
Abstract
Ongoing research has revealed that the existence of cancer stem cells (CSCs) is one of the biggest obstacles in the current cancer therapy. CSCs make an influential function in tumor progression, recurrence and chemoresistance due to their typical stemness characteristics. CSCs are preferentially distributed in niches, and those niche sites exhibit characteristics typical of the tumor microenvironment (TME). The complex interactions between CSCs and TME illustrate these synergistic effects. The phenotypic heterogeneity within CSCs and the spatial interactions with the surrounding tumor microenvironment led to increased therapeutic challenges. CSCs interact with immune cells to protect themselves against immune clearance by exploiting the immunosuppressive function of multiple immune checkpoint molecules. CSCs also can protect themselves against immune surveillance by excreting extracellular vesicles (EVs), growth factors, metabolites and cytokines into the TME, thereby modulating the composition of the TME. Therefore, these interactions are also being considered for the therapeutic development of anti-tumor agents. We discuss here the immune molecular mechanisms of CSCs and comprehensively review the interplay between CSCs and the immune system. Thus, studies on this topic seem to provide novel ideas for reinvigorating therapeutic approaches to cancer.
Similar content being viewed by others

Introduction
Many therapeutic modalities have been developed, which are currently used to treat cancer, such as surgery, radiation, chemotherapy and targeted therapies, but the risk of recurrence remains high [1, 2]. Studies have shown that the proliferation and spread of tumor cells are related to the presence of stem-like cells within the tumor, which are collectively referred to cancer stem cells (CSCs) [3]. The existence of this cell type was first reported in acute myeloid leukemia (AML) [1]; subsequently, the presence of CSCs is also reported in different types of solid tumors, containing brain, breast, lung, liver, pancreas, colon and prostate cancer [4, 5]. These cells are capable of differentiation, self-renewal, tumorigenesis and chemoresistance [6]. CSCs are also capable of controlling the role of immune cells, containing T cells, B cells, NK cells as well as macrophages [7]. The tumor microenvironment is resulted from the presence of immune checkpoint inhibitors, such as programmed death-1/programmed cell death ligand (PD-L1), cluster of differentiation 47 (CD47), T cell immunoglobulin and mucin-containing domain-3 (TIM3), lymphocyte activation gene 3 (LAG3) and cytotoxic T-lymphocyte antigen-4 (CTLA4) [8,9,10].
The contact between CSCs and immune cells is mediated not only through immune targets, but also through EVs that enable the transfer of large biomolecular cargos among different types of cells. CSCs regulate the composition of TME through the release of EVs and various soluble factors, including cytokines, chemokines, growth factors, metabolites and hormones [11,12,13]. Several factors are involved in the cross-talk between CSCs and the tumor microenvironment, such as interleukins (ILs) (IL-6, IL-8 and IL-1β), matrix metalloproteinases (MMPs), vascular endothelial growth factor (VEGF) as well as transforming growth factor beta 1 (TGF-β1) which can be freely released into the extracellular space or encapsulated in EVs [14,15,16,17,18,19,20]. Given the important immunomodulatory role of CSCs, further scientific studies are still required to evaluate the extent of the clinical impact of CSCs.
The current review primarily concentrates on the recent advances in the crosstalk between CSCs and immune cells, immune checkpoint molecules and EVs in the TME, together with the possible mechanisms of CSCs induced immune suppression in accordance with the above-mentioned interactions. In addition, we show the present understanding of the origins, activators, heterogeneity and plasticity of CSCs. In the end, we present major CSCs-based targeted immunotherapeutic strategies that can probably improve anti-tumor immunity in the TME and show several potential research directions in the future.
Development of cancer stem cells
Intrinsic features: genetic and epigenetic
Cancer stem cells (CSCs) stand for a small subpopulation of the tumor and possess self-renewal properties [2, 21,22,23]. CSCs undergo asymmetric division, giving rise to two different cell types with distinct cellular fates: one retains stem cell-like features keeping the capacity for self-renewal, whereras the other transforms into specialized progenitor cells with the capacity to generate proliferating tumor cells and populate the tumor mass [24]. Several surface markers of CSCs have been identified, including EpCAM, CD44 and CD133, which provide a possible identification method of CSCs in the tumor stroma (Table 1) [25,26,27,28,29,30,31,32,33,34].
Intrinsic heterogeneity includes genetic and epigenetic alterations that promote oncogenic activity [1, 35]. Genetic and epigenetic alterations make an integral function in promoting tumor development, progression, survival and therapeutic resistance to treatment. The plasticity of cancer stem cells allows phenotypic switching between CSCs and non-CSCs states in response to environmental signals and is ruled by intrinsic factors [36,37,38,39]. Maintenance of cancer stem cell plasticity makes a necessary role in stimulating the growth and survival of tumor cells. The maintenance of the cancer stem cell state can be controlled by genomic changes (chromosomal amplifications, deletions, rearrangements and DNA mutations), epigenetic modifications and microenvironmental cues [40]. In contrast to genetic changes, epigenetic reprogramming facilitates adaptation and resistance to treatments, thus, greatly influencing cellular fate decisions. Similarly, genetic and epigenetic modifications involved in the signaling pathways can promote the stemness of CSCs.
Cell signaling pathways regulating cancer stem cells
Numerous signaling pathways are activated in CSCs, including Wnt/β-catenin, Notch, Hedgehog (Hh), Nuclear factor kappa B (NF-κB), Yes-associated protein (YAP) and Integrins, making vital functions in controlling cell survival, growth, differentiation and self-renewal. Several components of the cell signaling pathways were found to be genetically altered in CSCs. All these genetic alterations lead to epigenetic reprogramming causing deregulation of many signaling pathways, which collectively determine the fate of CSCs present within the tumors.
The canonical Wnt/β-catenin signaling pathway is considered to be a vital regulator of tumor cell plasticity [41]. Activation of the canonical Wnt signaling pathway can be regulated by the transcription factor β-catenin [42, 43]. Wnt/β-catenin is a signaling pathway which regulates cell proliferation, differentiation, apoptosis and tissue homeostasis [44,45,46], whereas aberrant Wnt/β-catenin signaling enhances the expression of surface markers of CSCs and promotes self-renewal, localization within specialized niches and other related CSCs properties [47].
The Notch signaling pathway regulates stem cell differentiation and self-renewal [48,49,50]. Aberrant Notch signaling stimulates self-renewal of CSCs in ovarian, breast, and hepatocellular carcinoma (HCC) [38]. Epigenetic analysis of osteosarcoma cells indicates that leukemia inhibitory factor (LIF) is associated with the activation of NOTCH1 signaling through lysine 27 of histone H3 (H3K27 me3) demethylation, inducing the expression of "stemness" related genes, sphere formation, self-renewal as well as metastasis [51,52,53].
In CSCs, the hedgehog (Hh) signaling pathway has been engaged in driving tumor growth, invasion and tumor recurrence following therapeutic intervention [54, 55]. In colorectal cancer, cancer-initiating cells express the indian hedgehog (IHH) gene, which is present in a bivalent state and contributes to the maintenance of colorectal cancer-initiating cells [54, 56]. In gastric adenocarcinoma, increased promoter methylation of transcription factors CDX1/2 and KLF5, which are the downstream targets of the sonic hedgehog (SHH) signaling, caused reduced expression of CDX1 and KLF5 and elevated expression of CDX2. Elevated expression of CDX2 was related to lymph node metastases in patients [55]. Likewise, DNA hypermethylation of the CpG bank of the SHH gene leads to the loss of the expression of the SHH gene in invasive uroepithelial carcinoma [57].
Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding (TAZ) are transcriptional co-activators, which are upregulated in many cancer types [58,59,60,61]. A splice variant of the α6 cytoplasmic structural domain (α6β1) of integrin was discovered to be capable of activating TAZ, causing the transcription of genes related to self-renewal [59]. In prostate cancer, over-expression of α3 integrins in drug-resistant cancer cells led to the inhibition of metastasis, which occurred via the inhibition of Rho GTPase activity by Abl kinase in the Hippo oncogenic signaling pathway [62]. However, in glioblastoma, α3 expression was discovered to be associated with tumor invasion and metastasis via the activation of the extracellular signal-regulated kinase 1/2 (ERK1/2) signaling pathway [63].
Integrins refer to heterodimeric cell surface receptors that promote cell proliferation, differentiation, adhesion to extracellular matrix (ECM) and migration by sensing the cellular microenvironment [64,65,66,67]. Overexpression of integrins in different cancer types has been documented, and a variety of peptide ligands against integrins have been developed for targeted therapies [68, 69]. The alphavbeta3 (αVβ3) integrin has been implicated in developing resistance to receptor tyrosine kinase inhibitors [60]. Increased expression of αVβ3 integrin also was discovered in lung tumors that mediated resistance to erlotinib [70]. While in another research, integrin α6 was strongly denoted in glioblastoma cells [71]. Currently, α6 is used as a biomarker for the identification of cancer stem cells [72]. In addition, integrins can also regulate cellular signaling events facilitating extracellular signaling events.
Taken together, signaling pathways are deterministic in the establishment of stemness traits. Moreover, the survival of cancer stem cells can also be dependent on tumor microenvironment events in the local niche of the tumor that help them to remain in a quiescent state or switch to a proliferative state. Significantly, there exists a growing body of evidence showing that a favourable environment plays a vital role in dedifferentiating tumor cells into CSCs. Besides, further identification of more detailed microenvironmental signals supporting or determining the stemness is of paramount importance to propose better intervention strategies.
Immunomodulatory traits of CSCs and tumor microenvironment
In many cancer types, tumors consist of rare subpopulations of CSCs that differ in cellular phenotype, gene expression pattern and functional characteristics [73]. The tumor microenvironment can regulate the development of cancer stem cells [1, 74, 75], which includes the ECM and non-tumor cells present in the tumor stroma, like cancer-associated fibroblasts (CAF) and endothelial cells, exerts a vital function in the progression of the tumor. Stromal cells can regulate the activity of CSCs via paracrine signaling. For example, hepatocyte growth factor (HGF) secreted by myofibroblasts activates Wnt signaling pathway and induces dedifferentiation of non-CSCs into CSCs [76]. Likewise, the vasculature of the tumor microenvironment supports carcinogenesis and provides a specialized ecological niche for CSCs. It was shown that endothelial cells induce CSC phenotype in colon cancer by producing Notch ligand Delta-like ligand 4 (DLL4) [77]. Endothelial cells secrete growth factors that induce stem cell phenotype in glioblastoma [78, 79]. In addition, CSCs can even create their own ecological niche by trans-differentiation into endothelial progenitor cells [80, 81], providing CSCs with the necessary growth factors [80, 82]. Another perspective of the microenvironment possessing the power to influence CSCs behavior is the immune cells. Therefore, a better understanding of the interaction between CSCs and immune cells may provide potential new approaches to develop therapeutic interventions for tumors.
Immune cells-targeted immunotherapy for CSCs
Interaction between CSCs and immune cells in the TME
The CSCs niche maintains the state and plasticity of cancer cells and protects them from immune cell attack [26, 83,84,85,86,87,88,89,90,91,92]. The persistent interaction of cancer stem cells with the tumor microenvironment confers the ability to avoid recognition and eradication by immune cells, ensuring their survival and development [93, 94]. Therefore, understanding the capacity of cancer cells to circumvent immune evasion is a prerequisite to better understanding the immunobiology of CSC and thus developing more effective therapeutic approaches.
Dendritic cells (DCs) refer to the primary antigen-presenting cells (APCs), presenting tumor-associated antigens (TAAs) on major histocompatibility complex (MHC)-I molecules, thereby activating immune responses. CSCs can either impair the production of mature DCs or enhance the number of tolerogenic DCs by secreting TGF-β1 [95], leading to the downregulation of MHC-II expression as well as the production of CD80, CD86 costimulatory molecules [96,97,98]. CD105 expressing CSCs secrete EVs carrying MHC-I and human leukocyte antigen G (HLA-G), which impair the maturation of DCs through the signal transducer and activator of transcription 3 (STAT3) signaling pathway [99, 100]. The interaction between C-X-C motif chemokine ligand (CXCL)-12 on regulatory dendritic cells (DCregs) and C-X-C motif chemokine receptor (CXCR)-4 receptor on CSCs contributes to the maintenance of the self-renewal property of CSCs [101]. Furthermore, CXCL1+ DC-regs induce stemness signaling in CD133+ colon cancer cells to facilitate metastatic capacity [102].
The interaction of tumor-associated macrophages (TAMs) with CSCs confers the emergence of an immunosuppressive TME [103]. Ecotopes of CSCs are enriched in ILs, ECM, TGF-β and periostin that facilitate macrophage recruitment and macrophage polarization [85, 104]. The expression of periostin on the cell membrane of CSCs recruits monocytes from the vasculature [85] and converts monocytes into TAMs in the TME to support the activity and survival of CSCs. TGF-β1 promotes the generation of EpCAM+ CSCs, which facilitate HCC invasion and metastasis by triggering epithelial-mesenchymal transition (EMT) [105]. Furthermore, TAMs trigger the over-expression of CD47 on pancreatic [106], HCC [107] and leukemic [108] stem cells. CD47 on CSCs binds to SIRPα on macrophages protecting CSCs from immune cell-mediated phagocytosis. In addition to this, secreted factors from TAMs stimulate the expression of immune checkpoints, including PD-L1 [109]. Overall, the cross-talk between CSC and TAM induces the immunosuppressive TME, which supporting the survival of CSCs and complicates tumor eradication after immunotherapy.
Myeloid-derived suppressor cells (MDSCs) secrete cytokines and chemokines to reduce the efficacy of immunotherapy [110]. The mammalian target of rapamycin (mTOR) signaling in CSCs promotes the infiltration and aggregation of MDSCs at tumor sites [110]. In melanoma, CD133+ CSCs activate TGF-β1 expression and recruit immunosuppressive MDSCs in the tumor site [111]. In addition, TIM-3/Galectin 9 (Gal-9) expressed on the surface of leukemic stem cells (LSCs) elevates the number of infiltrating MDSCs and TAMs, leading to impaired anti-tumor immune responses [112]. Similarly, MDSCs induce stemness in CSCs through upregulation of piRNA-823 [113]. Moreover, MDSCs secrete exosome S100A9 that enhances the activity of signal transducer and activator of STAT3/noncanonical nuclear factor-kappaB (NF-κB) signaling [114] and the production of prostaglandin E2 (PEG-E2) [115], promoting cancer cell stemness and survival. These findings suggest that CSCs-MDSCs interactions reshape the stemness of CSCs, leading to tumor growth and progression.
