
Abstract
LINE1 retrotransposons are mobile DNA elements that copy and paste themselves into new sites in the genome. To ensure their evolutionary success, heritable new LINE-1 insertions accumulate in cells that can transmit genetic information to the next generation (i.e., germ cells and embryonic stem cells). It is our hypothesis that LINE1 retrotransposons, insertional mutagens that affect expression of genes, may be causal agents of early miscarriage in humans. The cell has evolved various defenses restricting retrotransposition-caused mutation, but these are occasionally relaxed in certain somatic cell types, including those of the early embryo. We predict that reduced suppression of L1s in germ cells or early-stage embryos may lead to excessive genome mutation by retrotransposon insertion, or to the induction of an inflammatory response or apoptosis due to increased expression of L1-derived nucleic acids and proteins, and so disrupt gene function important for embryogenesis. If correct, a novel threat to normal human development is revealed, and reverse transcriptase therapy could be one future strategy for controlling this cause of embryonic damage in patients with recurrent miscarriages.
Similar content being viewed by others


Background
Spontaneous abortion or miscarriage is defined as natural death of an embryo or fetus before the twentieth week of pregnancy (the term stillbirth is used after 20 weeks). Most miscarriages occur during the first 7 weeks when the embryonic trophoblast invades the endometrium in a process analogous to tumor invasion and metastasis. Among clinically confirmed pregnancies, the incidence of spontaneous miscarriage is about 15 percent. However, it is estimated that about 50 to 75 percent of total pregnancies are miscarried. Among these, most of the aborted embryos cease development soon after implantation, appearing as menorrhagia or delayed menstruation, and escape notice (reviewed in [1, 2]).
Numerous causes of spontaneous abortion have been identified, including maternal reproductive tract abnormalities, endocrine and immunological dysfunction, sperm issues, reproductive tract infections, cervical insufficiency, thrombophila, and chromosome abnormalities, among others [1, 3]. Abnormal chromosome karyotype is seen in about 50% of spontaneous abortion patients, with triploidy most common, followed by autosomal unbalanced translocation, and polyploidy, X monomer, autosomal monomer, chromosome balanced translocation, deletion, chimerism, inversion, overlap, and so on [4, 5]. During embryonic development a single lethal gene mutation may also lead to death of the embryo [6]. Furthermore, evidence suggests that epigenetic anomalies may lie behind some cases of early pregnancy loss [7]. Recently, the key role that the placenta exerts on embryo development has been uncovered, adding another layer of complexity to the miscarriage phenomenon [8]. However, in the case of recurrent pregnancy loss, defined as at least three consecutive miscarriages prior to 24 weeks gestation [9], cause can be identified in only about 50 percent of cases [10]. In general, the genetic causes of miscarriage are poorly understood: much more study is required.
Here we propose the hypothesis that Long Interspersed Element-1 (LINE-1 or L1) retrotransposon activity may be a previously unrecognized causal factor for some cases of spontaneous miscarriage in humans. We suggest that during the development of gametes or human embryos, increased LINE-1 genomic insertions may disrupt one or more genes critical for early human embryonic development leading to miscarriage. Retrotransposon insertions may also mediate chromosomal rearrangements and alter the local epigenetic environment, among other effects. Furthermore, as discussed below, there is increasing evidence that, apart from insertion mutation, elevated L1 expression, especially of its reverse transcriptase (RT) and endonuclease activities, may initiate DNA damage or an immune response [11, 12]. Such phenomena could lead to embryo damage.
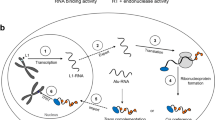
It has been estimated that over two-thirds of the human genome is repetitive DNA, most of this transposable elements (TEs) [13]. There are two main classes of TEs in genomes. Class II elements, the DNA transposons, replicate by a “cut and paste” mechanism, although no active transposons exist in humans. Class I elements, the retrotransposons, move by a “copy and paste” mechanism involving reverse transcription of an RNA intermediate and insertion of its cDNA copy at a new site in the genome. There are two major subgroups of Class I elements: long terminal repeat (LTR) and non-LTR retrotransposons. LTR retrotransposons include endogenous retroviruses (ERVs), relics of past rounds of germline infection by viruses that lost their ability to reinfect new cells. Human (H)ERVs compose 8% of our genome, although no remaining retrotransposition-competent HERVs have been identified. Nevertheless, genetic evidence suggests recent HERV activity in humans, and some HERV-K(HML-2) copies are polymorphic in the human population [14,15,16]. In humans the only autonomously active TE is LINE-1 (L1), a non-LTR retrotransposon with approximately half a million copies occupying about 17% of our genome [17]. L1s have also been responsible for the insertion in trans of over ten thousand processed pseudogenes and a million non-autonomous Short Interspersed Elements (SINEs), including Alu and SINE-VNTR-Alu (SVA) elements [18, 19]. A full-length active six kilobase bicistronic human L1 contains two non-overlapping open reading frames (ORFs) that encode the RNA-binding ORF1 protein (ORF1p) and the longer ORF2p, which functions both as a reverse transcriptase and DNA endonuclease (Fig. 1). Retrotransposition of a non-LTR retrotransposon is fundamentally different from that of an ERV, whose replication cycle involves reverse transcription of its genome in the cytoplasm. L1-encoded endonuclease nicks the bottom strand of target chromosomal DNA exposing a 3′-hydoxyl group that primes reverse transcription of the L1 RNA and synthesis of cDNA bound at the site of insertion, a process known as target primed reverse transcription (TPRT) [23].

The biology of a LINE-1 retrotransposon. The structure of a human L1 is shown. TSD: target site duplication; UTR: untranslated region; EN: endonuclease; RT: reverse transcriptase; C: carboxy-terminal segment; An: polyadenylation signal and tail. The LINE-1 replication cycle involves transcription and export of its RNA to the cytoplasm, which is translated and assembled in a ribonucleoprotein particle (RNP) together with L1 ORF1p and ORF2p. There is a strong cis-preference for L1 ORF1 and ORF2 proteins to bind their own encoding RNA in a retrotransposition-competent RNP. ORF1p binds L1 RNA as a trimer, however, it is unclear if it remains bound at the time of import of the RNP into the nucleus (denoted by ?) ) [20,21,22]. Reverse transcription of LINE-1 RNA to generate complementary (cDNA) occurs at the site of chromosomal insertion by TPRT [23]. L1s frequently become 5′-truncated when inserted in the genome
Most L1s are 5′ truncated and otherwise rearranged or mutated, and hence are incapable of retrotransposition. However, it is considered that about 100 LINE-1 sequences are full-length with intact ORFs and potentially active, although fewer than ten are considered to be “hot” and these consistently account for the bulk of new retrotransposition in humans [24,25,26,27]. Up to 5% of newborn children have a new retrotransposon insertion, and to date there have been 125 known human disease-causing germline non-LTR retrotransposon insertions [28,29,30,31,32]. The genomic revolution, including high-throughput (HT) sequencing analyses, has allowed estimates of the rates of L1 retrotransposition in mammals; indeed, recent studies indicate that a new L1 insertion may occur in 1 in 62 human births (1 in 40 births in the case of Alus), and 1 in 8 births in mice [33, 34]. The cell has evolved a battery of defenses to protect against unfettered retrotransposition (reviewed in [35, 36]). However, in some somatic cell types or under certain cellular conditions the defenses are lowered and retrotransposition increases.
Retrotransposon activity and its control in early embryonic development
In addition to the massive germ line expansion of L1s that occurred during mammalian evolution, recent investigations have documented ongoing retrotransposition in some somatic cell types, including neural progenitor cells, some tumors, stem cells, and notably early embryos (reviewed in [37,38,39,40,41,42,43,44,45,46,47,48,49]). Transgenic mouse and human studies demonstrated that somatic retrotransposition occurs in early-stage embryos causing somatic mosaicism [33, 50,51,52,53]. Cultured human embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs) express endogenous L1 RNA and proteins and support both retrotransposition of transfected reporter constructs [54,55,56,57,58,59,60] and modest levels of endogenous retrotransposition [61,62,63]. Recently, Muñoz-Lopez et al. [63] showed expression of non-LTR retrotransposons in the inner cell mass (ICM) and trophectoderm cells of pre-implantation human embryos and, using HT sequencing, de novo endogenous LINE-1 insertions within cells of the ICM as well as insertions restricted to the placenta. Thus, the cellular environment of early embryonic cells supports active retrotransposition. Of course, activity during early embryogenesis is beneficial for the evolutionary success of the L1, as new insertions have a high chance of being transmitted to the next generation.
