
Abstract
The number of software tools available for detecting transposable element insertions from whole genome sequence data has been increasing steadily throughout the last ~5 years. Some of these methods have unique features suiting them for particular use cases, but in general they follow one or more of a common set of approaches. Here, detection and filtering approaches are reviewed in the light of transposable element biology and the current state of whole genome sequencing. We demonstrate that the current state-of-the-art methods still do not produce highly concordant results and provide resources to assist future development in transposable element detection methods.
Similar content being viewed by others

Background
It has been 27 years since Haig Kazazian, Jr. published the seminal observation of active LINE-1 retrotransposition in humans [1], and 14 years since the initial publication of the assembled human genome reference sequence gave us a genome-wide view of human transposable element content, albeit largely from one individual [2]. Because LINEs, Alus, and SVAs are actively increasing in copy number at estimated rates of around 2-5 new insertions for every 100 live births for Alu [3–5], and around 0.5-1 in 100 for L1 [4–7], it stands to reason that the vast majority of transposable element insertions are not present in the reference genome assembly and are detectable as segregating structural variants in human populations.
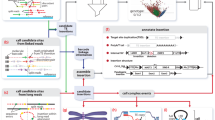
Identification of transposable element insertions (TEs) from the results of currently available high-throughput sequencing platforms is a challenge. A number of targeted methods are available to sequence junctions between TEs and their insertion sites, and have been reviewed elsewhere [8–10]. Similarly, there are several methods used for transposable element identification and annotation from genome assemblies, also reviewed elsewhere [11–15]. This review focuses on methods for discovering and/or genotyping transposable elements from whole genome sequence (WGS) data. The majority of the WGS data available today comes from Illumina platforms and consists of millions to billions of 100-150 bp reads in pairs, where each read in a pair represents the end of a longer fragment (Fig. 1a). Detection of small mutations, single-base or multiple-base substitutions, insertions, and deletions less than one read length, is achievable through accurate alignment to the reference genome followed by examination of aligned columns of bases for deviations from the reference sequence. Detection of structural variants is more difficult, principally because using current whole genome sequencing methods, the presence of rearrangements versus the reference genome must be inferred from short sequences that generally do not span the entire interval affected by a rearrangement. Typically, structural variant detection from short paired-end read data is solved through a combination of three approaches: 1. inference from discordant read-pair mappings, 2. clustering of ‘split’ reads sharing common alignment junctions, and 3. sequence assembly and re-alignment of assembled contigs [16].

Read mapping patterns typically associated with insertion detection. Panel a shows the read mapping patterns versus a reference TE sequence (grey rectangle, top) and the mapping of the same reads to a reference genome sequence (orange rectangle, bottom). Reads are represented as typical paired-end reads where the ends of each amplicon are represented as rectangles and the un-sequenced portion of the amplicons are represented as bars connecting the rectangles. Reads informative for identifying TE insertion locations are indicated by dashed boxes, other read mappings to the TE reference are shown in light blue boxes. Within the informative reads, reads or portions of reads mapping to the TE reference are coloured blue, and mappings to the reference genome sequence are coloured yellow. The exact location of this example insertion is indicated by the red triangle and the dashed line. Assembly of the reads supporting the two junction sequences is indicated to the right of the ‘consensus’ arrow, one example with a TSD and one without. If a TSD is present, the insertion breakends relative to the reference genome are staggered, and the overlap of reference-aligned sequence corresponds to the TSD. If a TSD is not present (and no bases are deleted upon insertion), the junctions obtained from the 5' end and the 3' end of the TE reference will match exactly. Panel b shows a typical pattern of discordant read mappings across a genome - the colored segments in circle represent chromosomes, each black link indicates a discordant read mapping supporting an insertion at the position indicated by the red triangle. The endpoints not corresponding to the insertion site map to TE elements at various locations in the reference genome
Transposable elements represent a majority of structural insertions longer than a few hundred base pairs [17], and require a further level of scrutiny on top of what is normally required for SV detection, which is informed by their insertion mechanism. This review is principally concerned with the detection of non-Long Terminal Repeat (LTR) retrotransposons in mammalian genomes, but many of the concepts should generalise to other transposable element types in other species. Regarding the mechanism of insertion, non-LTR retrotransposition in mammals is driven by the activity of Long INterspersed Elements (LINEs) which replicate through an mRNA-mediated series of events known as target-primed reverse transcription (TPRT) [18]. There are a number of important features of TPRT which one must be cognisant of when devising methods for detecting retrotransposon insertions. First, a message must be transcribed, and it seems that 3' polyadenylation is a necessary feature for recognition by poly-A binding proteins associated with the L1 Ribonuclear Particle (RNP) [19–22]. This does not necessarily mean that the message must be Pol II transcribed: for example, Alu elements are Pol III transcripts [23]. Insertions are processed transcripts: the cultured cell retrotransposition assay relies on this fact, as there is an intron in reverse-orientation to the reporter gene in these assays, which is spliced out when the construct is transcribed [24]. Additionally, the detection of processed pseudogenes uses the presence of splice junctions between coding exons as a defining feature [25, 26]. Polyadenylation at the 3' end of inserted L1 and SVA sequences is generally observed, and shorter A tails also exist on the 3' end of Alu insertions.