The cross-talk of Tregs with CSCs promotes the formation of immunosuppressive TME. PD-L1 and TGF-β1 expressed by CSCs mediate Tregs infiltration in glioblastoma [116]. Similarly, CSCs secrete CCL1 to recruit Tregs, producing TGF-β1 and IL-17 to stimulate the self-renewal capacity of CSCs [117,118,119]. Gastric CSCs facilitate the development of cancer stem cells through STAT3 signaling pathway while protecting CSCs from being recognized by T cells [120]. Tregs derive VEGF to maintain the survival, stemness and self-renewal of CSCs under hypoxia conditions [82]. Furthermore, Tregs secreted cyclooxygenase 2 (COX-2) hinders the function of effector T cells in a PEG-E2-dependent mechanism, verifying that the interactions between CSCs and Tregs promote immune escape, leading to the failure of cancer immunotherapy [121].
In general, T cells recognize TAAs on the surface of APCs as MHC-peptide complexes. However, CSCs can downregulate the expression of MHC-I [122] and TAAs [91], induce the expression of allelic variants of MHC-1 [123, 124] and upregulate the expression of immune checkpoints, including PD-L1 [125], to evade immune surveillance and recognition by anti-tumor immunity. Besides, downregulation of MHC-1 expression influences CD8+ T cell activation [126]. EMT/β-catenin signaling in CSCs regulates the glycosylation and stabilization of the immune checkpoint PD-L1, thus, evading T cell immune surveillance [127]. In a hypoxia environment, CSCs induce the expression of VEGF, PD-L1 and TIM-3 [128]. During the development and metastasis of human neural crest cells (HNCCs), CD276+ CSCs are found to be located at infiltrating tumor sites and evade anti-tumor immunity by hindering the infiltration of CD8+ T cells [129, 130]. Prostate CSCs inhibit T cell proliferation and cytokine production via Gal-3 expression, thus, protecting CSCs from cytotoxic T cells mediated lysis [131]. Furthermore, quiescent CSCs protect the ability of T cells to recognize and lysis of tumor cells by downregulating NLR family CARD domain containing 5 (NLRC5) trans-activator which belongs to the MHC class I mediated immune responses [132].
The activation of natural killer group 2 member D (NKG2D) receptor expressed on the surface of NK cells promote the lysis of MHC-I negative CSCs by a non-APC dependent mechanism [133]. NK cells expressing NKG2D can mediate the lysis of MHC-I negative colonic CD133+CD44+ CSCs [134], and NK cells expressing NKp30 and NKp44 can directly target and eradicate MHC-I negative CD24+ CSCs in ovarian cancer [135]. CSCs upregulate HLA-G expression, which interacts with the NK cell inhibitory ligands killer cell immunoglobulin-like receptor 2DL4 (KIR2DL4) and natural killer group 2 member A (NKG2A), making them become less sensitive to NK cell-mediated lysis by inhibiting NK cell activation [136,137,138]. In addition, CSCs expressing SOX2/SOX9 downregulate NKG2DL expression and protect them from NK cell-mediated immune clearance [139]. CSCs develop therapeutic resistance to NK cell-based immunotherapy by upregulating MHC-I molecules, which eventually leads to tumor recurrence [140]. Therefore, understanding the potential mechanism driving NK cell-mediated recognition and elimination of CSCs can probably provide opportunities for anti-CSC targeted immunotherapies (Fig. 1).

CSCs interfere with immune cell activity directly or through cytokines. CSCs suppress or evade antitumorigenic T cells in part by immune checkpoint (MHC-I, PD-L1 and CD80). CSCs reduce DCs mature and differentation via TGFβ and Ev (MHC-I, HLA-G). NKG2DL are able to kill MHC-I negative CSCs in an APC-independent manner. NK cells inhibitory ligands KIR2DL4 and NKG2A interact with HLA-G on CSCs and directly inhibits NK cells activation. CSCs further drive recruitment and polarization of TH17 cells and Treg cells by the combination of CCL-1, IL-2, IL-8, IL-10 and TGF-β1. Tregs produce TGF-β1 and IL-17 to promote self-renewal capacity, stem cell markers, and EMT toward tumor progression and invasion. CSCs also derived PD-L1 mediate the infiltration of Tregs. An additional layer of regulation of T cell activity is mediated indirectly by immunosuppressive myeloid cells, including macrophages and monocytic myeloid-derived suppressor cells (M-MDSCs). This effect partially depends on CSF1, CCL2, CCL5, TGF-β1 and PEG-E2 secreted by CSCs. The pathway of CSCs expressing TIM-3/Galectin 9 (Gal-9) expands the number of MDSCs. Exosome S100A9 enhances STAT3/NF-κB phosphorylation and production of prostaglandin E2 (PEG-E2) to promote CSCs. Collectively, these interactions reshape the tumour microenvironment and create a habitat where Treg cells and TH17 cells support CSCs, the latter via IL-17 production
Targeting CSC-immune cells therapy
From our perspective, CSCs and the tumor immune system an inextricable linked. CSCs can create their TME after cross-talk with immune cells, thus, promoting tumor immunosuppression and immune escape. This research demonstrates the therapeutic potential of focusing on CSC-TAM, CSC-T cell and CSC-MDSC cross-talk [141]. TAM can increase the increased expression of hyaluronic acid (HA) from CSCs in human neck squamous carcinoma (HNSCC); thus, targeting TAMs to inhibit CSC function is a viable option [142]. CSCs suppress T cell function by secreting cytokines (TGF-β1, CCL2 and Tenascin-C (TNC)) and exosomes to promote bone marrow-derived macrophage (BMDM) activation. MDSC promotes CSC stemness and inhibits T cell activation in breast cancer through the STAT3 signaling pathway [143]. MDSC also increases CSC stemness and PD-L1 expression in epithelial ovarian tumor cells by producing PGE2 [144]. MDSC also promotes the stemness of CSCs in ovarian cancer through triggering the CSF2/p-STAT3 signaling pathway. Therefore, it is considered that targeting MDSC and the CSF2/p-STAT3 signaling pathway can improve the efficacy of conventional therapies [145]. These preclinical studies show that targeting CSC-immune cell cross-talk has therapeutic potential in the treatment of cancer patients.
Targeting CSC-CAR-T cells therapy
TILs are isolated from a patient, cultured with IL-2, tested for their ability to recognize tumor-specific antigens, and then reinfused into the same patient [146]. T cells have been reprogrammed into chimeric antigen receptor (CAR) T cells through the use of artificially designed CARs and gene editing techniques, allowing T cells to more effectively lyse tumor cells. CAR-T cells first showed promise in hematological tumors, then in a variety of other solid tumors [147]. CAR-T cells currently lack unique and specific targets. Several issues concerning the effective concentration and persistence of CAR-T cells in the target region remain unresolved [148, 149]. Current CSC CAR-T cell therapy experimental studies primarily involve in vitro coculture systems and preclinical studies; more clinical studies are needed in the future to demonstrate its efficacy alone or in combination with other tumor-targeted therapies.
