
Abstract
Fundamental questions remain unresolved in diabetes: What is the actual mechanism of glucose toxicity? Why is there insulin resistance in type 2 diabetes? Why do diets rich in sugars or saturated fatty acids increase the risk of developing diabetes? Studying the C. elegans homologs of the anti-diabetic adiponectin receptors (AdipoR1 and AdipoR2) has led us to exciting new discoveries and to revisit what may be termed “The Membrane Theory of Diabetes”. We hypothesize that excess saturated fatty acids (obtained through a diet rich in saturated fats or through conversion of sugars into saturated fats via lipogenesis) leads to rigid cellular membranes that in turn impair insulin signalling, glucose uptake and blood circulation, thus creating a vicious cycle that contributes to the development of overt type 2 diabetes. This hypothesis is supported by our own studies in C. elegans and by a wealth of literature concerning membrane composition in diabetics. The purpose of this review is to survey this literature in the light of the new results, and to provide an admittedly membrane-centric view of diabetes.
Similar content being viewed by others


Background
Diabetes and new insights from C. elegans
The worldwide rise in the incidence of type 2 diabetes is a recent phenomenon that coincides with lifestyle changes during the 20th century. A diet of excess combined with an increasingly sedentary lifestyle clearly leads to an energy imbalance and the accumulation of fat depots. While there is no doubt that obesity and genetic variants are risk factors for developing type 2 diabetes, what, precisely, is the molecular and cell biology link between diet and diabetes? Many explanations have been proposed. Here, we revisit a “membrane-centric” view of diabetes because of some new results obtained with the small nematode worm C. elegans. Specifically, as little as 10 mM glucose is lethal to C. elegans mutants lacking a functional homolog of the mammalian adiponectin receptors [1–3]. This toxicity is accompanied by an increase in the abundance of saturated fatty acids (SFAs) in membrane phospholipids and a dramatic decrease in membrane fluidity. Given the proposed anti-diabetic activities of the adiponectin receptors [4–8], the C. elegans studies prompted us to examine the literature for possible connections between glucose toxicity, cellular membranes and diabetes.
Decreased membrane fluidity in diabetics
Red blood cells (RBCs) in diabetics are abnormally rigid. This fact is known since at least 1978 when purified RBCs were filmed as they deformed under different, quantifiable amounts of air pressure inside glass microcapillaries [9]. These findings were confirmed independently using a filtration rate assay [10], and more recently using high-speed filming of RBCs through microchannels [11]. The decreased deformability of RBCs is a likely source of shear stress that contributes to microcapillary hardening in diabetics, an idea proposed in 1978 by McMillan et al. [9]. Several methods were later used to show that the low deformability of RBCs in diabetics is caused by a reduced fluidity of the cellular membranes. Already in 1979, Baba et al. measured depolarization of a fluorescent probe and found reduced membrane fluidity in the RBCs of diabetics [12]. Similar findings were made in 1983 by Kamada and Otsuji, this time using spin labeling and electron spin resonance measurements [13]. Kamada et al. also showed that newly produced RBCs in diabetics start off with an already reduced fluidity, indicating that the low fluidity is not a result of faster decay of the RBCs in diabetics, but rather likely reflects a basic problem with the pool of fatty acids (FAs) available for membrane homeostasis [14]. Most studies of membrane properties are done on RBCs because of their easy availability. However, decreased membrane fluidity in diabetics has also been measured in several other cell types, including ileal enterocytes of the intestinal brush border [15, 16], the sarcolema of cardiac myocytes [17], leukocytes [18], synaptic vesicles in the cerebral cortex [19] and platelets [20, 21], and is likely affecting most cell types.
Abnormal phospholipid composition in diabetics
Phospholipid composition has a great influence on membrane properties. An excellent proof of this is homeoviscous adaptation in poikilotherms or deep water organisms: temperature [22, 23] and hydrostatic pressure [24–26] can have profound effects on membrane fluidity to which cells adapt by compensatory changes in lipid composition, a phenomenon termed “homeoviscous adaptation” [22, 23, 27]. Specifically, certain lipid types increase membrane fluidity (e.g., phospholipids containing unsaturated fatty acids (UFAs)), while others decrease it (e.g., cholesterol, ceramides and phospholipids containing saturated fatty acids (SFAs)) [28–33].
Several independent studies have found that the cellular membranes of diabetics are rich in rigidity-promoting lipids: excess cholesterol [34], excess sphingomyelin [35], and excess SFAs [36–39] have all been associated with diabetes. Even more tantalizing is the predictive power of lipid composition. At least two large longitudinal studies measured the phospholipid composition of RBCs in thousands of healthy subjects and followed them for several years [40–42]. In both studies, individuals with the highest proportion of SFAs were most likely to later develop type 2 diabetes. This suggests that low membrane fluidity may precede diabetes. However, like many other observations mentioned in this review, it remains to be seen to what degree this constitutes a “marker” or a “maker” of imminent diabetes.