Target-site duplication (TSD) is a feature of TPRT that is necessary to consider when detecting novel insertions. The ORF2 endonuclease cleavage is staggered, meaning there is some distance, typically 7-20 base-pairs [27], between the cut sites in the top strand and bottom strand. Some software tools have been developed specifically to detect TSDs [28, 29]. Once the insertion site is fully resolved at the end of TPRT through mechanisms that likely include host DNA repair but are incompletely understood, the sequence between the cut sites appears on either site of the new insertion. Although insertions without TSDs do occur due to co-occurring deletions at the target site (about 10 % of insertions) [30, 31], or via the endonuclease-independent pathway [32], the vast majority of new insertions occurring through TPRT have TSDs, and these can generally be readily identified through sequence analysis methods when identifying novel insertions.
Insertion of transduced sequences is another feature of transposable element insertions that may be detected computationally and is important to consider when applying or designing methods for insertion detection. When sequences immediately adjacent to the transposable elements are transcribed up- or down-stream as part of the TE message, both the TE RNA and non-TE RNA will be reverse transcribed and integrated into the insertion site as a DNA sequence [33–35]. As LINE insertions are often 5' truncated [36, 37], sometimes transduced sequences are all that is left of a message with a severe 5' truncation. As a result, in some instances an insertion may contain no recognizable transposable element sequence, but the mechanism can be surmised from the presence of the poly-A tail and TSDs [38].
Roughly 1 in 5 LINE insertions will have an inversion of the 5' end of the element due to a variant of the TPRT mechanism known as ‘twin-priming’, where two ORF2 molecules reverse-transcribe the L1 RNA from different directions, resulting in an insertion with a 5' end inversion. [39]. This is an important consideration when designing methods to identify insertions of these sequences, as the relative orientation of the 5' end is not predictable and filtering putative insertion sites without taking this into account may lead to a 20\% higher false negative rate for LINE detection from the 5' end.
Finally, maybe the most important feature of transposable element insertions that impacts methods used for their detection is simply their repetitive nature in the context of the reference genome: due to repeated copy-and-paste operations through TPRT, there are thousands of elements from each active class of transposable element present in the human genome. This is the key factor that makes accurate detection of transposable element insertions difficult: read pairs mapping to the insertion site will have paired ends that map to various locations throughout the reference genome where instances of the inserted element type are present (Fig. 1b). The presence of many copies of an element in the genome also confounds detection of new copies of that element by introducing false positives where what appears to be a novel insertion may actually just be a mapping artefact of an existing transposable element present in the reference genome.
Review
Given whole genome sequence (WGS) data, there are three basic approaches to looking for non-reference insertions that are often used together, integrating support from each approach: discordant read-pair clustering, split-read mapping, and sequence assembly. It bears mentioning that all of these are not applicable to every WGS method; read-pairs are not necessarily present depending on the library preparation method or sequencing technology. Currently, the most widespread approach to WGS is via Illumina HiSeq technology using paired-end reads. In the future, as methods for long-read sequencing mature, new computational methods for insertion detection may be required, or previous methods for detecting insertions from capillary sequence or comparative whole-genome assemblies [4] may be repurposed.
Discordant read-pair mapping
A discordant read pair is one that is inconsistent with the library preparation parameters. During library preparation, genomic DNA is sheared physically or chemically, and fragments of a specific size are selected for library preparation and sequencing. Given an expected fragment size distribution, anything significantly outside of that range may be considered discordant. What is significantly outside of the expected range of fragment sizes can be determined after sequencing and alignment based on the distribution of distances between paired reads. Additionally, given the library prep method and sequencing platform, the expected orientation of the ends of the read-pairs is known. For instance, Illumina read pairs are ‘forward-reverse’ meaning that relative to the reference genome, the first read in a pair will be in the ‘forward’ orientation and the second will be ‘reverse’. Reads inconsistent with this pattern may be considered discordant. Finally, reads pairs where one end maps to a different chromosome or contig than the other are considered discordant.
When using discordant read pairs to inform structural variant discovery, typically multiple pairs indicating the same non-reference junction must be present. For events between two regions of unique mappable sequence such as chromosome fusions, deletions, duplications, etc. the locations of both ends of the collection read pairs supporting an event should be consistent. As transposable elements exist in many copies dispersed throughout the genome, typically one end will be ‘anchored’ in unique sequence while the other may map to multiple distal locations located within various repeat elements throughout the genome (Fig. 1b). In general, there are two approaches to analysing discordant reads where one end maps to repeat sequence. One is to map all reads to a reference library of repeats, collect the reads where only one end in the pair aligns completely to the reference repeat sequences, and re-mapping the non-repeat end of these one-end-repeat pairs to the reference genome (Fig. 1a). A second approach is to use the repeat annotations available for the reference genome to note where one end of a pair maps to a repeat and the other does not (Fig. 1b). In either case, once ‘one-end-repeat’ reads have been identified, the non-repeat ends of the read pairs are clustered by genomic coordinate, and possibly filtered by various criteria concerning mapping quality, consistency in read orientations, underlying genomic features, and so forth. For example, TranspoSeq filters calls where greater than 30 % of clustered reads have a mapping quality of 0 [40], while Jitterbug excludes reads with a mapping quality score of less than 15 [41]. Most tools filter out insertion calls within a window around transposable element annotations in the reference genome. It is important to note that discordant read mapping alone does not yield exact junctions between the insertion and the reference sequence, therefore sites localised by discordant read mapping are typically refined through local sequence assembly and split-read mapping.