Various cellular mechanisms restrict retrotransposition in the germline and embryos. For example, small interfering RNA (siRNA)-mediated gene silencing is an ancient strategy for controlling activity of TEs. RNA interference acts at the post-transcriptional level by causing RNA degradation and loss of translation, or at the transcriptional level by causing epigenetic modifications, including de novo methylation of TE sequences. piRNAs are small RNAs found in testes as well as human fetal ovaries that specifically silence TEs in the germline ( [64]; reviewed in [65,66,67,68,69]). A large percentage of mouse prepachytene piRNAs derives from retrotransposon sequences [70, 71], and the importance of piRNA pathway proteins in repressing retrotransposon expression in prenatal gonad development and spermatogenesis has repeatedly been demonstrated in mutant mouse lines defective for piRNA pathway proteins (reviewed in [36]).
It has been proposed that DNA methylation of CpGs evolved primarily as a host defense mechanism against TEs [72, 73]. Indeed, the L1 promoter is a prototypical CpG island and L1 promoter methylation is inversely correlated with L1 expression [74, 75]. In early mouse embryogenesis, repression of retrotransposons is maintained by histone and DNA methylation. However, successive waves of demethylation occur in the developing embryo and open windows for increased retrotransposon activity [76,77,78]. The first wave occurs shortly after fertilization until the morula stage. Around E8.5, demethylation occurs again in post-implantation primordial germ cells (PGCs) and continues to around E13 when PGCs have colonized the genital ridges (summarized in [79,80,81,82]).
The promoters of young active L1 elements are hypomethylated in hESCs compared to differentiated cells, which accounts in part for their higher levels of expression [83, 84]. In the case of embryonic tissues, human L1 methylation status has mostly been studied for the placenta, and both hypermethylation and hypomethylation have been reported. According to one study, LINE-1 methylation is significantly decreased in third trimester compared with first trimester placentas, a trend not paralleled by change in global methylation [85]. Perrin et al. [86] found that, compared with unaffected individuals, LINE-1 hypermethylation during development and differentiation of the placenta is two-fold higher in human hydatidiform mole patients, a condition involving abnormal placental growth and spontaneous abortion; methylation of other repeats and global methylation did not differ. Vasil’ev et al. [87] observed increased LINE-1 methylation in placental tissues of spontaneous abortions having mosaic aneuploidy but not in miscarriages with complete aneuploidy or in induced abortions. On the other hand, in extraembryonic tissues of spontaneous abortions with normal karyotype, LINE-1s were excessively hypomethylated. LINE-1 hypomethylation can result in enhanced L1 activation and consequent mutational insertions. Consistent with this hypothesis, Sanchez-Luque et al. [84] recently uncovered the critical role for DNA methylation in controlling activity of “hot” L1s in humans.
Many genes are involved in early embyogenesis
In principle, a new L1 insertion into a lethal gene could initiate a cascade leading to fetal death, although our diploid nature limits such consequences. Many signaling pathways and genes are involved in the process of miscarriage and single gene mutations may cause spontaneous abortion [6]. Based on a study of 489 single gene knockout mouse models, White et al. [88] found 29 percent of the genes to be lethal and 13 percent sublethal. KIF7 (kinesin family member gene 7) was the first human gene associated with fetal lethality when it was found to cause hydrolethalus and acrocallosal syndromes [89], and since then many other candidate genes have been identified. A review of 50 human studies identified a range of possible causative gene and copy number variations (CNVs) for miscarriage, including CHRNA1 (cholinergic receptor, nicotinic, alpha polypeptide 1), DYNC2H1 (dynein, cytoplasmic 2, heavy chain 1), and RYR1 (ryanodine receptor 1), which were reported by multiple studies [6]. Several whole exome sequence analyses of euploid miscarriages have been conducted, including a study of 30 fetuses in which mutations in FGFR3 (fibroblast growth factor receptor 3), COL2A1 (collagen, type II, alpha 1), and OFD1 (oral-facial-digital syndrome 1) genes, in addition to structural variants, accounted for 10 percent of the cohort [90]. Fang et al. [91] found that expression of VEGF (vascular endothelial growth factor), part of the angiogenesis signaling pathway, was significantly decreased in missed abortion tissue and correlated with increased levels of VEGFR1 (Vascular Endothelial Growth Factor Receptor 1) and Notch-1. Adache et al. [92] reviewed the key role of the cyclooxygenase (COX)-1 and -2 signaling pathways for repeated failure of embryo implantation. Affected genes found in other studies include KIF14 (kinesin family member 14) [93], IFT122 (intraflagellar transport 122) [94], PLCD4 (phospholipase C delta 4), and OSBPL5 (protein-like 5) [95]. In the case of recurrent miscarriage, cytokine gene polymorphisms, novel HLA alelles, and mutations in inflammatory factors and synaptonemal complex protein 3 (SYCP3) have been implicated. SYCP3 encodes an essential structural component of the synaptonemal complex and its mutation may result in chromosome abnormalities [96,97,98,99]. Thus, it is increasingly evident that mutation of any of many cellular pathway genes can initiate miscarriage.
Studies have demonstrated that healthy humans carry many mutated gene alleles [100]: elevated L1 retrotransposition during early embryogenesis could contribute to this mutation burden. It is possible that during early development epigenetic change or loss of a retrotransposon inhibiting factor could trigger derepression of active retrotransposons increasing the likelihood of an L1 inserting into a lethal gene. Recent studies have revealed the complexity of cellular factors and pathways regulating the activity of human retrotransposons. To date about 80 factors have been identified that limit expression or insertion of retrotransposons in cell culture or mouse models ([101]; reviewed in [36]). For example, knockout of DNA Methyltransferase 3 Like (DNMT3L) protein in mouse germ cells was accompanied by epigenetic change, reactivation of retrotransposons, and meiotic collapse [77]. Loss of TEX19.1 in mice leads to placental growth retardation, increased embryonic lethality, and derepressed retrotransposon expression in placenta and hypomethylated trophectoderm-derived cells, and its loss in mouse pluripotent embryonic stem cells increases retrotransposition of engineered L1 constructs [60, 102]. To cite another example, using a digital droplet PCR detection strategy, a startling 70-fold increase in retrotransposition of an L1 reporter transgene in a mouse deficient for MOV10L1, a piRNA pathway protein, was claimed by Newkirk et al. [103].
The impacts of retrotransposons on gene integrity extend beyond simple mutation by insertion: these have been the subjects of many reviews [18, 32, 104,105,106,107]. Ongoing retrotransposition events salt genomes with novel splice sites, polyadenylation signals, promoters, and transcription factor binding sites that can alter gene expression. Recombination between retrotransposons causes deletions, duplications, or rearrangements of gene sequence, and this is especially true for Alus [108]. L1-mediated retrotransposition insertion can also cause deletions up to a megabase at their sites of insertion [18, 105, 109,110,111,112]: one example is the deletion of an entire HLA-A gene caused by an SVA insertion that resulted in leukemia [113]. Retrotransposons are also associated with segmental duplications [114]; significantly, CNVs have also been linked with human miscarriage [115, 116]. Even more dramatic non-LTR retrotransposon-mediated genomic rearrangements may occur. L1 endonuclease activity and SVA retrotransposition leading to multiple DNA breaks was proposed as causal for one case of human germline chromothripsis [117], a phenomenon involving numerous chromosomal rearrangements in a single event, and one that has also been linked with severe congenital defects [118]. In summary, the mutagenic potential of active human retrotransposons can be significant.
A possible role for misregulation of retrotransposon expression in embryonic failure
Apart from insertion mutation, various studies have proposed physiological roles for retrotransposon expression, and these roles may turn pathological when expression is misregulated. Significant research has centered on the cellular effects of reverse transcriptase with implications for the developing embryo.
