
Abstract
Lung lipid metabolism participates both in infant and adult pulmonary disease. The lung is composed by multiple cell types with specialized functions and coordinately acting to meet specific physiologic requirements. The alveoli are the niche of the most active lipid metabolic cell in the lung, the type 2 cell (T2C). T2C synthesize surfactant lipids that are an absolute requirement for respiration, including dipalmitoylphosphatidylcholine. After its synthesis and secretion into the alveoli, surfactant is recycled by the T2C or degraded by the alveolar macrophages (AM). Surfactant biosynthesis and recycling is tightly regulated, and dysregulation of this pathway occurs in many pulmonary disease processes. Alveolar lipids can participate in the development of pulmonary disease from their extracellular location in the lumen of the alveoli, and from their intracellular location in T2C or AM. External insults like smoke and pollution can disturb surfactant homeostasis and result in either surfactant insufficiency or accumulation. But disruption of surfactant homeostasis is also observed in many chronic adult diseases, including chronic obstructive pulmonary disease (COPD), and others. Sustained damage to the T2C is one of the postulated causes of idiopathic pulmonary fibrosis (IPF), and surfactant homeostasis is disrupted during fibrotic conditions. Similarly, surfactant homeostasis is impacted during acute respiratory distress syndrome (ARDS) and infections. Bioactive lipids like eicosanoids and sphingolipids also participate in chronic lung disease and in respiratory infections. We review the most recent knowledge on alveolar lipids and their essential metabolic and signaling functions during homeostasis and during some of the most commonly observed pulmonary diseases.
Similar content being viewed by others


Introduction
The lung is seldom considered a lipid metabolic organ. However, it does sustain active lipid metabolism, especially in the alveolar area, where surfactant homeostasis is exquisitely regulated to ensure continuous optimal function in each respiration cycle. Surfactant is a lipoprotein complex, composed mostly of phospholipid, and it is an absolute requirement for gas exchange. It has been known for a long time that the cause of infant respiratory distress syndrome (IRDS) and death of premature infants is surfactant insufficiency, and exogenous therapy is nowadays a standard of care in these cases (Table 1). Both animal derived and synthetic surfactants provide clinical benefits and decrease mortality of preterm infants with IRDS. For expanding on the lipid pathology of IRDS, the reader is referred to the existing revisions in the literature ([1] and references herein).
Beyond their essential roles as surfactant, energy storage and structural components, different lipids can also exert different signaling functions during physiological and pathophysiological processes. In the lung, lipids were intensely studied in the context of surfactant metabolism during the second half of the twentieth century and are now garnering new interest in multiple disease contexts, partly owing to the latest development of sophisticated and sensitive methods for detection and data analysis. Currently ongoing research on pulmonary fibrosis and electronic cigarette-induced lung injury highlights the relevance of pulmonary lipids during disease.
In this review, we focus on adult pulmonary disease to give a consolidated view of the most updated literature on alveolar lipids. Reviewing the roles of all lipid species in all pulmonary cell types in every form of disease would be excessively lengthy and exceed our scope. Instead, this paper is focused on the alveolar area, which is the most studied and where the major pulmonary lipid metabolic cells reside. However, we are convinced that lipid metabolism will reveal itself of interest in any other lung region and cell type considered.
Alveolar cellular environment and surfactant biology
The alveolar epithelium is composed of alveolar type 1 (T1C) and type 2 cells (T2C). Type 1 cells cover most of the alveolar surface and are highly specialized in performing the gas exchange between blood and air. T1C also participate in interactions with alveolar macrophages, and modulation of fibrotic responses [2, 3]. At the junctions of the alveolar sacs reside the T2C, whose main function is the production of surfactant.
Pulmonary surfactant is probably the best-known lipid complex in the lung, and it is an absolute requirement for respiration. Surfactant reduces surface tension during inspiration and prevents alveolar collapse at the end of expiration. Alveolar T2C are the major lipid metabolic cells of the lung because they need to orchestrate a complex set of lipid metabolic pathways to effectively adjust surfactant synthesis, secretion and recycling in different physiologic situations. Pulmonary surfactant is a lipoprotein complex, with 90% of its mass being lipid and the remaining 10% proteins that are in many cases specific of the alveolar compartment. Amongst the 90% lipid, the large majority is phospholipid (PL), especially phosphatidylcholine (PC) and more specifically dipalmitoyl phosphatidylcholine (DPPC), the main lipid species responsible for the surface tension-reduction properties of the surfactant mixture. Phosphatidylglycerol is capable of modulating macrophage function and it is used as a marker of lung maturity [4]. Palmitoyl-oleoyl-phosphatidylglycerol and phosphatidylinositol (PI) can antagonize Toll-like receptor (TLR) activation. TLR activation is a crucial step in the virulence of certain viruses such as Influenza A and Respiratory Syncytial Virus [5], underscoring the role of phosphatidylglycerol and PI in controlling viral infections and the associated inflammatory cascades [5]. Other lipids of smaller abundance in alveolar surfactant include cholesterol, sphingolipids and plasmalogen phospholipids [6, 7].
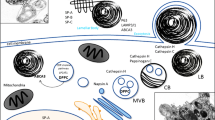
Surfactant lipid metabolic genes are transcriptionally regulated by the same factors governing lipid metabolism and lipogenesis in other tissues [8], and in T2C, surfactant lipid synthesis and secretion are coupled with lipid availability [9]. T2C obtain lipid precursors from the plasma using proteins capable of transporting and interacting with lipids, such as CD36 and glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein 1 (GPIHBP1) [9,10,11,12,13,14,15,16]. De novo lipogenesis can also contribute to the intracellular pool of fatty acids (FA) in T2C. As in other eukaryotic cells, the synthesis of PC in T2C occurs mainly through the Kennedy pathway (Fig. 1). The rate limiting enzyme of this pathway is CTP:phosphocholine cytidyltransferase alpha (CCTα). Newly synthesized PC usually contains a monounsaturated FA in position sn-2, and incorporated PC can have heterogeneous acyl composition. Hence, PC needs to undergo remodeling by phospholipase A2 (PLA2) and lysophosphatidylcholine acyltransferase 1 (LPCAT1) through the Lands cycle to render DPPC. Intracellular DPPC is then transported into a specialized organelle, the lamellar body, through the specific transporter ABCA3 and stored there until secretion to the alveoli [17,18,19]. Mouse models with loss of function for ATP-binding cassette A1, G1, or A3 (ABCA1, ABCG1, ABCA3) or CCTα all show surfactant insufficiency, pulmonary intracellular accumulation of lipid, and inflammation [20,21,22,23,24,25,26]. In humans, impairment of lipid metabolism in pulmonary T2C causes surfactant insufficiency resulting in deficient pulmonary function. For example, ABCA3 loss of function results in neonatal respiratory distress syndrome and defective lamellar body synthesis [27].

Surfactant lipid synthesis and exocytosis. Simplified scheme of intracellular pathways leading to de novo synthesis of DPPC and its routing to lamellar bodies, from where it will be released into the alveolar lumen, where it will be used, recycled and degraded. For the synthesis, CDP-choline and diacylglycerol are coupled to form PC. A large proportion of PC is remodeled to render DPPC. Surfactant is stored in lamellar bodies until secretion to the alveolar lumen, where it organizes in bilayers and monolayers (see text for further detail). The surfactant life cycle is completed by its recycling by T2C or degradation by AM. T1C: type 1 cell; T2C: type 2 cell, AM: alveolar macrophage; LB: lamellar body; SP: surfactant protein; PC: phosphatidylcholine; DPPC: dipalmitoylphosphatidylcholine; CCTα: CTP:phosphocholine cytidylyltransferase alpha; PLA2: phospholipase A2; LPCAT1: lysophosphatidylcholine acyltransferase 1.
In the alveolar lumen, surfactant reduces the surface tension in the alveolar walls from 70 mN/m to nearly 1mN/m [28]. The biophysical properties and extracellular metabolism of secreted surfactant are complex and the exact mechanisms for surfactant ability to reduce surface tension during alveolar expansion and to support high levels of pressure during lateral compression at the end of expiration are still undergoing intense research [29, 30]. Surfactant secretion by T2C occurs by exocytosis, with lamellar bodies fusing their limiting membrane with the apical plasma membrane and releasing their content into the alveolar lumen (reviewed in [31]). The content of the lamellar bodies is initially detected in the alveolar spaces as densely packed lamellar body-like particles that are thought to organize in multiple phospholipid bilayers and monolayers when they reach the alveolar surface [32]. In vitro and ex vivo experiments with clinically used surfactant and with simplified model phospholipid mixtures have shown the coexistence of two domains in the pulmonary surfactant film at physiological temperatures: bilayers enriched in unsaturated phospholipids and monolayers mainly composed of DPPC. A “squeeze-out” mechanism has been proposed. Compression of the film during expiration would result in the formation of multilayers of phospholipids that would re-spread during inspiration. These reversible transitions between bilayer and monolayer formations seem to be enabled by the hydrophobic surfactant proteins B and C (SP-B and SP-C) [33,34,35]. The interactions between lipids and proteins are complex and have been discussed in detail elsewhere [36].
Surfactant-specific proteins account for ~ 10% of the lipoprotein complex by mass, but they affect the behavior of the surfactant lipids. The most abundant and studied proteins are Surfactant Proteins A, B, C and D (SP-A, SP-B, SP-C and SP-D). SP-A and SP-D are hydrophilic and participate in the immune function roles of surfactant, as they can bind exogenous pathogens and facilitate their clearance by alveolar macrophages. Indeed, SP-A and SP-D knockout mice are more susceptible to infections and other lung diseases [37,38,39,40]. In contrast, SP-B and SP-C are hydrophobic and participate in surfactant lipid film dynamics. Once it is secreted into the alveolar lumen with the rest of the components of the lamellar bodies, SP-B is adsorbed in the air liquid interface. Extracellularly, SP-B participates in stabilizing the lipid film at all stages of the respiratory cycle [41]. SP-C is an exclusive constituent of pulmonary surfactant. SP-C also acts at the lipid film and participates in enabling the lipid to be compressed in a manner compatible with subsequent re-spreading during inspiration [42, 43]. SP-C knockout models develop profound alterations in surfactant lipid metabolism, discussed in the following pages in the context of disease [44,45,46].
Cholesterol comprises 5–10% of surfactant lipid. Experiments using native surfactant material showed that cholesterol was required for effective spreading properties of the lipid monolayer, as well as maintenance of the lateral structure, with differentiated lipid domains [47]. It was initially thought that cholesterol impaired the surface tension activity of surfactant, leading to its exclusion in the initial preparations clinically used for premature infants. However, subsequent experiments showed that cholesterol inhibits film compression and surface tension activity only at 20% and higher concentrations in the surfactant [48]. Elevated levels of cholesterol in the surfactant mixture has been proposed to be a mechanism for ventilator-induced lung injury [49] and cholesterol crystals have been found in the bronchoalveolar lavage (BAL) of idiopathic pulmonary fibrosis (IPF) patients [50]. Patients with acute respiratory distress syndrome (ARDS) also showed increased neutral lipid amounts in their BAL and this elevation persisted after exogenous surfactant administration. In vitro, neutral lipid supplementation of a clinically used or natural surfactant reduced surface tension properties, with monoglycerides and cholesterol exhibiting the greatest inhibitory effects [51]. Multiple experiments have consistently shown the ability of neutral lipids to disrupt surfactant function [48, 49, 52, 53]. Neutral lipids may function as barriers to surfactant therapy efficacy, hence further contributing to pathology. In a clinical study, the therapeutic benefits of exogenous surfactant administration in ARDS were hampered by accumulation of cholesterol in the alveolar space [51].
Extracellular surfactant can be degraded by alveolar macrophages (AM) or recycled by T2C during physiological conditions (Fig. 1) [17]. AM make up 90 to 95% of the cells collected by BAL [54], and they degrade surfactant lipids [55]. This function can be essential also during pathological conditions when the lung is exposed to oxidizing agents that can induce oxidation of surfactant proteins and lipid, as well as lipid aggregation [56, 57]. Granulocyte macrophage colony stimulating factor (GM-CSF) is required for macrophages’ ability to clear surfactant, and deficiency in GM-CSF can lead to excessive surfactant accumulation and pulmonary alveolar proteinosis (PAP) [58]. Indeed, PAP is characterized by abnormal accumulation of surfactant in the alveoli and the terminal airways. Quantitative lipidomics of BAL of patients with PAP showed a significant increase in both free cholesterol and cholesterol esters. Other surfactant components, including sphingolipids, ceramides, PE, PS, PC, LPC and the surfactant proteins were also increased [6]. The clinical course is heterogeneous, ranging from spontaneous resolution to death due to respiratory failure [59]. In the absence of GM-CSF signaling, AM massively accumulate intracellular cholesterol-rich lipid droplets and extracellular surfactant also showed an increased proportions of cholesterol. This points out at defects in GM-CSF signaling and AM cholesterol clearance as the primary drivers of PAP [60,61,62].
The most effective and proven treatment for PAP is whole lung lavage [63], and GM–CSF can be used as inhalation therapy, or subcutaneous injection if whole lung lavage fails or is contraindicated. Experimentally, inhibition of cholesterol synthesis by statins showed positive therapeutic value. Since the main driver of PAP is defective cholesterol clearance by AM and not increased synthesis, the mechanism for the observed effects is not clear [64].
On the other end of the spectrum is surfactant deficiency, the cause of neonatal IRDS [65, 66]. But deficiency and alterations in surfactant lipids and proteins have long been described also in adult pulmonary diseases, including IPF, adult onset PAP, ARDS and lung cancer [27, 67,68,69]. Genetic alterations in components of the surfactant metabolism pathway have detrimental effects on pulmonary function and disease pathogenesis. Mutations in the genes encoding SP–A, SFTPA1 and SFTPA2, are associated with interstitial lung disease and increased susceptibility to adenocarcinoma of the lung [70, 71]. Autosomal recessive mutations in the gene encoding SP–B, SFTPB, were among the earliest described [72, 73]. SP–B–deficient humans and mice develop respiratory distress and respiratory failure independent of surfactant lipid composition [74,75,76]. SP–B knockout mice show aberrant lamellar body formation, and incorrect SP–C intracellular processing [75]. The lack of SP–B disturbs lamellar body formation in T2C, impairs processing of other surfactant proteins, and disrupts surfactant recycling [77]. Deficiency of ABCA3 also recapitulates this phenotype and causes respiratory failure [78, 79]. Consistently, mutations in the homeodomain–containing transcription factor TTF–1, master regulator of surfactant protein and ABCA3 expression, were also associated with respiratory failure [80]. Finally, SP–C mutations were also associated with increased susceptibility to pneumonitis and emphysema due to decreased SP–C–mediated alveolar surfactant spreading [45]. Other genetic abnormalities or deficiencies in SP–C lead to cholesterol accumulation in the alveoli and are described in detail in the following pages [46, 81].
Although we focus on the lipid aspects of alveolar cell function in this review, it is worth mentioning that alveolar cells partake in other processes. T2C contribute to other essential roles for pulmonary homeostasis and alveolar repair [82], as well as in immune defense processes [83] and inflammation [84, 85]. Interstitial macrophages are also crucial in regulating and suppressing unwanted or excessive immune reactions [86]. During pathogenic processes, fibroblasts recruit inflammatory cells, and partake in remodeling and regenerating the extracellular matrix after tissue injury. Excessive activation of fibroblasts can lead to fibrosis and scarring of the lung [87]. The role of lipid mediators in the development of fibrosis and other adult and chronic pulmonary diseases is discussed in the following pages.