CAR-T therapies have a distinct structure, the single-stranded variable fragment (scFv), which recognizes cell surface antigens directly and specifically without relying on MHC down-regulation [150]. The identification of CSC surface markers such as EpCAM, CD44, and CD133 has caused the identification of specific therapeutic targets for inhibiting tumor recurrence and metastasis [151]. Furthermore, CSC expressed molecular markers like epidermal growth factor receptor variant III (EGFRvIII), human epidermal growth factor receptor 2 (HER2) as well as chondroitin proteoglycan sulfate 4 (CSPG4) provide therapeutic targets for inhibiting tumor recurrence and metastasis [152,153,154]. CAR-T cell development targeting CSC molecular markers has so far demonstrated therapeutic efficacy. As shown in Table 2, CD133, EpCAM and ALDH have been adopted for CSC-directed immunotherapy, and the majority of them are recruited. Because the presence of CSCs in TME prevents autologous cells and T cells receiving CAR-T therapy from directly destroying tumor cells, a combination of CSCs-targeted CAR-T therapy and CSCs-targeted TME strategies may improve prognosis. Current research indicates that increased PD-L1 expression in CSCs promotes the occurrence and progression of TME [155]. The binding of PD-L1 to PD-1 on activated T cells can inhibit CAR-T cell function, resulting in CAR-T cell failure [156]. Therefore, CSCs targeted therapies combined with FDA-approved PD-1/PD-L1 checkpoint inhibitors [157] or dual CTLA-4 blockade provided significant anti-tumor effects and CSC eradication [157, 158]. Therefore, combining a-PD-L1 and a-CTLA-4 inhibitors with CAR-T cells that target CSCs may become an efficient immunotherapeutic strategy for treating cancer patients.
Targeting CSC-NK cells therapy
The targeting of NK cells to CSCs highlights the translational potential of NK immunotherapy as a treatment for solid malignancies [159, 160]. Moreover, CD34+ AML stem cells suppress NKG2DL expression via poly-ADP-ribose polymerase 1 (PARP1), implying that NKG2DL mediates immune evasion of NK cell depletion and that genetic or pharmacological inhibition of PARP1 inhibits NKG2DL expression in CD34+ AML stem cells. This causes NKG2DL re-expression on the surface of AML stem cells, making them re-sensitive to NK cells [161]. Melanoma CCR7+ CSCs have increased NKp30/NKp46 ligand expression while decreasing MHC-I expression, making them become vulnerable to NK cell-mediated cytotoxicity [162].
The combination of autologous NK cell enhancement and engineered CAR-NK cells can target CSCs with increased affinity. Activation of NK cells by cytokines induction into killer (CIK) cells can resensitize NK-resistant CSCs, but the cytokine dose must be adjusted to avoid the expansion of immunosuppressive Tregs [163]. CIKs with anti-tumor activity recognize NKG2D and kill CSCs [164], combining CIK-mediated tumor cell killing with artificially engineered CAR cells. CAR-CIKs can be created to target CSC antigens including CD44v6 and CSPG4 [165, 166]. These CAR-CIKs are effective at eliminating CSCs both in vitro and in vivo, but more clinical trials are required to assess the synergistic effect with other therapeutic strategies. The therapeutic effect of breast CSCs has been significantly improved by using CAR-NK cells to eliminate the EGFR in the mouse in vivo model [167]. The same cytokine IL-15 can induce CAR-NK cell expansion in vivo and has a high affinity for EpCAM+ CSC [168]. Understanding the underlying mechanisms of NK cell-mediated CSC recognition and clearance may thus lay the groundwork for a new generation of CSC-targeted immunotherapy.
Targeting CSC-DC vaccines therapy
Tumor vaccines can stimulate the human immune system, inhibiting tumor growth or eliminating tumor cells. Patients can be immunized by delivering tumor antigens through various established methods. DC-based vaccines are effective against CSCs in a variety of cancers. ALDH+ CSC-DC vaccines can directly target ALDH upregulated by CSCs, indicating the potential for adjuvant therapy in cancer patients [158, 169]. MUC1, a transmembrane glycoprotein, is involved in CSC stemness maintenance, and CSC vaccines targeting MUC1 have been developed, primarily by activating humoral immunity to inhibit CD133+ CSCs [170]. At the moment, a strategy of combining CSC-DC vaccine with chemotherapeutic drugs has been proposed to make targeting CSCs more effective and safe [171], and more clinical trials are needed to prove this.
CSCs targeted by Oncolytic Viro Therapy (OVT)
OVT has an anti-CSC effect by inducing tumor cell death and activating T cells. OVT has been shown to mediate IFN-γ release, angiogenesis inhibition and a decrease in the number of regulatory T cells in the tumor [172]. Oncolytic viruses that target specific CSC markers and signaling pathways can potentially be used as CSC therapeutics. Herpes simplex virus (HSV), adenovirus (Ads), measles virus (MV), retrovirus and vaccinia virus (VACV) have all been used in clinical trials to target CSCs. HSV has received much attention for its ability to kill tumor cells [173], with oncolytic HSV (oHSV), G207, being used in clinical advanced glioma trials, and CD133+ CSCs glioma cells being susceptible to tumor lysis by HSV [174]. oHSV modified by interleukin IL-12, on the other hand, will switch from a pro-tumor T helper (Th) -2 response to an anti-tumor Th-1 response [175]. Ads have the ability to infect both dividing and non-dividing tumor cells [176]. To evaluate the killing effect of conditional replication Ad (CRAd) on breast cancer, CD44+CD24− CSCs extracted from the pleural effusion of patients with metastatic breast cancer and injected into the fat pad of SCID mice decreased after tumor formation, which may contribute to the differentiation and proliferation of CSCs to form solid tumors. Five weeks after intratumoral injection, CRAd treatment demonstrated significant anti-tumor effects [177]. MVS form syncytial bodies in neighboring cells via viral protein binding and receptor protein fusion, so oncolytic MVS (oMVS) are used to induce syncytial formation in CSCs to ensure complete tumor eradication [178, 179]. After chemotherapy, the proportion of CD44+CD24– CSCs increased significantly, whereas oMV infection caused apoptosis of CD44+CD24– CSCs [180]. The oncolytic potential of VACV is realized via susceptibility and oncolytic action. In the breast cancer model, mice were injected into the left and right fat pads with tumor implants containing CD44+CD24– and CD44+CD24+ CSCs, respectively. After post-orbital delivery of VACV, the left and right breast tumors were generally suppressed, indicating that VACV could be used for systemic treatment of breast cancer [181]. At the moment, oncolytic viruses combined with standard chemotherapy have been shown to be feasible and effective in the treatment of CSCs [182]. Furthermore, the sensitivity and susceptibility of oncolytic viruses to host tumor cells remains a critical issue for oncolytic virus engineering.
Immune checkpoint-targeted immunotherapy for CSCs
Cancer stem cells and innate immune checkpoint
A leading conundrum is how it is probable that even a subset of patients can yield a spontaneous CD8+ T cell response against tumor-associated antigens, obviously in the lack of pathogen involvement. Moreover, this can narrow to a question of mechanisms of sterile immunity and indicate the likely participation of stress-associated or damage-associated molecular patterns triggering innate immune activation [183]. CD47 is a transmembrane protein belonging to the immunoglobulin superfamily [106, 184,185,186,187]. The binding of CD47 to SIRPα generates a "do not eat me" signal [188,189,190,191,192,193]. Increased CD47 expression in tumors to evade immune surveillance by macrophages has also been associated with poor clinical prognosis [194]. Blockade of CD47-SIRPα interaction in cancer induces the activity of the innate immune system and increases phagocytosis of CSCs by macrophages [195]. By extending the potential clinical application of CD47 blockade combined with CAR-T cells to a wider range of malignancies [195], these treatment modalities can reduce the survival of CSCs and thereby prevent tumor recurrence. Therefore, targeting CD47 have emerged as an effective therapeutic strategy for cancer.