Functional RT activity has been reported in mature spermatozoa and pre-implantation embryos of mice [119,120,121]. Treatment of early stage mouse embryos with either antisense L1 oligonucleotides, an antibody to RT, or the RT inhibitor nevirapine reportedly arrested preimplantation development at the 2- to 4-cell stage, perhaps by altering levels of cellular cDNA synthesized by RT [120, 122]. (However, It should be noted, non-nucleoside reverse transciptase inhibitors like nevirapine, while they inhibit ERVs, were subsequently shown to not inhibit L1 cell culture retrotransposition [123,124,125]).
More recently, using antisense oligonucleotides to deplete L1 transcripts, Percharde et al. [126, 127] presented evidence that LINE1 expression plays a role in mouse embryonic exit from the 2-cell stage by recruiting nucleolin and Kap1 to repress the master transcriptional regulator Dux and activate rRNA synthesis. Furthermore, Jachowicz et al. [128] reported that LINE-1 activation after fertilization regulates global chromatin accessibility, and that artificial prolongation of L1 transcription in mouse embryos interferes with their development. Thus, both teams obtained comparable results after altering LINE-1 expression in mouse embryos, suggesting that proper functioning of a potential mutagen paradoxically also plays a role in embryonic development.
Elevated expression of an L1 transgene in mice null for Maelstrom, a piRNA pathway gene, was associated with increased meiotic prophase I defects, DNA damage, and fetal oocyte attrition [129, 130]. Oocyte attrition is a mysterious process involving loss of about two-thirds of human meiotic prophase oocytes [131]. The fact that treating mice with a nucleoside analog blocked oocyte attrition suggests roles for retrotransposon RT and perhaps endonuclease activities. As a normal part of TPRT, L1 ORF2 endonuclease generates dsDNA breaks that recruit repair proteins to the site of element insertion. However, transient transfection of an L1 in cell culture has been reported to induce DNA breaks manyfold in excess of what would be expected for TPRT-mediated insertions alone, and DNA damage caused by overexpression of ORF2p can induce genotoxic stress and cell death [132,133,134].
Recent evidence suggests that cellular conditions that stimulate increased expression of L1s, and therefore ORF2 protein and its RT, may generate ectopic retrotransposon cDNAs not engaged in TPRT at the site of genome integration. For example, aged cells and mice accumulate cytoplasmic L1-derived cDNAs, triggering an interferon response as a result of misidentification of these self-derived nucleic acids as non-self, while treatment with reverse transcriptase inhibitors reduces inflammation and increases viability and lifespan [135, 136]. Thomas et al. [137] also reported an interferon response and toxicity associated with accumulation of extrachomosomal L1-related single-stranded DNAs in neurons derived from hESCs lacking TREX1, a DNA exonuclease mutated in patients with Aicardi-Goutières syndrome (AGS), a rare childhood Type I interferonopathy involving loss of brain white matter [138].
While some studies have suggested that interferons play crucial roles in mammalian pregnancy, abnormal inflammatory reactions have also been associated with early pregnancy loss (reviewed in [139, 140]). Higher levels of Th1-type or pro-inflammatory cytokines, including IFNγ, were found in women with recurrent miscarriage when compared with women with normal pregnancies [141, 142]. Whether increased expression of retrotransposon-encoded RT can induce an interferon response in the developing embryo remains to be tested.
Testing the hypothesis
Recent years have seen the development of various HT sequencing strategies that could be applied to detection of de novo non-LTR retrotransposon insertions in genomic DNA of miscarriage samples. These include hybridization-based enrichment methods (including RC-seq [143]), selective PCR amplification (including ATLAS-Seq, L1-Seq, TIP-seq, and other methods [144,145,146,147,148,149,150]), and algorithms to analyze whole genome sequence (including The Transposable Element Analyzer (Tea), TEBreak, The Mobile Element Locator Tool (MELT), and others (https://github.com/adamewing/tebreak; [31, 151,152,153,154,155,156]). Candidate insertions are compared with insertions detected in the reference human genome, databases of non-reference polymorphic retrotansposons (such as dbRIP and euL1db [157, 158]), and parental blood DNA sequence to ascertain that the insertions occurred during development of the embryo or within the parental germline. One should further validate the insertions by site-specific PCR and Sanger sequencing of the amplicons to confirm the exact location of the 3′ and 5′ junctions. The best candidate tissues for initial testing for retrotransposon-caused defects may be recurrent miscarriages, which affect 1 to 2 percent of couples and for which cause can be identified in only half of cases [10, 159,160,161]. If available fetal tissue amounts are limited, primary cell lines may be derived and expanded in culture. Alternatively, and despite significant challenges [162], single cell genomics may be used to identify new L1 insertions in miscarriage samples. Of course, studies to assess retrotransposon insertions in early human embryonic development may be frustrated by access to tissues, so alternatively transgenic mouse models for L1 retrotransposition can be useful [51, 53, 163,164,165].
L1 RNA expression in miscarriage-related samples may be assessed by RT-qPCR, Northern blotting, RNA FISH, and RNA-Seq methods. A number of papers discuss the analysis algorithms, special protocols, difficulties, and caveats to be considered when analyzing expression of high-copy number retrotransposon loci with highly similar sequences [42, 83, 165,166,167,168,169,170]. Changes in L1 protein levels or patterns of subcellular distribution may be assayed using immunohistochemistry and Western blotting. Many labs have developed effective L1 α-ORF1p antibodies; we recommend the 4H1 α-ORF1p antibody available from MilliporeSigma [171]. Endogenous L1 ORF2p is expressed at very low levels and few effective antibodies have been reported [172,173,174].
If increased retroelement mRNA and proteins are detected in miscarriage samples, one would predict an increase in RT activity with possible consequences for the cell, as noted above. Various assays have been established to detect RT activity in cells, whether deriving from L1 ORF2p or HERV pol genes [175,176,177]. Using RT-qPCR to assay changes in expression of interferon-stimulated genes may also reveal autoinflammatory effects of retrotransposon misregulation, as described above for AGS and some other autoimmune conditions [137, 178,179,180].
If this hypothesis is supported, that retrotransposon activity significantly contributes to fetal damage in some patients, ameliorative options are conceivable. Administration of low doses of RT inhibitor to such patients could reduce the incidence of future retrotransposition and miscarriage. In cell culture experiments, L1 retrotransposition is strongly inhibited by nucleoside reverse transcriptase inhibitors (NRTIs) and recent studies have identified NRTIs that limit L1s and/or HERVs, including drugs widely used against HIV-1 infection [123,124,125]. Of interest, pilot clinical trials using NRTI inhibitors to reduce retrotransposon activity have begun for amyotrophic lateral sclerosis (ClinicalTrials.gov Identifiers NCT02437110, NCT02868580, [181]) and AGS (NCT02363452, NCT03304717). One of the AGS trials, now completed, reported reduction in interferon-stimulated gene expression in treated patients [182].
In summary, we propose that increased LINE-1 activity may be one cause of spontaneous miscarriage. This concept is reasonable according to the points outlined above, and especially considering the reported involvement L1 RNAs in proper preimplantation embryo development [126, 128] and the increased activity of L1s in early human embryos [63]. Deleterious cell effects of elevated retrotransposon activity may involve L1-mediated gene disruption by insertion mutation or the initiation of inflammatory or DNA damage responses. However, as for oocyte attrition in mice [129], it is possible that human embryos typically clear damaged embryonic cells by apoptosis and related mechanisms. If active L1s are indeed involved in miscarriage, it would increase understanding of spontaneous miscarriage mechanisms and have clinical significance for pregnant women. LINE-1 insertions may become a new reason given to miscarriage patients, and such knowledge could be used to develop novel preventive measures.
Availability of data and materials
Not applicable.