COPD and other smoking–related diseases
The pathophysiology of chronic obstructive pulmonary disease (COPD), which involves emphysematous destruction of alveolar sacs and airway remodeling, is highly dependent on environmental and genetic factors. Cigarette smoking is among the most prevalent pulmonary public health concerns worldwide and is a leading cause of COPD in smokers and former smokers, although other causes, including exposure to environmental pollutants, also contribute significantly to this burden.
COPD patients have both decreased surfactant amount and altered surfactant composition, potentially making it less effective at reducing the surface tension (Table 1) [6, 7, 88]. Our group recently reported the decrease in total surfactant lipid, as well as specific lipid species, in patients with COPD [7]. Decreases in total BAL lipid, total PL, PC 30:0, PC 32:0, and total cholesterol, amongst other lipids, strongly correlated with decreased lung function. The main surfactant lipid changes observed in COPD patients were replicated in a mouse model exposed to 6 months of second–hand smoke, which enables future mechanistic research. This study is well aligned with prior ones showing that smoking reduced BAL PL content in humans [89]. Surfactant replacement therapy provided pulmonary function improvement in a small clinical trial in individuals with stable bronchitis, often a component of COPD [90]. However, the mechanism for this improvement, as well as the roles of surfactant lipids in COPD, are not clear [67, 91].
COPD and emphysema can result in decreased alveolar T2C or premature senescence, potentially impacting lipid metabolism (Table 1) [92, 93]. Cigarette smoking can affect lung lipid homeostasis through direct and indirect mechanisms. The mechanisms for smoke exposure–induced damage to the T2C include inflammation, oxidative stress, dysfunctional DNA repair mechanisms, and proteolysis–antiproteolysis imbalance, amongst others [94,95,96]. Indeed, T2C of individuals with emphysema have increased reactive oxygen species generation and DNA damage when compared to those of healthy controls [97]. In T2C–derived human A549 cells, cigarette smoke induced apoptosis, inhibited proliferation, and spurred epithelial–mesenchymal transition (EMT) [98].
Smoke exposure damaged T2C and caused alterations of surfactant secretion and composition in multiple animal models [95, 99,100,101,102,103,104]. Most studies report decreased surfactant lipid availability after chronic exposures to smoke, a common model of COPD. Rats exposed to 60 weeks of nose–only smoke showed significant decreases in BAL DPPC content when compared to room air controls [100]. BAL from smoke–exposed rats had excessive surfactant compressibility and reduced re–spreadability. A mechanism for smoke decreasing surfactant levels can be direct damage to the T2C, including alterations in cell adhesion, proliferation and direct cell lysis.
At the molecular level, there are many potential mechanisms of smoke–mediated disruption of surfactant metabolism. Smoke was found to directly interfere with DPPC synthesis by reducing PLA2 activity by more than 50% [101], and both in humans and mice, smoking caused alterations in pulmonary expression of proteins implicated in reverse lipid transport, including ABCA1, ABCG1, ApoE and SRB1 [105, 106]. Cultured T2C acutely exposed to cigarette smoke extract showed inhibition of stimulated PC secretion, while basal PC secretion was not affected [99]. Another mechanism by which cigarette smoke may interrupt surfactant metabolism is by the generation of free radicals, and enhancing oxidative stress, which in turn can elevate the levels of TNFα, and other proinflammatory markers [107, 108] [103]. In addition, nicotine can induce excessive expression of SP–A and SP–C in an embryonic mouse lung culture [109]. In addition to T2C damage, cigarette smoke exposure also affect the whole lung parenchyma. In addition to alveolar congestion following smoke exposure, one study reports diffuse alveolar collapse, septal hypertrophy, and other gross lung abnormalities [104].
AM are also affected by cigarette smoke (Table 1). Tran et al. [110] showed that AM exposed to cigarette smoke had less efficient efferocytosis, and this was attributed to impaired sphingosine kinase (SPK) activity. Similarly, in vivo instillation of ceramide impaired AM efferocytosis, and this effect was reproducible by sphingosine treatment, suggesting the involvement of ceramidase activity in this impairment [111]. Efferocytosis is necessary for elimination of airway apoptotic cells that if not removed, can eventually necrotize and lead to chronic inflammation [112]. Both cigarette smoking and electronic cigarettes disrupted the endothelial barrier by affecting intracellular ceramides, mitogen activated protein kinase (MAPK) activation and myosin light chain phosphorylation [113].
The use of electronic cigarettes and other “vaping” devices has become popular in recent years. Electronic cigarette use can have deleterious effects on lung lipid metabolism regardless of nicotine content (Table 1). The use in mice of an electronic nicotine delivery system (ENDS) during 4 months lead to increased deposition of surfactant in the alveoli without increase in inflammatory markers or emphysema, and interestingly, there were no differences between the nicotine–containing and vehicle–only groups when compared to air–exposed mice. Exposure to ENDS increased cytoplasmic lipid droplets in AM and increased the number of lamellar bodies with disrupted architecture in T2C. In addition, the expression of ABCA1 and ABCG1 was significantly decreased, together with the expression of surfactant proteins SP–A and SP–D. When challenged with influenza infection, ENDS–treated mice showed disrupted innate immunity and enhanced inflammation, with persistent expression of interferon–γ (IFN–γ) and tumor necrosis factor–α (TNF–α) [114].
In November 2019, a public health concern was raised following the report of multiple cases of e–cigarette and vaping associated lung injury requiring hospitalization with some cases being fatal, and with high incidence amongst teenagers and young adults. The symptoms included dyspnea and hypoxemia with no associated infection, and in multiple cases the BAL showed increased accumulation of neutral lipids in AM. While the mechanisms for the lung injury have not been deciphered at the moment of writing this article, multiple cases seem to be associated with vaping of cannabinoid and other terpene–rich oils that may have been customized by the final users [115,116,117,118,119,120]. In a histopathologic study of lung biopsies obtained from vaping–induced lung injury cases, acute lung injury was confirmed and foamy AM and pneumocyte vacuolization were observed in all the samples [118]. The potential effects of lipid–vapor inhalation in alveolar lipid metabolism are still largely unknown.
Other particulate and gaseous environmental agents have also been linked to altered surfactant lipids. Chronic inhalation of silica dust causes silicosis, which is characterized by foamy macrophages. These AM have higher intracellular levels of neutral lipids and show elevated mRNA levels of the lipid receptor CD36 and transcription factor PPARγ [121]. Inhaled ozone (O3) generated from nitrogen oxides and volatile hydrocarbons alter lung function and cause inflammation [122]. Once inhaled, approximately 80% of O3 remains in the lungs and may react with the carbon–carbon double bonds of surfactant phospholipids and cholesterol, a process termed “lipid ozonation” [122]. In addition, common household dust mites have also been linked to the generation of proinflammatory eicosanoids and cytokines in alveolar macrophages [123].
Lipids in interstitial lung disease and idiopathic pulmonary fibrosis
IPF is a rare disease that has attracted attention lately due to the approval of the first few drugs for its treatment. IPF patients experience a progressive decline in pulmonary forced vital capacity (FVC), with their lung parenchyma and airways showing extensive remodeling, fibroblast proliferation, excessive extracellular matrix deposition and loss of compliance. Together, the clinical manifestation is shortness of breath and difficulty in lung inflation. The progression after diagnosis is fast, with death occurring in 3–10 years unless a transplant is provided. The currently approved drugs decelerate the disease progression, but there is no regression.
A main difficulty in IPF treatment is the lack of markers for early disease detection. In an attempt to identify a biomarker, a small study showed increased LysoPC in the serum of IPF patients, and this provided a rationale for the development of autotaxin inhibitor drugs [124]. However, there are no specific pulmonary biomarkers. Another difficulty in IPF treatment lies on the current lack of knowledge on early triggers and clear mechanisms for disease progression before the end–stage, when patients normally present extensive remodeling.
Recent studies using single cell RNA sequencing have shed some light on the pathobiology of IPF. Reyfman et al. [125] detected over 500 genes differentially expressed in patients with IPF. Interestingly, in T2C the top 500 genes downregulated in IPF were all included in lipid metabolic pathways, including “sterol/steroid biosynthetic process”, “cholesterol metabolic process” and “lipid metabolism process”. AMs also showed profound alterations in lipid metabolism, with downregulated pathways including “cellular response to fatty acids” and “positive regulation of lipid metabolic processes”. It is not clear if this loss of ability to handle lipid metabolism by alveolar cells constitutes a cause or a consequence of the disease. The data were confirmed with further qPCR analysis and by other studies [126,127,128], and these pathways actually comprised the IPF RNA signature of the whole lung when different cell types were not grouped separately.
Naturally occurring mutations can confer susceptibility to fibrosis development later in life. Mutations in ABCA3, like the most common missense mutation E292V, result in IPF development. Mice carrying this mutation showed chronic surfactant insufficiency, with decreased PL in BAL. T2C showed increased number of lamellar bodies, but of smaller volume, and decreased alveolar surface area. BAL cells, mostly AMs, were increased and there were numerous tissue morphological changes, including peribronchial immune infiltrates and a combination of fibrotic and emphysematous regions. Bleomycin instillation, a common model of experimental fibrosis, generated worse fibrosis and higher morbidity in mutant than in WT mice [129]. Mutations in surfactant protein C (SP–C) also result in IPF development. Nureki et al. [81] generated tamoxifen–inducible knock–in mice expressing the substitution of isoleucine by threonine at codon 73, a mutation associated with familial IPF (SP–CI73T mice). SP–CI73T mice had overexpression of SP–C and developed premature fibrosis that recapitulated the human phenotype, with collagen deposition, T2C hyperplasia, fibroblast proliferation, and decreased compliance. Intracellular lipid metabolism and surfactant lipids were not assessed in this model, and it remains unknown so far if the ability of T2C from SP–CI73T mice to sustain normal lipid homeostasis is affected. In addition to the findings above, SP–C was recently reported to modulate alveolar lipid homeostasis during development of fibrosis. SP–C knockout mice, aside from to developing spontaneous lung fibrosis, also showed AM cholesterol accumulation. In vitro, addition of SP–C to cholesterol–containing vesicles in an AM cell line increased expression of genes involved in cholesterol metabolism and transport [46]. These are the first models of spontaneous lung fibrosis and are valuable tools for the study of IPF pathogenesis.
In agreement with the observations in human IPF, bleomycin–induced fibrosis caused decrease in mRNA of lipogenic genes in whole lung and specifically in T2C [130]. Intracellular lipids including cholesterol, free FA, triglycerides and PL were decreased, as well as ABCA3 mRNA expression. However, BAL lipids were increased. Consistently with a decreased intracellular lipogenic program, T2C showed increased phosphorylation of AMP kinase and acetyl–CoA carboxylase, and they were more glycolytic, with decreased intracellular ATP levels and increased lactic acid production. In vitro experiments with T2C showed that bleomycin induced lipid secretion and increased extracellular ATP, a known surfactant secretagogue, while surfactant lipid reuptake was impaired. Treatment of cultured AM with lipid extracts from BAL of bleomycin–treated mice resulted in foam cell formation. Treatment with oxidized PC was by itself capable of increasing mRNA expression of transforming growth factor beta (TGFβ) and M2 markers in cultured macrophages. These observations led the authors to propose a model where the increase in TGFβ1 and collagen deposition secondary to bleomycin injections causes accumulation of abnormal lipid surfactant in the alveolar space. Accumulated PC can then become oxidized and induce AM transformation into foam cells, further contributing to the profibrotic phenotype generating a feedforward loop. The extent of the overlap of this mechanism with the observed lipid metabolic RNA signature of human IPF patients remains to be determined, but in any case, this work shows a dysregulation of lipid homeostasis that integrates three different alveolar compartments, T2C, alveolar space and alveolar macrophages, during the pathogenesis of pulmonary fibrosis.
Experimental bleomycin exposure results in alterations in alveolar lipids, but there is no consensus on the specific changes and directions in different models of experimental fibrosis. Similarly, human studies have reported seemingly contradictory data regarding the direction and magnitude of change in surfactant lipids during disease, and there is no agreement on a mechanism for the observed alterations [67] (Table 1). In some studies, increased levels of PC, cholesterol and bis monoacyl phosphoglycerate (BMP) in whole lung tissue and of all lipids in BAL up to 21 days after bleomycin have been reported [131]. Intratracheally administered bleomycin increased cholesterol and free FA in BAL in rats, and this was associated with increased collagen deposition and epithelial cell proliferation, elastic recoil and surface tension of the BAL. In these studies, the acyl composition of the cholesterol esters in BAL was prominently 16:0, 18:2, 18:1 and 18:0 and it did not change with bleomycin treatment [132, 133].
Pulmonary fibrosis induced by administration of the antiarrhythmic compound amiodarone caused hyperplasic T2C that accumulated PL, BMP and surfactant proteins, and this was associated with ER stress and enhanced pro–apoptotic response. In contrast to bleomycin treatments, amiodarone–induced fibrosis decreased extracellular surfactant DPPC and proportionally increased unsaturated species of PC. Similarly, phosphatidylglycerol decreased and plasmalogen phosphatidylethanolamine increased in BAL [134].
DNA damage and telomere shortening in T2C induced pulmonary fibrosis and decrease survival in mice [135] (Table 1). T2C genetic deletion of telomeric repeat factor 1 (TRF1), a protector of telomere ends, induced pulmonary fibrosis by increasing cellular senescence and apoptosis. In addition, bleomycin treatment in the setting of telomerase–deficiency also recapitulated this fibrotic phenotype [135]. Mice overexpressing telomerase in T2C showed increased T2C proliferation and downregulation of fibrotic and inflammatory gene expression [136]. Aberrant telomere shortening has previously been associated with alveolar stem cell dysfunction [137], and these new data show how telomere dysfunction and subsequent T2C–depletion could be used as a model of IPF.
Bleomycin, silica and radiation exposures all showed deranged lipid metabolism in AMs [130] (Table 1). They accumulated neutral lipid as well as phospholipid, and showed increased mRNA expression of lipid transporters CD36, scavenger receptor A (SRA), ABCA1, ABCG1 and its upstream regulator LXRα. There was an increase in oxidized PC both in BAL and intracellularly in the alveolar macrophages. Simultaneously with these events (14 days after bleomycin treatment), terminal airspaces started to show macrophage infiltration, and progressively increased mRNA expression of TGFβ1 and collagen 1a1. Chronologically, histochemical and biological onset of fibrosis occurred after AM lipid accumulation had started. Fibrosis subsequent to nitrogen mustard exposure triggered the transformation of AM into foam cells [138], and in this case, lipid–laden pulmonary macrophages also showed altered lipid handling pathways as analyzed by RNAseq.
Despite the uncertain direction of the BAL lipid changes during IPF, administration of extracellular surfactant lipid was attempted to improve pulmonary compliance. In mice with bleomycin–induced fibrosis, surfactant replacement therapy rescued compliance and inspiratory capacity, and the number of open alveoli was strongly correlated with static compliance [139]. Overexpression of TGFβ1, which occurs naturally during IPF progression, was associated with loss of apical membrane in T2C during experimental fibrosis [140]. Pretreatment with commercially available surfactant (Curosurf) improved lung mechanics and tissue elastance, increased the number of open alveoli, and preserved the apical membrane surface in T2C. This raises the question of what is the mechanism of alveolar surfactant to protect against the deleterious effects of TGFβ1 treatment. Decreased BAL surfactant proteins, as well as their intracellular mRNA and that of the lipid transporter ABCA3 preceded morphological remodeling of TGFβ1–treated mice, pointing at the role of intracellular T2C lipid metabolism regulation in the development of fibrosis [141]. Through a different mechanism, DPPC and Survanta for 24 and 48 h increased eicosanoid synthesis and inhibited thromboxane A2 synthesis in silica dust–treated AMs [142].