Split-read mapping
Split reads are where one segment maps to some location in the reference genome, and the remaining segment maps to one or more locations distal from the first, or is unmapped (i.e. does not match anything in the reference). This term may also refer to a longer assembled contig which can be split into multiple mapped locations distal from one another. The ability to detect split reads is highly dependent on the choice of aligner. Some short read aligners (e.g. BWA MEM [42]) have the ability to partially align (‘soft’ or ‘hard’ clip) reads and give alternate mapping locations for the clipped portion as secondary or supplementary alignments. Aligners intended for lower throughput and longer reads (BLAT [43], LAST [44], BLAST [45]) are natural choices for detecting split reads, especially from longer assembled sequences. Since split reads are the means for identifying the exact insertion location at base-pair resolution, analysis of split reads is critical for identifying features indicative of TPRT activity including transductions, target site duplications, endonuclease cleavage site, and the addition of untemplated bases. Additionally, it is possible to take advantage of overlaps between reads supporting an insertion and use sequence assembly in an attempt to generate longer contigs of sequence that better resolve the junctions between the insertion and the reference genome, essentially creating very long split reads which have the potential to span both the 5' and 3' junctions of an inserted sequence. This is particularly useful for elucidating transduced sequences and studying untemplated base incorporation at the junctions in detail. In general, it is highly advisable that TE detection methods incorporate split-read analysis as this is the primary means to detect 5' and 3' junctions with nucleotide resolution, and thus the primary means to detecting many hallmarks of TE insertion necessary both for filtering false positives and for biological inferences.
Filtering putative insertions
Given the challenge associated with detecting structural variants from short-read data, compounded with the difficulty of detecting insertions of sequences into a background that already contains thousands of similar interspersed copies, any scheme purporting to detect transposable element insertions with reasonable sensitivity must implement filters to control for false positives.
Most methods use the number of reads supporting an insertion as a first cutoff - either as a parameter or as a function of local sequence depth. For WGS data, split reads and discordant read support may be considered independently when filtering insertions. The target allele fraction (i.e. fraction of cells in which an insertion is expected to be present) is an important consideration: somatic insertions arising later in the history of a tissue or a tumour may be supported by fewer reads than germline insertions expected to be present in 1-2 copies per mononucleated cell. In addition to the quantity of reads, the quality of reads should be considered both in terms of their alignment and base quality. Base quality (e.g. phred score) over clipped bases is particularly important when considering soft clipped read mappings: if the clipped bases have poor quality, it is likely they do not represent transposable element sequence and can be ignored. Mappings of high-quality sequence with a high number (e.g. > 5 %) of mismatches versus either the genome around the insertion site or versus the consensus transposable element are often associated with false positives, but this cutoff should be implemented according to the expected divergence of the TE insertions with respect to the reference TE sequence: if the available TE reference is not a good representation of the expected insertions (e.g. the reference is constructed from a different species) this filter should be relaxed.
A second major consideration when filtering transposable element insertions is the nature of the genome at the insertion site. As with any attempt at annotation or mutation detection versus a reference genome, the concept of mappability (or alignability) is important [46, 47]. A sequence is considered ‘mappable’ (or ‘alignable’) if it aligns to one and only one location. For a given segment of the reference genome, mappability can be calculated by considering the number of uniquely mapping k-mers (i.e. sequences of length k) corresponding to commonly encountered read lengths (e.g. 35 bp, 50 bp, 100 bp), possibly allowing for some number of mismatches. Filtering insertions that overlap annotated transposable elements is often done and may serve as a proxy for mappability as TE sequences often have relatively fewer unique k-mers relative to the non-repeat genome.
As mentioned, it is usually advisable to filter TE insertions that map onto the coordinates of TEs of the same subfamily represented in the reference genome. This is due to low mappability over recent transposable element insertions due to their similarity to the active consensus element, which can be addressed using a mappability filter as described, and it also guards against artefacts due to similarity between the insertion site and the inserted element. Finally, in instances where the goal is detection of somatic or novel germline insertions, a good database of known non-reference insertion sites is essential. Existing published resources to this end include dbRIP [48] and euL1db [49]. As the former has not been updated in some years and the latter only considers L1 insertions, a simple listing of reported non-reference insertion coordinates derived from the supplementary tables associated with most current studies reporting non-reference human retrotransposon insertions is included as Additional file 1: Table S1 (see Addtitional file 1 for table legend).
Considerations for analyses in non-humans
Many of the methods listed in Table 1 have been successfully applied to species other than human, and to transposable element varieties other than the non-LTR elements focused on in this review so far. For example Retroseq [50] has been applied to mouse genomes to detect LTR elements such as IAP and MusD in addition to the mouse varieties of LINE (L1Md) and SINE (B1/B2) elements [51]. T-lex [52] and T-lex2 [53] have been applied to Drosophila genomes, detecting a wide variety of different TE families. While non-LTR TEs in human have a consensus insertion site preference that is widespread in the human genome, other TE families have more specific integration site preferences. For example, the Ty1 LTR retroelement strongly prefers integration near Pol III transcribed tRNA genes and seems to associate with nucleosomes [54], while Tf1 elements (also LTRs) prefer nucleosome-free regions near Pol II promoters [55]. Hermes elements (a type of DNA transposon) also prefer nucleosome-free regions and have a characteristic TSD sequence motif (nTnnnnAn) [56]. Non-LTR retroelements can also have strong insertions site preferences as well, a prominent example being the R1 and R2 elements from Bombyx mori, which target 28S ribosomal genes [57] and have been used to dissect the biochemical steps involved in non-LTR integration [18]. These various propensities to insert proximal to genomic features and have defined sequence characteristics at the insertion site could be used to filter insertion detections from WGS data for these TE families in non-human species, in combination with the general approaches already covered for non-LTR elements that have weaker insertion site preferences. Additionally, some of the characteristics of non-LTR retrotransposition presented so far may not apply to other TE classes and families and could lead to false negatives if putative insertions are inappropriately filtered against certain characteristics. For example, some DNA transposons (e.g. Spy) do not create target site duplications, so software that requires TSD will miss these [58]. Other TEs have fixed TSD lengths, e.g. the Ac/Ds transposons in maize, famously initially described by McClintock in the 1950s [59], create an 8 bp TSD [60, 61], so a detector that allows Ac/Ds predictions with other TSD sizes might be more prone to false positives.