Abbreviations
- AGS:
-
Aicardi-Goutières syndrome
- AZT:
-
azidothymidine (zidovudine)
- CNV:
-
copy number variation
- ESC:
-
embryonic stem cell
- HERV:
-
human endogenous retrovirus
- HT:
-
high-throughput
- ICM:
-
inner cell mass
- iPSC:
-
induced pluripotent stem cell
- LINE-1:
-
Long Interspersed Element-1
- LTR:
-
long terminal repeat
- NRTI:
-
nucleoside reverse transcriptase inhibitor
- ORF:
-
open reading frame
- PGC:
-
primordial germ cell
- RNP :
-
ribonucleoprotein particle
- SINE:
-
Short Interspersed Element
- RT:
-
reverse transcriptase
- TPRT:
-
target primed reverse transcription
References
Larsen EC, Christiansen OB, Kolte AM, Macklon N. New insights into mechanisms behind miscarriage. BMC Med. 2013;11:154.
Pinar MH, Gibbins K, He M, Kostadinov S, Silver R. Early pregnancy losses: review of nomenclature, histopathology, and possible etiologies. Fetal Pediatr Pathol. 2018;37(3):191–209.
Homer HA. Modern management of recurrent miscarriage. Aust N Z J Obstet Gynaecol. 2019;59(1):36–44.
Hassold T, Chen N, Funkhouser J, Jooss T, Manuel B, Matsuura J, Matsuyama A, Wilson C, Yamane JA, Jacobs PA. A cytogenetic study of 1000 spontaneous abortions. Ann Hum Genet. 1980;44(2):151–78.
Daniely M, Aviram-Goldring A, Barkai G, Goldman B. Detection of chromosomal aberration in fetuses arising from recurrent spontaneous abortion by comparative genomic hybridization. Hum Reprod. 1998;13(4):805–9.
Colley E, Hamilton S, Smith P, Morgan NV, Coomarasamy A, Allen S. Potential genetic causes of miscarriage in euploid pregnancies: a systematic review. Hum Reprod Update. 2019;25(4):452–72.
Yin LJ, Zhang Y, Lv PP, He WH, Wu YT, Liu AX, Ding GL, Dong MY, Qu F, Xu CM, et al. Insufficient maintenance DNA methylation is associated with abnormal embryonic development. BMC Med. 2012;10:26.
Perez-Garcia V, Fineberg E, Wilson R, Murray A, Mazzeo CI, Tudor C, Sienerth A, White JK, Tuck E, Ryder EJ, et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature. 2018;555(7697):463–8.
Ford HB, Schust DJ. Recurrent pregnancy loss: etiology, diagnosis, and therapy. Rev Obstet Gynecol. 2009;2(2):76–83.
Practice Committee of the American Society for Reproductive Medicine. Evaluation and treatment of recurrent pregnancy loss: a committee opinion. Fertil Steril. 2012;98(5):1103–11.
Wallace NA, Belancio VP, Deininger PL. L1 mobile element expression causes multiple types of toxicity. Gene. 2008;419(1-2):75–81.
Saleh A, Macia A, Muotri AR. Transposable elements, inflammation, and neurological disease. Front Neurol. 2019;10:894.
de Koning AP, Gu W, Castoe TA, Batzer MA, Pollock DD. Repetitive elements may comprise over two-thirds of the human genome. PLoS Genet. 2011;7(12):e1002384.
Marchi E, Kanapin A, Magiorkinis G, Belshaw R. Unfixed endogenous retroviral insertions in the human population. J Virol. 2014;88(17):9529–37.
Naveira H, Bello X, Abal-Fabeiro JL, Maside X. Evidence for the persistence of an active endogenous retrovirus (ERVE) in humans. Genetica. 2014;142(5):451–60.
Wildschutte JH, Williams ZH, Montesion M, Subramanian RP, Kidd JM, Coffin JM. Discovery of unfixed endogenous retrovirus insertions in diverse human populations. Proc Natl Acad Sci. 2016;113(16):E2326–34.
Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921.
Richardson SR, Doucet AJ, Kopera HC, Moldovan JB, Garcia-Perez JL, Moran JV. The Influence of LINE-1 and SINE retrotransposons on mammalian genomes. Microbiol Spectr. 2015;3(2):MDNA3-0061-2014.
Frankish A, Diekhans M, Ferreira AM, Johnson R, Jungreis I, Loveland J, Mudge JM, Sisu C, Wright J, Armstrong J, et al. GENCODE reference annotation for the human and mouse genomes. Nucleic Acids Res. 2019;47(D1):D766–d73.
Wei W, Gilbert N, Ooi SL, Lawler JF, Ostertag EM, Kazazian HH, Boeke JD, Moran JV. Human L1 retrotransposition: cis preference versus trans complementation. Mol Cell Biol. 2001;21(4):1429–39.
Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat Genet. 2000;24(4):363–7.
Martin SL, Branciforte D, Keller D, Bain DL. Trimeric structure for an essential protein in L1 retrotransposition. Proc Natl Acad Sci U S A. 2003;100(24):13815–20.
Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72(4):595–605.
Brouha B, Meischl C, Ostertag E, de Boer M, Zhang Y, Neijens H, Roos D, Kazazian HH Jr. Evidence consistent with human L1 retrotransposition in maternal meiosis I. Am J Hum Genet. 2002;71(2):327–36.
Seleme MC, Vetter MR, Cordaux R, Bastone L, Batzer MA, Kazazian HH Jr. Extensive individual variation in L1 retrotransposition capability contributes to human genetic diversity. Proc Natl Acad Sci U S A. 2006;103(17):6611–6.
Beck CR, Collier P, Macfarlane C, Malig M, Kidd JM, Eichler EE, Badge RM, Moran JV. LINE-1 retrotransposition activity in human genomes. Cell. 2010;141(7):1159–70.
Yang L, Metzger GA, McLaughlin RN. Characterization of LINE-1 transposons in a human genome at allelic resolution. bioRxiv. 2019. https://doi.org/10.1101/594200.
Kazazian HH Jr. An estimated frequency of endogenous insertional mutations in humans. Nat Genet. 1999;22(2):130.
Li X, Scaringe WA, Hill KA, Roberts S, Mengos A, Careri D, Pinto MT, Kasper CK, Sommer SS. Frequency of recent retrotransposition events in the human factor IX gene. Hum Mutat. 2001;17(6):511–9.
Cordaux R, Hedges DJ, Herke SW, Batzer MA. Estimating the retrotransposition rate of human Alu elements. Gene. 2006;373:134–7.
Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, Zhou Q, Kirkness EF, Levy S, Batzer MA, et al. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009;19(9):1516–26.
Hancks DC, Kazazian HH. Roles for retrotransposon insertions in human disease. Mob DNA. 2016;7(1):1.
Richardson SR, Gerdes P, Gerhardt DJ, Sanchez-Luque FJ, Bodea GO, Munoz-Lopez M, Jesuadian JS, Kempen MHC, Carreira PE, Jeddeloh JA, et al. Heritable L1 retrotransposition in the mouse primordial germline and early embryo. Genome Res. 2017;27(8):1395–405.
Feusier J, Watkins WS, Thomas J, Farrell A, Witherspoon DJ, Baird L, Ha H, Xing J, Jorde LB. Pedigree-based estimation of human mobile element retrotransposition rates. Genome Res. 2019;29(10):1567-77.
Crichton JH, Dunican DS, MacLennan M, Meehan RR, Adams IR. Defending the genome from the enemy within: mechanisms of retrotransposon suppression in the mouse germline. Cell Mol Life Sci. 2014;71(9):1581–605.
Goodier JL. Restricting retrotransposons: a review. Mob DNA. 2016;7:16.
Coufal NG, Garcia-Perez JL, Peng GE, Yeo GW, Mu Y, Lovci MT, Morell M, O’Shea KS, Moran JV, Gage FH. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460(7259):1127–31.
Thomas CA, Paquola AC, Muotri AR. LINE-1 retrotransposition in the nervous system. Annu Rev Cell Dev Biol. 2012;28:555–73.
Reilly MT, Faulkner GJ, Dubnau J, Ponomarev I, Gage FH. The role of transposable elements in health and diseases of the central nervous system. J Neurosci. 2013;33(45):17577–86.
Carreira PE, Richardson SR, Faulkner GJ. L1 retrotransposons, cancer stem cells and oncogenesis. FEBS J. 2014;281(1):63–73.
Erwin JA, Marchetto MC, Gage FH. Mobile DNA elements in the generation of diversity and complexity in the brain. Nat Rev Neurosci. 2014;15(8):497–506.