Intracellular lipid mismanagement also partakes in fibrosis development (Table 1). IPF patients and mice treated with bleomycin show decreased expression and activity of multiple lipid metabolic enzymes. Elongation of very long chain fatty acids protein 6 (Elovl6) is one of them. Elovl6 catalyzes the elongation of C16 fatty acids to longer acyl chains and renders unsaturated fatty acyl chains. Elovl6−/− mice are protected from HFD–induced hepatic steatosis and fibrosis, potentially due to their higher palmitoleic/palmitic acid ratio. Alveolar T2C also express Elovl6, and when the Elovl6−/− mice were treated with bleomycin, they developed worse fibrosis, with more collagen deposition and increased mortality [143]. The pulmonary FA composition was altered in Elovl6−/− mice, with a higher proportion of palmitic acid C16:0, in detriment of palmitoleic C16:1(n–9), and this effect was more pronounced after bleomycin. Treatment of cultures of a T2C cell line with palmitic acid triggered apoptosis and increased TGFβ1 expression, both of which were attenuated by treatment with unsaturated fatty acids oleic or linoleic acid. The authors proposed that the increase in palmitic acid content resulted in profibrotic events such as increased TGFβ and apoptosis through increased intracellular generation of reactive oxygen species. Increased FA content, including palmitic acid has been encountered in IPF lungs [144, 145], and treatment of cell cultures with palmitic acid triggered ER stress and apoptotic responses. In vivo, mice pretreated with 2–weeks of high fat diet (HFD) had higher collagen content upon bleomycin treatment. However, it is difficult to assess the contribution of increased extracellular availability of lipid and subsequent pulmonary lipid metabolism to this phenotype, since HFD is known to trigger multiple systemic responses, including low–grade inflammation prior to the onset of obesity [146].
ER stress can serve as a priming event to pulmonary fibrosis by affecting intracellular lipid events. For example, enhanced autophagy in Golgin A2 (GOLGA2)−/− mice limited the subcellular availability of functional mitochondria and lamellar bodies, and this was associated with decreased DPPC and a mild increase in extracellular matrix (ECM) deposition in both lungs and liver [147]. Intranasal tunicamycin increased ER stress, as well as expression of lipogenic enzymes fatty acid synthase (FAS), stearoyl–CoA desaturase 1 (SCD1) and diglyceride acyltransferase (DGAT), their upstream regulator SREBP1, and intracellular triglyceride and PL content [148]. Loss of mitochondrial mitofusin 1 or 2, as well as inhibition of FAS in T2C, worsened bleomycin–induced fibrosis and was associated with perturbed surfactant lipid metabolism [149]. Silica treatment also resulted in a similar lipid synthetic signature that could be rescued by treatment with LXR agonist TO901317, which has been shown to be antifibrotic in other studies [150]. Dysregulated intracellular lipid metabolism can be a cause and a consequence of ER stress. The sole inhibition of SCD1 was enough to induce ER stress and collagen deposition. It was not determined if the potentially increased proportion of saturated fatty acids, including palmitic, resulting of SCD1 inhibition could contribute to this ER stress [148, 151]. Lipid metabolic pathways are often challenging to interpret unless direct metabolite measurements are performed, since same fatty acid substrates can result in different products, with different implications in metabolic and inflammatory pathways, highlighting the relevance and complexity of metabolic fluxes in different cellular conditions.
Activation of PLA2 and its action on membrane PC can release arachidonic acid (AA), which serves as a precursor for eicosanoids, potent signaling lipids. AA can be further processed by three different pathways. The cyclooxygenase pathway leads to the generation of prostaglandin H and its derived prostaglandins and thromboxanes, collectively called prostanoids. AA metabolism by the lipoxygenase (12/15 LOX) pathway generates leukotrienes and lipoxins, amongst other lipids, and metabolism by the epoxygenase P–450 pathway generates epoxyeicosatetraenoic acids. The functions of eicosanoids in IPF have been previously summarized in the literature [152, 153] and here we will only briefly mention the latest updates.
Prostaglandins (PGs) and other cyclooxygenase 2 (COX2)–derived prostanoids seem to be protective against experimental fibrosis. In mice, COX2 but not COX1 deletion worsened the fibrotic phenotype induced by bleomycin [154]. The specific PG downstream of COX2 and responsible for these observed effects seems to be cell–type specific. Hematopoietic cells express PGD, and PGD synthase knockout mice had higher degree of collagen deposition and increased mRNA expression of TNFα and other pro–inflammatory mediators [155]. Fibroblasts from IPF patients synthesize lower amounts of PGE2 than control subjects, and they are also less responsive to treatments with PGE2 [156]. The mechanisms for the antifibrotic effects of PGE2 are unclear, with different studies having shown even contradictory conclusions in some cases. Mice knockout for PGE synthase 1 (PGES1) were unable to increase PGE2 amounts after bleomycin. However, no differences were observed in inflammation, fibrosis and pulmonary loss of function between WT and PGES1−/− mice. Similarly, knocking out PGE2 or its receptors EP2 or EP4 did not alter the course of bleomycin–induced fibrosis. In contrast, knocking out the receptor for PGI2 phenocopied COX2−/− mice, suggesting that the antifibrotic effects downstream COX2 are attributable to PGI2 and not PGE2. However, other experiments have shown that the responsiveness to PGE2 differs according to fibrosis etiology, and that there is significant inter–patient variability [157]. This could be related to the ability of the cells to successfully trigger local signal events through PGE2 and its receptor EP2. Fibroblasts from human and murine fibrotic lungs showed lower expression of EP2 and this was associated to its promoter’s hypermethylation [158]. In vivo, administration of PGE2 prior to bleomycin–induced fibrosis offered protective effects against decreased pulmonary function and increased collagen production. However, there was no therapeutic effect, as experimental fibrosis developed equally in mice treated with saline or PGE2 after fibrosis had been induced with bleomycin [159, 160]. TGFβ stimulation of a human fibroblast cell line altered the expression of over 1000 genes, and treatment with PGE2 reversed multiple of these changes, especially those involved in the development of a myofibroblast phenotype [161, 162]. These effects were reproducible in a cell line of fetal fibroblasts and seemed to be mediated by receptor EP2.
A different mechanism for the antifibrotic effects of PGE2 involves the activation of plasminogen and plasminogen activator system [163]. PAI–1−/− mice showed increased production of PGE2 in the lung. Treating primary fibroblasts from control and bleomycin–treated mice with both uPA and plasminogen together, but not when separated, increased PGE2 secretion and COX2 expression. The authors suggested an axis plasminogen/plasmin/extracellular hepatocyte growth factor (HGF)/HGF receptor as antifibrotic mechanism. But other mechanisms independent of HGF have also been proposed [164]. Plasminogen activation and plasmin enhanced protein kinase A (PKA) signaling by decreasing protein phosphatase 2A (PP2A) activity, thus leading to sustained phosphorylated status of PKA substrates. Suppression of PP2A activity in IPF–patient derived cell lines of fibroblasts helped overcome the resistance to PGE2 treatment. Finally, it was concluded that PAI–1−/− mice are resistant to experimental fibrosis because of a sustained activation of plasminogen and enhanced proteolytic activity of uPA and downstream activation of plasmin.
Leukotrienes are lipid metabolites also derived from arachidonic acid and with signaling properties in pulmonary fibrosis. Leukotriene–deficient 5–lipooxigenase knockout mice (5–LO−/− mice) were resistant to FITC–triggered experimental fibrosis potentially due to their inability to trigger receptor–mediated proliferation of basal fibrocytes [165].
Lipoxins, resolvins, protectins and maresins are other eicosanoid lipids with different chemical structures grouped under the umbrella term “resolving mediators” due to their roles in the resolution of inflammation. Resolvins and maresins derive from docosahexaenoic acid (DHA). In mice, resolvins ameliorated the bleomycin–induced increases in BAL cellularity and profibrotic cytokines, they improved Aschroft fibrosis score and also restored the levels of MMP9 to pre–bleomycin levels [166]. Maresin 1, which is produced by activated macrophages during inflammation, suppressed EMT by suppressing Smad2/3 and Akt signaling in vivo [167]. In vitro, Maresin 1 prevented TGFβ1–induced fibroblast proliferation, migration and differentiation into myofibroblast [168].
Amongst sphingolipids, Sphingosine–1–phosphate (S1P) is perhaps the best studied lipid in pulmonary fibrosis. Sphingolipids are essential constituents of plasma membranes and regulate important cellular functions, including apoptosis and proliferation. The balance of intracellular sphingomyelin and ceramide is crucial in inflammatory conditions, and the roles of ceramides and sphingolipids in chronic lung disease have been reviewed recently [169, 170]. S1P is synthesized by phosphorylation of sphingosine by sphingosine kinase 1 (SPK–1) and Sphingosine Kinase 2 (SPK–2). S1P can be secreted as a potent water–soluble signaling lipid capable of activating G–proteins coupled receptors in the target cells. Signaling can be stopped by degradation of S1P through the action of S1P phosphatases and S1P lyase [171].
S1P and SPK–1 were higher in BAL of IPF patients, and the expression of SPK1 inversely correlated with pulmonary function measures such as diffusing capacity for carbon monoxide (DLCO), forced expiratory volume in 1 s (FEV1), and FVC. Bleomycin–induced fibrosis in animal models showed consistent phenotypes, and genetic deletion of SPK1 improved pulmonary fibrosis, while deletion of S1PL worsened it [172, 173]. In vitro studies to seek the mechanism showed that fibroblast treatment with TGFβ increased S1P as well as expression of S1PL through SMAD3 activation. Overexpression of S1PL restored intracellular S1P levels through modulation of autophagy. Together, these data highlight the relevance of fine regulation of S1P signaling during disease (Table 1) [174].
Role of alveolar lipids during pulmonary infections
The lungs are constantly exposed to microbes that enter the respiratory tract by aspiration. An effective pulmonary host defense is able to tolerate a low level of microbial invasion. However, the development of respiratory infections may occur in the event of defect in host defense, an overwhelming inoculum, or exposure to a virulent microorganism. Respiratory infections can be broadly categorized into upper respiratory tract infections, affecting the mouth, nose, sinuses, throat, larynx and trachea, and lower respiratory tract infections, affecting the lower airways, bronchi and alveoli. Upper respiratory tract infections typically present as common colds, influenzas, epiglottitis, sinusitis, and pharyngitis. Lower respiratory tract infections typically include bronchitis, bronchiolitis and pneumonia. Some of the deadliest infections, including influenza, pneumonia, and tuberculosis, exploit properties of lipids to enhance their propagation and pathogenicity, making lipid metabolism a critical player in the pathogenesis of pulmonary infections.
Pneumonia encompasses an umbrella of conditions that may arise from many etiologies, including bacterial, viral, mycoplasmal, fungal, lipoid, and aspiration of other exogenous substances. The most common form of pneumonia is bacterial pneumonia, and it alters host lipid composition and transport. BAL from human subjects with bacterial pneumonia showed mild decrease in total surfactant PL, marked decrease in glycerophospholipid and increase in phosphatidylinositol and sphingomyelin amounts (Table 1) [67]. While much remains to be uncovered regarding the consequences of surfactant alterations, these changes in the surfactant lipidome were thought to interfere with the surface tension reduction and antimicrobial functions [67, 175]. In addition to surfactant lipids, other BAL lipids also changed during pneumonia. Cardiolipin, a mitochondrial–specific lipid, was significantly elevated in BAL from infected humans and mice, and this markedly increased alveolar surface tension, decreasing lung compliance and increased IL–10 and BAL protein concentration [176]. Enrichment of cardiolipin in BAL was also correlated with decreased BAL surfactant proteins SP–A and SP–C and disruption of alveolar architecture. One proposed mechanism was that cardiolipin interferes with the packaging of surfactant DPPC, thereby increasing surface tension. The amount of cardiolipin in the alveolar lumen is regulated by the cardiolipin transporter ATP8b1, which internalizes and sequesters cardiolipin from the extracellular space. Mice bearing a missense mutant form of ATP8b1 present in many humans showed increased susceptibility to infection and infection–induced lung injury [176]. These increases in BAL cardiolipin content are also consistent with other types of acute lung injury [177, 178].
Interestingly, not only does bacterial pneumonia influence host surfactant composition, but exposure of bacteria to specific surfactant lipids was found to alter bacterial transcriptomics suggesting novel mechanisms of host–pathogen interaction. K. pneumoniae MGH78578 exposed to purified PC and cholesterol in vitro showed increased transcriptional levels of genes involved in capsule synthesis, lipopolysaccharide modification, antibiotic resistance, biofilm formation, and metabolism [179]. This increase in virulence gene expression may be especially relevant in cases of PAP and associated surfactant lipid accumulation. Mechanistic studies are needed in this field to determine the potential roles of the different surfactant lipids in different types of infection.
Lipopolysaccharide (LPS) is a major immunogenic constituent of the Gram–negative bacterial cell membrane. Accordingly, LPS is recognized by the host immune system, including TLRs, and triggers the cellular release of pro–inflammatory cytokines, eicosanoids, and potent vasodilators. Structurally, LPS is composed of Lipid A and two different oligosaccharides. Lipid A contains multiple fatty acid chains, and can interact with other hydrophobic lipids. Indeed, LPS interacts with pulmonary surfactant and inactivates it. LPS–surfactant complexing was shown in pneumonia and was proposed to contribute to its pathophysiology [180, 181]. Re–LPS, the minimal form of LPS required for bacterial growth in vitro, interacted with DPPC and caused DPPC monolayers to disperse and fluidize, altering their surface tension reducing properties [182]. Whole LPS also exerted this fluidizing effect on films in vitro, preventing lipid packing when they were compressed [183] and also prevented cholesterol packing in vitro [184].
LPS can also interact with SP–A and SP–D. In fact, SP–A specifically recognizes LPS lipid A [185, 186]. SP–A and SP–D play an important role in the innate immune response to pathogen–associated molecular patterns (PAMPs), and they can modulate the host response to LPS challenge by altering host cytokine release [187,188,189] and by scavenging LPS, minimizing LPS–mediated surfactant clumping [183]. In addition, SP–A and SP–D also destabilized the bacterial cell membrane [190].
Lipoid pneumonia is a rare condition characterized by the accumulation of endogenous or exogenous lipids in the alveoli and has been described as a precursor for other respiratory conditions, including PAP [191]. Lipoid pneumonia often presents with sudden onset of nonspecific respiratory symptoms and may be diagnostically confirmed by the demonstration of lipid–engorged macrophages in BAL, sputum, or lung tissue. Treatment for this rare condition is ill–defined, but whole lung lavage and corticosteroid administration have been described as potential treatments for advanced or recurrent lipoid pneumonia [192]. Endogenous lipoid pneumonia may occur following damage to alveolar cells that causes release of lipids into the alveolar lumen [193]. The influx of cellular lipids into the airspace and compensatory uptake by AM activates an immune response that often leads to the progression of pulmonary disease. Exogenous lipoid pneumonia, on the other hand, has traditionally been linked to the aspiration of oily substances; however, this condition is now being linked to e–cigarette vaping, which has an increasing incidence [194, 195]. In a case study, vaping–associate lipoid pneumonia was associated to lung accumulation of vegetable glycerin, a major e–cigarette liquid component, causing dysregulation of AM lipid metabolism [196]. Moreover, the dysregulation of endogenous and exogenous alveolar lipid uptake by macrophages disrupts surfactant clearing and induces an immune response.