Low membrane fluidity as a cause of diabetes
Optimal membrane properties are essential for numerous cellular processes that are often defective in diabetics: vesicular trafficking (including insulin secretion by beta cells [43]), glucose transport [44], endocytosis [45, 46], regulation of metabolic rate [47], platelet aggregation [20], etc. Of special interest in the context of diabetes is the importance of membrane fluidity on the function of membrane proteins. Several studies have shown that insulin receptor signaling is impaired by low membrane fluidity, probably because lateral diffusion and localization to membrane microdomains is important for ligand binding and signaling [44, 48–50]. This is particularly important for two reasons: 1) it has long been known that insulin signaling activates FA desaturases in the liver and is thus important for regulating membrane fluidity [51–54]; and 2) insulin signaling is important for inducing the transport of GLUT4 to the plasma membranes of muscle cells and adipocytes, which is essential for quick clearance of blood glucose [55]. Also important is that GLUT4 transport to the plasma membrane is itself impaired by decreased membrane fluidity, which further exacerbates the problems of glucose clearance [50, 56]. Defects in phospholipid membrane composition of adipocytes is also responsible for inflammation and limits the insulin-induced expansion of adipose tissues in obese human [57]. Thus, a state of low membrane fluidity is very much diabetes-prone since it impairs insulin signaling and response.
Effect of diet on membrane composition
Several studies have shown that the composition of membrane phospholipids is influenced by the fatty acid composition of the diet. For example, fish oil supplements lead to an increased abundance of polyunsaturated fatty acids (PUFAs) in RBC membranes [58, 59]. Also, rats fed with diets differing in their FA composition (i.e., a range of SFA: UFA ratios) show phospholipid compositions that reflects the dietary fats [60, 61]. Similar findings were made with human subjects assigned to diets differing in their FA composition [62]. These results show that dietary fatty acids can be directly incorporated into phospholipids. Consequently, a diet rich in SFAs will tend to reduce the fluidity of cellular membranes. This is also true of a carbohydrate-rich diet since glucose can readily be converted into SFAs via de novo lipogenesis (DNL) in liver and adipocytes, which can then be made available throughout the body via the bloodstream as lipids transported in lipoproteins, or as free fatty acids [63]. The so-called “Western diet” therefore may promote diabetes by lowering membrane fluidity, hence impairing insulin signaling and other processes. Conversely, improvements in membrane fluidity may explain the insulin-sensitizing benefits of PUFA-rich diets [64, 65].
Incidentally, DNL is tightly associated with desaturation of the newly synthesized SFAs so as to create a balanced composition of the FA pool. This is evidenced from the observation that supplementing cultivated adipocytes with the SFA palmitate activates a membrane-protective desaturase activity that is paradoxically accompanied by the coordinated activation of the entire DNL pathway, which produces even more palmitate [66]. However, while the enzymes for DNL are restricted to a few tissues, most cells express one or two desaturases [67–69]. Why? Probably so that each cell can locally adjust its mix of SFAs, monounsaturated fatty acids (MUFAs) and PUFAs available for membrane turnover, as we shall now discuss.
Regulation of membrane fluidity
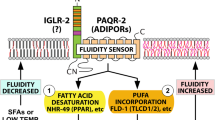
Regulatory mechanisms must exist within each cell to adjust membrane composition and maintain near-optimal properties. This is evident from the fact that substantial changes in dietary fatty acid composition are usually required for relatively small changes in membrane composition. To put it bluntly: without such regulatory mechanisms, some of us would be butter-like solids at room temperature while other would be oil-like liquids, depending on whether our diets are rich in animal fats or vegetable oils. In spite of their obvious importance, it is only in recent years that molecular regulators of membrane composition have been identified. First came the discovery of the bacterial fluidity regulator DesK, a kinase that activates a fatty acid desaturase upon reduced membrane fluidity; this helps restore membrane fluidity during homeoviscous adaptation to low temperature [70–75]. In the yeast Saccharomyces cerevisiae, the transmembrane protein Mga2 was found to act as a sensor for endoplasmic reticulum (ER) membrane lipid saturation: it is cleaved when ER membranes become too rigid, thus releasing a transcription factor domain that activates expression of a Δ9 fatty acid desaturase, hence restoring membrane fluidity [76]. Finally, a plasma membrane fluidity regulator was recently identified in the nematode C. elegans and consists of at least two proteins, PAQR-2 and IGLR-2 that are homologs of the ubiquitously expressed human adiponectin receptors and LRIG-type proteins, respectively [1–3, 77]. The mechanism of fluidity sensing is not known for PAQR-2/IGLR-2. However, our work in C. elegans suggests that PAQR-2 improves membrane fluidity by causing the upregulation of FA desaturases, likely via ligand-regulated transcription factors such as NHR-49 (a functional ortholog of the mammalian PPARα) and SBP-1 (an ortholog of the mammalian SREBP). Based on sequence homology and structural considerations, we and others suspect that PAQR-2 and the mammalian adiponectin receptors are hydrolases acting on a lipid substrate to release a ligand that regulates downstream transcription factors [2, 77–79]. An alternative explanation, inspired by studies on a yeast homolog, is that the adiponectin receptors act as ceramidases that deplete fluidity-lowering ceramides and release the signaling molecule sphingosine-1-phosphate [6, 80].
