cancer Ther. 2013;12:2541–58. https://doi.org/10.1158/1535-7163.mct-13-0170.
Tian Y, et al. Anexelekto (AXL) increases resistance to EGFR-TKI and activation of AKT and ERK1/2 in non-small cell lung Cancer cells. Oncol Res. 2016;24:295–303. https://doi.org/10.3727/096504016x14648701447814.
Sen T, et al. Targeting AXL and mTOR pathway overcomes primary and acquired resistance to WEE1 inhibition in small-cell lung Cancer. Clin Cancer Res. 2017;23:6239–53. https://doi.org/10.1158/1078-0432.ccr-17-1284.
Chiang JY, Jang IK, Hodes R, Gu H. Ablation of Cbl-b provides protection against transplanted and spontaneous tumors. J Clin Invest. 2007;117:1029–36. https://doi.org/10.1172/jci29472.
Wohlfert EA, Callahan MK, Clark RB. Resistance to CD4+CD25+ regulatory T cells and TGF-beta in Cbl-b−/− mice. J Immunol (Baltimore, Md : 1950). 2004;173:1059–65. https://doi.org/10.4049/jimmunol.173.2.1059.
Gheibi Hayat SM, Bianconi V, Pirro M, Sahebkar A. Efferocytosis: molecular mechanisms and pathophysiological perspectives. Immunol Cell Biol. 2019;97:124–33. https://doi.org/10.1111/imcb.12206.
Flannagan RS, Canton J, Furuya W, Glogauer M, Grinstein S. The phosphatidylserine receptor TIM4 utilizes integrins as coreceptors to effect phagocytosis. Mol Biol Cell. 2014;25:1511–22. https://doi.org/10.1091/mbc.E13-04-0212.
Zhang Q, et al. TIM-4 promotes the growth of non-small-cell lung cancer in a RGD motif-dependent manner. Br J Cancer. 2015;113:1484–92. https://doi.org/10.1038/bjc.2015.323.
Baghdadi M, et al. TIM-4 glycoprotein-mediated degradation of dying tumor cells by autophagy leads to reduced antigen presentation and increased immune tolerance. Immunity. 2013;39:1070–81. https://doi.org/10.1016/j.immuni.2013.09.014.
Tan X, Zhang Z, Yao H, Shen L. Tim-4 promotes the growth of colorectal cancer by activating angiogenesis and recruiting tumor-associated macrophages via the PI3K/AKT/mTOR signaling pathway. Cancer Lett. 2018;436:119–28. https://doi.org/10.1016/j.canlet.2018.08.012.
Sainaghi PP, et al. Gas6 induces proliferation in prostate carcinoma cell lines expressing the Axl receptor. J Cell Physiol. 2005;204:36–44. https://doi.org/10.1002/jcp.20265.
Loges S, et al. Malignant cells fuel tumor growth by educating infiltrating leukocytes to produce the mitogen Gas6. Blood. 2010;115:2264–73. https://doi.org/10.1182/blood-2009-06-228684.
Oba J, et al. Expression of milk fat globule epidermal growth factor-VIII may be an indicator of poor prognosis in malignant melanoma. Br J Dermatol. 2011;165:506–12. https://doi.org/10.1111/j.1365-2133.2011.10409.x.
Fens MH, et al. Angiogenic endothelium shows lactadherin-dependent phagocytosis of aged erythrocytes and apoptotic cells. Blood. 2008;111:4542–50. https://doi.org/10.1182/blood-2007-06-094763.
Li BZ, Zhang HY, Pan HF, Ye DQ. Identification of MFG-E8 as a novel therapeutic target for diseases. Expert Opin Ther Targets. 2013;17:1275–85. https://doi.org/10.1517/14728222.2013.829455.
Jia M, et al. Prognostic correlation between MFG-E8 expression level and colorectal Cancer. Arch Med Res. 2017;48:270–5. https://doi.org/10.1016/j.arcmed.2017.06.003.
Yu L, et al. MFG-E8 overexpression is associated with poor prognosis in breast cancer patients. Pathol Res Pract. 2019;215:490–8. https://doi.org/10.1016/j.prp.2018.12.036.
Kanemura T, et al. Immunoregulatory influence of abundant MFG-E8 expression by esophageal cancer treated with chemotherapy. Cancer Sci. 2018;109:3393–402. https://doi.org/10.1111/cas.13785.