Goodier JL. Retrotransposition in tumors and brains. Mob DNA. 2014;5(1):11.
Richardson SR, Morell S, Faulkner GJ. L1 retrotransposons and somatic mosaicism in the brain. Annu Rev Genet. 2014;48:1–27.
Kemp JR, Longworth MS. Crossing the LINE toward genomic instability: LINE-1 retrotransposition in cancer. Front Chem. 2015;3:68.
Gerdes P, Richardson SR, Mager DL, Faulkner GJ. Transposable elements in the mammalian embryo: pioneers surviving through stealth and service. Genome Biol. 2016;17(1):1.
Burns KH. Transposable elements in cancer. Nat Rev Cancer. 2017;17(7):415–24.
Scott EC, Devine SE. The role of somatic L1 retrotransposition in human cancers. Viruses. 2017;9(6):E131.
Faulkner GJ, Garcia-Perez JL. L1 mosaicism in mammals: extent, effects, and evolution. Trends Genet. 2017;33(11):802–16.
Richardson SR, Faulkner GJ. Heritable L1 retrotransposition events during development: understanding their origins: examination of heritable, endogenous L1 retrotransposition in mice opens up exciting new questions and research directions. Bioessays. 2018;40(6):e1700189.
Babushok DV, Ostertag EM, Courtney CE, Choi JM, Kazazian HH Jr. L1 integration in a transgenic mouse model. Genome Res. 2006;16(2):240–50.
An W, Han JS, Wheelan SJ, Davis ES, Coombes CE, Ye P, Triplett C, Boeke JD. Active retrotransposition by a synthetic L1 element in mice. Proc Natl Acad Sci. 2006;103(49):18662–7.
van den Hurk JA, Meij IC, Seleme MC, Kano H, Nikopoulos K, Hoefsloot LH, Sistermans EA, de Wijs IJ, Mukhopadhyay A, Plomp AS, et al. L1 retrotransposition can occur early in human embryonic development. Hum Mol Genet. 2007;16(13):1587–92.
Kano H, Godoy I, Courtney C, Vetter MR, Gerton GL, Ostertag EM, Kazazian HH. L1 retrotransposition occurs mainly in embryogenesis and creates somatic mosaicism. Genes Dev. 2009;23(11):1303–12.
Garcia-Perez JL, Marchetto MC, Muotri AR, Coufal NG, Gage FH, O’Shea KS, Moran JV. LINE-1 retrotransposition in human embryonic stem cells. Hum Mol Genet. 2007;16(13):1569–77.
Garcia-Perez JL, Morell M, Scheys JO, Kulpa DA, Morell S, Carter CC, Hammer GD, Collins KL, O’Shea KS, Menendez P, et al. Epigenetic silencing of engineered L1 retrotransposition events in human embryonic carcinoma cells. Nature. 2010;466(7307):769–73.
Macia A, Muñoz-Lopez M, Cortes JL, Hastings RK, Morell S, Lucena-Aguilar G, Marchal JA, Badge RM, Garcia-Perez JL. Epigenetic control of retrotransposon expression in human embryonic stem cells. Mol Cell Biol. 2011;31(2):300–16.
Wissing S, Montano M, Garcia-Perez JL, Moran JV, Greene WC. Endogenous APOBEC3B restricts LINE-1 retrotransposition in transformed cells and human embryonic stem cells. J Biol Chem. 2011;286(42):36427–37.
Wissing S, Muñoz-Lopez M, Macia A, Yang Z, Montano M, Collins W, Garcia-Perez JL, Moran JV, Greene WC. Reprogramming somatic cells into iPS cells activates LINE-1 retroelement mobility. Hum Mol Genet. 2012;21(1):208–18.
Friedli M, Turelli P, Kapopoulou A, Rauwel B, Castro-Díaz N, Rowe HM, Ecco G, Unzu C, Planet E, Lombardo A, et al. Loss of transcriptional control over endogenous retroelements during reprogramming to pluripotency. Genome Res. 2014;24(8):1251–9.
MacLennan M, Garcia-Canadas M, Reichmann J, Khazina E, Wagner G, Playfoot CJ, Salvador-Palomeque C, Mann AR, Peressini P, Sanchez L, et al. Mobilization of LINE-1 retrotransposons is restricted by Tex19.1 in mouse embryonic stem cells. Elife. 2017;6:e26152.
Arokium H, Kamata M, Kim S, Kim N, Liang M, Presson AP, Chen IS. Deep sequencing reveals low incidence of endogenous LINE-1 retrotransposition in human induced pluripotent stem cells. PLoS One. 2014;9(10):e108682.
Klawitter S, Fuchs NV, Upton KR, Muñoz-Lopez M, Shukla R, Wang J, Garcia-Cañadas M, Lopez-Ruiz C, Gerhardt DJ, Sebe A, et al. Reprogramming triggers endogenous L1 and Alu retrotransposition in human induced pluripotent stem cells. Nat Commun. 2016;7:10286.
Muñoz-Lopez M, Vilar R, Philippe C, Rahbari R, Richardson SR, Andres-Anton M, Widmann T, Cano D, Cortes JL, Rubio-Roldan A, et al. LINE-1 retrotransposition impacts the genome of human pre-implantation embryos and extraembryonic tissues. bioRxiv. 2019. https://doi.org/10.1101/522623.
Williams Z, Morozov P, Mihailovic A, Lin C, Puvvula PK, Juranek S, Rosenwaks Z, Tuschl T. Discovery and Characterization of piRNAs in the human fetal ovary. Cell Rep. 2015;13(4):854–63.
O’Donnell KA, Boeke JD. Mighty Piwis defend the germline against genome intruders. Cell. 2007;129(1):37–44.
Obbard DJ, Gordon KHJ, Buck AH, Jiggins FM. The evolution of RNAi as a defence against viruses and transposable elements. Philos Trans R Soc B: Biol Sci. 2009;364(1513):99–115.
Siomi MC, Sato K, Pezic D, Aravin AA. PIWI-interacting small RNAs: the vanguard of genome defence. Nat Rev Mol Cell Biol. 2011;12(4):246–58.
Yang Z, Chen K-M, Pandey Radha R, Homolka D, Reuter M, Janeiro Bruno Kotska R, Sachidanandam R, Fauvarque M-O, McCarthy Andrew A, Pillai Ramesh S. PIWI Slicing and EXD1 drive biogenesis of nuclear piRNAs from cytosolic targets of the mouse piRNA pathway. Mol Cell. 2016;61(1):138–52.
Russell SJ, Stalker L, LaMarre J. PIWIs, piRNAs and retrotransposons: complex battles during reprogramming in gametes and early embryos. Reprod Domest Anim. 2017;52(Suppl 4):28–38.
Aravin AA, Sachidanandam R, Girard A, Fejes-Toth K, Hannon GJ. Developmentally regulated piRNA clusters implicate MILI in transposon control. Science. 2007;316(5825):744–7.
Aravin AA, Sachidanandam R, Bourc’his D, Schaefer C, Pezic D, Toth KF, Bestor T, Hannon GJ. A piRNA pathway primed by individual transposons is linked to de novo DNA methylation in mice. Mol Cell. 2008;31(6):785–99.
Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13(8):335–40.
Bestor TH. Cytosine methylation mediates sexual conflict. Trends Genet. 2003;19(4):185–90.
Crowther P, Doherty J, Linsenmeyer M, Williamson M, Woodcock D. Revised genomic consensus for the hypermethylated CpG island region of the human L1 transposon and integration sites of full length L1 elements from recombinant clones made using methylation-tolerant host strains. Nucleic Acids Res. 1991;19(9):2395–401.
Hata K, Sakaki Y. Identification of critical CpG sites for repression of L1 transcription by DNA methylation. Gene. 1997;189(2):227–34.
Sanford JP, Clark HJ, Chapman VM, Rossant J. Differences in DNA methylation during oogenesis and spermatogenesis and their persistence during early embryogenesis in the mouse. Genes Dev. 1987;1(10):1039–46.
Bourc’his D, Bestor TH. Meiotic catastrophe and retrotransposon reactivation in male germ cells lacking Dnmt3L. Nature. 2004;431(7004):96–9.