Interestingly, worms lacking PAQR-2 or IGLR-2 are the most glucose-intolerant C. elegans mutants identified so far: they succumb in the presence of as little as 10 mM glucose [3]. In these mutants, glucose causes a dramatic accumulation of SFAs in membranes with a concomitant loss of membrane fluidity, and this is likely due to the conversion of glucose into SFAs via DNL. This shows that the PAQR-2/IGLR-2 complex is essential for homeoviscous adaptation in the presence of glucose, with obvious implications for diabetes. Many membrane-related phenotypes of the PAQR-2 or IGLR-2 deficient worms, including cold and glucose intolerance, can be suppressed by the inclusion of small amounts of non-ionic detergents, such as NP-40 or Triton X-100, which act as membrane fluidizers. This is especially interesting because metformin, an antihyperglycemic agent already commonly used to treat diabetes, may also act by improving membrane fluidity [81–84]. It will therefore be very interesting to define the roles of the mammalian homologs of PAQR-2/IGLR-2 in the context of the carbohydrate and SFA-rich Western diet, and diabetes. Specifically: is the mammalian homolog of the PAQR-2/IGLR-2 complex constantly “playing catch-up” to compensate for the fluidity-lowering effects of the Western diet? Could the well-documented diabetes-preventing effects of adiponectin and its receptors be explained by their roles in membrane homeostasis?
Genetics
Like all human traits, propensity to develop type 2 diabetes is influenced by genetic variation [85]. Several loci likely to have an impact on membrane fluidity have been linked to type 2 diabetes. In particular, several studies have linked polymorphisms in desaturase activity to abnormal fatty acid composition and type 2 diabetes risk [86–88]. Several polymorphisms in adiponectin or its receptors have also been linked to insulin resistance [89–93]. One study of special interest established a provocative correlation between certain single-nucleotide polymorphorphisms in adiponectin and AdipoR1 with the levels of plasma SFAs and insulin resistance [89]. The authors of this study concluded that “Personalized dietary advice to decrease SFA consumption in these individuals may be recommended as a possible therapeutic measure to improve insulin sensitivity.” There is also genetic evidence for the “flip side” of the membrane fluidity coin: Greenland Inuits with a highly fluidity-promoting omega-3 fat-rich diet show strong signs of positive selection for reduced-activity variants of delta-5 and delta-6 desaturases [94], which essentially reduces their ability to generate excessively fluid membranes.
Conclusions
Here then is a bite-sized theory that attempts to weave together the observations listed above into a “Membrane Theory of Diabetes”. SFAs obtained from the diet or via lipogenesis in the liver and adipocytes pose a relentless challenge to fluidity-sustaining systems, even more so in genetically predisposed individuals. This is likely exacerbated by unnatural fats of various types generated during the production of margarines or superheated vegetable oils used for frying much of our (fast) foods, and which may not be handled efficiently by the cellular machinery [95–97]. Chronic low fluidity in our membranes has several diabetes-promoting consequences, including impairing insulin secretion and signaling, reduced efficacy of GLUT4 localization to membranes and hardening of blood vessels. The idea that low membrane fluidity is an important component of diabetes pathophysiology is an old one that has been reviewed a few times [50, 56, 88, 98–100]. However, the recent identification of eukaryotic regulators of membrane fluidity should revive interest in this subject since they open novel experimental and therapeutic avenues.