Yamada K, et al. MFG-E8 drives melanoma growth by stimulating mesenchymal stromal cell-induced angiogenesis and M2 polarization of tumor-associated macrophages. Cancer Res. 2016;76:4283–92. https://doi.org/10.1158/0008-5472.can-15-2812.
Soki FN, et al. Polarization of prostate cancer-associated macrophages is induced by milk fat globule-EGF factor 8 (MFG-E8)-mediated efferocytosis. J Biol Chem. 2014;289:24560–72. https://doi.org/10.1074/jbc.M114.571620.
Chalasani P, et al. A phase I clinical trial of bavituximab and paclitaxel in patients with HER2 negative metastatic breast cancer. Cancer Med. 2015;4:1051–9. https://doi.org/10.1002/cam4.447.
Digumarti R, et al. Bavituximab plus paclitaxel and carboplatin for the treatment of advanced non-small-cell lung cancer. Lung Cancer (Amsterdam, Netherlands). 2014;86:231–6. https://doi.org/10.1016/j.lungcan.2014.08.010.
Gerber DE, et al. Phase I safety and pharmacokinetic study of bavituximab, a chimeric phosphatidylserine-targeting monoclonal antibody, in patients with advanced solid tumors. Clin Cancer Res. 2011;17:6888–96. https://doi.org/10.1158/1078-0432.ccr-11-1074.
McDaniel NK, et al. MERTK mediates intrinsic and adaptive resistance to AXL-targeting agents. Mol Cancer Ther. 2018;17:2297–308. https://doi.org/10.1158/1535-7163.mct-17-1239.
Duan Y, et al. A novel human anti-AXL monoclonal antibody attenuates tumour cell migration. Scand J Immunol. 2019;90:e12777. https://doi.org/10.1111/sji.12777.
Costa M, Bellosta P, Basilico C. Cleavage and release of a soluble form of the receptor tyrosine kinase ARK in vitro and in vivo. J Cell Physiol. 1996;168:737–44. https://doi.org/10.1002/(sici)1097-4652(199609)168:3<737::aid-jcp27>3.0.co;2-u.
Lee YJ, Kim SY, Lee C. Axl is a novel target of celastrol that inhibits cell proliferation and migration, and increases the cytotoxicity of gefitinib in EGFR mutant nonsmall cell lung cancer cells. Mol Med Rep. 2019;19:3230–6. https://doi.org/10.3892/mmr.2019.9957.
Paccez JD, et al. Dihydroartemisinin inhibits prostate cancer via JARID2/miR-7/miR-34a-dependent downregulation of Axl. Oncogenesis. 2019;8:14. https://doi.org/10.1038/s41389-019-0122-6.
Zhao JY, et al. Down-regulation of MFG-E8 by RNA interference combined with doxorubicin triggers melanoma destruction. Clin Exp Med. 2015;15:127–35. https://doi.org/10.1007/s10238-014-0277-6.
Banerjee S, et al. Intracellular HMGB1 negatively regulates efferocytosis. J Immunol (Baltimore, Md : 1950). 2011;187:4686–94. https://doi.org/10.4049/jimmunol.1101500.
Friggeri A, et al. HMGB1 inhibits macrophage activity in efferocytosis through binding to the alphavbeta3-integrin. Am J Phys Cell Phys. 2010;299:C1267–76. https://doi.org/10.1152/ajpcell.00152.2010.
Kim D, et al. Anti-CD47 antibodies promote phagocytosis and inhibit the growth of human myeloma cells. Leukemia. 2012;26:2538–45. https://doi.org/10.1038/leu.2012.141.
Chao MP, et al. Extranodal dissemination of non-Hodgkin lymphoma requires CD47 and is inhibited by anti-CD47 antibody therapy. Blood. 2011;118:4890–901. https://doi.org/10.1182/blood-2011-02-338020.
Yang M, Liu J, Piao C, Shao J, Du J. ICAM-1 suppresses tumor metastasis by inhibiting macrophage M2 polarization through blockade of efferocytosis. Cell Death Dis. 2015;6:e1780. https://doi.org/10.1038/cddis.2015.144.
Gerke V, Creutz CE, Moss SE. Annexins: linking Ca2+ signalling to membrane dynamics. Nat Rev Mol Cell Biol. 2005;6:449–61. https://doi.org/10.1038/nrm1661.
Munoz LE, et al. The influence on the immunomodulatory effects of dying and dead cells of Annexin V. J Leukoc Biol. 2007;81:6–14. https://doi.org/10.1189/jlb.0306166.