Cancer stem cells and adaptive immune checkpoint
CSCs avoid immune attacks by reducing the expression of adaptive immune checkpoints, which can directly contribute to immune activation. PD-1-PD-L1 axis refers to one of the immune checkpoints that can enable tumor cells to evade immune attack from PD-1+ T cells [196]. Following interaction with PD-L1 and PD-L2, PD-1 inhibits T cells mediated immune responses and subsequently induces IL-10 production by the tumor [197,198,199,200,201,202]. PD-L1 expression has also been detected in CSCs [203,204,205,206,207]. Activation of PI3K/AKT and mTOR signaling pathways by PD-L1 is a key cellular process that maintains the pluripotency of CSCs and detects the differentiation fate of CSCs [208]. Activation of the EMT/STAT3 signaling axis induces PD-L1 expression on CSCs, enabling them to circumvent immune attacks [209]. Therefore, specific targeting of the PD-1-PD-L1 axis with monoclonal antibodies may serve as a potential therapeutic intervention for CSCs [127, 210].
Cytotoxic T lymphocyte-associated protein 4 (CTLA-4) is a member of the immunoglobulin superfamily and encodes a protein that inhibits the overactivation of T cells [211, 212]. Upregulation of CTLA-4 on Tregs plays an immunomodulatory role in suppressing overreactive T cells and protecting tissues from immune-mediated damage [213, 214]. CD28 on T cells interacts with CD80 and CD86 on the surface of APC and can offer a costimulatory signal for T cell activation [214]. Monoclonal antibody targeting CTLA-4 can trigger an anti-tumor immune response [215]. Combination therapy with specific antibodies to CTLA-4 and PD-1 may be an effective way to treat patients with tumors [216].
T cell immunoglobulin mucin receptor 3 (TIM-3) regulates immune responses mediated by various kinds of immune cells, like CD8+ T cells, Foxp3+ Treg cells and macrophages [217,218,219,220,221,222]. TIM-3 plays the role of an immune checkpoint on T cells driving immune tolerance, and thus the defective expression of this checkpoint contributes to the development of autoimmune diseases and tumors [223,224,225]. TIM-3 is overexpressed on AML LSCs [226, 227] and CSCs in many solid tumors [112, 228,229,230,231,232,233]. TIM-3+Foxp3+ Treg cells express IL-10 and upregulate CTLA-4 and PD-1 expression, and these cells display more tumor suppressive function than TIM-3–Foxp3+ Treg cells [234, 235]. IL-12 and IL-18 mediate the expression of TIM-3 on NK cells and inhibit the anti-tumor activity of NK cells [236,237,238]. There is evidence that TIM-3 expressed on T cells interacts with Gal-9 on CD11b+Ly6G+ MDSCs to induce the proliferation of MDSCs, creating an immunosuppressive environment to regulate immune responses [239]. Treatment of Th-1 cells by TIM-3 monoclonal antibodies induces immune responses against tumor cells by modulating the ERK signaling pathway in Th-1 cells [240].
Lymphocyte-activation protein 3 (LAG-3), an immunoglobin (Ig) superfamily protein, is denoted on NK cells, activated CD4+ and CD8+ T cells and Treg cells [241]. Galectin-3, a carbohydrate-binding protein, is highly expressed in breast, gastric, colorectal and ovarian cancers [242]. Interaction of LAG-3 with MHC class II molecules can inhibit the function of melanoma-infiltrating lymphocytes and enables tumors to escape recognition and lysis by immune cells [243, 244]. Interaction of Galectin-3 on tumor cells with LAG-3 on CD8+ T cells inhibits anti-tumor immune responses [245]. In glioblastoma multiforme (GBM), expression of galectin-3 on CSCs mediates immunosuppression by inducing T cell apoptosis [246]. CSCs mediated activation of LAG-3 and PD-1/PD-L1 signaling pathways synergistically hinders IFN-γ and TNF secretion from CD8+ T cells; therefore, combined blockade of LAG-3 and PD-1 is likely to activate T cells more potently in clinical settings [247, 248] (Fig. 2).

Immune checkpoint targeting CSCs. Administrated NK cells or CAR NK cells target TAAs on CSCs. Ex vivo maturation of DCs exposed to CSCs-lysate/TAAs/peptides produce a vaccine that after administration arm the cytotoxic T cells in an MHC-1-TCR-dependent manner for targeting specifc CSCs. Antibodies targeting immune checkpoint molecules such as PD1/PDL1, CD276, and CTLA4 could improve the anticancer immune responses. Anti-CD47 antibody sensitizes CSCs to cell-mediated phagocytosis. FASL, FAS ligand; mDC, mature DC; TRAIL, TNF-related apoptosis-inducing ligand
Targeting CSCs on the efficacy of immune checkpoint inhibitor therapy
Evaluating CSCs regulation with immune checkpoints and their relationship to tumor recurrence is an issue that needs to be addressed further. Alternative checkpoints, such as v-domain immunoglobulin inhibitory T-cell activation (VISTA) and indoleamine 2, 3-dioxygenase 1 (IDO1), inhibit the tumor-killing function of T cells in addition to PD-L1 and CTLA-4 [249]. The intrinsic mechanism of tumor resistance caused by alternative checkpoints and PD-1 treatment must be clarified [250]. Anti-PD-L1 antibodies have limited specificity, and PD-L1 heterogeneity is caused by differences in affinity or target epitopes [251]. On the other hand, patients with androgen receptor prostate cancer did not express PD-1, PD-L1 or CTLA-4, whereas the B7-H3 is highly expressed [252] and inhibits cytotoxic T cell activity [253]. Immune checkpoint heterogeneity will influence immune checkpoint inhibitor (ICI) therapy response and can be applied to be a tool to identify appropriate targeted checkpoints in different tumor types. In addition, based on a better understanding of CSC surface biomarkers, obvious progress has been made in the development of antibodies that target CSCs (Table 3).
In terms of anti-tumor and immunotherapy efficacy, CSCs represent a novel target for cancer treatment. Because CSCs can continue to develop into drug-resistant tumors even after conventional treatment. CSC therapy can therefore be combined with immune checkpoint inhibitor (ICI) therapy to produce a more potent antitumor effect. B-lymphoma Mo-MLV insertion region 1 (BMI1) is a critical component of polycomb reactive complex 1, which coordinates immune escape in CSCs [254]. The proportion of BMI1+ CSCs in HNSCC increased significantly after anti-PD-1 and cisplatin combination therapy, whereas BMI1 inhibition resulted in the elimination of these CSCs and a significant increase in CD8+ T cell infiltration. Depletion of BMI1+ CSCs may thereby be an efficient strategy for improving anti-PD-1 therapy efficacy and preventing tumor recurrence [255]. Depletion of BMI1+ CSCs may thus be an effective strategy for improving anti-PD-1 therapy efficacy and preventing tumor recurrence [255]. Metformin directly kills cancer stem cells [256] while improving anti-PD-1 therapy efficacy [257]. In addition, the two functions are linked.
TME complex components contribute to CSC dedifferentiation, causing them to intervene in tumor immunogenicity rather than tumor immunosuppression [258]. There exists a significant positive relationship between tumor immunogenicity and ICI therapy efficacy, but more research is needed in this area, particularly in different types of cancer. The concept of tumor heterogeneity leading to a low immune response to tumors is important in clinical evaluation [259]. Intertumor or intratumor heterogeneity is thought to be an impediment to tumor targeted therapy [260]. Tumor heterogeneity and cancer stem cell plasticity are linked, and it is thought to be an emerging marker related to cancer invasion [261]. Tumor heterogeneity is caused by the state of the complex tumor immune microenvironment [262], tumor mutation reflects immune characteristics [263] and represents tumor sensitivity to anti-PD-1 treatment [264].