Abbreviations
- DNL:
-
De novo lipogenesis
- ER:
-
Endoplasmic reticulum
- FAs:
-
Fatty acids
- FRAP:
-
Fluorescence recovery after photobleaching
- MUFAs:
-
Monounsaturated fatty acids
- PUFAs:
-
Polyunsaturated fatty acids
- RBCs:
-
Red blood cells
- SFAs:
-
Saturated fatty acids
- UFAs:
-
Unsaturated fatty acids
References
Svensson E, Olsen L, Mörck C, Brackmann C, Enejder A, Faergeman NJ, et al. The adiponectin receptor homologs in C. elegans promote energy utilization and homeostasis. PLoS ONE. 2011;6:e21343. Public Library of Science.
Svensk E, Ståhlman M, Andersson C-H, Johansson M, Borén JN, Pilon M. PAQR-2 regulates fatty acid desaturation during cold adaptation in C. elegans. PLoS Genet. 2013;9:e1003801. Ashrafi K, editor.
Svensk E, Devkota R, Ståhlman M, Ranji P, Rauthan M, Magnusson F, et al. Caenorhabditis elegans PAQR-2 and IGLR-2 protect against glucose toxicity by modulating membrane lipid composition. PLoS Genet. 2016;12:e1005982. Ashrafi K, editor.
Yamauchi T, Nio Y, Maki T, Kobayashi M, Takazawa T, Iwabu M, et al. Targeted disruption of AdipoR1 and AdipoR2 causes abrogation of adiponectin binding and metabolic actions. Nat Med. 2007;13:332–9.
Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–9. Nature Publishing Group.
Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. 2011;17:55–63. Nature Publishing Group.
Yamauchi T, Kadowaki T. Adiponectin receptor as a key player in healthy longevity and obesity-related diseases. Cell Metab. 2013;17:185–96.
Wang ZV, Scherer PE. Adiponectin, the past two decades. J Mol Cell Biol. 2016;8:93–100. Oxford University Press.
McMillan DE, Utterback NG, La Puma J. Reduced erythrocyte deformability in diabetes. Diabetes. 1978;27:895–901. American Diabetes Association.
Garnier M, Attali JR, Valensi P, Delatour-Hanss E, Gaudey F, Koutsouris D. Erythrocyte deformability in diabetes and erythrocyte membrane lipid composition. Metab Clin Exp. 1990;39:794–8.
Tsukada K, Sekizuka E, Oshio C, Minamitani H. Direct measurement of erythrocyte deformability in diabetes mellitus with a transparent microchannel capillary model and high-speed video camera system. Microvasc Res. 2001;61:231–9.
Baba Y, Kai M, Kamada T, Setoyama S, Otsuji S. Higher levels of erythrocyte membrane microviscosity in diabetes. Diabetes. 1979;28:1138–40. American Diabetes Association.
Kamada T, Otsuji S. Lower levels of erythrocyte membrane fluidity in diabetic patients: a spin label study. Diabetes. 1983;32:585–91. American Diabetes Association.
Kamada T, McMillan DE, Yamashita T, Otsuji S. Lowered membrane fluidity of younger erythrocytes in diabetes. Diabetes Res Clin Pract. 1992;16:1–6.
Brasitus TA, Dudeja PK. Correction of abnormal lipid fluidity and composition of rat ileal microvillus membranes in chronic streptozotocin-induced diabetes by insulin therapy. J Biol Chem. 1985;260:12405–9. American Society for Biochemistry and Molecular Biology.
Bhor VM, Sivakami S. Regional variations in intestinal brush border membrane fluidity and function during diabetes and the role of oxidative stress and non-enzymatic glycation. Mol Cell Biochem. 2003;252:125–32.
Ziegelhöffer-Mihalovicová B, Waczulíková I, Sikurová L, Styk J, Cársky J, Ziegelhöffer A. Remodelling of the sarcolemma in diabetic rat hearts: the role of membrane fluidity. Mol Cell Biochem. 2003;249:175–82. Kluwer Academic Publishers.
Masuda M, Murakami T, Egawa H, Murata K. Decreased fluidity of polymorphonuclear leukocyte membrane in streptozocin-induced diabetic rats. Diabetes. 1990;39:466–70.
Kamboj SS, Chopra K, Sandhir R. Hyperglycemia-induced alterations in synaptosomal membrane fluidity and activity of membrane bound enzymes: beneficial effect of N-acetylcysteine supplementation. Neuroscience. 2009;162:349–58.
Winocour PD, Bryszewska M, Watala C, Rand ML, Epand RM, Kinlough-Rathbone RL, et al. Reduced membrane fluidity in platelets from diabetic patients. Diabetes. 1990;39:241–4. American Diabetes Association.
Caimi G, Presti Lo R, Montana M, Canino B, Ventimiglia G, Romano A, et al. Membrane fluidity, membrane lipid pattern, and cytosolic Ca2+ content in platelets from a group of type II diabetic patients with macrovascular complications. Diabetes Care. 1995;18:60–3.