Stach CM, et al. Treatment with annexin V increases immunogenicity of apoptotic human T-cells in Balb/c mice. Cell Death Differ. 2000;7:911–5. https://doi.org/10.1038/sj.cdd.4400715.
DeRose P, Thorpe PE, Gerber DE. Development of bavituximab, a vascular targeting agent with immune-modulating properties, for lung cancer treatment. Immunotherapy. 2011;3:933–44. https://doi.org/10.2217/imt.11.87.
Ran S, Downes A, Thorpe PE. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002;62:6132–40.
Huang X, Bennett M, Thorpe PE. A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumors in mice. Cancer Res. 2005;65:4408–16. https://doi.org/10.1158/0008-5472.can-05-0031.
Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1:256–68. https://doi.org/10.1158/2326-6066.cir-13-0073.
Gerber DE, et al. Docetaxel combined with Bavituximab in previously treated, advanced nonsquamous non-small-cell lung Cancer. Clin Lung Cancer. 2016;17:169–76. https://doi.org/10.1016/j.cllc.2016.02.003.
Onken J, et al. Inhibiting receptor tyrosine kinase AXL with small molecule inhibitor BMS-777607 reduces glioblastoma growth, migration, and invasion in vitro and in vivo. Oncotarget. 2016;7:9876–89. https://doi.org/10.18632/oncotarget.7130.
Balaji K, et al. AXL inhibition suppresses the DNA damage response and sensitizes cells to PARP inhibition in multiple cancers. Mol Cancer Res. 2017;15:45–58. https://doi.org/10.1158/1541-7786.mcr-16-0157.
Liu L, et al. Novel mechanism of lapatinib resistance in HER2-positive breast tumor cells: activation of AXL. Cancer Res. 2009;69:6871–8. https://doi.org/10.1158/0008-5472.can-08-4490.
Weinger JG, et al. Loss of the receptor tyrosine kinase Axl leads to enhanced inflammation in the CNS and delayed removal of myelin debris during experimental autoimmune encephalomyelitis. J Neuroinflammation. 2011;8:49. https://doi.org/10.1186/1742-2094-8-49.
Miller MA, et al. Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov. 2016;6:382–99. https://doi.org/10.1158/2159-8290.cd-15-0933.
Kariolis MS, et al. An engineered Axl ‘decoy receptor’ effectively silences the Gas6-Axl signaling axis. Nat Chem Biol. 2014;10:977–83. https://doi.org/10.1038/nchembio.1636.
Sawyers CL. Opportunities and challenges in the development of kinase inhibitor therapy for cancer. Genes Dev. 2003;17:2998–3010. https://doi.org/10.1101/gad.1152403.
Leconet W, et al. Preclinical validation of AXL receptor as a target for antibody-based pancreatic cancer immunotherapy. Oncogene. 2014;33:5405–14. https://doi.org/10.1038/onc.2013.487.
Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157:220–33. https://doi.org/10.1111/j.1476-5381.2009.00190.x.
Lu Q, Lemke G. Homeostatic regulation of the immune system by receptor tyrosine kinases of the tyro 3 family. Science (New York, NY). 2001;293:306–11. https://doi.org/10.1126/science.1061663.
Ksantini M, Lafont E, Bocquet B, Meunier I, Hamel CP. Homozygous mutation in MERTK causes severe autosomal recessive retinitis pigmentosa. Eur J Ophthalmol. 2012;22:647–53. https://doi.org/10.5301/ejo.5000096.
Chen FL, Xia W, Spector NL. Acquired resistance to small molecule ErbB2 tyrosine kinase inhibitors. Clin Cancer Res. 2008;14:6730–4. https://doi.org/10.1158/1078-0432.ccr-08-0581.
Mancini M, et al. Combining three antibodies nullifies feedback-mediated resistance to erlotinib in lung cancer. Sci Signal. 2015;8:ra53. https://doi.org/10.1126/scisignal.aaa0725.
Yamaguchi H, Chang SS, Hsu JL, Hung MC. Signaling cross-talk in the resistance to HER family receptor targeted therapy. Oncogene. 2014;33:1073–81. https://doi.org/10.1038/onc.2013.74.
Kawada I, et al. Dramatic antitumor effects of the dual MET/RON small-molecule inhibitor LY2801653 in non-small cell lung cancer. Cancer Res. 2014;74:884–95. https://doi.org/10.1158/0008-5472.can-12-3583.
Kasikara C, et al. Phosphatidylserine sensing by TAM receptors regulates AKT-dependent Chemoresistance and PD-L1 expression. Mol Cancer Res. 2017;15:753–64. https://doi.org/10.1158/1541-7786.mcr-16-0350.