CSCs can transition from epithelial to mesenchymal cells [265], which is due to their epithelial-mesenchymal plasticity (EMP) [266]. The phenotype of EMT is strongly associated with elevated levels of immune checkpoint expression (PD-1, PD-L1, CTLA-4 and TIM-3). Therefore, EMT characteristics have been proposed as predictors of response to ICI therapy [267]. Zinc finger E-box binding homeobox 1 (ZEB1) is a critical transcription factor in EMT that connects CSCs to EMT [268]. ZEB1 was also linked to increased PD-L1 expression and tumor killing by T cells [269]. PD-L1 on the surface of CSCs is downregulated after they transform to the MET phenotype, resulting in increased sensitivity to TIM-3 targeted therapy.
CSCs make a critical function in the promotion of angiogenesis in solid tumors. Anti-angiogenic inhibitors, like cabozantinib and regorafenib, are currently approved for the treatment of HCC after sorafenib failure [270, 271]. Ramucirumab, an anti-VEGF antibody, has also been approved for patients undergoing unresectable HCC who have failed sorafenib treatment [272, 273]. These anti-vascular therapies, when combined, may have anti-tumor effects by targeting CSCs. Furthermore, the relationship between CSCs and angiogenesis promotes tumor cell immune escape. Therefore, immunotherapy combined with a VEGF antagonist is a novel approach with clinical potential [274]. A phase III trial (IMbrave150) recently found that combining atezolizumab (a PD-L1 inhibitor) and bevacizumab (an anti-VEGF antibody) led to improved overall survival and progression-free survival in patients undergoing unresectable HCC (NCT03434379) [275]. Therefore, the FDA approved atezolizumab and bevacizumab as the most recent first-line systemic treatment for patients with unresectable HCC [275]. Furthermore, the REGONIVO trial (NCT03406871) showed that combining nivolumab (a PD-L1 inhibitor) and regorafenib (an anti-VEGFR antibody) resulted in responses in patients suffering from gastric and colorectal cancer [276]. Finally, the combination of immune checkpoint inhibitors and anti-angiogenic inhibitors may result in CSC depletion.
Secretome-targeted immunotherapy for CSCs
CSCs and their EVs are essential for the progression of cancer
Cell-to-cell communication occurs through different pathways, such as tunneling, microtubules reorganization and direct intercellular connections created by connexin channels; while extracellular vesicles (EVs) are increasingly recognized as an important mediator of intercellular communication [277]. EVs mediate intercellular transport of biomolecular cargo, such as non-coding nucleic acids, mRNA, proteins, metabolites and intact organelles [278]. EVs can influence the proliferation and energy metabolism of cancer cells as well as the components of the tumor microenvironment [279, 280]. EVs also result in the dedifferentiation of cancer cells into the CSC state.
Given the considerable heterogeneity of CSCs and EVs in various cancers, the impact of these cells and EVs secreted by these cells is also widespread, yet CSCs share some properties with cancer cells that help develop resistance to immunotherapy by evading immune surveillance [281]. Numerous cellular processes contribute to the maintenance of the specific functions of CSCs, including autophagy and EVs secretion [282], with autophagy contributing to the transport of cellular proteins as well as the secretion of EVs [282, 283]. Targeting EV secretion could become a possible therapeutic strategy for anti-tumor therapy [282, 284]. EV-mediated communication between non-CSCs and CSCs are essential for adaptation to the ecological niches [285, 286]. CSCs-derived EVs are engaged in tumor metastasis, resistance to therapy, angiogenesis, maintenance of stemness and immunosuppression [287, 288]. The fusion of CSC-derived EVs with macrophages and other immune cells mediates immunosuppression through the release of proteins and miRNAs [289,290,291,292,293,294].
The EVs: the role of CSCs and immune cells
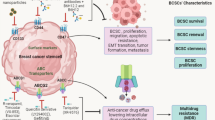
Cell-to-cell interaction in the TME contributes to carcinogenesis [295]. The interaction between CSCs and immune cells is mediated not only through immune targets, but also through EVs that enable the transfer of large biomolecular cargos among different types of cells [295, 296]. Exosomes, with an average diameter of -100 nm, are a subset of EVs. Interaction of exosomal tenascin C with integrins α5β1 and αvβ6 on T cells attenuates p-mTOR signaling [297, 298]. CSCs-derived exosomes can also suppress T cell function by inducing bone marrow-derived myeloid cells [299]. EVs released from CD105+ CSCs inhibit dendritic cell maturation and T cell-mediated immune responses [99]. Furthermore, the transfer of CSCs-derived exosomes into monocytes triggers monocyte agonist protein reorganization, induces monocyte differentiation into immunosuppressive M2 macrophages, and increases PD-L1 expression on CSCs via the STAT3 signaling pathway [300]. Glioblastoma is infiltrated with numerous microglia, and cross-talk between glioblastoma and microglia induces immunosuppressive TME in tumor mass [301]. Following coculture with microglia cells, glioma CSCs release exosomes carrying lncRNA MALAT1, which induces secretion of IL-6 and TNF-α from LPS-stimulated microglia cells. Colorectal CSCs derived exosomes activate NF-κB signaling cascade in neutrophils inducing IL-1β expression [302, 303] (Fig. 3).

Crosstalk between CSCs and TME cells. CSCs secrete exosome regulated infltrating immune cells (IICs), MDSC, DC, macrophage and neutrophils to the TME. Cell–cell interactions in TME contribute to the development of cancer. The mechanism of interaction between CSCs and tumor-infiltrating immune cells is not only through immune targets, but can likewise be through exosomes that enable a large exchange
EVs-based therapeutic strategies for targeting CSCs
Tumor therapy mediated by exosomes targeting CSCs has been revealed to be extremely efficient in the clinical trials. More precise targeted therapy can be achieved by improving existing exosome engineering technology to target the unique markers of CSCs. CD44 is highly expressed in metastatic HCC CSCs, and anti-CD44 antibody-coated liposomes can directly deliver doxorubicin to CSCs [304]. The anti-CD44 antibody can cause apoptosis in CD90+ HCC CSCs. Similarly, anti-CD44 antibody-coated exosomes can cause CSCs to die [305]. In addition, anti-CD44 antibody-coated exosomes can also be used for drug delivery. As a result, other CSC markers including EpCAM, CD133 and CD24 can be applied as targeting candidates to enhance the efficacy of engineered exosomes targeting CSCs. Because CSCs surface markers may be denoted on normal cells, antibody-coated exosomes must be engineered to enhance their targeting efficiency to cancer stem cells and thus reduce side effects on normal cells.
Compared to synthetic nanoparticles, nanotechnology-based drug delivery systems are more biocompatible, biodegradable, less toxic and immunogenic [306,307,308,309]. Thus, exosome-based nanocarrier drug delivery technologies with advanced targeting capabilities have been developed, and they show great promise in targeting CSCs [310,311,312]. The development of exosome-nanoparticle technology based on EVs as a drug delivery vehicle targeting CSCs will aid in improving the anti-tumor immune response [313, 314]. A recent study found that biocompatible tumor cell-exocytosed exosomes encapsulated doxorubicin-loaded mimetic porous silica nanoparticles (PSiNPs) have the potential to be enriched inside CSCs, resulting in CSC eradication [315]. Finally, exosome engineering approaches are likely to improve the efficacy of CSCs targeted therapies.