Hazel JR. Thermal adaptation in biological membranes: is homeoviscous adaptation the explanation? Annu Rev Physiol. 1995;57:19–42.
Crockett EL. The cold but not hard fats in ectotherms: consequences of lipid restructuring on susceptibility of biological membranes to peroxidation, a review. J Comp Physiol B. 2008;178:795–809. Springer-Verlag.
DeLong EF, Yayanos AA. Adaptation of the membrane lipids of a deep-sea bacterium to changes in hydrostatic pressure. Science. 1985;228:1101–3. American Association for the Advancement of Science. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.3992247.
Somero GN. Adaptations to high hydrostatic pressure. Annu Rev Physiol. 1992;54:557–77.
Avrova NF. The effect of natural adaptations of fishes to environmental temperature on brain ganglioside fatty acid and long chain base composition. Comp Biochem Physiol B Comp Biochem Pergamon. 1984;78:903–9.
Guschina IA, Harwood JL. Mechanisms of temperature adaptation in poikilotherms. FEBS Lett. 2006;580:5477–83.
Stubbs CD, Smith AD. The modification of mammalian membrane polyunsaturated fatty acid composition in relation to membrane fluidity and function. Biochim Biophys Acta. 1984;779:89–137.
Ramstedt B, Slotte JP. Sphingolipids and the formation of sterol-enriched ordered membrane domains. Biochim Biophys Acta. 2006;1758:1945–56.
Dawaliby R, Trubbia C, Delporte C, Noyon C, Ruysschaert J-M, Van Antwerpen P, et al. Phosphatidylethanolamine is a Key regulator of membrane fluidity in eukaryotic cells. J Biol Chem. 2016;291:3658–67. American Society for Biochemistry and Molecular Biology.
Holthuis JCM, Menon AK. Lipid landscapes and pipelines in membrane homeostasis. Nature. 2014;510:48–57. Nature Research.
McIntosh TJ, Simon SA. Roles of bilayer material properties in function and distribution of membrane proteins. Annu Rev Biophys Biomol Struct. 2006;35:177–98. Annual Reviews.
van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nature Rev Mol Cell Biol. 2008;9:112–24. Nature Publishing Group.
Bryszewska M, Watala C, Torzecka W. Changes in fluidity and composition of erythrocyte membranes and in composition of plasma lipids in Type I diabetes. Br J Haematol. 1986;62:111–6. Blackwell Publishing Ltd.
Watała C, Jóźwiak Z. The phospholipid composition of erythrocyte ghosts and plasma lipoproteins in diabetes type 1 in children. Clin Chim Acta. 1990;188:211–9.
Ruiz-Gutierrez V, Stiefel P, Villar J, García-Donas MA, Acosta D, Carneado J. Cell membrane fatty acid composition in Type 1 (insulin-dependent) diabetic patients: relationship with sodium transport abnormalities and metabolic control. Diabetologia. 1993;36:850–6. Springer-Verlag.
Borkman M, Storlien LH, Pan DA, Jenkins AB, Chisholm DJ, Campbell LV. The relation between insulin sensitivity and the fatty-acid composition of skeletal-muscle phospholipids. N Engl J Med. 1993;328:238–44. Massachusetts Medical Society.
Clifton PM, Nestel PJ. Relationship between plasma insulin and erythrocyte fatty acid composition. Prostaglandins Leukot Essent Fatty Acids. 1998;59:191–4.
Bakan E, Yildirim A, Kurtul N, Polat MF, Dursun H, Cayir K. Effects of type 2 diabetes mellitus on plasma fatty acid composition and cholesterol content of erythrocyte and leukocyte membranes. Acta Diabetol. 2006;43:109–13. Springer-Verlag.
Kröger J, Zietemann V, Enzenbach C, Weikert C, Jansen EH, Döring F, et al. Erythrocyte membrane phospholipid fatty acids, desaturase activity, and dietary fatty acids in relation to risk of type 2 diabetes in the European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Am J Clin Nutr. 2011;93:127–42. American Society for Nutrition.
Kröger J, Jacobs S, Jansen EHJM, Fritsche A, Boeing H, Schulze MB. Erythrocyte membrane fatty acid fluidity and risk of type 2 diabetes in the EPIC-Potsdam study. Diabetologia. 2015;58:282–9. Springer Berlin Heidelberg.
Forouhi NG, Koulman A, Sharp SJ, Imamura F, Kröger J, Schulze MB, et al. Differences in the prospective association between individual plasma phospholipid saturated fatty acids and incident type 2 diabetes: the EPIC-InterAct case-cohort study. Lancet Diabetes Endocrinol. 2014;2:810–8.