A special example of alterations in pulmonary lipid metabolism by an external hydrophobic agent is Amiodarone, a highly effective anti–arrhythmic drug that has potential serious side effects and toxicities, with pulmonary toxicity incidence being around 7% [197]. Apoptosis of alveolar T2C is a major contributor in amiodarone induced lung injury. In a murine model of amiodarone treatment, electron microscopy showed T2C hyperplasia and extensive lung fibrosis. Surfactant phospholipids and proteins accumulated intracellularly over time [134]. Using polarized light microscopy, Haller et al. showed that amiodarone induced alterations in lamellar bodies, leading to impaired pulmonary surfactant packing and function [198]. High resolution subcellular imaging also showed amiodarone accumulation in lysosomes of lung macrophages [199]. Other mechanisms are immune–related and activation of the renin angiotensin system [200].
Viral infections can alter a myriad of metabolic pathways in the host. In a recent untargeted metabolomic study of serum from a cohort of adult subjects infected with the influenza virus, 26 different host metabolites showed differential alterations upon infection. The metabolic pathways affected included FA biosynthesis and oxidation, PL metabolism, steroid hormone metabolism, and nucleotide and amino acid synthesis (Table 1). These data point at the effects a pulmonary infection can exert in circulating lipids [201].
Influenza infections course with inflammation and NSAIDs are a common first line of treatment. The efficacy of additional anti–inflammatory candidates is currently being tested for the treatment of influenza. PGE2 and its synthase PGES–1 are attractive targets for the development of new drugs. In mice, one of the evaluated compounds successfully decreased the expression of cytokines and other pro–inflammatory genes and provided improvement in infection resolution [202]. Protectin D1 (PD1) is a DHA–derived pro–resolving mediator synthesized by the 12/15–LOX pathway that potently inhibits viral replication by inhibition of RNA export from the nucleus of infected cells. In mice, deletion of 12/15–LOX increased viral replication and disease propagation [203]. Indeed, 12/15–LOX and its metabolites were protective during inflammation resolution after influenza infection [204].
The virulence of the influenza virus can be modulated by interactions between the host lipids and the viral proteins. After initial infection and proliferation within the host cell, influenza viral particle assembly is a critical step. Viral particle assembly is orchestrated through the viral matrix protein M1, which must contact plasma membrane lipids for effective viral packaging and release. The lipid composition of the inner and outer leaflets of the plasma membrane was essential for influenza virulence. Specifically, M1 bound phosphatidylserine with high affinity and facilitated viral assembly [205]. The influenza protein M2 also takes advantage of cholesterol in the plasma membrane, which can bind the amphipathic helices of M2 to stabilize the protein and induce a conformational change. This conformational change confers an increased ability of M2 to induce the membrane curvature required for viral budding [206]. Thus, influenza increases its virulence through manipulation of host alveolar lipids.
Influenza not only coopts host lipids to its advantage, it also utilizes its own lipid packaging to enhance its virulence. Efforts to profile the lipid composition of influenza envelope have found a high degree of structural flexibility; this flexibility, conferred by the pathogen’s lipid profile was found to substantially protect the virus, and increase puncturing capacity of target cells [207].
Tuberculosis (TB), a leading cause of death worldwide, causes an estimated 1.2 million deaths and 10 million incident cases in 2018 according to the World Health Organization (WHO) [208]. TB transmission occurs through the inhalation of Mycobacterium tuberculosis (Mtb)–containing aerosolized liquid droplets by the new host [209]. One third of the world’s population is estimated to harbor the latent Mtb pathogen, but reactivation of the pathogen and development of active TB occurs only in approximately 5–10% of these individuals with latent TB, frequently as a result of immunodepression.
The lipids present in the host environment are important factors contributing to Mtb pathogenesis. Mtb H37Rv cultured in lipid–rich media showed increased expression of 368 genes, many of which are involved in conferring drug resistance and increasing the pathogen’s longevity [210]. In fact, Mtb preferentially metabolizes host–derived lipids, namely triglycerides and cholesterol, in order to perform at optimum virulence capacity. Defects in the bacterial catabolism pathways of these fuels constrains Mtb development [211]. In addition, eicosanoids also modulate host responses to Mtb infection. Infected PGE2 receptor–deficient mice accumulate higher pathogen loads than WT mice, suggesting that the host’s PGE2 is protective against Mtb infection [212, 213]. In contrast, Lipoxin A4 (LXA4) and other 5–lipoxygenase products enhance Mtb propagation in the host [213, 214].
Interestingly, over 250 genes in the Mtb genome encode proteins that participate in lipid metabolism, representing a vast proportion of the pathogen’s genome, and many of the lipids in these metabolic pathways are part of the bacterial cell wall [215]. A study profiling the evolution of Mtb strains comparing modern and ancestral bacteria, showed that modern Mtb contains more apolar cell–surface lipids, with decreased proportions of exposed polar lipids [216]. The effect of these changes is an enhanced capacity of the pathogen for aerosolized transmission. The cell wall lipid phthiocerol dimycocerosates (PDIM) was one of the first virulence factors to be identified in M. tuberculosis, and it is ubiquitously expressed in patient isolates. PDIM–deficient H37Rv mutants demonstrated significantly attenuated virulence in guinea pigs [217]. Two Mtb cell wall lipids, diacylated sulfoglycolipids and the phosphatidyl–myo–inositol dimannosides are currently being used as antigens in preclinical trials for a vaccine development for TB [218]. Altogether, these findings illustrate the important roles that the host and viral lipids play in the development of respiratory infection with various pathogens.
While this paper was under revision, an outbreak of a new infectious respiratory illness, named COVID–19, was declared Public Health emergency as it reached pandemic levels. The knowledge on the biology of COVID–19 is extremely limited at this moment [219]. The transmission seems to occur through droplets and symptoms generally included a high fever, headache, cough, fatigue, and respiratory distress that can quickly evolve to pneumonia and ARDS. The virus responsible for this new disease was named SARS–CoV–2, for its genetic similarity to SARS–CoV, the cause of the severe acute respiratory syndrome (SARS) outbreak first reported in February of 2003. Studies of human lung tissues from individuals with COVID–19 and SARS reported similar cell tropism for the two viruses: alveolar T1C, T2C and AM [220, 221]. Biopsied SARS and COVID–19–infected lungs document diffuse alveolar damage as a major hallmark of the diseases, as well as gross organizational changes in the alveoli and interstitial fibrosis by immunofluorescence [222, 223]. SARS infects T2C and eventually induces their apoptosis, spreading to adjacent alveoli, and it has been proposed that SARS–CoV–2 follows a similar path [224]. COVID–19–infected lung tissue shows T2C hyperplasia and erosion of the T1C epithelial lining [222], and autopsied lungs also confirm these findings, with clear T2C proliferation and alveolar barrier break down [221]. At the moment, there is no knowledge on the long term–effects of SARS–CoV–2 infection on the T2C functions of surfactant homeostasis and of alveolar repair.
Lipids during acute lung injury and acute respiratory distress syndrome
Animal–derived surfactant therapy was first used clinically in 1980 to treat 10 preterm infants with IRDS [66]. Since then, a number of studies and randomized control trials have deemed the use of animal–derived surfactants, most commonly bovine– or porcine–derived, successful [65, 225,226,227,228,229,230,231,232,233,234,235,236]. The positive therapeutic effect of these surfactants in the treatment of IRDS is unambiguous, but concerns about the potential infectivity and antigenicity of animal–derived surfactant, as well as production and cost have encouraged the development of synthetic options, which could potentially expand the therapeutic applications to adult patients with ARDS.
Therapeutic surfactant formulations have evolved over the past few decades to yield improved therapeutic benefit to neonate patients. Early clinical trials using nebulized synthetic DPPC for the treatment of IRDS showed negative results [237, 238], and these were attributed to limited delivery of DPPC to the alveoli. Nearly 20 years later, a multicenter trial successfully piloted the use of pumactant, a synthetic surfactant preparation composed of a combination of DPPC and phosphatidylglycerol [239]. Since then, additional synthetic surfactant preparations have been formulated. Colfosceril palmitate was an FDA–approved protein–free surfactant that showed positive results in randomized control trials [240]. However, a meta–analysis comparing synthetic and animal–derived surfactants reported an inferiority of synthetic surfactants owing to their lack of SP–B and SP–C, and for this reason colfosceril palmitate is no longer used [241].
Second–generation synthetic surfactants incorporated molecules that mimic surfactant protein function. A preparation containing recombinant SP–C showed moderate symptomatic improvement in adults with ALI, but did not improve survival [242]. Other synthetic preparations contain different compounds designed to mimic SP–B activity, and have improved stability and resistance to inactivation [243]. Lucinactant was FDA– approved in 2012 and contains an SP–B–like peptide. The efficacy of these second–generation, protein–containing synthetic surfactants were shown in randomized clinical trials to be comparable to that of animal–derived surfactants for the treatment of IRDS [244, 245]. Additional synthetic surfactant preparations are currently being investigated and optimized for resisting the surfactant inhibitory conditions in the alveolar microenviroment of the patient with IRDS [246].
Indeed, decreased pulmonary compliance and increased edema are major pathophysiological findings also in ARDS, and decreased surfactant PL as well as incorporation of PL into polymerized fibrin contribute to this pathophysiology (Table 1) [247]. BAL phosphatidylglycerol was also decreased in ARDS with increased BAL surface tension [69]. Experimental supplementation with phosphatidylglycerol in a neonatal piglet ARDS model reduced IL–6 and alveolar apoptosis, and preserved the alveolar–capillary barrier, thus decreasing pulmonary edema [248].
The role of sphingolipids in acute lung injury (ALI) and ARDS is still unclear and controversial. S1P has been shown to have protective effects against ALI, but the downstream effects of SPK1 and SPK2 in inflammation and lung injury seem to be related to the type of initial insult. SPK1−/− mice were more susceptible to lipopolysaccharide (LPS)–induced lung injury than WT mice, showing increased neutrophil infiltration and endothelial leakage, as well as increased inflammatory cell numbers in BAL [249]. However, Escherichia coli lung infection resulted in enhanced progression of disease in SPK2 but not SPK1−/− mice, independently of neutrophil recruitment and effector functions [250]. In mice with S1PL inhibition or genetic deletion and subsequent increase in S1P levels, LPS challenge had low efficiency at eliciting lung injury and inflammation. Moreover, down–regulation of S1PL expression in human lung endothelial cells decreased LPS–induced endothelial barrier disruption and IL–6 production, suggesting that S1PL might be a potential therapeutic target in ALI and ARDS [251]. Further studies are required to evaluate these hypotheses.
The efficacy of aerosolized surfactant therapy has been piloted in clinical trials for ARDS. In a randomized prospective control trial piloting the use of exogenous surfactant therapy to treat chronic bronchitis, aerosolized surfactant therapy was found to improve subject pulmonary function and improve sputum transport by respiratory cilia [90]. The potential of surfactant for the treatment of ARDS is still unclear.
Summary and conclusions
The lung parenchyma consists of multiple cell types with specific structures and functions, conferring a remarkable complexity in the study of the pathophysiology of pulmonary disease (Fig. 2).

Alveolar lipids in pulmonary homeostasis. Schematic representation of alveolar cell types and the main lipids that partake in multiple functions during pulmonary homeostasis and pathophysiological conditions.
Alveolar surfactant is essential for respiratory function and it is mostly composed of phospholipid, with minor but very specific amounts of other lipids and surfactant proteins. Qualitative and quantitative changes in the surfactant lipids are involved in multiple adult pulmonary pathologies (Table 1). COPD, exposure to pollution and smoke, and the use of cigarettes result in detrimental changes of the surfactant lipids, the surfactant–synthesizing T2C and the surfactant–clearing alveolar macrophages. IPF can arise as a result of prolonged damage to the T2C, and the cell–specific transcriptomic signature of this disease shows profound alterations in intracellular lipid metabolism in T2C and in macrophages. Not only are surfactant lipid metabolic pathways impacted, but bioactive sphingolipids and prostaglandins also show mechanistic involvement in IPF models. Alveolar lipids also partake in the pathophysiology of infectious diseases and ARDS.
The current limitations on the study of pulmonary lipids include the vast complexity that lipid metabolism can quickly acquire. Mass spectrometry now allows sensitive detection of specific lipid species, allowing for more detailed analysis, and the new challenge is the interpretation of such lipidomic data, taking into account that lipids are often metabolites, and as such, they may not accumulate and instead be in a state of flux. Another important limitation is the cellular heterogeneity of the lung. Different cell types may regulate lipid metabolism differently according to their specific functions, despite cells interacting with each other and residing in the same niche. Specific modulation of metabolism in specific cell populations is a challenge yet unresolved. For analytical goals, techniques like single cell sequencing allow transcriptomic assessment of different cell populations, but for lipid biology the techniques are restricted to whole tissue assessment, or cell–separations by combinations of differential centrifugation and surface marker–based selection.
Much work remains to be done to elucidate the details of lung lipid metabolism and signaling with the ultimate goal of developing new therapies, but this is a promising field that will likely expand in the years to come.
Availability of data and materials
Not applicable.
Abbreviations
- AA:
-
Arachidonic acid
- ABC:
-
ATP–binding cassette
- AM:
-
Alveolar macrophage
- ARDS:
-
Acute respiratory distress syndrome
- BMP:
-
Bis monoacyl phosphoglycerate
- BAL:
-
Bronchoalveolar lavage
- CCTα:
-
CTP:phosphocholine cytidyltransferase alpha
- COPD:
-
Chronic obstructive pulmonary disease
- COX:
-
Cyclooxygenase
- DLCO:
-
Diffusing capacity for carbon monoxide
- DGAT:
-
Diglyceride acyltransferase
- DHA:
-
Docosahexaenoic acid
- DPPC:
-
Dipalmitoyl phosphatidylcholine
- ECM:
-
Extracellular matrix
- Elovl6:
-
Elongation of very long chain fatty acids protein 6
- EMT:
-
Epithelial–mesenchymal transition
- ENDS:
-
Electronic nicotine delivery system
- FA:
-
Fatty acid
- FAS:
-
Fatty acid synthase
- FEV1 :
-
Forced expiratory volume in 1 s
- FVC:
-
Forced vital capacity
- GPIHBP1:
-
Glycosylphosphatidylinositol–anchored high density lipoprotein–binding protein 1
- GOLGA2:
-
Golgin A2
- GM–CSF:
-
Granulocyte macrophage colony stimulating factor
- HFD:
-
High fat diet
- IL:
-
Interleukin
- IFN–γ:
-
Interferon–γ
- IPF:
-
Idiopathic pulmonary fibrosis
- IRDS:
-
Infant respiratory distress syndrome
- LFA:
-
Lymphocyte function–associated antigen
- LPCAT1:
-
Lysophosphatidylcholine acyltransferase 1
- LpL:
-
Lipoprotein lipase
- LPS:
-
Lipopolysaccharide
- LysoPC:
-
Lysophosphatidylcholine
- LOX:
-
Lipoxygenase
- LXA4:
-
Lipoxin A4
- MAPK:
-
Mitogen–activated protein kinase
- PAI–1:
-
Plasminogen activator inhibitor
- PGES1:
-
PGE synthase 1
- PC:
-
Phosphatidylcholine
- PD1:
-
Protectin D1
- PDIM:
-
Phthiocerol dimycocerosates
- PG:
-
Prostaglandin
- PKA:
-
Protein kinase A
- PL:
-
Phospholipid
- PLA2 :
-
Phospholipase A2
- POG:
-
Palmitoyl–oleoyl–phosphatidylglycerol
- PP2A:
-
Protein phosphatase 2A
- S1P:
-
Sphingosine–1–phosphate
- SCD1:
-
Stearoyl–CoA desaturase 1
- SP:
-
Surfactant protein
- SPK:
-
Sphingosine kinase
- SRA:
-
Scavenger receptor A
- T1C:
-
Alveolar type 1 cells
- T2C:
-
Alveolar type 2 cells
- TB:
-
Tuberculosis
- TGFβ:
-
Transforming growth factor beta
- TLR:
-
Toll–like receptor
- TNF–α:
-
Tumor necrosis factor–α
- TX:
-
Thromboxane
- uPA:
-
Urokinase–type plasminogen activator
- URI:
-
Upper respiratory infection
- WT:
-
Wild–type
References
Jeon GW. Surfactant preparations for preterm infants with respiratory distress syndrome: past, present, and future. Korean J Pediatr. 2019;62:155–61.