Conclusions
Cancer immunotherapy is adopted to either suppress tumor growth or remove tumor cells through activating the immune system; consequently, cancer immunotherapy shows great potential in treating malignant diseases from different cancer types. CSCs can suppress the immune response by recruiting immunosuppressive cells (TAM and Tregs); thus, promoting the establishment of an immunosuppressive TME. CSCs can also impair NK cell function by expressing specific ligands. In this outlook, we demonstrate the mechanism by which CSCs communicate with immune cells in the tumor microenvironment in a variety of cancer types. Therefore, there is a need to find new strategies to target CSCs through immunotherapeutic approaches.
CSCs evade immune surveillance through various immune checkpoints, which are expressed at higher levels in CSCs. CSCs express CD47, CTLA4, PD-L1, TIM-3 and LAG3, which promote immune evasion in the malignant environment and maintain tumor survival. In addition, CSCs orchestrate the tumor microenvironment by releasing immunosuppressive cytokines and growth factors. CSCs can also modulate the immune microenvironment of tumors through the excretion of EVs; thus, further understanding of the molecular mechanism driving anti-tumor immune response is a prerequisite to develop new anti-tumor therapies with higher efficacy.
To achieve desired therapeutic goals, CSC protocols need to be optimized in immunocompetent preclinical models, and the contact of CSCs with the immune system is needed to be studied using models that rigorously validate functional and phenotypic characteristics of CSCs. Traditional two-dimensional coculture experiments have been performed to clarify the mechanisms promoting CSC characteristics in the TME; unfortunately, two-dimensional models do not allow for the observation of dynamic cellular interactions in real time. Three-dimensional coculture systems enable us to better visualize the complex interactions between CSCs and immune effectors. The current emerging 3D cell coculture models are represented by organoids that closely resemble tumor microenvironments, including ecological niches that nurture host CSCs. With the rapid development of single-cell spatial analysis, it would be possible to visualize the complex interactions involving different types of immune cells and CSCs.
Availability of data and materials
Not applicable.
Abbreviations
- CSCs:
-
Cancer stem cells
- TME:
-
Tumor microenvironment
- EVs:
-
Extracellular vesicles
- TIME:
-
Tumor immune microenvironment
- AML:
-
Acute myeloid leukemia
- PD-L1:
-
Programmed death-1/programmed cell death ligand
- CD47:
-
Cluster of differentiation 47
- TIM3:
-
T cell immunoglobulin and mucin-containing domain-3
- LAG3:
-
Lymphocyte activation gene 3
- CTLA4:
-
Cytotoxic T-lymphocyte antigen-4
- ILs:
-
Interleukins
- MMPs:
-
Matrix metalloproteinases
- VEGF:
-
Vascular endothelial growth factor
- TGF-β1:
-
Transforming growth factor beta 1
- Hh:
-
Hedgehog
- NF-κB:
-
Nuclear factor kappa B
- YAP:
-
Yes-associated protein
- HCC:
-
Hepatocellular carcinoma
- LIF:
-
Leukemia inhibitory factor
- H3K27 me3:
-
Lysine 27 of histone H3
- IHH:
-
Indian hedgehog
- SHH:
-
Sonic hedgehog
- TAZ:
-
Transcriptional coactivator with PDZ-binding
- ERK1/2:
-
Extracellular signal-regulated kinase 1/2
- ECM:
-
Extracellular matrix
- αVβ3:
-
Alphavbeta3
- CAF:
-
Cancer-associated fibroblasts
- HGF:
-
Hepatocyte growth factor
- DLL4:
-
Notch ligand Delta-like ligand 4
- DCs:
-
Dendritic cells
- APCs:
-
Antigen-presenting cells
- TAAs:
-
Tumor-associated antigens
- MHC:
-
Major histocompatibility complex
- HLA-G:
-
Human leukocyte antigen G
- STAT3:
-
Signal transducer and activator of transcription 3
- CXCL:
-
C-X-C motif chemokine ligand
- DCregs:
-
Regulatory dendritic cells
- CXCR:
-
C-X-C motif chemokine receptor
- TAMs:
-
Tumor-associated macrophages
- EMT:
-
Epithelial-mesenchymal transition
- MDSCs:
-
Myeloid-derived suppressor cells
- mTOR:
-
Mammalian target of rapamycin
- Gal-9:
-
Galectin 9
- LSCs:
-
Leukemic stem cells
- NF-κB:
-
Noncanonical nuclear factor-kappaB
- PEG-E2:
-
Prostaglandin E2
- COX-2:
-
Cyclooxygenase 2
- HNCCs:
-
Human neural crest cells
- NLRC5:
-
NLR Family CARD Domain Containing 5
- NKG2D:
-
Natural killer group 2 member D
- KIR2DL4:
-
Killer cell immunoglobulin-like receptor 2DL4
- NKG2A:
-
Natural killer group 2 member A
- HA:
-
Hyaluronic acid
- HNSCC:
-
Human neck squamous carcinoma
- TNC:
-
Tenascin-C
- BMDM:
-
Bone marrow-derived macrophage
- CAR:
-
Chimeric antigen receptor
- scFv:
-
Single-stranded variable fragment
- EGFRvIII:
-
Epidermal growth factor receptor variant III
- HER2:
-
Human epidermal growth factor receptor 2
- CSPG4:
-
Chondroitin proteoglycan sulfate 4
- PARP1:
-
Poly-ADP-ribose polymerase 1
- CIK:
-
Cytokines induction into killer
- OVT:
-
Oncolytic Viro Therapy
- HSV:
-
Herpes simplex virus
- Ads:
-
Adenovirus
- MV:
-
Measles virus
- VACV:
-
Vaccinia virus
- oHSV:
-
Oncolytic HSV
- Th:
-
T helper
- CRAd:
-
Conditional replication Ad
- oMVS:
-
Oncolytic MVS
- TCR:
-
T cell receptor
- Ig:
-
Immunoglobin
- GBM:
-
Glioblastoma multiforme
- VISTA:
-
V-domain immunoglobulin inhibitory T-cell activation
- IDO1:
-
Indoleamine 2, 3-dioxygenase 1
- ICI:
-
Immune checkpoint inhibitor
- BMI1:
-
B-lymphoma Mo-MLV insertion region 1
- EMP:
-
Epithelial-mesenchymal plasticity
- ZEB1:
-
Zinc finger E-box binding homeobox 1
- PSiNPs:
-
Porous silica nanoparticles
References
Albini A, Bruno A, Gallo C, Pajardi G, Noonan DM, Dallaglio K. Cancer stem cells and the tumor microenvironment: interplay in tumor heterogeneity. Connect Tissue Res. 2015;56(5):414–25.
Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. 2017;23(10):1124–34.
Huang T, Song X, Xu D, Tiek D, Goenka A, Wu B, Sastry N, Hu B, Cheng SY. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics. 2020;10(19):8721–43.
Li JJ, Shen MM. Prostate Stem Cells and Cancer Stem Cells. Cold Spring Harb Perspect Med. 2019;9(6):a030395.
Munro MJ, Wickremesekera SK, Peng L, Tan ST, Itinteang T. Cancer stem cells in colorectal cancer: a review. J Clin Pathol. 2018;71(2):110–6.
Nio K, Yamashita T, Kaneko S. The evolving concept of liver cancer stem cells. Mol Cancer. 2017;16(1):4.
Shi M, Liu ZW, Wang FS. Immunomodulatory properties and therapeutic application of mesenchymal stem cells. Clin Exp Immunol. 2011;164(1):1–8.
Zhang D, Tang DG, Rycaj K. Cancer stem cells: Regulation programs, immunological properties and immunotherapy. Semin Cancer Biol. 2018;52(Pt 2):94–106.