Marius Walter AT, Pérez-Palacios R, Bourc’his D. An epigenetic switch ensures transposon repression upon dynamic loss of DNA methylation in embryonic stem cells. eLife Sci. 2016;5:e11418.
Seki Y, Yamaji M, Yabuta Y, Sano M, Shigeta M, Matsui Y, Saga Y, Tachibana M, Shinkai Y, Saitou M. Cellular dynamics associated with the genome-wide epigenetic reprogramming in migrating primordial germ cells in mice. Development. 2007;134(14):2627–38.
Bao J, Yan W. Male germline control of transposable elements. Biol Reprod. 2012;86(5):162–14.
Saitou M, Kagiwada S, Kurimoto K. Epigenetic reprogramming in mouse pre-implantation development and primordial germ cells. Development. 2012;139(1):15–31.
Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48(6):849–62.
Munoz-Lopez M, Garcia-Canadas M, Macia A, Morell S, Garcia-Perez JL. Analysis of LINE-1 expression in human pluripotent cells. Methods Mol Biol. 2012;873:113–25.
Sanchez-Luque FJ, Kempen MHC, Gerdes P, Vargas-Landin DB, Richardson SR, Troskie RL, Jesuadian JS, Cheetham SW, Carreira PE, Salvador-Palomeque C, et al. LINE-1 evasion of epigenetic repression in humans. Mol Cell. 2019;75(3):590–604.e12.
He ZM, Li J, Hwa YL, Brost B, Fang Q, Jiang SW. Transition of LINE-1 DNA methylation status and altered expression in first and third trimester placentas. PLoS One. 2014;9(5):e96994.
Perrin D, Ballestar E, Fraga MF, Frappart L, Esteller M, Guerin JF, Dante R. Specific hypermethylation of LINE-1 elements during abnormal overgrowth and differentiation of human placenta. Oncogene. 2007;26(17):2518–24.
Vasil’ev SA, Tolmacheva EN, Kashevarova AA, Sazhenova EA, Lebedev IN. Methylation status of line-1 retrotransposon in chromosomal mosaicism during the early stages of human embryonic development. Mol Biol (Mosk). 2015;49(1):165–74.
White JK, Gerdin AK, Karp NA, Ryder E, Buljan M, Bussell JN, Salisbury J, Clare S, Ingham NJ, Podrini C, et al. Genome-wide generation and systematic phenotyping of knockout mice reveals new roles for many genes. Cell. 2013;154(2):452–64.
Putoux A, Thomas S, Coene KL, Davis EE, Alanay Y, Ogur G, Uz E, Buzas D, Gomes C, Patrier S, et al. KIF7 mutations cause fetal hydrolethalus and acrocallosal syndromes. Nat Genet. 2011;43(6):601–6.
Carss KJ, Hillman SC, Parthiban V, McMullan DJ, Maher ER, Kilby MD, Hurles ME. Exome sequencing improves genetic diagnosis of structural fetal abnormalities revealed by ultrasound. Hum Mol Genet. 2014;23(12):3269–77.
Fang Y, Yu S, Ma Y, Sun P, Ma D, Ji C, Kong B. Association of Dll4/notch and HIF-1a -VEGF signaling in the angiogenesis of missed abortion. PLoS One. 2013;8(8):e70667.
Achache H, Tsafrir A, Prus D, Reich R, Revel A. Defective endometrial prostaglandin synthesis identified in patients with repeated implantation failure undergoing in vitro fertilization. Fertil Steril. 2010;94(4):1271–8.
Filges I, Nosova E, Bruder E, Tercanli S, Townsend K, Gibson WT, Rothlisberger B, Heinimann K, Hall JG, Gregory-Evans CY, et al. Exome sequencing identifies mutations in KIF14 as a novel cause of an autosomal recessive lethal fetal ciliopathy phenotype. Clin Genet. 2014;86(3):220–8.
Tsurusaki Y, Yonezawa R, Furuya M, Nishimura G, Pooh RK, Nakashima M, Saitsu H, Miyake N, Saito S, Matsumoto N. Whole exome sequencing revealed biallelic IFT122 mutations in a family with CED1 and recurrent pregnancy loss. Clin Genet. 2014;85(6):592–4.
Filges I, Manokhina I, Penaherrera MS, McFadden DE, Louie K, Nosova E, Friedman JM, Robinson WP. Recurrent triploidy due to a failure to complete maternal meiosis II: whole-exome sequencing reveals candidate variants. Mol Hum Reprod. 2015;21(4):339–46.
Bolor H, Mori T, Nishiyama S, Ito Y, Hosoba E, Inagaki H, Kogo H, Ohye T, Tsutsumi M, Kato T, et al. Mutations of the SYCP3 gene in women with recurrent pregnancy loss. Am J Hum Genet. 2009;84(1):14–20.
Rull K, Nagirnaja L, Laan M. Genetics of recurrent miscarriage: challenges, current knowledge, future directions. Front Genet. 2012;3:34.
Suzumori N, Sugiura-Ogasawara M. Genetic factors as a cause of miscarriage. Curr Med Chem. 2010;17(29):3431–7.
Aruna M, Nagaraja T, Andal Bhaskar S, Tarakeswari S, Reddy AG, Thangaraj K, Singh L, Reddy BM. Novel alleles of HLA-DQ and -DR loci show association with recurrent miscarriages among South Indian women. Hum Reprod. 2011;26(4):765–74.
Xue Y, Chen Y, Ayub Q, Huang N, Ball EV, Mort M, Phillips AD, Shaw K, Stenson PD, Cooper DN, et al. Deleterious- and disease-allele prevalence in healthy individuals: insights from current predictions, mutation databases, and population-scale resequencing. Am J Hum Genet. 2012;91(6):1022–32.
Liu N, Lee CH, Swigut T, Grow E, Gu B, Bassik MC, Wysocka J. Selective silencing of euchromatic L1s revealed by genome-wide screens for L1 regulators. Nature. 2018;553(7687):228–32.
Reichmann J, Reddington JP, Best D, Read D, Ollinger R, Meehan RR, Adams IR. The genome-defence gene Tex19.1 suppresses LINE-1 retrotransposons in the placenta and prevents intra-uterine growth retardation in mice. Hum Mol Genet. 2013;22(9):1791–806.
Newkirk SJ, Lee S, Grandi FC, Gaysinskaya V, Rosser JM, Vanden Berg N, Hogarth CA, Marchetto MCN, Muotri AR, Griswold MD, et al. Intact piRNA pathway prevents L1 mobilization in male meiosis. Proc Natl Acad Sci U S A. 2017;114(28):E5635–e44.
Goodier JL, Kazazian HH. Retrotransposons revisited: the restraint and rehabilitation of parasites. Cell. 2008;135(1):23–35.
Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12(1):187–215.
Huang CR, Burns KH, Boeke JD. Active transposition in genomes. Annu Rev Genet. 2012;46:651–75.
Mita P, Boeke JD. How retrotransposons shape genome regulation. Curr Opin Genet Dev. 2016;37:90–100.
Kim S, Cho CS, Han K, Lee J. Structural variation of alu element and human disease. Genomics Inform. 2016;14(3):70–7.
Gilbert N, Lutz-Prigge S, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110(3):315–25.
Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110(3):327–38.
Mine M, Chen JM, Brivet M, Desguerre I, Marchant D, de Lonlay P, Bernard A, Ferec C, Abitbol M, Ricquier D, et al. A large genomic deletion in the PDHX gene caused by the retrotranspositional insertion of a full-length LINE-1 element. Hum Mutat. 2007;28(2):137–42.
Lee J, Ha J, Son SY, Han K. Human genomic deletions generated by SVA-associated events. Comp Funct Genomics. 2012;2012:807270.
Takasu M, Hayashi R, Maruya E, Ota M, Imura K, Kougo K, Kobayashi C, Saji H, Ishikawa Y, Asai T, et al. Deletion of entire HLA-A gene accompanied by an insertion of a retrotransposon. Tissue Antigens. 2007;70(2):144–50.
Bailey JA, Liu G, Eichler EE. An Alu transposition model for the origin and expansion of human segmental duplications. Am J Hum Genet. 2003;73(4):823–34.