Guillot L, Nathan N, Tabary O, Thouvenin G, Le Rouzic P, Corvol H, Amselem S, Clement A. Alveolar epithelial cells: master regulators of lung homeostasis. Int J Biochem Cell Biol. 2013;45:2568–73.
Kasper M, Barth K. Potential contribution of alveolar epithelial type I cells to pulmonary fibrosis. Biosci Rep. 2017;37:BSR20171301.
Bernhard W. Lung surfactant: Function and composition in the context of development and respiratory physiology. Ann Anat. 2016;208:146–50.
Voelker DR, Numata M. Phospholipid regulation of innate immunity and respiratory viral infection. J Biol Chem. 2019;294:4282–9.
Griese M, Bonella F, Costabel U, de Blic J, Tran NB, Liebisch G. Quantitative Lipidomics in Pulmonary Alveolar Proteinosis. Am J Respir Crit Care Med. 2019;200:881–7.
Agudelo CW, Kumley BK, Area–Gomez E, Xu Y, Dabo AJ, Geraghty P, Campos M, Foronjy R, Garcia–Arcos I. Decreased surfactant lipids correlate with lung function in chronic obstructive pulmonary disease (COPD). PLoS One. 2020;15:e0228279.
Xu Y, Zhang M, Wang Y, Kadambi P, Dave V, Lu LJ, Whitsett JA. A systems approach to mapping transcriptional networks controlling surfactant homeostasis. BMC Genomics. 2010;11:451.
Mallampalli RK, Salome RG, Bowen SL, Chappell DA. Very low density lipoproteins stimulate surfactant lipid synthesis in vitro. J Clin Invest. 1997;99:2020–9.
Engelmann B, Wiedmann MK. Cellular phospholipid uptake: flexible paths to coregulate the functions of intracellular lipids. Biochim Biophys Acta. 1801;2010:609–16.
Garcia–Arcos I, Hiyama Y, Drosatos K, Bharadwaj KG, Hu Y, Son NH, O'Byrne SM, Chang CL, Deckelbaum RJ, Takahashi M, et al. Adipose–specific lipoprotein lipase deficiency more profoundly affects brown than white fat biology. J Biol Chem. 2013;288:14046–58.
Goetzman ES, Alcorn JF, Bharathi SS, Uppala R, McHugh KJ, Kosmider B, Chen R, Zuo YY, Beck ME, McKinney RW, et al. Long–chain acyl–CoA dehydrogenase deficiency as a cause of pulmonary surfactant dysfunction. J Biol Chem. 2014;289:10668–79.
Guthmann F, Haupt R, Looman AC, Spener F, Rustow B. Fatty acid translocase/CD36 mediates the uptake of palmitate by type II pneumocytes. Am J Physiol. 1999;277:L191–6.
Olafsen T, Young SG, Davies BS, Beigneux AP, Kenanova VE, Voss C, Young G, Wong KP, Barnes RH 2nd, Tu Y, et al. Unexpected expression pattern for glycosylphosphatidylinositol–anchored HDL–binding protein 1 (GPIHBP1) in mouse tissues revealed by positron emission tomography scanning. J Biol Chem. 2010;285:39239–48.
Ryan AJ, McCoy DM, Mathur SN, Field FJ, Mallampalli RK. Lipoprotein deprivation stimulates transcription of the CTP:phosphocholine cytidylyltransferase gene. J Lipid Res. 2000;41:1268–77.
Voyno–Yasenetskaya TA, Dobbs LG, Erickson SK, Hamilton RL. Low density lipoprotein– and high density lipoprotein–mediated signal transduction and exocytosis in alveolar type II cells. Proc Natl Acad Sci U S A. 1993;90:4256–60.
Agassandian M, Mallampalli RK. Surfactant phospholipid metabolism. Biochim Biophys Acta. 1831;2013:612–25.
Andreeva AV, Kutuzov MA, Voyno–Yasenetskaya TA. Regulation of surfactant secretion in alveolar type II cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L259–71.
Mulugeta S, Nureki S, Beers MF. Lost after translation: insights from pulmonary surfactant for understanding the role of alveolar epithelial dysfunction and cellular quality control in fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2015;309:L507–25.
Fitzgerald ML, Xavier R, Haley KJ, Welti R, Goss JL, Brown CE, Zhuang DZ, Bell SA, Lu N, McKee M, et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J Lipid Res. 2007;48:621–32.
Hammel M, Michel G, Hoefer C, Klaften M, Muller–Hocker J, de Angelis MH, Holzinger A. Targeted inactivation of the murine Abca3 gene leads to respiratory failure in newborns with defective lamellar bodies. Biochem Biophys Res Commun. 2007;359:947–51.
Besnard V, Matsuzaki Y, Clark J, Xu Y, Wert SE, Ikegami M, Stahlman MT, Weaver TE, Hunt AN, Postle AD, Whitsett JA. Conditional deletion of Abca3 in alveolar type II cells alters surfactant homeostasis in newborn and adult mice. Am J Physiol Lung Cell Mol Physiol. 2010;298:L646–59.
Tian Y, Zhou R, Rehg JE, Jackowski S. Role of phosphocholine cytidylyltransferase alpha in lung development. Mol Cell Biol. 2007;27:975–82.
Bates SR, Tao JQ, Collins HL, Francone OL, Rothblat GH. Pulmonary abnormalities due to ABCA1 deficiency in mice. Am J Physiol Lung Cell Mol Physiol. 2005;289:L980–9.
Baldan A, Tarr P, Vales CS, Frank J, Shimotake TK, Hawgood S, Edwards PA. Deletion of the transmembrane transporter ABCG1 results in progressive pulmonary lipidosis. J Biol Chem. 2006;281:29401–10.
de Aguiar Vallim TQ, Lee E, Merriott DJ, Goulbourne CN, Cheng J, Cheng A, Gonen A, Allen RM, Palladino END, Ford DA, et al. ABCG1 regulates pulmonary surfactant metabolism in mice and men. J Lipid Res. 2017;58:941–54.
Whitsett JA, Wert SE, Weaver TE. Diseases of pulmonary surfactant homeostasis. Annu Rev Pathol. 2015;10:371–93.
Schurch S, Bachofen H, Possmayer F. Surface activity in situ, in vivo, and in the captive bubble surfactometer. Comp Biochem Physiol A Mol Integr Physiol. 2001;129:195–207.
Autilio C, Perez–Gil J. Understanding the principle biophysics concepts of pulmonary surfactant in health and disease. Arch Dis Child Fetal Neonatal Ed. 2019;104:F443–51.
Lopez–Rodriguez E, Perez–Gil J. Structure–function relationships in pulmonary surfactant membranes: from biophysics to therapy. Biochim Biophys Acta. 2014;1838:1568–85.
Dietl P, Haller T. Exocytosis of lung surfactant: from the secretory vesicle to the air–liquid interface. Annu Rev Physiol. 2005;67:595–621.
Haller T, Dietl P, Stockner H, Frick M, Mair N, Tinhofer I, Ritsch A, Enhorning G, Putz G. Tracing surfactant transformation from cellular release to insertion into an air–liquid interface. Am J Physiol Lung Cell Mol Physiol. 2004;286:L1009–15.
Keating E, Zuo YY, Tadayyon SM, Petersen NO, Possmayer F, Veldhuizen RA. A modified squeeze–out mechanism for generating high surface pressures with pulmonary surfactant. Biochim Biophys Acta. 2012;1818:1225–34.
Bernardino de la Serna J, Oradd G, Bagatolli LA, Simonsen AC, Marsh D, Lindblom G, Perez–Gil J. Segregated phases in pulmonary surfactant membranes do not show coexistence of lipid populations with differentiated dynamic properties. Biophys J. 2009;97:1381–9.
Blanco O, Cruz A, Ospina OL, Lopez–Rodriguez E, Vazquez L, Perez–Gil J. Interfacial behavior and structural properties of a clinical lung surfactant from porcine source. Biochim Biophys Acta. 2012;1818:2756–66.
Serrano AG, Perez–Gil J. Protein–lipid interactions and surface activity in the pulmonary surfactant system. Chem Phys Lipids. 2006;141:105–18.
Crouch E, Wright JR. Surfactant proteins a and d and pulmonary host defense. Annu Rev Physiol. 2001;63:521–54.
Wert S, Jones T, Korfhagen T, Fisher J, Whitsett J. Spontaneous emphysema in surfactant protein D gene–targeted mice. Chest. 2000;117:248S.
McCormack FX, Whitsett JA. The pulmonary collectins, SP–A and SP–D, orchestrate innate immunity in the lung. J Clin Invest. 2002;109:707–12.
LeVine AM, Bruno MD, Huelsman KM, Ross GF, Whitsett JA, Korfhagen TR. Surfactant protein A–deficient mice are susceptible to group B streptococcal infection. J Immunol. 1997;158:4336–40.
Cruz A, Vazquez L, Velez M, Perez–Gil J. Influence of a fluorescent probe on the nanostructure of phospholipid membranes: dipalmitoylphosphatidylcholine interfacial monolayers. Langmuir. 2005;21:5349–55.
Wang Z, Hall SB, Notter RH. Roles of different hydrophobic constituents in the adsorption of pulmonary surfactant. J Lipid Res. 1996;37:790–8.
Malcharek S, Hinz A, Hilterhaus L, Galla HJ. Multilayer structures in lipid monolayer films containing surfactant protein C: effects of cholesterol and POPE. Biophys J. 2005;88:2638–49.
Glasser SW, Burhans MS, Korfhagen TR, Na CL, Sly PD, Ross GF, Ikegami M, Whitsett JA. Altered stability of pulmonary surfactant in SP–C–deficient mice. Proc Natl Acad Sci U S A. 2001;98:6366–71.
Glasser SW, Detmer EA, Ikegami M, Na CL, Stahlman MT, Whitsett JA. Pneumonitis and emphysema in sp–C gene targeted mice. J Biol Chem. 2003;278:14291–8.
Ruwisch J, Sehlmeyer K, Roldan N, Garcia–Alvarez B, Perez–Gil J, Weaver TE, Ochs M, Knudsen L, Lopez–Rodriguez E. Air Space Distension Precedes Spontaneous Fibrotic Remodeling and Impaired Cholesterol Metabolism in the Absence of Surfactant Protein C. Am J Respir Cell Mol Biol. 2020;62:466–78.
Bernardino de la Serna J, Perez–Gil J, Simonsen AC, Bagatolli LA. Cholesterol rules: direct observation of the coexistence of two fluid phases in native pulmonary surfactant membranes at physiological temperatures. J Biol Chem. 2004;279:40715–22.
Gunasekara L, Schurch S, Schoel WM, Nag K, Leonenko Z, Haufs M, Amrein M. Pulmonary surfactant function is abolished by an elevated proportion of cholesterol. Biochim Biophys Acta. 1737;2005:27–35.
Vockeroth D, Gunasekara L, Amrein M, Possmayer F, Lewis JF, Veldhuizen RA. Role of cholesterol in the biophysical dysfunction of surfactant in ventilator–induced lung injury. Am J Physiol Lung Cell Mol Physiol. 2010;298:L117–25.
Fireman E, Spitzer S, Grief J, Kivity S, Topilsky M. Cholesterol crystals in BAL fluid from patients with idiopathic pulmonary fibrosis. Respir Med. 1996;90:361–3.
Markart P, Ruppert C, Wygrecka M, Colaris T, Dahal B, Walmrath D, Harbach H, Wilhelm J, Seeger W, Schmidt R, Guenther A. Patients with ARDS show improvement but not normalisation of alveolar surface activity with surfactant treatment: putative role of neutral lipids. Thorax. 2007;62:588–94.
Discher BM, Maloney KM, Grainger DW, Hall SB. Effect of neutral lipids on coexisting phases in monolayers of pulmonary surfactant. Biophys Chem. 2002;101–102:333–45.
Discher BM, Maloney KM, Grainger DW, Sousa CA, Hall SB. Neutral lipids induce critical behavior in interfacial monolayers of pulmonary surfactant. Biochemistry. 1999;38:374–83.
Morales–Nebreda L, Misharin AV, Perlman H, Budinger GR. The heterogeneity of lung macrophages in the susceptibility to disease. Eur Respir Rev. 2015;24:505–9.
Trapnell BC, Whitsett JA. Gm–CSF regulates pulmonary surfactant homeostasis and alveolar macrophage–mediated innate host defense. Annu Rev Physiol. 2002;64:775–802.
Kuzmenko AI, Wu H, Bridges JP, McCormack FX. Surfactant lipid peroxidation damages surfactant protein A and inhibits interactions with phospholipid vesicles. J Lipid Res. 2004;45:1061–8.
Rodriguez–Capote K, Manzanares D, Haines T, Possmayer F. Reactive oxygen species inactivation of surfactant involves structural and functional alterations to surfactant proteins SP–B and SP–C. Biophys J. 2006;90:2808–21.
Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol. 2010;135:223–35.
Suzuki T, Trapnell BC. Pulmonary Alveolar Proteinosis Syndrome. Clin Chest Med. 2016;37:431–40.
Sallese A, Suzuki T, McCarthy C, Bridges J, Filuta A, Arumugam P, Shima K, Ma Y, Wessendarp M, Black D, et al. Targeting cholesterol homeostasis in lung diseases. Sci Rep. 2017;7:10211.
Kitamura T, Tanaka N, Watanabe J. Uchida, Kanegasaki S, Yamada Y, Nakata K: Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony–stimulating factor. J Exp Med. 1999;190:875–80.
Suzuki T, Maranda B, Sakagami T, Catellier P, Couture CY, Carey BC, Chalk C, Trapnell BC. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur Respir J. 2011;37:201–4.
Seymour JF, Presneill JJ. Pulmonary alveolar proteinosis: progress in the first 44 years. Am J Respir Crit Care Med. 2002;166:215–35.
McCarthy C, Lee E, Bridges JP, Sallese A, Suzuki T, Woods JC, Bartholmai BJ, Wang T, Chalk C, Carey BC, et al. Statin as a novel pharmacotherapy of pulmonary alveolar proteinosis. Nat Commun. 2018;9:3127.
Fujiwara T, Konishi M, Chida S, Okuyama K, Ogawa Y, Takeuchi Y, Nishida H, Kito H, Fujimura M, Nakamura H, et al. Surfactant replacement therapy with a single postventilatory dose of a reconstituted bovine surfactant in preterm neonates with respiratory distress syndrome: final analysis of a multicenter, double–blind, randomized trial and comparison with similar trials. The Surfactant–TA Study Group. Pediatrics. 1990;86:753–64.
Fujiwara T, Maeta H, Chida S, Morita T, Watabe Y, Abe T. Artificial surfactant therapy in hyaline–membrane disease. Lancet. 1980;1:55–9.
Griese M. Pulmonary surfactant in health and human lung diseases: state of the art. Eur Respir J. 1999;13:1455–76.
Schmidt R, Meier U, Yabut–Perez M, Walmrath D, Grimminger F, Seeger W, Gunther A. Alteration of fatty acid profiles in different pulmonary surfactant phospholipids in acute respiratory distress syndrome and severe pneumonia. Am J Respir Crit Care Med. 2001;163:95–100.
Gunther A, Siebert C, Schmidt R, Ziegler S, Grimminger F, Yabut M, Temmesfeld B, Walmrath D, Morr H, Seeger W. Surfactant alterations in severe pneumonia, acute respiratory distress syndrome, and cardiogenic lung edema. Am J Respir Crit Care Med. 1996;153:176–84.