Comparing methods
When it comes to detecting mutations, especially somatic mutations, different methods and/or different parametrisations yield markedly different results [62–64], and transposable element detection is no exception [5]. Publications presenting new tools often include comparisons where a number of competing methods are run by the authors of the new tool. While valuable, these experiments may not reflect optimal parametrisations of the competing tools for the dataset used as a basis of comparison, whereas by virtue of having developed a novel method, the authors will have better parametrisations of their own tools, leading to the usual outcome of the new tool outperforming previously published methods.
To illustrate the extent of the differences in TE insertion calls from different methods run on the same data, we present comparisons between somatic TE detections from three recent studies. In each case, two different methods were used to call mutations on the same data, yielding substantial overlap and an equally if not more substantial amount of non-overlap. Importantly, these calls were generated by the developers of their respective TE detection methods. Coordinates and sample identities were obtained from the supplemental information of the respective studies, and one [65] needed to be converted from hg18 to hg19 coordinates via liftOver. Insertion coordinates were padded by +/- 100 bp and compared via BEDTools v2.23. Lee et al. [65] (Tea) and Helman et al. [40] (TranspoSeq) share 7 samples, Tubio et al. [66] (TraFiC) and Helman et al. (TranspoSeq) share 15 samples. No samples are shared between Lee et al. and Helman et al. The overall Jaccard distance between TranspoSeq and Tea results across shared samples was 0.573 (Additional file 2 and Additional file 3: Table S2a), and between TranspoSeq and TraFiC the distance was 0.741 (Additional file 2 and Additional file 3: Table S2b), indicating that TranspoSeq and Tea seem to yield more similar results than between TranspoSeq and TraFiC. Summing counts for intersected insertion calls and method-specific calls yields the overlaps shown in Fig. 2. While this comparison is somewhat cursory and high-level, it is clear there is a substantial amount of difference in the results of these methods: in both comparisons, more insertions are identified by a single program than by both programs. Given that all three studies report a high validation rate (greater than 94 %) where samples were available for validation, this may reflect a difficulty in tuning methods for high sensitivity while maintaining high specificity. This also suggests that perhaps an ensemble approach combining calls across all three (or more) methods may be preferable where high sensitivity is required.

Intersections between somatic insertion detection methods. Overlap and non-overlap between insertion sites from reported in publications using the indicated software tools on the same data. The number of samples included in the comparison shared between the each pair of studies is indicated
In addition to the tools already highlighted, a rapidly increasing number of tools exist with the common goal of detecting transposable element insertions from WGS data. As indicated in Table 1, these include purpose-built methods aimed specifically at transposable elements in addition to more general methods that identify a wide variety of structural alterations versus a reference genome, transposable element insertions included. Table 1 is not intended to represent an exhaustive listing of currently existing methods - the OMICtools website (http://omictools.com/) currently supports an up-to-date database of TE detection tools, and the Bergman lab website also hosts a list of transposable element detection tools which includes tools aimed at a wide variety of applications, a subset of which are relevant for TE detection from WGS data [11].
Conclusions
Transposable element insertions are a subset of structural variants that can be identified from WGS data. Although generalised SV discovery methods sometimes support TE detection, specialised software is often used by those interested in studying the specific peculiarities of the insertion mechanism and mitigating the false positives associated with their high copy number. TE discovery methods developed in the last 5 years are predominantly aimed at short-read paired-end WGS data, most often generated on Illumina platforms, and use a combination of paired-end, split-read, and sequence assembly approaches to identify insertions. Technological and methodological developments will change how the ascertainment of transposable element insertion sites is carried out. Long-read sequencing has the potential to both improve resolution of TE insertions, especially those located in repetitive regions [67], and to improve the information available regarding the sequence of the insertion itself. Currently this technology has been successful for de novo assembly of microbial genomes [68], but for human genomes, high sequence coverage [69] and a combination multiple sequencing approaches [70] and sophisticated error correction models [71] may be required to get a good consensus sequence given the currently high error rates associated with long-read sequencing technologies. Over time, it is expected that throughput will increase and error rate will decrease, making this a viable option. Even if relatively higher error rates for long-read single-molecule sequencing approaches persist, the key may be to obtain good whole-genome assemblies of individual genomes accomplished through higher throughput. Methodologically, new software tools will be published when new sequencing technologies or new alignment methods and formats attain widespread acceptance. Additional new software tools utilising current sequencing technology will also continue to be developed and published - that said, it is important that new methods offer some demonstrable, substantial improvement over the many existing methods, and there does appear to be room for improvement given the low concordance currently observed between different tools on the same data. For those seeking to develop additional methods, an improved focus on software engineering and usability would also be welcome. The subfield of transposable element insertion detection from WGS data currently lacks standards against which authors of new tools can benchmark their methods. Some recent tools have been tested on high-coverage trios e.g. NA12878/NA12891/NA12892 which is probably a step in the right direction as these are high-quality and readily available. Establishing or extending standardised datasets such as those already developed for variant calling [72, 73] would be a further step in the right direction. Going beyond this, a “living benchmark” similar to what exists for protein structure prediction through CASP [74] or more topically what currently exists through the ICGC-TCGA DREAM Somatic Mutation Calling Challenge [64] would provide a publicly available “proving ground” for existing and novel TE insertion detection methods.