Bagheri H, Mercier E, Qiao Y, Stephenson MD, Rajcan-Separovic E. Genomic characteristics of miscarriage copy number variants. Mol Hum Reprod. 2015;21(8):655–61.
Wen J, Hanna CW, Martell S, Leung PC, Lewis SM, Robinson WP, Stephenson MD, Rajcan-Separovic E. Functional consequences of copy number variants in miscarriage. Mol Cytogenet. 2015;8:6.
Nazaryan-Petersen L, Bertelsen B, Bak M, Jonson L, Tommerup N, Hancks DC, Tumer Z. Germline chromothripsis driven by L1-mediated retrotransposition and Alu/Alu homologous recombination. Hum Mutat. 2016;37(4):385–95.
de Pagter MS, van Roosmalen MJ, Baas AF, Renkens I, Duran KJ, van Binsbergen E, Tavakoli-Yaraki M, Hochstenbach R, van der Veken LT, Cuppen E, et al. Chromothripsis in healthy individuals affects multiple protein-coding genes and can result in severe congenital abnormalities in offspring. Am J Hum Genet. 2015;96(4):651–6.
Giordano R, Magnano AR, Zaccagnini G, Pittoggi C, Moscufo N, Lorenzini R, Spadafora C. Reverse transcriptase activity in mature spermatozoa of mouse. J Cell Biol. 2000;148(6):1107–13.
Pittoggi C, Sciamanna I, Mattei E, Beraldi R, Lobascio AM, Mai A, Quaglia MG, Lorenzini R, Spadafora C. Role of endogenous reverse transcriptase in murine early embryo development. Mol Reprod Dev. 2003;66(3):225–36.
Sciamanna I, Vitullo P, Curatolo A, Spadafora C. Retrotransposons, reverse transcriptase and the genesis of new genetic information. Gene. 2009;448(2):180–6.
Beraldi R, Pittoggi C, Sciamanna I, Mattei E, Spadafora C. Expression of LINE-1 retroposons is essential for murine preimplantation development. Mol Reprod Dev. 2006;73(3):279–87.
Jones RB, Garrison KE, Wong JC, Duan EH, Nixon DF, Ostrowski MA. Nucleoside analogue reverse transcriptase inhibitors differentially inhibit human LINE-1 retrotransposition. PLoS One. 2008;3(2):e1547.
Dai L, Huang Q, Boeke JD. Effect of reverse transcriptase inhibitors on LINE-1 and Ty1 reverse transcriptase activities and on LINE-1 retrotransposition. BMC Biochem. 2011;12:18.
Banuelos-Sanchez G, Sanchez L, Benitez-Guijarro M, Sanchez-Carnerero V, Salvador-Palomeque C, Tristan-Ramos P, Benkaddour-Boumzaouad M, Morell S, Garcia-Puche JL, Heras SR, et al. Synthesis and characterization of specific reverse transcriptase inhibitors for mammalian LINE-1 retrotransposons. Cell Chem Biol. 2019;26(8):1095–109.e14.
Percharde M, Lin CJ, Yin Y, Guan J, Peixoto GA, Bulut-Karslioglu A, Biechele S, Huang B, Shen X, Ramalho-Santos M. A LINE1-nucleolin partnership regulates early development and ESC identity. Cell. 2018;174(2):391–405.e19.
Honson DD, Macfarlan TS. A lncRNA-like role for LINE1s in development. Dev Cell. 2018;46(2):132–4.
Jachowicz JW, Bing X, Pontabry J, Boskovic A, Rando OJ, Torres-Padilla ME. LINE-1 activation after fertilization regulates global chromatin accessibility in the early mouse embryo. Nat Genet. 2017;49(10):1502–10.
Malki S, van der Heijden Godfried W, O’Donnell Kathryn A, Martin Sandra L, Bortvin A. A role for retrotransposon LINE-1 in fetal oocyte attrition in mice. Dev Cell. 2014;29(5):521–33.
Chuma S. LINE-1 of evidence for fetal oocyte attrition by retrotransposon. Dev Cell. 2014;29(5):501–2.
Morita Y, Tilly JL. Oocyte apoptosis: like sand through an hourglass. Dev Biol. 1999;213(1):1–17.
Haoudi A, Semmes OJ, Mason JM, Cannon RE. Retrotransposition-competent human LINE-1 induces apoptosis in cancer cells with intact p53. J Biomed Biotechnol. 2004;2004(4):185–94.
Belgnaoui SM, Gosden RG, Semmes OJ, Haoudi A. Human LINE-1 retrotransposon induces DNA damage and apoptosis in cancer cells. Cancer Cell Int. 2006;6(1):1.
Gasior SL, Wakeman TP, Xu B, Deininger PL. The human LINE-1 retrotransposon creates DNA double-strand breaks. J Mol Biol. 2006;357(5):1383–93.
De Cecco M, Ito T, Petrashen AP, Elias AE, Skvir NJ, Criscione SW, Caligiana A, Brocculi G, Adney EM, Boeke JD, et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature. 2019;566(7742):73–8.
Simon M, Van Meter M, Ablaeva J, Ke Z, Gonzalez RS, Taguchi T, De Cecco M, Leonova KI, Kogan V, Helfand SL, et al. LINE1 derepression in aged wild-type and SIRT6-deficient mice drives inflammation. Cell Metab. 2019;29(4):871–85.e5.
Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, Macia A, Crow YJ, Muotri AR. Modeling of TREX1-dependent autoimmune disease using human stem cells highlights L1 accumulation as a source of neuroinflammation. Cell Stem Cell. 2017;21(3):319–31.e8.
Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC, et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. 2006;38(8):917-20.
Chaouat G, Menu E, Mognetti B, Moussa M, Cayol V, Mostefaoui Y, Dubanchet S, Khadel P, Volumenie JL, Rongieres CB, et al. Immunopathology of early pregnancy. Infect Dis Obstet Gynecol. 1997;5(2):73–92.
Micallef A, Grech N, Farrugia F, Schembri-Wismayer P, Calleja-Agius J. The role of interferons in early pregnancy. Gynecol Endocrinol. 2014;30(1):1–6.
Polgar K, Hill JA. Identification of the white blood cell populations responsible for Th1 immunity to trophoblast and the timing of the response in women with recurrent pregnancy loss. Gynecol Obstet Investig. 2002;53(1):59–64.
Marzi M, Vigano A, Trabattoni D, Villa ML, Salvaggio A, Clerici E, Clerici M. Characterization of type 1 and type 2 cytokine production profile in physiologic and pathologic human pregnancy. Clin Exp Immunol. 1996;106(1):127–33.
Baillie JK, Barnett MW, Upton KR, Gerhardt DJ, Richmond TA, De Sapio F, Brennan PM, Rizzu P, Smith S, Fell M, et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011;479(7374):534–7.
Badge RM, Alisch RS, Moran JV. ATLAS: a system to selectively identify human-specific L1 insertions. Am J Hum Genet. 2003;72(4):823–38.
Iskow RC, McCabe MT, Mills RE, Torene S, Pittard WS, Neuwald AF, Van Meir EG, Vertino PM, Devine SE. Natural mutagenesis of human genomes by endogenous retrotransposons. Cell. 2010;141(7):1253–61.
Ewing AD, Kazazian HH Jr. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res. 2010;20(9):1262–70.
Evrony G, Cai X, Lee E, Hills LB, Elhosary PC, Lehmann Hillel S, Parker JJ, Atabay Kutay D, Gilmore Edward C, Poduri A, et al. Single-neuron sequencing analysis of L1 retrotransposition and somatic mutation in the human brain. Cell. 2012;151(3):483–96.
Rahbari R, Badge RM. Combining amplification typing of L1 active subfamilies (ATLAS) with high-throughput sequencing. Methods Mol Biol. 2016;1400:95–106.
Philippe C, Vargas-Landin DB, Doucet AJ, van Essen D, Vera-Otarola J, Kuciak M, Corbin A, Nigumann P, Cristofari G. Activation of individual L1 retrotransposon instances is restricted to cell-type dependent permissive loci. Elife. 2016;5:e13926.