Tsutsumida H, Goto M, Kitajima S, Kubota I, Hirotsu Y, Yonezawa S. Combined status of MUC1 mucin and surfactant apoprotein A expression can predict the outcome of patients with small–size lung adenocarcinoma. Histopathology. 2004;44:147–55.
Garcia CK. Idiopathic pulmonary fibrosis: update on genetic discoveries. Proc Am Thorac Soc. 2011;8:158–62.
Nogee LM, Garnier G, Dietz HC, Singer L. Murphy AM, deMello DE, Colten HR: A mutation in the surfactant protein B gene responsible for fatal neonatal respiratory disease in multiple kindreds. J Clin Invest. 1994;93:1860–3.
Pantelidis P, Veeraraghavan S, du Bois RM. Surfactant gene polymorphisms and interstitial lung diseases. Respiratory Research. 2001;3:14.
Klein JM, Thompson MW, Snyder JM, George TN, Whitsett JA, Bell EF, McCray PB Jr, Nogee LM. Transient surfactant protein B deficiency in a term infant with severe respiratory failure. J Pediatr. 1998;132:244–8.
Clark JC, Wert SE, Bachurski CJ, Stahlman MT, Stripp BR, Weaver TE, Whitsett JA. Targeted disruption of the surfactant protein B gene disrupts surfactant homeostasis, causing respiratory failure in newborn mice. Proc Natl Acad Sci U S A. 1995;92:7794–8.
Tokieda K, Whitsett JA, Clark JC, Weaver TE, Ikeda K, McConnell KB, Jobe AH, Ikegami M, Iwamoto HS. Pulmonary dysfunction in neonatal SP–B–deficient mice. Am J Physiol. 1997;273:L875–82.
Whitsett JA. The molecular era of surfactant biology. Neonatology. 2014;105:337–43.
Hayes D Jr, Lloyd EA, Fitch JA, Bush A. ABCA3 transporter deficiency. Am J Respir Crit Care Med. 2012;186:807.
Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N Engl J Med. 2004;350:1296–303.
Bohinski RJ, Di Lauro R, Whitsett JA. The lung–specific surfactant protein B gene promoter is a target for thyroid transcription factor 1 and hepatocyte nuclear factor 3, indicating common factors for organ–specific gene expression along the foregut axis. Mol Cell Biol. 1994;14:5671–81.
Nureki SI, Tomer Y, Venosa A, Katzen J, Russo SJ, Jamil S, Barrett M, Nguyen V, Kopp M, Mulugeta S, Beers MF. Expression of mutant Sftpc in murine alveolar epithelia drives spontaneous lung fibrosis. J Clin Invest. 2018;128:4008–24.
Fehrenbach H. Alveolar epithelial type II cell: defender of the alveolus revisited. Respiratory research. 2001;2:33.
Mason RJ. Biology of alveolar type II cells. Respirology. 2006;11:S12–5.
Guadiz G, Sporn LA, Goss RA, Lawrence SO, Marder VJ, Simpson–Haidaris PJ. Polarized secretion of fibrinogen by lung epithelial cells. Am J Respir Cell Mol Biol. 1997;17:60–9.
Simon RH, Gross TJ, Edwards JA, Sitrin RG. Fibrin degradation by rat pulmonary alveolar epithelial cells. Am J Physiol. 1992;262:L482–8.
Reddy SP, Mehta D. Lung Interstitial Macrophages Redefined: It Is Not That Simple Anymore. Am J Respir Cell Mol Biol. 2017;57:135–6.
Barron L, Gharib SA, Duffield JS. Lung Pericytes and Resident Fibroblasts: Busy Multitaskers. Am J Pathol. 2016;186:2519–31.
Telenga ED, Hoffmann RF. Ruben tK, Hoonhorst SJ, Willemse BW, van Oosterhout AJ, Heijink IH, van den Berge M, Jorge L, Sandra P, et al: Untargeted lipidomic analysis in chronic obstructive pulmonary disease. Uncovering sphingolipids. Am J Respir Crit Care Med. 2014;190:155–64.
Finley TN, Ladman AJ. Low yield of pulmonary surfactant in cigarette smokers. N Engl J Med. 1972;286:223–7.
Anzueto A, Jubran A, Ohar JA, Piquette CA, Rennard SI, Colice G, Pattishall EN, Barrett J, Engle M, Perret KA, Rubin BK. Effects of aerosolized surfactant in patients with stable chronic bronchitis: a prospective randomized controlled trial. JAMA. 1997;278:1426–31.
Devendra G, Spragg RG. Lung surfactant in subacute pulmonary disease. Respir Res. 2002;3:19.
Otto–Verberne CJ, Ten Have–Opbroek AA, Willems LN, Franken C, Kramps JA, Dijkman JH. Lack of type II cells and emphysema in human lungs. Eur Respir J. 1991;4:316–23.
Tsuji T, Aoshiba K, Nagai A. Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med. 2006;174:886–93.
Bagdonas E, Raudoniute J, Bruzauskaite I, Aldonyte R. Novel aspects of pathogenesis and regeneration mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 2015;10:995–1013.
Aoshiba K, Nagai A. Oxidative stress, cell death, and other damage to alveolar epithelial cells induced by cigarette smoke. Tob Induc Dis. 2003;1:219–26.
Mizumura K, Justice MJ, Schweitzer KS, Krishnan S, Bronova I, Berdyshev EV, Hubbard WC, Pewzner–Jung Y, Futerman AH, Choi AMK, Petrache I. Sphingolipid regulation of lung epithelial cell mitophagy and necroptosis during cigarette smoke exposure. FASEB J. 2018;32:1880–90.
Kosmider B, Lin CR, Vlasenko L, Marchetti N, Bolla S, Criner GJ, Messier E, Reisdorph N, Powell RL, Madesh M, et al. Impaired non–homologous end joining in human primary alveolar type II cells in emphysema. Sci Rep. 2019;9:920.
Liu Y, Gao W, Zhang D. Effects of cigarette smoke extract on A549 cells and human lung fibroblasts treated with transforming growth factor–beta1 in a coculture system. Clin Exp Med. 2010;10:159–67.
Wirtz HR, Schmidt M. Acute influence of cigarette smoke on secretion of pulmonary surfactant in rat alveolar type II cells in culture. Eur Respir J. 1996;9:24–32.
Subramaniam S, Bummer P, Gairola CG. Biochemical and biophysical characterization of pulmonary surfactant in rats exposed chronically to cigarette smoke. Fundam Appl Toxicol. 1995;27:63–9.
Oulton M, Moores HK, Scott JE, Janigan DT, Hajela R. Effects of smoke inhalation on surfactant phospholipids and phospholipase A2 activity in the mouse lung. Am J Pathol. 1991;138:195–202.
Scott JE. The pulmonary surfactant: impact of tobacco smoke and related compounds on surfactant and lung development. Tob Induc Dis. 2004;2:3–25.
Kimura T, Shibata Y, Yamauchi K, Igarashi A, Inoue S, Abe S, Fujita K, Uosaki Y, Kubota I. Oxidized phospholipid, 1–palmitoyl–2–(9′–oxo–nonanoyl)–glycerophosphocholine (PON–GPC), produced in the lung due to cigarette smoking, impairs immune function in macrophages. Lung. 2012;190:169–82.
Matulionis DH. Pulmonary tissue and cigarette smoke. 2. Parenchymal response. Environ Res. 1983;31:176–88.
Jubinville E, Talbot M, Berube JC, Hamel–Auger M, Maranda–Robitaille M, Beaulieu MJ, Aubin S, Pare ME, Kallend DG, Arsenault B, et al. Interplay between cigarette smoking and pulmonary reverse lipid transport. Eur Respir J. 2017;50:1700681.
Sonett J, Goldklang M, Sklepkiewicz P, Gerber A, Trischler J, Zelonina T, Westerterp M, Lemaitre V, Okada Y, D'Armiento J. A critical role for ABC transporters in persistent lung inflammation in the development of emphysema after smoke exposure. FASEB J. 2018;32:fj201701381.
Church DF, Pryor WA. Free–radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–26.
Rahman I, Li XY, Donaldson K, Harrison DJ, MacNee W. Glutathione homeostasis in alveolar epithelial cells in vitro and lung in vivo under oxidative stress. Am J Physiol. 1995;269:L285–92.
Wuenschell CW, Zhao J, Tefft JD, Warburton D. Nicotine stimulates branching and expression of SP–A and SP–C mRNAs in embryonic mouse lung culture. Am J Physiol. 1998;274:L165–70.
Tran HB, Barnawi J, Ween M, Hamon R, Roscioli E, Hodge G, Reynolds PN, Pitson SM, Davies LT, Haberberger R, Hodge S. Cigarette smoke inhibits efferocytosis via deregulation of sphingosine kinase signaling: reversal with exogenous S1P and the S1P analogue FTY720. J Leukoc Biol. 2016;100:195–202.
Petrusca DN, Gu Y, Adamowicz JJ, Rush NI, Hubbard WC, Smith PA, Berdyshev EV, Birukov KG, Lee CH, Tuder RM, et al. Sphingolipid–mediated inhibition of apoptotic cell clearance by alveolar macrophages. J Biol Chem. 2010;285:40322–32.
Hodge S, Hodge G, Scicchitano R, Reynolds PN, Holmes M. Alveolar macrophages from subjects with chronic obstructive pulmonary disease are deficient in their ability to phagocytose apoptotic airway epithelial cells. Immunol Cell Biol. 2003;81:289–96.
Schweitzer KS, Chen SX, Law S, Van Demark M, Poirier C, Justice MJ, Hubbard WC, Kim ES, Lai X, Wang M, et al. Endothelial disruptive proinflammatory effects of nicotine and e–cigarette vapor exposures. Am J Physiol Lung Cell Mol Physiol. 2015;309:L175–87.
Madison MC, Landers CT, Gu BH, Chang CY, Tung HY, You R, Hong MJ, Baghaei N, Song LZ, Porter P, et al. Electronic cigarettes disrupt lung lipid homeostasis and innate immunity independent of nicotine. J Clin Invest. 2019;129:4290.
Blount BC, Karwowski MP, Morel–Espinosa M, Rees J, Sosnoff C, Cowan E, Gardner M, Wang L, Valentin–Blasini L, Silva L, et al. Evaluation of Bronchoalveolar Lavage Fluid from Patients in an Outbreak of E–cigarette, or Vaping, Product Use–Associated Lung Injury – 10 States, August–October 2019. MMWR Morb Mortal Wkly Rep. 2019;68:1040–1.
Abeles M, Popofsky S, Wen A, Valsamis C, Webb A, Halaby C, Pirzada M. Vaping–associated lung injury caused by inhalation of cannabis oil. Pediatr Pulmonol. 2019.
Blagev DP, Harris D, Dunn AC, Guidry DW, Grissom CK, Lanspa MJ. Clinical presentation, treatment, and short–term outcomes of lung injury associated with e–cigarettes or vaping: a prospective observational cohort study. Lancet. 2019.
Butt YM, Smith ML, Tazelaar HD, Vaszar LT, Swanson KL, Cecchini MJ, Boland JM, Bois MC, Boyum JH, Froemming AT, et al. Pathology of Vaping–Associated Lung Injury. N Engl J Med. 2019;381:1780–1.
Pambuccian SE. Testing for lipid–laden macrophages in bronchoalveolar lavage fluid to diagnose vaping–associated pulmonary injury. Are we there yet? J Am Soc Cytopathol. 2019.
Kalininskiy A, Bach CT, Nacca NE, Ginsberg G, Marraffa J, Navarette KA, McGraw MD, Croft DP. E–cigarette, or vaping, product use associated lung injury (EVALI): case series and diagnostic approach. Lancet Respir Med. 2019.
Hou X, Summer R, Chen Z, Tian Y, Ma J, Cui J, Hao X, Guo L, Xu H, Wang H, Liu H. Lipid Uptake by Alveolar Macrophages Drives Fibrotic Responses to Silica Dust. Sci Rep. 2019;9:399.
Bromberg PA. Mechanisms of the acute effects of inhaled ozone in humans. Biochim Biophys Acta. 1860;2016:2771–81.
Henkel FDR, Friedl A, Haid M, Thomas D, Bouchery T, Haimerl P, de Los Reyes Jimenez M, Alessandrini F, Schmidt–Weber CB, Harris NL, et al. House dust mite drives proinflammatory eicosanoid reprogramming and macrophage effector functions. Allergy. 2019;74:1090–101.
Rindlisbacher B, Schmid C, Geiser T, Bovet C, Funke–Chambour M. Serum metabolic profiling identified a distinct metabolic signature in patients with idiopathic pulmonary fibrosis – a potential biomarker role for LysoPC. Respir Res. 2018;19:7.
Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie–Pimentel AC, Chiu S, Fernandez R, Akbarpour M, Chen CI, Ren Z, et al. Single–Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019;199:1517–36.
Lv T, Jiang K, Wang J, Tang N, Dai H, Wang C. Single–cell RNA sequencing profiling of the effects of aging on alveolar stem cells. Science China Life Sciences. 2019;62:1028–37.
Sivakumar P, Thompson JR, Ammar R, Porteous M, McCoubrey C, Cantu E 3rd, Ravi K, Zhang Y, Luo Y, Streltsov D, et al. RNA sequencing of transplant–stage idiopathic pulmonary fibrosis lung reveals unique pathway regulation. ERJ Open Res. 2019;5.
Schiller HB, Montoro DT, Simon LM, Rawlins EL, Meyer KB, Strunz M, Vieira Braga FA, Timens W, Koppelman GH, Budinger GRS, et al. The Human Lung Cell Atlas: A High–Resolution Reference Map of the Human Lung in Health and Disease. Am J Respir Cell Mol Biol. 2019;61:31–41.
Beers MF, Knudsen L, Tomer Y, Maronn J, Zhao M, Ochs M, Mulugeta S. Aberrant lung remodeling in a mouse model of surfactant dysregulation induced by modulation of the Abca3 gene. Ann Anat. 2017;210:135–46.
Romero F, Shah D, Duong M, Penn RB, Fessler MB, Madenspacher J, Stafstrom W, Kavuru M, Lu B, Kallen CB, et al. A pneumocyte–macrophage paracrine lipid axis drives the lung toward fibrosis. Am J Respir Cell Mol Biol. 2015;53:74–86.
Saito K, Tanaka N, Ikari J, Suzuki M, Anazawa R, Abe M, Saito Y, Tatsumi K. Comprehensive lipid profiling of bleomycin–induced lung injury. J Appl Toxicol. 2019;39:658–71.
Swendsen CL, Skita V, Thrall RS. Alterations in surfactant neutral lipid composition during the development of bleomycin–induced pulmonary fibrosis. Biochim Biophys Acta. 1996;1301:90–6.
Horiuchi T, Ikegami M, Cherniack RM, Mason RJ. Increased surface tension of the lung and surfactant in bleomycin–induced pulmonary fibrosis in rats. Am J Respir Crit Care Med. 1996;154:1002–5.
Mahavadi P, Henneke I, Ruppert C, Knudsen L, Venkatesan S, Liebisch G, Chambers RC, Ochs M, Schmitz G, Vancheri C, et al. Altered surfactant homeostasis and alveolar epithelial cell stress in amiodarone–induced lung fibrosis. Toxicol Sci. 2014;142:285–97.
Povedano JM, Martinez P, Flores JM, Mulero F, Blasco MA. Mice with Pulmonary Fibrosis Driven by Telomere Dysfunction. Cell Rep. 2015;12:286–99.