Abbreviations
- L1:
-
LINE-1/Long Interspersed Element-1
- LTR:
-
Long Terminal Repeat
- RNP:
-
Ribonuclear Particle
- SV:
-
Structural Variant
- SVA:
-
SINE VNTR ALU
- TE:
-
Transposable Element
- TPRT:
-
Target-primed Reverse Transcription
- TSD:
-
Target Site Duplication
- VNTR:
-
Variable Number of Tandem Repeats
- WGS:
-
Whole Genome Sequencing
References
Kazazian HH, Wong C, Youssoufian H, Scott AF, Phillips DG, Antonarakis SE. Haemophilia A resulting from de novo insertion of L1 sequences represents a novel mechanism for mutation in man. Nature. 1988;332(6160):164–6. doi:10.1038/332164a0.
International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome. Nature. 2001;409(6822):860–921. doi:10.1038/35057062.
Cordaux R, Hedges DJ, Herke SW, Batzer MA. Estimating the retrotransposition rate of human Alu elements. Gene. 2006;373:134–7. doi:10.1016/j.gene.2006.01.019.
Xing J, Zhang Y, Han K, Salem AH, Sen SK, Huff CD, et al. Mobile elements create structural variation: analysis of a complete human genome. Genome Res. 2009;19(9):1516–26. doi:10.1101/gr.091827.109.
Stewart C, Kural D, Strömberg MP, Walker JA, Konkel MK, Stütz AM, et al. 1000 Genomes Project: A comprehensive map of mobile element insertion polymorphisms in humans. PLoS Genet. 2011;7(8):1002236. doi:10.1371/journal.pgen.1002236.
Ewing AD, Kazazian HH. High-throughput sequencing reveals extensive variation in human-specific L1 content in individual human genomes. Genome Res. 2010;20(9):1262–70. doi:10.1101/gr.106419.110.
Huang CRL, Schneider AM, Lu Y, Niranjan T, Shen P, Robinson MA, et al. Mobile interspersed repeats are major structural variants in the human genome. Cell. 2010;141(7):1171–82. doi:10.1016/j.cell.2010.05.026.
Beck CR, Garcia-Perez JL, Badge RM, Moran JV. LINE-1 elements in structural variation and disease. Annu Rev Genomics Hum Genet. 2011;12:187–215. doi:10.1146/annurev-genom-082509-141802.
Faulkner GJ. Retrotransposons: mobile and mutagenic from conception to death. FEBS Lett. 2011;585(11):1589–94. doi:10.1016/j.febslet.2011.03.061.
Xing J, Witherspoon DJ, Jorde LB. Mobile element biology: new possibilities with high-throughput sequencing. Trends Genet. 2013;29(5):280–9. doi:10.1016/j.tig.2012.12.002.
Bergman CM, Quesneville H. Discovering and detecting transposable elements in genome sequences. Brief Bioinformatics. 2007;8(6):382–92. doi:10.1093/bib/bbm048.
Saha S, Bridges S, Magbanua ZV, Peterson DG. Empirical comparison of ab initio repeat finding programs. Nucleic Acids Res. 2008;36(7):2284–94. doi:10.1093/nar/gkn064.
Cordaux R, Sen SK, Konkel MK, Batzer MA. Computational methods for the analysis of primate mobile elements. Methods Mol Biol. 2010;628:137–51. doi:10.1007/978-1-60327-367-1.
Lerat E. Identifying repeats and transposable elements in sequenced genomes: how to find your way through the dense forest of programs. Heredity. 2010;104(6):520–33. doi:10.1038/hdy.2009.165.
Hoen DR, Hickey G, Bourque G, Casacuberta J, Cordaux R, Feschotte C, et al. A call for benchmarking transposable element annotation methods. Mob DNA. 2015;6:13. doi:10.1186/s13100-015-0044-6.
Alkan C, Coe BP, Eichler EE. Genome structural variation discovery and genotyping. Nat Rev Genet. 2011;12(5):363–76. doi:10.1038/nrg2958.
Korbel JO, Urban AE, Affourtit JP, Godwin B, Grubert F, Simons JF, et al. Paired-end mapping reveals extensive structural variation in the human genome. Science. 2007;318(5849):420–6. doi:10.1126/science.1149504.
Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72(4):595–605. doi:10.1016/0092-8674(93)90078-5.
Roy-Engel AM, Salem A-H, Oyeniran OO, Deininger L, Hedges DJ, Kilroy GE, et al. Active Alu Element “A-Tails”: Size Does Matter. Genome Res. 2002;12(9):1333–44. doi:10.1101/gr.384802.
Dewannieux M, Heidmann T. Role of poly(A) tail length in Alu retrotransposition. Genomics. 2005;86(3):378–81. doi:10.1016/j.ygeno.2005.05.009.
Dai L, Taylor MS, O’Donnell KA, Boeke JD. Poly(A) binding protein C1 is essential for efficient L1 retrotransposition and affects L1 RNP formation. Mol Cell Biol. 2012;32(21):4323–36. doi:10.1128/MCB.06785-11.