Pradhan B, Cajuso T, Katainen R, Sulo P, Tanskanen T, Kilpivaara O, Pitkanen E, Aaltonen LA, Kauppi L, Palin K. Detection of subclonal L1 transductions in colorectal cancer by long-distance inverse-PCR and Nanopore sequencing. Sci Rep. 2017;7(1):14521.
Ewing AD, Kazazian HH Jr. Whole-genome resequencing allows detection of many rare LINE-1 insertion alleles in humans. Genome Res. 2011;21(6):985–90.
Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, Lohr JG, Harris CC, Ding L, Wilson RK, et al. Landscape of somatic retrotransposition in human cancers. Science. 2012;337(6097):967–71.
Tubio JMC, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, Gundem G, Pipinikas CP, Zamora J, Raine K, et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345(6196):1251343.
Helman E, Lawrence MS, Stewart C, Sougnez C, Getz G, Meyerson M. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014;24(7):1053–63.
Ewing AD. Transposable element detection from whole genome sequence data. Mob DNA. 2015;6(1):24.
Gardner EJ, Lam VK, Harris DN, Chuang NT, Scott EC, Pittard WS, Mills RE, Devine SE. The Mobile Element Locator Tool (MELT): population-scale mobile element discovery and biology. Genome Res. 2017;27(11):1916–29.
Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat. 2006;27(4):323–9.
Mir AA, Philippe C, Cristofari G. euL1db: the European database of L1HS retrotransposon insertions in humans. Nucleic Acids Res. 2015;43(Database issue):D43–7.
Katz VL, Kuller JA. Recurrent miscarriage. Am J Perinatol. 1994;11(6):386–97.
Li TC, Makris M, Tomsu M, Tuckerman E, Laird S. Recurrent miscarriage: aetiology, management and prognosis. Hum Reprod Update. 2002;8(5):463–81.
Rai R, Regan L. Recurrent miscarriage. Lancet. 2006;368(9535):601–11.
Evrony GD, Lee E, Park PJ, Walsh CA. Resolving rates of mutation in the brain using single-neuron genomics. eLife. 2016;5:e12966.
Ostertag EM, DeBerardinis RJ, Goodier JL, Zhang Y, Yang N, Gerton GL, Kazazian HH Jr. A mouse model of human L1 retrotransposition. Nat Genet. 2002;32(4):655–60.
Okudaira N, Goto M, Yanobu-Takanashi R, Tamura M, An A, Abe Y, Kano S, Hagiwara S, Ishizaka Y, Okamura T. Involvement of retrotransposition of long interspersed nucleotide element-1 in skin tumorigenesis induced by 7,12-dimethylbenz[a]anthracene and 12-O-tetradecanoylphorbol-13-acetate. Cancer Sci. 2011;102(11):2000–6.
O’Donnell KA, An W, Schrum CT, Wheelan SJ, Boeke JD. Controlled insertional mutagenesis using a LINE-1 (ORFeus) gene-trap mouse model. Proc Natl Acad Sci U S A. 2013;110(29):E2706–13.
Deininger P, Morales ME, White TB, Baddoo M, Hedges DJ, Servant G, Srivastav S, Smither ME, Concha M, DeHaro DL, et al. A comprehensive approach to expression of L1 loci. Nucleic Acids Res. 2017;45(5):e31.
Jin Y, Hammell M. Analysis of RNA-Seq data using TEtranscripts. Methods Mol Biol. 2018;1751:153–67.
Jeong HH, Yalamanchili HK, Guo C, Shulman JM, Liu Z. An ultra-fast and scalable quantification pipeline for transposable elements from next generation sequencing data. Pac Symp Biocomput. 2018;23:168–79.
Navarro FC, Hoops J, Bellfy L, Cerveira E, Zhu Q, Zhang C, Lee C, Gerstein MB. TeXP: Deconvolving the effects of pervasive and autonomous transcription of transposable elements. PLoS Comput Biol. 2019;15(8):e1007293.
Bendall ML, de Mulder M, Iñiguez LP, Lecanda-Sánchez A, Pérez-Losada M, Ostrowski MA, et al. Telescope: characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput Biol. 2019;15(9):e1006453.
Rodić N, Sharma R, Sharma R, Zampella J, Dai L, Taylor MS, Hruban RH, Iacobuzio-Donahue CA, Maitra A, Torbenson MS, et al. Long interspersed element-1 protein expression is a hallmark of many human cancers. Am J Pathol. 2014;184(5):1280–6.
Chen L, Dahlstrom JE, Chandra A, Board P, Rangasamy D. Prognostic value of LINE-1 retrotransposon expression and its subcellular localization in breast cancer. Breast Cancer Res Treat. 2012;136(1):129–42.
Ergun S, Buschmann C, Heukeshoven J, Dammann K, Schnieders F, Lauke H, Chalajour F, Kilic N, Stratling WH, Schumann GG. Cell type-specific expression of LINE-1 open reading frames 1 and 2 in fetal and adult human tissues. J Biol Chem. 2004;279(26):27753–63.
Ardeljan D, Wang X, Oghbaie M, Taylor MS, Husband D, Deshpande V, Steranka JP, Gorbounov M, Yang WR, Sie B, et al. LINE-1 ORF2p expression is nearly imperceptible in human cancers. Mob DNA. 2019;11:1.
MacGowan DJ, Scelsa SN, Imperato TE, Liu KN, Baron P, Polsky B. A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology. 2007;68(22):1944–6.
Kulpa DA, Moran JV. Cis-preferential LINE-1 reverse transcriptase activity in ribonucleoprotein particles. Nat Struct Mol Biol. 2006;13(7):655–60.
Viollet S, Doucet AJ, Cristofari G. Biochemical Approaches to Study LINE-1 Reverse Transcriptase Activity In Vitro. Methods Mol Biol. 2016;1400:357–76.
Carter V, LaCava J, Taylor MS, Liang SY, Mustelin C, Ukadike KC, Bengtsson A, Lood C, Mustelin T. High prevalence and disease correlation of autoantibodies against p40 encoded by long interspersed nuclear elements (LINE-1) in systemic lupus erythematosus. Arthritis Rheumatol. 2019. https://doi.org/10.1002/art.41054.
Heinrich MJ, Purcell CA, Pruijssers AJ, Zhao Y, Spurlock CF 3rd, Sriram S, Ogden KM, Dermody TS, Scholz MB, Crooke PS 3rd, et al. Endogenous double-stranded Alu RNA elements stimulate IFN-responses in relapsing remitting multiple sclerosis. J Autoimmun. 2019;100:40–51.
Tam OH, Ostrow LW, Gale Hammell M. Diseases of the nERVous system: retrotransposon activity in neurodegenerative disease. Mob DNA. 2019;10:32.
Gold J, Rowe DB, Kiernan MC, Vucic S, Mathers S, van Eijk RPA, Nath A, Garcia Montojo M, Norato G, Santamaria UA, et al. Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: the lighthouse trial. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20(7-8):595–604.
Rice GI, Meyzer C, Bouazza N, Hully M, Boddaert N, Semeraro M, Zeef LAH, Rozenberg F, Bondet V, Duffy D, et al. Reverse-transcriptase inhibitors in the Aicardi–Goutières Syndrome. N Engl J Med. 2018;379(23):2275–7.
Acknowledgements
The authors thank Jose Luis García-Pérez, University of Edinburgh, for critical reading of the manuscript and Alisha O. Soares for assistance with manuscript preparation.
Funding
JLG was supported by a grant from the NIH National Institute of Aging (5R21AG056840-02).
Author information
Authors and Affiliations
Contributions
CL, JLG, and RQ conceived and wrote the manuscript. All authors read and approved the final manuscript.
Authors’ information
Chao Lou - Attending Physician of Genetics, MD, Ph.D.
John L. Goodier, M.Sc., Ph.D.
Rong Qiang - Chief Physician of Genetics, MD, Ph.D.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
All authors approved to publish.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Lou, C., Goodier, J.L. & Qiang, R. A potential new mechanism for pregnancy loss: considering the role of LINE-1 retrotransposons in early spontaneous miscarriage. Reprod Biol Endocrinol 18, 6 (2020). https://doi.org/10.1186/s12958-020-0564-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12958-020-0564-x