Povedano JM, Martinez P, Serrano R, Tejera A, Gomez–Lopez G, Bobadilla M, Flores JM, Bosch F, Blasco MA. Therapeutic effects of telomerase in mice with pulmonary fibrosis induced by damage to the lungs and short telomeres. Elife. 2018;7.
Alder JK, Barkauskas CE, Limjunyawong N, Stanley SE, Kembou F, Tuder RM, Hogan BL, Mitzner W, Armanios M. Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A. 2015;112:5099–104.
Venosa A, Smith LC, Murray A, Banota T, Gow AJ, Laskin JD, Laskin DL. Regulation of Macrophage Foam Cell Formation during Nitrogen Mustard (NM)–Induced Pulmonary Fibrosis by Lung Lipids. Toxicological Sciences. 2019.
Steffen L, Ruppert C, Hoymann HG, Funke M, Ebener S, Kloth C, Muhlfeld C, Ochs M, Knudsen L, Lopez–Rodriguez E. Surfactant replacement therapy reduces acute lung injury and collapse induration–related lung remodeling in the bleomycin model. Am J Physiol Lung Cell Mol Physiol. 2017;313:L313–27.
Beike L, Wrede C, Hegermann J, Lopez–Rodriguez E, Kloth C, Gauldie J, Kolb M, Maus UA, Ochs M, Knudsen L. Surfactant dysfunction and alveolar collapse are linked with fibrotic septal wall remodeling in the TGF–beta1–induced mouse model of pulmonary fibrosis. Lab Invest. 2019;99:830–52.
Lopez–Rodriguez E, Boden C, Echaide M, Perez–Gil J, Kolb M, Gauldie J, Maus UA, Ochs M, Knudsen L. Surfactant dysfunction during overexpression of TGF–beta1 precedes profibrotic lung remodeling in vivo. Am J Physiol Lung Cell Mol Physiol. 2016;310:L1260–71.
Kuhn DC, Demers LM. Effect of surfactant on basal and silica–induced eicosanoid production by the alveolar macrophage. Am J Physiol. 1995;269:L165–70.
Sunaga H, Matsui H, Ueno M, Maeno T, Iso T, Syamsunarno MR, Anjo S, Matsuzaka T, Shimano H, Yokoyama T, Kurabayashi M. Deranged fatty acid composition causes pulmonary fibrosis in Elovl6–deficient mice. Nat Commun. 2013;4:2563.
Chu SG, Villalba JA, Liang X, Xiong K, Tsoyi K, Ith B, Ayaub EA, Tatituri RV, Byers DE, Hsu FF, et al. Palmitic Acid–Rich High Fat Diet Exacerbates Experimental Pulmonary Fibrosis by Modulating Endoplasmic Reticulum Stress. Am J Respir Cell Mol Biol. 2019.
Summer R, Mora AL. Lipid Metabolism: a New Player in the Conundrum of Lung Fibrosis. Am J Respir Cell Mol Biol. 2019.
Duan Y, Zeng L, Zheng C, Song B, Li F, Kong X, Xu K. Inflammatory Links Between High Fat Diets and Diseases. Front Immunol. 2018;9:2649.
Park S, Kim S, Kim MJ, Hong Y, Lee AY, Lee H, Tran Q, Kim M, Cho H, Park J, et al. GOLGA2 loss causes fibrosis with autophagy in the mouse lung and liver. Biochem Biophys Res Commun. 2018;495:594–600.
Romero F, Hong X, Shah D, Kallen CB, Rosas I, Guo Z, Schriner D, Barta J, Shaghaghi H, Hoek JB, et al. Lipid Synthesis Is Required to Resolve Endoplasmic Reticulum Stress and Limit Fibrotic Responses in the Lung. Am J Respir Cell Mol Biol. 2018;59:225–36.
Chung KP, Hsu CL, Fan LC, Huang Z, Bhatia D, Chen YJ, Hisata S, Cho SJ, Nakahira K, Imamura M, et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat Commun. 2019;10:3390.
Shichino S, Ueha S, Hashimoto S, Otsuji M, Abe J, Tsukui T, Deshimaru S, Nakajima T, Kosugi–Kanaya M, Shand FH, et al. Transcriptome network analysis identifies protective role of the LXR/SREBP–1c axis in murine pulmonary fibrosis. JCI Insight. 2019;4.
Kamp DW. Mitigating Lung Fibrosis by Targeting Dysfunctional Alveolar Epithelial Cell Lipid Metabolism. Am J Respir Cell Mol Biol. 2018;59:139–40.
Moore BB, Peters–Golden M. Opposing roles of leukotrienes and prostaglandins in fibrotic lung disease. Expert Rev Clin Immunol. 2006;2:87–100.
Charbeneau RP, Peters–Golden M. Eicosanoids: mediators and therapeutic targets in fibrotic lung disease. Clin Sci (Lond). 2005;108:479–91.
Lovgren AK, Jania LA, Hartney JM, Parsons KK, Audoly LP, Fitzgerald GA, Tilley SL, Koller BH. COX–2–derived prostacyclin protects against bleomycin–induced pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2006;291:L144–56.
Kida T, Ayabe S, Omori K, Nakamura T, Maehara T, Aritake K, Urade Y, Murata T. Prostaglandin D2 Attenuates Bleomycin–Induced Lung Inflammation and Pulmonary Fibrosis. PLoS One. 2016;11:e0167729.
Marinkovic A, Liu F, Tschumperlin DJ. Matrices of physiologic stiffness potently inactivate idiopathic pulmonary fibrosis fibroblasts. Am J Respir Cell Mol Biol. 2013;48:422–30.
Huang SK, Wettlaufer SH, Hogaboam CM, Flaherty KR, Martinez FJ, Myers JL, Colby TV, Travis WD, Toews GB, Peters–Golden M. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am J Respir Crit Care Med. 2008;177:66–74.
Huang SK, Fisher AS, Scruggs AM, White ES, Hogaboam CM, Richardson BC, Peters–Golden M. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol. 2010;177:2245–55.
Dackor RT, Cheng J, Voltz JW, Card JW, Ferguson CD, Garrett RC, Bradbury JA, DeGraff LM, Lih FB, Tomer KB, et al. Prostaglandin E (2) protects murine lungs from bleomycin–induced pulmonary fibrosis and lung dysfunction. Am J Physiol Lung Cell Mol Physiol. 2011;301:L645–55.
Lacy SH, Woeller CF, Thatcher TH, Pollock SJ, Small EM, Sime PJ, Phipps RP. Activated Human Lung Fibroblasts Produce Extracellular Vesicles with Antifibrotic Prostaglandins. Am J Respir Cell Mol Biol. 2019;60:269–78.
Wettlaufer SH, Scott JP, McEachin RC, Peters–Golden M, Huang SK. Reversal of the Transcriptome by Prostaglandin E2 during Myofibroblast Dedifferentiation. Am J Respir Cell Mol Biol. 2016;54:114–27.
Kolodsick JE, Peters–Golden M, Larios J, Toews GB, Thannickal VJ, Moore BB. Prostaglandin E2 inhibits fibroblast to myofibroblast transition via E. prostanoid receptor 2 signaling and cyclic adenosine monophosphate elevation. Am J Respir Cell Mol Biol. 2003;29:537–44.
Bauman KA, Wettlaufer SH, Okunishi K, Vannella KM, Stoolman JS, Huang SK, Courey AJ, White ES, Hogaboam CM, Simon RH, et al. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J Clin Invest. 2010;120:1950–60.
Okunishi K, Sisson TH, Huang SK, Hogaboam CM, Simon RH, Peters–Golden M. Plasmin overcomes resistance to prostaglandin E2 in fibrotic lung fibroblasts by reorganizing protein kinase A signaling. J Biol Chem. 2011;286:32231–43.
Vannella KM, McMillan TR, Charbeneau RP, Wilke CA, Thomas PE, Toews GB, Peters–Golden M, Moore BB. Cysteinyl leukotrienes are autocrine and paracrine regulators of fibrocyte function. J Immunol. 2007;179:7883–90.
Yatomi M, Hisada T, Ishizuka T, Koga Y, Ono A, Kamide Y, Seki K, Aoki–Saito H, Tsurumaki H, Sunaga N, et al. 17(R)–resolvin D1 ameliorates bleomycin–induced pulmonary fibrosis in mice. Physiol Rep. 2015;3.
Wang Y, Li R, Chen L, Tan W, Sun Z, Xia H, Li B, Yu Y, Gong J, Tang M, et al. Maresin 1 Inhibits Epithelial–to–Mesenchymal Transition in Vitro and Attenuates Bleomycin Induced Lung Fibrosis in Vivo. Shock. 2015;44:496–502.
Sun Q, Wu Y, Zhao F, Wang J. Maresin 1 inhibits transforming growth factor–beta1–induced proliferation, migration and differentiation in human lung fibroblasts. Mol Med Rep. 2017;16:1523–9.
Koike K, Berdyshev EV, Bowler RP, Scruggs AK, Cao D, Schweitzer KS, Serban KA, Petrache I. Bioactive Sphingolipids in the Pathogenesis of Chronic Obstructive Pulmonary Disease. Ann Am Thorac Soc. 2018;15:S249–52.
Petrache I, Berdyshev EV. Ceramide Signaling and Metabolism in Pathophysiological States of the Lung. Annu Rev Physiol. 2016;78:463–80.
Uhlig S, Gulbins E. Sphingolipids in the lungs. Am J Respir Crit Care Med. 2008;178:1100–14.
Milara J, Navarro R, Juan G, Peiró T, Serrano A, Ramón M, Morcillo E, Cortijo J. Sphingosine–1–phosphate is increased in patients with idiopathic pulmonary fibrosis and mediates epithelial to mesenchymal transition. Thorax. 2012;67:147–56.
Huang LS, Natarajan V. Sphingolipids in pulmonary fibrosis. Adv Biological Reg. 2015;57:55–63.
Huang LS, Berdyshev EV, Tran JT, Xie L, Chen J, Ebenezer DL, Mathew B, Gorshkova I, Zhang W, Reddy SP, et al. Sphingosine–1–phosphate lyase is an endogenous suppressor of pulmonary fibrosis: role of S1P signalling and autophagy. Thorax. 2015;70:1138–48.
Banaschewski BJ, Veldhuizen EJ, Keating E, Haagsman HP, Zuo YY, Yamashita CM, Veldhuizen RA. Antimicrobial and biophysical properties of surfactant supplemented with an antimicrobial peptide for treatment of bacterial pneumonia. Antimicrob Agents Chemother. 2015;59:3075–83.
Ray NB, Durairaj L, Chen BB, McVerry BJ, Ryan AJ, Donahoe M, Waltenbaugh AK, O'Donnell CP, Henderson FC, Etscheidt CA, et al. Dynamic regulation of cardiolipin by the lipid pump Atp8b1 determines the severity of lung injury in experimental pneumonia. Nat Med. 2010;16:1120–7.
Liau DF, Barrett CR, Bell AL, Cernansky G, Ryan SF. Diphosphatidylglycerol in experimental acute alveolar injury in the dog. J Lipid Res. 1984;25:678–83.
Ksenzenko SM, Davidson SB, Saba AA, Franko AP, Raafat AM, Diebel LN, Dulchavsky SA. Effect of triiodothyronine augmentation on rat lung surfactant phospholipids during sepsis. J Appl Physiol (1985). 1997;82:2020–7.
Willsey GG, Ventrone S, Schutz KC, Wallace AM, Ribis JW, Suratt BT, Wargo MJ. Pulmonary Surfactant Promotes Virulence Gene Expression and Biofilm Formation in Klebsiella pneumoniae. Infect Immun. 2018;86.
Brogden KA, Cutlip RC, Lehmkuhl HD. Complexing of bacterial lipopolysaccharide with lung surfactant. Infect Immun. 1986;52:644–9.
DeLucca AJ 2nd, Brogden KA, Engen R. Enterobacter agglomerans lipopolysaccharide–induced changes in pulmonary surfactant as a factor in the pathogenesis of byssinosis. J Clin Microbiol. 1988;26:778–80.
Garcia–Verdugo I, Canadas O, Taneva SG, Keough KM, Casals C. Surfactant protein A forms extensive lattice–like structures on 1,2–dipalmitoylphosphatidylcholine/rough–lipopolysaccharide–mixed monolayers. Biophys J. 2007;93:3529–40.
Canadas O, Keough KM, Casals C. Bacterial lipopolysaccharide promotes destabilization of lung surfactant–like films. Biophys J. 2011;100:108–16.
Hiansen JQ, Keating E, Aspros A, Yao LJ, Bosma KJ, Yamashita CM, Lewis JF, Veldhuizen RA. Cholesterol–mediated surfactant dysfunction is mitigated by surfactant protein A. Biochim Biophys Acta. 1848;2015:813–20.
Van Iwaarden JF, Pikaar JC, Storm J, Brouwer E, Verhoef J, Oosting RS, van Golde LM, van Strijp JA. Binding of surfactant protein A to the lipid A moiety of bacterial lipopolysaccharides. Biochem J. 1994;303(Pt 2):407–11.
Sano H, Chiba H, Iwaki D, Sohma H, Voelker DR, Kuroki Y. Surfactant proteins A and D bind CD14 by different mechanisms. J Biol Chem. 2000;275:22442–51.
Gardai SJ, Xiao YQ, Dickinson M, Nick JA, Voelker DR, Greene KE, Henson PM. By binding SIRPalpha or calreticulin/CD91, lung collectins act as dual function surveillance molecules to suppress or enhance inflammation. Cell. 2003;115:13–23.
Bufler P, Schmidt B, Schikor D, Bauernfeind A, Crouch EC, Griese M. Surfactant protein A and D differently regulate the immune response to nonmucoid Pseudomonas aeruginosa and its lipopolysaccharide. Am J Respir Cell Mol Biol. 2003;28:249–56.
Borron P, McIntosh JC, Korfhagen TR, Whitsett JA, Taylor J, Wright JR. Surfactant–associated protein A inhibits LPS–induced cytokine and nitric oxide production in vivo. Am J Physiol Lung Cell Mol Physiol. 2000;278:L840–7.
Wu H, Kuzmenko A, Wan S, Schaffer L, Weiss A, Fisher JH, Kim KS, McCormack FX. Surfactant proteins A and D inhibit the growth of Gram–negative bacteria by increasing membrane permeability. J Clin Invest. 2003;111:1589–602.
Antoon JW, Hernandez ML, Roehrs PA, Noah TL, Leigh MW, Byerley JS. Endogenous lipoid pneumonia preceding diagnosis of pulmonary alveolar proteinosis. Clin Respir J. 2016;10:246–9.
Lau C, Abdelmalak BB, Farver CF, Culver DA. Whole lung lavage for lipoid pneumonia. Thorax. 2016;71:1066–7.
Harris K, Chalhoub M, Maroun R, Abi–Fadel F, Zhao F. Lipoid pneumonia: a challenging diagnosis. Heart Lung. 2011;40:580–4.
Davidson K, Brancato A, Heetderks P, Mansour W, Matheis E, Nario M, Rajagopalan S, Underhill B, Wininger J, Fox D. Outbreak of Electronic–Cigarette–Associated Acute Lipoid Pneumonia – North Carolina, July–August 2019. MMWR Morb Mortal Wkly Rep. 2019;68:784–6.
Dicpinigaitis PV, Trachuk P, Fakier F, Teka M, Suhrland MJ. Vaping–Associated Acute Respiratory Failure Due to Acute Lipoid Pneumonia. Lung; 2019.
Viswam D, Trotter S, Burge PS, Walters GI. Respiratory failure caused by lipoid pneumonia from vaping e–cigarettes. BMJ Case Rep. 2018;2018.
Vasic NR, Milenkovic BA, Pesut DP, Stevic RS, Jovanovic DM. Drug induced lung disease––amiodarone in focus. Med Pregl. 2014;67:334–7.