Doucet A, Wilusz J, Miyoshi T, Liu Y, Moran J. A 3’ poly(a) tract is required for line-1 retrotransposition. Mol Cell. 2015. doi:10.1016/j.molcel.2015.10.012.
Fuhrman SA, Deininger PL, LaPorte P, Friedmann T, Geiduschek EP. Analysis of transcription of the human Alu family ubiquitous repeating element by eukaryotic RNA polymerase III. Nucleic Acids Res. 1981;9(23):6439–56. doi:10.1093/nar/9.23.6439.
Moran JV, Holmes SE, Naas TP, DeBerardinis RJ, Boeke JD, Kazazian HH. High frequency retrotransposition in cultured mammalian cells. Cell. 1996;87(5):917–27.
Esnault C, Maestre J, Heidmann T. Human LINE retrotransposons generate processed pseudogenes. Nat Gen. 2000;24(4):363–7. doi:10.1038/74184.
Zhang Z, Harrison PM, Liu Y, Gerstein M. Millions of years of evolution preserved: a comprehensive catalog of the processed pseudogenes in the human genome. Genome Res. 2003;13(12):2541–58. doi:10.1101/gr.1429003.
Kazazian HH, Moran JV. The impact of L1 retrotransposons on the human genome. Nat Genet. 1998;19(1):19–24. doi:10.1038/ng0598-19.
Szak ST, Pickeral OK, Makalowski W, Boguski MS, Landsman D, Boeke JD. Molecular archeology of L1 insertions in the human genome. Genome Biol. 2002;3(10):0052.
Lucier J-F, Perreault J, Noël J-F, Boire G, Perreault J-P. RTAnalyzer: a web application for finding new retrotransposons and detecting L1 retrotransposition signatures. Nucleic Acids Res. 2007;35 suppl 2:269–74. doi:10.1093/nar/gkm313.
Gilbert N, Lutz-Prigge S, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110(3):315–25. doi:10.1016/S0092-8674(02)00828-0.
Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, et al. Human l1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110(3):327–38. doi:10.1016/S0092-8674(02)00839-5.
Morrish TA, Garcia-Perez JL, Stamato TD, Taccioli GE, Sekiguchi J, Moran JV. Endonuclease-independent LINE-1 retrotransposition at mammalian telomeres. Nature. 2007;446(7132):208–12. doi:10.1038/nature05560.
Moran JV, DeBerardinis RJ, Kazazian HH. Exon shuffling by L1 retrotransposition. Science. 1999;283(5407):1530–4. doi:10.1126/science.283.5407.1530.
Goodier JL, Ostertag EM, Kazazian HH. Transduction of 3’-flanking sequences is common in L1 retrotransposition. Hum Mol Genet. 2000;9(4):653–7. doi:10.1093/hmg/9.4.653.
Pickeral OK, Makalowski W, Boguski MS, Boeke JD. Frequent human genomic DNA transduction driven by LINE-1 retrotransposition. Genome Res. 2000;10(4):411–5. doi:10.1101/gr.10.4.411.
Grimaldi G, Skowronski J, Singer MF. Defining the beginning and end of KpnI family segments. EMBO J. 1984;3(8):1753–9.
Pavlı́cek A, Paces J, Zı́ka R, Hejnar J. Length distribution of long interspersed nucleotide elements (LINEs) and processed pseudogenes of human endogenous retroviruses: implications for retrotransposition and pseudogene detection. Gene. 2002;300(1-2):189–94. doi:10.1016/S0378-1119(02)01047-8.
Solyom S, Ewing AD, Hancks DC, Takeshima Y, Awano H, Matsuo M, et al. Pathogenic orphan transduction created by a nonreference LINE-1 retrotransposon. Hum Mutat. 2012;33(2):369–71. doi:10.1002/humu.21663.
Ostertag EM, Kazazian HH. Twin priming: a proposed mechanism for the creation of inversions in L1 retrotransposition. Genome Res. 2001;11(12):2059–65. doi:10.1101/gr.205701.
Helman E, Lawrence MS, Stewart C, Sougnez C, Getz G, Meyerson M. Somatic retrotransposition in human cancer revealed by whole-genome and exome sequencing. Genome Res. 2014;24(7):1053–63. doi:10.1101/gr.163659.113.
Hénaff E, Zapata L, Casacuberta JM, Ossowski S. Jitterbug: somatic and germline transposon insertion detection at single-nucleotide resolution. BMC genomics. 2015;16(1):768. doi:10.1186/s12864-015-1975-5.
Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997 [q-bio]. 2013; arXiv: 1303.3997
Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. doi:10.1101/gr.229202.
Kielbasa SM, Wan R, Sato K, Horton P, Frith MC. Adaptive seeds tame genomic sequence comparison. Genome Res. 2011;21(3):487–93. doi:10.1101/gr.113985.110.
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215(3):403–10. doi:10.1016/S0022-2836(05)80360-2.
Derrien T, Estellé J, Marco Sola S, Knowles DG, Raineri E, Guigó R, et al. Fast computation and applications of genome mappability. PLoS ONE. 2012;7(1):30377. doi:10.1371/journal.pone.003037.
Lee H, Schatz MC. Genomic dark matter: the reliability of short read mapping illustrated by the genome mappability score. Bioinformatics. 2012;28(16):2097–105. doi:10.1093/bioinformatics/bts330.
Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat. 2006;27(4):323–9. doi:10.1002/humu.20307.
Mir AA, Philippe C, Cristofari G. euL1db: the European database of L1hs retrotransposon insertions in humans. Nucleic Acids Res. 2015;43(Database issue):43–7. doi:10.1093/nar/gku1043.