MacDonald MJ, Ade L, Ntambi JM, Ansari I-UH, Stoker SW. Characterization of phospholipids in insulin secretory granules and mitochondria in pancreatic beta cells and their changes with glucose stimulation. J Biol Chem. 2015;290:11075–92. American Society for Biochemistry and Molecular Biology.
Ginsberg BH, Jabour J, Spector AA. Effect of alterations in membrane lipid unsaturation on the properties of the insulin receptor of Ehrlich ascites cells. Biochimica et Biophysica Acta (BBA) - Biomembranes. 1982;690:157–64. Elsevier.
Krischer J, Gilbert A, Gorden P, Carpentier JL. Endocytosis is inhibited in hepatocytes from diabetic rats. Diabetes. 1993;42:1303–9.
Illinger D, Poindron P, Kuhry JG. Membrane fluidity aspects in endocytosis; a study with the fluorescent probe trimethylamino-diphenylhexatriene in L929 cells. Biol Cell. 1991;71:293–6.
Hulbert AJ, Else PL. Mechanisms underlying the cost of living in animals. Annu Rev Physiol. 2000;62:207–35.
Winter PW, Van Orden AK, Roess DA, Barisas BG. Actin-dependent clustering of insulin receptors in membrane microdomains. Biochim Biophys Acta. 1818;2012:467–73.
Ginsberg BH, Brown TJ, Simon I, Spector AA. Effect of the membrane lipid environment on the properties of insulin receptors. Diabetes. 1981;30:773–80. American Diabetes Association.
Elmendorf JS. Fluidity of insulin action. Mol Biotechnol [Internet]. 2004;27:127–38. Available from: http://link.springer.com/10.1385/MB:27:2:127.
Mercuri O, Peluffo RO, Brenner RR. Effect of insulin on the oxidative desaturation ofa-linolenic, oleic and palmitic acids. Lipids. 1967;2:284–5. Springer-Verlag.
Eck MG, Wynn JO, Carter WJ, Faas FH. Fatty acid desaturation in experimental diabetes mellitus. Diabetes. 1979;28:479–85. American Diabetes Association.
Rimoldi OJ, Finarelli GS, Brenner RR. Effects of diabetes and insulin on hepatic delta6 desaturase gene expression. Biochem Biophys Res Commun. 2001;283:323–6.
Mauvoisin D, Rocque G, Arfa O, Radenne A, Boissier P, Mounier C. Role of the PI3-kinase/mTor pathway in the regulation of the stearoyl CoA desaturase (SCD1) gene expression by insulin in liver. J Cell Commun Signal. 2007;1:113–25. Springer Netherlands.
Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–77.
Weijers RNM. Lipid composition of cell membranes and its relevance in type 2 diabetes mellitus. Curr Diabetes Rev. 2012;8:390–400. Bentham Science Publishers.
Pietiläinen KH, Róg T, Seppänen-Laakso T, Virtue S, Gopalacharyulu P, Tang J, et al. Association of lipidome remodeling in the adipocyte membrane with acquired obesity in humans. PLoS Biol. 2011;9:e1000623. Tieleman DP, editor. Public Library of Science.
Kamada T, Yamashita T, Baba Y, Kai M, Setoyama S, Chuman Y, et al. Dietary sardine oil increases erythrocyte membrane fluidity in diabetic patients. Diabetes. 1986;35:604–11. American Diabetes Association.
Lund EK, Harvey LJ, Ladha S, Clark DC, Johnson IT. Effects of dietary fish oil supplementation on the phospholipid composition and fluidity of cell membranes from human volunteers. Ann Nutr Metab. 2000;43:290–300. Karger Publishers.
Pan DA, Storlien LH. Dietary lipid profile is a determinant of tissue phospholipid fatty acid composition and rate of weight gain in rats. J Nutr. 1993;123:512–9.
Abbott SK, Else PL, Atkins TA, Hulbert AJ. Fatty acid composition of membrane bilayers: importance of diet polyunsaturated fat balance. Biochim Biophys Acta. 1818;2012:1309–17.
Andersson A, Nälsén C, Tengblad S, Vessby B. Fatty acid composition of skeletal muscle reflects dietary fat composition in humans. Am J Clin Nutr. 2002;76:1222–9. American Society for Nutrition.
Kersten S. Mechanisms of nutritional and hormonal regulation of lipogenesis. EMBO Rep. 2001;2:282–6. EMBO Press.
Summers LKM, Fielding BA, Bradshaw HA, Ilic V, Beysen C, Clark ML, et al. Substituting dietary saturated fat with polyunsaturated fat changes abdominal fat distribution and improves insulin sensitivity. Diabetologia. 2002;45:369–77. Springer-Verlag.