Haller T, Cerrada A, Pfaller K, Braubach P, Felder E. Polarized light microscopy reveals physiological and drug–induced changes in surfactant membrane assembly in alveolar type II pneumocytes. Biochimica et Biophysica Acta (BBA)–Biomembranes. 2018;1860:1152–61.
Jiang H, Passarelli MK, Munro PM, Kilburn MR, West A, Dollery CT, Gilmore IS, Rakowska PD. High–resolution sub–cellular imaging by correlative NanoSIMS and electron microscopy of amiodarone internalisation by lung macrophages as evidence for drug–induced phospholipidosis. Chemical Communications. 2017;53:1506–9.
Papiris SA, Triantafillidou C, Kolilekas L, Markoulaki D, Manali ED. Amiodarone: review of pulmonary effects and toxicity. Drug Saf. 2010;33:539–58.
Cui L, Fang J, Ooi EE, Lee YH. Serial Metabolome Changes in a Prospective Cohort of Subjects with Influenza Viral Infection and Comparison with Dengue Fever. J Proteome Res. 2017;16:2614–22.
Park JH, Park EB, Lee JY, Min JY. Identification of novel membrane–associated prostaglandin E synthase–1 (mPGES–1) inhibitors with anti–influenza activities in vitro. Biochem Biophys Res Commun. 2016;469:848–55.
Morita M, Kuba K, Ichikawa A, Nakayama M, Katahira J, Iwamoto R, Watanebe T, Sakabe S, Daidoji T, Nakamura S, et al. The lipid mediator protectin D1 inhibits influenza virus replication and improves severe influenza. Cell. 2013;153:112–25.
Tam VC, Quehenberger O, Oshansky CM, Suen R, Armando AM, Treuting PM, Thomas PG, Dennis EA, Aderem A. Lipidomic profiling of influenza infection identifies mediators that induce and resolve inflammation. Cell. 2013;154:213–27.
Bobone S, Hilsch M, Storm J, Dunsing V, Herrmann A, Chiantia S. Phosphatidylserine Lateral Organization Influences the Interaction of Influenza Virus Matrix Protein 1 with Lipid Membranes. J Virol. 2017;91.
Veglia G, Nelson SE, Gopinath T. Allosteric Regulation of the M2 Protein from Influenza A by Cholesterol. Biophys J. 2016;110:1207–8.
Li S, Eghiaian F, Sieben C, Herrmann A, Schaap IAT. Bending and puncturing the influenza lipid envelope. Biophys J. 2011;100:637–45.
World Health Organization (WHO). Global Tuberculosis Report 2019. 17 October 2019 edition: World Health Organization; 2019.
Forrellad MA, Klepp LI, Gioffre A, y Sabio GJ, Morbidoni HR, de la Paz Santangelo M, Cataldi AA, Bigi F. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4:3–66.
Aguilar–Ayala DA, Tilleman L, Van Nieuwerburgh F, Deforce D, Palomino JC, Vandamme P, Gonzalez YMJA, Martin A. The transcriptome of Mycobacterium tuberculosis in a lipid–rich dormancy model through RNAseq analysis. Sci Rep. 2017;7:17665.
VanderVen BC, Fahey RJ, Lee W, Liu Y, Abramovitch RB, Memmott C, Crowe AM, Eltis LD, Perola E, Deininger DD, et al. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium's metabolism is constrained by the intracellular environment. PLoS Pathog. 2015;11:e1004679.
Kaul V, Bhattacharya D, Singh Y, Van Kaer L, Peters–Golden M, Bishai WR, Das G. An important role of prostanoid receptor EP2 in host resistance to Mycobacterium tuberculosis infection in mice. J Infect Dis. 2012;206:1816–25.
Chen M, Divangahi M, Gan H, Shin DS, Hong S, Lee DM, Serhan CN, Behar SM, Remold HG. Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death. J Exp Med. 2008;205:2791–801.
Bafica A, Scanga CA, Serhan C, Machado F, White S, Sher A, Aliberti J. Host control of Mycobacterium tuberculosis is regulated by 5–lipoxygenase–dependent lipoxin production. J Clin Invest. 2005;115:1601–6.
Neyrolles O, Guilhot C. Recent advances in deciphering the contribution of Mycobacterium tuberculosis lipids to pathogenesis. Tuberculosis (Edinb). 2011;91:187–95.
Jankute M, Nataraj V, Lee OY, Wu HHT, Ridell M, Garton NJ, Barer MR, Minnikin DE, Bhatt A, Besra GS. The role of hydrophobicity in tuberculosis evolution and pathogenicity. Sci Rep. 2017;7:1315.
Goren MB, Brokl O, Schaefer WB. Lipids of putative relevance to virulence in Mycobacterium tuberculosis: phthiocerol dimycocerosate and the attenuation indicator lipid. Infect Immun. 1974;9:150–8.
Larrouy–Maumus G, Layre E, Clark S, Prandi J, Rayner E, Lepore M, de Libero G, Williams A, Puzo G, Gilleron M. Protective efficacy of a lipid antigen vaccine in a guinea pig model of tuberculosis. Vaccine. 2017;35:1395–402.
Sun P, Lu X, Xu C, Sun W, Pan B. Understanding of COVID–19 based on current evidence. J Med Virol. 2020.
Chu H, Chan JF, Wang Y, Yuen TT, Chai Y, Hou Y, Shuai H, Yang D, Hu B, Huang X, et al. Comparative replication and immune activation profiles of SARS–CoV–2 and SARS–CoV in human lungs: an ex vivo study with implications for the pathogenesis of COVID–19. Clin Infect Dis. 2020. https://doi.org/10.1093/cid/ciaa410. [Epub ahead of print].
Yao XH, Li TY, He ZC, Ping YF, Liu HW, Yu SC, Mou HM, Wang LH, Zhang HR. Fu WJ, et al: [A pathological report of three COVID–19 cases by minimally invasive autopsies]. Zhonghua Bing Li Xue Za Zhi. 2020;49:E009.
Zhang H, Zhou P, Wei Y, Yue H, Wang Y, Hu M, Zhang S, Cao T, Yang C, Li M, et al. Histopathologic Changes and SARS–CoV–2 Immunostaining in the Lung of a Patient With COVID–19. Ann Intern Med. 2020;43(0):E040;43(0):E040. https://doi.org/10.3760/cma.j.cn112147-20200311-00312. [Epub ahead of print].
Zhang T, Sun LX. Feng RE: [Comparison of clinical and pathological features between severe acute respiratory syndrome and coronavirus disease 2019]. Zhonghua Jie He He Hu Xi Za Zhi. 2020;43:E040.
Mason RJ. Pathogenesis of COVID–19 from a cell biologic perspective. European Respiratory Journal. 2020;2000607.
Chen JY. Exogenous surfactant for treatment of respiratory distress syndrome in premature infants. J Formos Med Assoc. 1990;89:110–4.
Gitlin JD, Soll RF, Parad RB, Horbar JD, Feldman HA, Lucey JF, Taeusch HW. Randomized controlled trial of exogenous surfactant for the treatment of hyaline membrane disease. Pediatrics. 1987;79:31–7.
Gortner L, Bartmann P, Pohlandt F, Bernsau U, Porz F, Hellwege HH, Seitz RC, Hieronimi G, Bremer C, Jorch G, et al. Early treatment of respiratory distress syndrome with bovine surfactant in very preterm infants: a multicenter controlled clinical trial. Pediatr Pulmonol. 1992;14:4–9.
Gortner L, Bernsau U, Hellwege HH, Hieronimi G, Jorch G, Reiter HL. A multicenter randomized controlled clinical trial of bovine surfactant for prevention of respiratory distress syndrome. Lung. 1990;168(Suppl):864–9.
Hallman M, Merritt TA, Jarvenpaa AL, Boynton B, Mannino F, Gluck L, Moore T, Edwards D. Exogenous human surfactant for treatment of severe respiratory distress syndrome: a randomized prospective clinical trial. J Pediatr. 1985;106:963–9.
Horbar JD, Soll RF, Schachinger H, Kewitz G, Versmold HT, Lindner W, Duc G, Mieth D, Linderkamp O, Zilow EP, et al. A European multicenter randomized controlled trial of single dose surfactant therapy for idiopathic respiratory distress syndrome. Eur J Pediatr. 1990;149:416–23.
Horbar JD, Soll RF, Sutherland JM, Kotagal U, Philip AG, Kessler DL, Little GA, Edwards WH, Vidyasagar D, Raju TN, et al. A multicenter randomized, placebo–controlled trial of surfactant therapy for respiratory distress syndrome. N Engl J Med. 1989;320:959–65.
Lang MJ, Hall RT, Reddy NS, Kurth CG, Merritt TA. A controlled trial of human surfactant replacement therapy for severe respiratory distress syndrome in very low birth weight infants. J Pediatr. 1990;116:295–300.
Liechty EA, Donovan E, Purohit D, Gilhooly J, Feldman B, Noguchi A, Denson SE, Sehgal SS, Gross I, Stevens D, et al. Reduction of neonatal mortality after multiple doses of bovine surfactant in low birth weight neonates with respiratory distress syndrome. Pediatrics. 1991;88:19–28.
Raju TN, Vidyasagar D, Bhat R, Sobel D, McCulloch KM, Anderson M, Maeta H, Levy PS, Furner S. Double–blind controlled trial of single–dose treatment with bovine surfactant in severe hyaline membrane disease. Lancet. 1987;1:651–6.
Svenningsen N, Robertson B, Andreason B, Berggren P, Jonson B, Lindroth M. Endotracheal administration of surfactant in very low birth weight infants with respiratory distress syndrome. Crit Care Med. 1987;15:918–22.
Vaucher YE, Merritt TA, Hallman M, Jarvenpaa AL, Telsey AM, Jones BL. Neurodevelopmental and respiratory outcome in early childhood after human surfactant treatment. Am J Dis Child. 1988;142:927–30.
Chu J, Clements JA, Cotton EK, Klaus MH, Sweet AY, Tooley WH, Bradley BL, Brandorff LC. Neonatal pulmonary ischemia. I. Clinical and physiological studies. Pediatrics. 1967;40(Suppl):709–82.
Robillard E, Alarie Y, Dagenais–Perusse P, Baril E, Guilbeault A. Microaerosol Administration of Synthetic Beta–Gamma–Dipalmitoyl–L–Alpha–Lecithin in the Respiratory Distress Syndome: A Preliminary Report. Can Med Assoc J. 1964;90:55–7.
Ten Center Study Group. Ten centre trial of artificial surfactant (artificial lung expanding compound) in very premature babies. Ten Centre Study Group. Br Med J (Clin Res Ed). 1987;294:991–6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1246154/?page=1.
Soll RF. Prophylactic synthetic surfactant for preventing morbidity and mortality in preterm infants. Cochrane Database Syst Rev. 2000:CD001079.
Soll RF, Blanco F. Natural surfactant extract versus synthetic surfactant for neonatal respiratory distress syndrome. Cochrane Database Syst Rev. 2001:CD000144.
Spragg RG, Lewis JF, Walmrath HD, Johannigman J, Bellingan G, Laterre PF, Witte MC, Richards GA, Rippin G, Rathgeb F, et al. Effect of recombinant surfactant protein C–based surfactant on the acute respiratory distress syndrome. N Engl J Med. 2004;351:884–92.
Manalo E, Merritt TA, Kheiter A, Amirkhanian J, Cochrane C. Comparative effects of some serum components and proteolytic products of fibrinogen on surface tension–lowering abilities of beractant and a synthetic peptide containing surfactant KL4. Pediatr Res. 1996;39:947–52.
Moya FR, Gadzinowski J, Bancalari E, Salinas V, Kopelman B, Bancalari A, Kornacka MK, Merritt TA, Segal R, Schaber CJ, et al. A multicenter, randomized, masked, comparison trial of lucinactant, colfosceril palmitate, and beractant for the prevention of respiratory distress syndrome among very preterm infants. Pediatrics. 2005;115:1018–29.
Sinha SK, Lacaze–Masmonteil T, Valls i Soler A, Wiswell TE, Gadzinowski J, Hajdu J, Bernstein G, Sanchez–Luna M, Segal R, Schaber CJ, et al. A multicenter, randomized, controlled trial of lucinactant versus poractant alfa among very premature infants at high risk for respiratory distress syndrome. Pediatrics. 2005;115:1030–8.
Echaide M, Autilio C, Lopez–Rodriguez E, Cruz A, Perez–Gil J. In Vitro Functional and Structural Characterization of A Synthetic Clinical Pulmonary Surfactant with Enhanced Resistance to Inhibition. Sci Rep. 2020;10:1385.
Seeger W, Gunther A, Walmrath HD, Grimminger F, Lasch HG. Alveolar surfactant and adult respiratory distress syndrome. Pathogenetic role and therapeutic prospects. Clin Investig. 1993;71:177–90.
Spengler D, Winoto–Morbach S, Kupsch S, Vock C, Blochle K, Frank S, Rintz N, Diekotter M, Janga H, Weckmann M, et al. Novel therapeutic roles for surfactant–inositols and –phosphatidylglycerols in a neonatal piglet ARDS model: a translational study. Am J Physiol Lung Cell Mol Physiol. 2018;314:L32–l53.
Wadgaonkar R, Patel V, Grinkina N, Romano C, Liu J, Zhao Y, Sammani S, Garcia JG, Natarajan V. Differential regulation of sphingosine kinases 1 and 2 in lung injury. Am J Physiol Lung Cell Mol Physiol. 2009;296:L603–13.
Zemann B, Urtz N, Reuschel R, Mechtcheriakova D, Bornancin F, Badegruber R, Baumruker T, Billich A. Normal neutrophil functions in sphingosine kinase type 1 and 2 knockout mice. Immunol Lett. 2007;109:56–63.
Zhao Y, Gorshkova IA, Berdyshev E, He D, Fu P, Ma W, Su Y, Usatyuk PV, Pendyala S, Oskouian B. Protection of LPS–induced murine acute lung injury by sphingosine–1–phosphate lyase suppression. Am J Resp Cell Mol Biol. 2011;45:426–35.
Haller T, Cerrada A, Pfaller K, Braubach P, Felder E. Polarized light microscopy reveals physiological and drug–induced changes in surfactant membrane assembly in alveolar type II pneumocytes. Biochim Biophys Acta Biomembr. 1860;2018:1152–61.
Jiang H, Passarelli MK, Munro PM, Kilburn MR, West A, Dollery CT, Gilmore IS, Rakowska PD. High–resolution sub–cellular imaging by correlative NanoSIMS and electron microscopy of amiodarone internalisation by lung macrophages as evidence for drug–induced phospholipidosis. Chem Commun (Camb). 2017;53:1506–9.
Acknowledgements
The authors would like to thank all the investigators that conducted the original work in the topic, as well as the funding agencies.
Funding
This manuscript was supported with grants from American Heart Association 17GRNT33700252 (I.G.A), the Flight Attendant Medical Research Institute YCSA13005 (I.G.A), and the National Heart, Lung, And Blood Institute of the National Institutes of Health under Award Number R56HL148774 (I.G.A). The content is solely the responsibility of the authors and does not represent the official views of the National Institutes of Health.
Author information
Authors and Affiliations
Contributions
CWA designed the outline, reviewed the literature, wrote the manuscript and edited the drafts. GS designed the outline, reviewed the literature and wrote the manuscript. IGA designed the outline, reviewed the literature, wrote the manuscript and edited the drafts. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Agudelo, C.W., Samaha, G. & Garcia-Arcos, I. Alveolar lipids in pulmonary disease. A review. Lipids Health Dis 19, 122 (2020). https://doi.org/10.1186/s12944-020-01278-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-020-01278-8