Jinushi M, et al. Milk fat globule EGF-8 promotes melanoma progression through coordinated Akt and twist signaling in the tumor microenvironment. Cancer Res. 2008;68:8889–98. https://doi.org/10.1158/0008-5472.can-08-2147.
Jinushi M, et al. MFG-E8-mediated uptake of apoptotic cells by APCs links the pro- and antiinflammatory activities of GM-CSF. J Clin Invest. 2007;117:1902–13. https://doi.org/10.1172/jci30966.
Neutzner M, et al. MFG-E8/lactadherin promotes tumor growth in an angiogenesis-dependent transgenic mouse model of multistage carcinogenesis. Cancer Res. 2007;67:6777–85. https://doi.org/10.1158/0008-5472.can-07-0165.
Sugano G, et al. Milk fat globule--epidermal growth factor--factor VIII (MFGE8)/lactadherin promotes bladder tumor development. Oncogene. 2011;30:642–53. https://doi.org/10.1038/onc.2010.446.
Hanayama R, et al. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–7. https://doi.org/10.1038/417182a.
Akitake-Kawano R, et al. Inhibitory role of Gas6 in intestinal tumorigenesis. Carcinogenesis. 2013;34:1567–74. https://doi.org/10.1093/carcin/bgt069.
Bosurgi L, et al. Paradoxical role of the proto-oncogene Axl and Mer receptor tyrosine kinases in colon cancer. Proc Natl Acad Sci U S A. 2013;110:13091–6. https://doi.org/10.1073/pnas.1302507110.
Ravichandran KS. Find-me and eat-me signals in apoptotic cell clearance: progress and conundrums. J Exp Med. 2010;207:1807–17. https://doi.org/10.1084/jem.20101157.
Willingham SB, et al. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci U S A. 2012;109:6662–7. https://doi.org/10.1073/pnas.1121623109.
Jaiswal S, Chao MP, Majeti R, Weissman IL. Macrophages as mediators of tumor immunosurveillance. Trends Immunol. 2010;31:212–9. https://doi.org/10.1016/j.it.2010.04.001.
Wyckoff JB, et al. Direct visualization of macrophage-assisted tumor cell intravasation in mammary tumors. Cancer Res. 2007;67:2649–56. https://doi.org/10.1158/0008-5472.can-06-1823.
Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–5. https://doi.org/10.1016/s0962-8924(00)01906-1.
Majeti R, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–99. https://doi.org/10.1016/j.cell.2009.05.045.
Obeid M, et al. Calreticulin exposure is required for the immunogenicity of gamma-irradiation and UVC light-induced apoptosis. Cell Death Differ. 2007;14:1848–50. https://doi.org/10.1038/sj.cdd.4402201.
Obeid M, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. https://doi.org/10.1038/nm1523.
Clynes RA, Towers TL, Presta LG, Ravetch JV. Inhibitory fc receptors modulate in vivo cytotoxicity against tumor targets. Nat Med. 2000;6:443–6. https://doi.org/10.1038/74704.
Staunton DE, Marlin SD, Stratowa C, Dustin ML, Springer TA. Primary structure of ICAM-1 demonstrates interaction between members of the immunoglobulin and integrin supergene families. Cell. 1988;52:925–33. https://doi.org/10.1016/0092-8674(88)90434-5.
Hubbard AK, Rothlein R. Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radic Biol Med. 2000;28:1379–86. https://doi.org/10.1016/s0891-5849(00)00223-9.
Hamai A, et al. ICAM-1 has a critical role in the regulation of metastatic melanoma tumor susceptibility to CTL lysis by interfering with PI3K/AKT pathway. Cancer Res. 2008;68:9854–64. https://doi.org/10.1158/0008-5472.can-08-0719.
Acknowledgements
Wish the global epidemic of 2019 novel coronavirus pneumonia ends soon.
Funding
This work was funded by China Postdoctoral Science Foundation (2017 M612010) and National Natural Science Foundation of China (81701144).
Author information
Authors and Affiliations
Contributions
YXZ, YHY, AWS drafted the manuscript; YCD revised the manuscript; YXZ and AWS reviewed and modified the manuscript. All authors agreed on the final version.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Zhou, Y., Yao, Y., Deng, Y. et al. Regulation of efferocytosis as a novel cancer therapy. Cell Commun Signal 18, 71 (2020). https://doi.org/10.1186/s12964-020-00542-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12964-020-00542-9