Keane TM, Wong K, Adams DJ. RetroSeq: transposable element discovery from next-generation sequencing data. Bioinformatics. 2013;29(3):389–90. doi:10.1093/bioinformatics/bts697.
Nellåker C, Keane TM, Yalcin B, Wong K, Agam A, Belgard TG, et al. The genomic landscape shaped by selection on transposable elements across 18 mouse strains. Genome Biol. 2012;13(6):45. doi:10.1186/gb-2012-13-6-r45.
Fiston-Lavier A-S, Carrigan M, Petrov DA, González J. T-lex: a program for fast and accurate assessment of transposable element presence using next-generation sequencing data. Nucleic Acids Res. 2011;39(6):36. doi:10.1093/nar/gkq1291.
Fiston-Lavier A-S, Barrón MG, Petrov DA, González J. T-lex2: genotyping, frequency estimation and re-annotation of transposable elements using single or pooled next-generation sequencing data. Nucleic Acids Res. 2015;43(4):22. doi:10.1093/nar/gku1250.
Mularoni L, Zhou Y, Bowen T, Gangadharan S, Wheelan SJ, Boeke JD. Retrotransposon Ty1 integration targets specifically positioned asymmetric nucleosomal DNA segments in tRNA hotspots. Genome Res. 2012;22(4):693–703. doi:10.1101/gr.129460.111.
Guo Y, Levin HL. High-throughput sequencing of retrotransposon integration provides a saturated profile of target activity in Schizosaccharomyces pombe. Genome Res. 2010;20(2):239–48. doi:10.1101/gr.099648.109.
Gangadharan S, Mularoni L, Fain-Thornton J, Wheelan SJ, Craig NL. DNA transposon Hermes inserts into DNA in nucleosome-free regions in vivo. Proc Nat Acad Sci. 2010;107(51):21966–72. doi:10.1073/pnas.1016382107.
Xiong Y, Eickbush TH. The site-specific ribosomal DNA insertion element R1bm belongs to a class of non-long-terminal-repeat retrotransposons. Mol Cell Biol. 1988;8(1):114–23. doi:10.1128/MCB.8.1.114.
Han M-J, Xu H-E, Zhang H-H, Feschotte C, Zhang Z. Spy: a new group of eukaryotic DNA transposons without target site duplications. Genome Biol Evol. 2014;6(7):1748–57. doi:10.1093/gbe/evu140.
McClintock B. The origin and behavior of mutable loci in maize. Proc Nat Acad Sci USA. 1950;36(6):344–55.
Sutton WD, Gerlach WL, Peacock WJ, Schwartz D. Molecular analysis of ds controlling element mutations at the adh1 locus of maize. Science (New York, NY). 1984;223(4642):1265–8. doi:10.1126/science.223.4642.1265.
Döring HP, Tillmann E, Starlinger P. DNA sequence of the maize transposable element Dissociation. Nature. 1984;307(5947):127–30. doi:10.1038/307127a0.
O’Rawe J, Jiang T, Sun G, Wu Y, Wang W, Hu J, et al. Low concordance of multiple variant-calling pipelines: practical implications for exome and genome sequencing. Genome Med. 2013;5(3):28. doi:10.1186/gm432.
Kim SY, Speed TP. Comparing somatic mutation-callers: beyond Venn diagrams. BMC Bioinformatics. 2013;14:189. doi:10.1186/1471-2105-14-189.
Ewing AD, Houlahan KE, Hu Y, Ellrott K, Caloian C, Yamaguchi TN, et al. ICGC-TCGA DREAM Somatic Mutation Calling Challenge participants, Kellen, M.R., Norman, T.C., Haussler, D., Friend, S.H., Stolovitzky, G., Margolin, A.A., Stuart, J.M., Boutros, P.C.: Combining tumor genome simulation with crowdsourcing to benchmark somatic single-nucleotide-variant detection. Nat Methods. 2015;12(7):623–30. doi:10.1038/nmeth.3407.
Lee E, Iskow R, Yang L, Gokcumen O, Haseley P, Luquette LJ, et al. Cancer Genome Atlas Research Network: Landscape of somatic retrotransposition in human cancers. Science. 2012;337(6097):967–71. doi:10.1126/science.1222077.
Tubio JMC, Li Y, Ju YS, Martincorena I, Cooke SL, Tojo M, et al. Mobile DNA in cancer. Extensive transduction of nonrepetitive DNA mediated by L1 retrotransposition in cancer genomes. Science. 2014;345(6196):1251343. doi:10.1126/science.1251343.
Chaisson MJP, Huddleston J, Dennis MY, Sudmant PH, Malig M, Hormozdiari F, et al. Resolving the complexity of the human genome using single-molecule sequencing. Nature. 2015;517(7536):608–11. doi:10.1038/nature13907.
Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods. 2015;12(8):733–5. doi:10.1038/nmeth.3444.
Ammar R, Paton TA, Torti D, Shlien A, Bader GD. Long read nanopore sequencing for detection of HLA and CYP2d6 variants and haplotypes. F1000Research. 2015. doi:10.12688/f1000research.6037.2.
Madoui M-A, Engelen S, Cruaud C, Belser C, Bertrand L, Alberti A, et al. Genome assembly using Nanopore-guided long and error-free DNA reads. BMC Genomics. 2015;16:327. doi:10.1186/s12864-015-1519-z.