Tishinsky JM, Gulli RA, Mullen KL, Dyck DJ, Robinson LE. Fish oil prevents high-saturated fat diet-induced impairments in adiponectin and insulin response in rodent soleus muscle. Am J Physiol Regul Integr Comp Physiol. 2012;302:R598–605. American Physiological Society.
Collins JM, Neville MJ, Hoppa MB, Frayn KN. De novo lipogenesis and stearoyl-CoA desaturase are coordinately regulated in the human adipocyte and protect against palmitate-induced cell injury. J Biol Chem. 2010;285:6044–52. American Society for Biochemistry and Molecular Biology.
Dridi S, Taouis M, Gertler A, Decuypere E, Buyse J. The regulation of stearoyl-CoA desaturase gene expression is tissue specific in chickens. J Endocrinol BioScientifica. 2007;192:229–36.
Ntambi JM. Stearoyl-CoA Desaturase Genes in Lipid Metabolism. New York: Springer New York; 2013.
Ntambi JM, Miyazaki M, Dobrzyn A. Regulation of stearoyl-CoA desaturase expression. Lipids. 2004;39:1061–5.
Cybulski LE, Ballering J, Moussatova A, Inda ME, Vazquez DB, Wassenaar TA, et al. Activation of the bacterial thermosensor DesK involves a serine zipper dimerization motif that is modulated by bilayer thickness. Proc Natl Acad Sci U S A. 2015;112:6353–8. National Acad Sciences.
Inda ME, Vandenbranden M, Fernández A, de Mendoza D, Ruysschaert J-M, Cybulski LE. A lipid-mediated conformational switch modulates the thermosensing activity of DesK. Proc Natl Acad Sci U S A. 2014;111:3579–84. National Acad Sciences.
Cybulski LE, Martín M, Mansilla MC, Fernández A, de Mendoza D. Membrane thickness cue for cold sensing in a bacterium. Curr Biol. 2010;20:1539–44.
Albanesi D, Mansilla MC, de Mendoza D. The membrane fluidity sensor DesK of Bacillus subtilis controls the signal decay of its cognate response regulator. J Bacteriol. 2004;186:2655–63.
Aguilar PS, Hernandez-Arriaga AM, Cybulski LE, Erazo AC, de Mendoza D. Molecular basis of thermosensing: a two-component signal transduction thermometer in Bacillus subtilis. EMBO J. 2001;20:1681–91. EMBO Press.
Mansilla MC, Cybulski LE, Albanesi D, de Mendoza D. Control of membrane lipid fluidity by molecular thermosensors. J Bacteriol. 2004;186:6681–8. American Society for Microbiology.
Covino R, Ballweg S, Stordeur C, Michaelis JB, Puth K, Wernig F, et al. A eukaryotic sensor for membrane lipid saturation. Mol Cell. 2016;63:49–59.
Pilon M, Svensk E. PAQR-2 may be a regulator of membrane fluidity during cold adaptation. Worm. 2013;2. e27123.
Pei J, Millay DP, Olson EN, Grishin NV. CREST--a large and diverse superfamily of putative transmembrane hydrolases. Biol Direct. 2011;6:37.
Tanabe H, Fujii Y, Okada-Iwabu M, Iwabu M, Nakamura Y, Hosaka T, et al. Crystal structures of the human adiponectin receptors. Nature. 2015;520:312–6. Nature Publishing Group.
Villa NY, Kupchak BR, Garitaonandia I, Smith JL, Alonso E, Alford C, et al. Sphingolipids function as downstream effectors of a fungal PAQR. Mol Pharmacol. 2009;75:866–75.
Muller S, Denet S, Candiloros H, Barrois R, Wiernsperger N, Donner M, et al. Action of metformin on erythrocyte membrane fluidity in vitro and in vivo. Eur J Pharmacol. 1997;337:103–10.
Wiernsperger NF. Membrane physiology as a basis for the cellular effects of metformin in insulin resistance and diabetes. Diabetes Metab. 1999;25:110–27.
Viollet B, Guigas B, Garcia NS, Leclerc J, Foretz M, Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clin Sci. 2012;122:253–70. Portland Press Limited.
Pernicova I, Korbonits M. Metformin--mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–56. Nature Publishing Group.
Ahlqvist E, Ahluwalia TS, Groop L. Genetics of type 2 diabetes. Clin Chem. 2011;57:241–54. American Association for Clinical Chemistry.
Kröger J, Schulze MB. Recent insights into the relation of Δ5 desaturase and Δ6 desaturase activity to the development of type 2 diabetes. Curr Opin Lipidol. 2012;23:4–10.