Jain M, Fiddes IT, Miga KH, Olsen HE, Paten B, Akeson M. Improved data analysis for the MinION nanopore sequencer. Nat Meth. 2015;12(4):351–6. doi:10.1038/nmeth.3290.
Talwalkar A, Liptrap J, Newcomb J, Hartl C, Terhorst J, Curtis K, et al. SMaSH: a benchmarking toolkit for human genome variant calling. Bioinformatics. 2014;30(19):2787–95. doi:10.1093/bioinformatics/btu345.
Zook JM, Catoe D, McDaniel J, Vang L, Spies N, Sidow A, et al. Extensive sequencing of seven human genomes to characterize benchmark reference materials. bioRxiv. 2015; 026468. doi:10.1101/026468
Moult J, Fidelis K, Kryshtafovych A, Schwede T, Tramontano A. Critical assessment of methods of protein structure prediction (CASP) — round x. Proteins: Struct Funct Bioinf. 2014;82:1–6. doi:10.1002/prot.24452.
Wu J, Lee W-P, Ward A, Walker JA, Konkel MK, Batzer MA, et al. Tangram: a comprehensive toolbox for mobile element insertion detection. BMC Genomics. 2014;15:795. doi:10.1186/1471-2164-15-795.
Hormozdiari F, Hajirasouliha I, Dao P, Hach F, Yorukoglu D, Alkan C, et al. Next-generation VariationHunter: combinatorial algorithms for transposon insertion discovery. Bioinformatics. 2010;26(12):350–7. doi:10.1093/bioinformatics/btq216.
Hormozdiari F, Alkan C, Ventura M, Hajirasouliha I, Malig M, Hach F, et al. Alu repeat discovery and characterization within human genomes. Genome Res. 2011;21(6):840–9. doi:10.1101/gr.115956.110.
Ewing AD, Ballinger TJ, Earl D, Broad Institute Genome Sequencing and Analysis Program and Platform, Harris CC, Ding L, et al. Retrotransposition of gene transcripts leads to structural variation in mammalian genomes. Genome Biol. 2013;14(3):22. doi:10.1186/gb-2013-14-3-r22.
Quinlan AR, Clark RA, Sokolova S, Leibowitz ML, Zhang Y, Hurles ME, et al. Genome-wide mapping and assembly of structural variant breakpoints in the mouse genome. Genome Res. 2010;20(5):623–35. doi:10.1101/gr.102970.109.
Robb SMC, Lu L, Valencia E, Burnette JM, Okumoto Y, Wessler SR, et al. The use of RelocaTE and unassembled short reads to produce high-resolution snapshots of transposable element generated diversity in rice. G3 (Bethesda). 2013;3(6):949–57. doi:10.1534/g3.112.005348.
Jiang C, Chen C, Huang Z, Liu R, Verdier J. ITIS, a bioinformatics tool for accurate identification of transposon insertion sites using next-generation sequencing data. BMC Bioinformatics. 2015;16:72. doi:10.1186/s12859-015-0507-2.
Linheiro RS, Bergman CM. Whole genome resequencing reveals natural target site preferences of transposable elements in Drosophila melanogaster. PLoS ONE. 2012;7(2):30008. doi:10.1371/journal.pone.0030008.
Platzer A, Nizhynska V, Long Q. TE-Locate: A Tool to Locate and Group Transposable Element Occurrences Using Paired-End Next-Generation Sequencing Data. Biology (Basel). 2012;1(2):395–410. doi:10.3390/biology1020395.
Chen K, Chen L, Fan X, Wallis J, Ding L, Weinstock G. TIGRA: a targeted iterative graph routing assembler for breakpoint assembly. Genome Res. 2014;24(2):310–7. doi:10.1101/gr.162883.113.
Thung DT, de Ligt J, Vissers LEM, Steehouwer M, Kroon M, de Vries P, et al. Mobster: accurate detection of mobile element insertions in next generation sequencing data. Genome Biol. 2014;15(10):488. doi:10.1186/s13059-014-0488-x.
Zhuang J, Wang J, Theurkauf W, Weng Z. TEMP: a computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Res. 2014;42(11):6826–38. doi:10.1093/nar/gku323.
Gilly A, Etcheverry M, Madoui M-A, Guy J, Quadrana L, Alberti A, et al. TE-Tracker: systematic identification of transposition events through whole-genome resequencing. BMC Bioinformatics. 2014;15:377. doi:10.1186/s12859-014-0377-z.
Kroon M, Lameijer EW, Lakenberg N, Hehir-Kwa JY, Thung DT, Slagboom PE, et al. Detecting dispersed duplications in high-throughput sequencing data using a database-free approach. Bioinformatics. 2015; 621. doi:10.1093/bioinformatics/btv621
Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, et al. An integrated map of structural variation in 2,504 human genomes. Nature. 2015;526:75–81. doi:10.1038/nature15394.
Acknowledgements
I would like to acknowledge support from the Australian Research Council (DE150101117) and from the Mater Foundation, and thank Geoff Faulkner for critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that they have no competing interests.
Additional files
Additional file 1:
Supplemental Table Legends. Description and references for supplemental tables 1 and 2. (PDF 48 kb)
Additional file 2: Table S1.
List of known or predicted germline insertions derived from several publications. (BED 1239 kb)
Additional file 3: Table S2.
Sample-level examination of intersections between TE detection methods. (XLSX 6 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Ewing, A.D. Transposable element detection from whole genome sequence data. Mobile DNA 6, 24 (2015). https://doi.org/10.1186/s13100-015-0055-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13100-015-0055-3