Warensjö E, Ingelsson E, Lundmark P, Lannfelt L, Syvänen A-C, Vessby B, et al. Polymorphisms in the SCD1 gene: associations with body fat distribution and insulin sensitivity. Obesity (Silver Spring, Md). 2007;15:1732–40. Blackwell Publishing Ltd.
Dobrzyn P, Jazurek M, Dobrzyn A. Stearoyl-CoA desaturase and insulin signaling--what is the molecular switch? Biochim Biophys Acta. 2010;1797:1189–94.
Ferguson JF, Phillips CM, Tierney AC, Pérez-Martínez P, Defoort C, Helal O, et al. Gene—nutrient interactions in the metabolic syndrome: single nucleotide polymorphisms in ADIPOQ and ADIPOR1 interact with plasma saturated fatty acids to modulate insulin resistance. Am J Clin Nutr. 2010;91:794–801. American Society for Nutrition.
Vaxillaire M, Dechaume A, Vasseur-Delannoy V, Lahmidi S, Vatin V, Leprêtre F, et al. Genetic analysis of ADIPOR1 and ADIPOR2 candidate polymorphisms for type 2 diabetes in the Caucasian population. Diabetes. 2006;55:856–61.
Ruchat S-M, Loos RJF, Rankinen T, Vohl M-C, Weisnagel SJ, Després J-P, et al. Associations between glucose tolerance, insulin sensitivity and insulin secretion phenotypes and polymorphisms in adiponectin and adiponectin receptor genes in the Quebec Family Study. Diabet Med. 2008;25:400–6. Blackwell Publishing Ltd.
Stefan N, Machicao F, Staiger H, Machann J, Schick F, Tschritter O, et al. Polymorphisms in the gene encoding adiponectin receptor 1 are associated with insulin resistance and high liver fat. Diabetologia. 2005;48:2282–91. Springer-Verlag.
Kim JT, Kim Y, Cho YM, Koo BK, Lee EK, Shin HD, et al. Polymorphisms of ADIPOR1 and ADIPOR2 are associated with phenotypes of type 2 diabetes in Koreans. Clin Endocrinol (Oxf). 2009;70:66–74. Blackwell Publishing Ltd.
Fumagalli M, Moltke I, Grarup N, Racimo F, Bjerregaard P, Jørgensen ME, et al. Greenlandic Inuit show genetic signatures of diet and climate adaptation. Science. 2015;349:1343–7. American Association for the Advancement of Science.
Ibrahim A, Natrajan S, Ghafoorunissa R. Dietary trans-fatty acids alter adipocyte plasma membrane fatty acid composition and insulin sensitivity in rats. Metab Clin Exp. 2005;54:240–6.
Roach C, Feller SE, Ward JA, Shaikh SR, Zerouga M, Stillwell W. Comparison of cis and trans fatty acid containing phosphatidylcholines on membrane properties. Biochemistry. 2004;43:6344–51.
Mozaffarian D, Katan MB, Ascherio A, Stampfer MJ, Willett WC. Trans fatty acids and cardiovascular disease. N Engl J Med. 2006;354:1601–13.
Murphy MG. Dietary fatty acids and membrane protein function. J Nutr Biochem. 1990;1:68–79.
Hulbert AJ, Turner N, Storlien LH, Else PL. Dietary fats and membrane function: implications for metabolism and disease. Biol Rev Camb Philos Soc. 2005;80:155–69.
Storlien LH, Hulbert AJ, Else PL. Polyunsaturated fatty acids, membrane function and metabolic diseases such as diabetes and obesity. Curr Opin Clin Nutr Metab Care. 1998;1:559–63.
Acknowledgements
This work was funded by the following Swedish agencies: Vetenskaprådet, Cancerfonden, Carl Trygger Stiftelse and Diabetesfonden. I wish to thank Peter Carlsson, Jan Oscarsson and members of the Pilon group for discussions and comments on the manuscript, as well as our collaborators Jan Borén and Marcus Ståhlman for their insights on lipids and for inducing me to review the mammalian literature on this topic.
Funding
Vetenskaprådet (612-2012-2152), Cancerfonden (15 0326), Carl Tryggers Stiftelse (CTS 15:391) and Diabetesfonden (DIA2015-007) funded the author during the writin of this review.
Authors’ contributions
MP reviewed the literature and wrote the manuscript.
Competing interests
The author declares that he has no competing interests.
Consent for publication
Not applicable.
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Pilon, M. Revisiting the membrane-centric view of diabetes. Lipids Health Dis 15, 167 (2016). https://doi.org/10.1186/s12944-016-0342-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12944-016-0342-0