Maccalli C, Rasul KI, Elawad M, Ferrone S. The role of cancer stem cells in the modulation of anti-tumor immune responses. Semin Cancer Biol. 2018;53:189–200.
Schwarzenbacher D, Balic M, Pichler M. The role of microRNAs in breast cancer stem cells. Int J Mol Sci. 2013;14(7):14712–23.
Capece D, Verzella D, Tessitore A, Alesse E, Capalbo C, Zazzeroni F. Cancer secretome and inflammation: The bright and the dark sides of NF-κB. Semin Cell Dev Biol. 2018;78:51–61.
Boccellato F, Woelffling S, Imai-Matsushima A, Sanchez G, Goosmann C, Schmid M, Berger H, Morey P, Denecke C, Ordemann J, et al. Polarised epithelial monolayers of the gastric mucosa reveal insights into mucosal homeostasis and defence against infection. Gut. 2019;68(3):400–13.
Abels ER, Breakefield XO. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell Mol Neurobiol. 2016;36(3):301–12.
Ekström EJ, Bergenfelz C, von Bülow V, Serifler F, Carlemalm E, Jönsson G, Andersson T, Leandersson K. WNT5A induces release of exosomes containing pro-angiogenic and immunosuppressive factors from malignant melanoma cells. Mol Cancer. 2014;26(13):88.
Skog J, Würdinger T, van Rijn S, Meijer DH, Gainche L, Sena-Esteves M, Curry WT Jr, Carter BS, Krichevsky AM, Breakefield XO. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol. 2008;10(12):1470–6.
Taraboletti G, D’Ascenzo S, Giusti I, Marchetti D, Borsotti P, Millimaggi D, Giavazzi R, Pavan A, Dolo V. Bioavailability of VEGF in tumor-shed vesicles depends on vesicle burst induced by acidic pH. Neoplasia. 2006;8(2):96–103.
Chen X, Liang H, Zhang J, Zen K, Zhang CY. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012;22(3):125–32.
Webber J, Steadman R, Mason MD, Tabi Z, Clayton A. Cancer exosomes trigger fibroblast to myofibroblast differentiation. Cancer Res. 2010;70(23):9621–30.
Gu J, Qian H, Shen L, Zhang X, Zhu W, Huang L, Yan Y, Mao F, Zhao C, Shi Y, et al. Gastric cancer exosomes trigger differentiation of umbilical cord derived mesenchymal stem cells to carcinoma-associated fibroblasts through TGF-β/Smad pathway. PLoS ONE. 2012;7(12):e52465.
Aga M, Bentz GL, Raffa S, Torrisi MR, Kondo S, Wakisaka N, Yoshizaki T, Pagano JS, Shackelford J. Exosomal HIF1α supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene. 2014;33(37):4613–22.
Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–8.
Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr Opin Biotechnol. 2007;18(5):460–6.
O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16(12):3113–20.
Najafi M, Mortezaee K, Ahadi R. Cancer stem cell (a)symmetry & plasticity: Tumorigenesis and therapy relevance. Life Sci. 2019;15(231):116520.
Park DJ, Sung PS, Kim JH, Lee GW, Jang JW, Jung ES, Bae SH, Choi JY, Yoon SK. EpCAM-high liver cancer stem cells resist natural killer cell-mediated cytotoxicity by upregulating CEACAM1. J Immunother Cancer. 2020;8(1):e000301.
Chikamatsu K, Takahashi G, Sakakura K, Ferrone S, Masuyama K. Immunoregulatory properties of CD44+ cancer stem-like cells in squamous cell carcinoma of the head and neck. Head Neck. 2011;33(2):208–15.
Zhang C, Wang H, Wang X, Zhao C, Wang H. CD44, a marker of cancer stem cells, is positively correlated with PD-L1 expression and immune cells infiltration in lung adenocarcinoma. Cancer Cell Int. 2020;20(1):583.
Hou YC, Chao YJ, Hsieh MH, Tung HL, Wang HC, Shan YS. Low CD8+ T Cell Infiltration and High PD-L1 Expression Are Associated with Level of CD44+/CD133+ Cancer Stem Cells and Predict an Unfavorable Prognosis in Pancreatic Cancer. Cancers (Basel). 2019;11(4):541.
Kursunel MA, Taskiran EZ, Tavukcuoglu E, Yanik H, Demirag F, Karaosmanoglu B, Ozbay FG, Uner A, Esendagli D, Kizilgoz D, et al. Small cell lung cancer stem cells display mesenchymal properties and exploit immune checkpoint pathways in activated cytotoxic T lymphocytes. Cancer Immunol Immunother. 2022;71(2):445–59.
Shi J, Lu P, Shen W, He R, Yang MW, Fang Y, Sun YW, Niu N, Xue J. CD90 highly expressed population harbors a stemness signature and creates an immunosuppressive niche in pancreatic cancer. Cancer Lett. 2019;1(453):158–69.
Li X, Bu W, Meng L, Liu X, Wang S, Jiang L, Ren M, Fan Y, Sun H. CXCL12/CXCR4 pathway orchestrates CSC-like properties by CAF recruited tumor associated macrophage in OSCC. Exp Cell Res. 2019;378(2):131–8.
El-Ashmawy NE, Salem ML, Abd El-Fattah EE, Khedr EG. Targeting CD166+ lung cancer stem cells: Molecular study using murine dendritic cell vaccine. Toxicol Appl Pharmacol. 2021;15(429):115699.
Miller TJ, McCoy MJ, Hemmings C, Bulsara MK, Iacopetta B, Platell CF. The prognostic value of cancer stem-like cell markers SOX2 and CD133 in stage III colon cancer is modified by expression of the immune-related markers FoxP3, PD-L1 and CD3. Pathology. 2017;49(7):721–30.
Noh KH, Kim BW, Song KH, Cho H, Lee YH, Kim JH, Chung JY, Kim JH, Hewitt SM, Seong SY, et al. Nanog signaling in cancer promotes stem-like phenotype and immune evasion. J Clin Invest. 2012;122(11):4077–93.
Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. 2017;50(3):117–25.
Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–67.
Fearon ER. Molecular genetics of colorectal cancer. Annu Rev Pathol. 2011;6:479–507.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Lessard J, Sauvageau G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature. 2003;423(6937):255–60.
Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015;347(6217):78–81.
Wend P, Holland JD, Ziebold U, Birchmeier W. Wnt signaling in stem and cancer stem cells. Semin Cell Dev Biol. 2010;21(8):855–63.
Shang Z, Zhao J, Zhang Q, Cao C, Tian S, Zhang K, Liu L, Shi L, Yu N, Yang S. USP9X-mediated deubiquitination of B-cell CLL/lymphoma 9 potentiates Wnt signaling and promotes breast carcinogenesis. J Biol Chem. 2019;294(25):9844–57.
Chen Y, Fang R, Yue C, Chang G, Li P, Guo Q, Wang J, Zhou A, Zhang S, Fuller GN, et al. Wnt-Induced Stabilization of KDM4C Is Required for Wnt/β-Catenin Target Gene Expression and Glioblastoma Tumorigenesis. Cancer Res. 2020;80(5):1049–63.
Regel I, Eichenmüller M, Mahajan UM, Hagl B, Benitz S, Häberle B, Vokuhl C, von Schweinitz D, Kappler R. Downregulation of SFRP1 is a protumorigenic event in hepatoblastoma and correlates with beta-catenin mutations. J Cancer Res Clin Oncol. 2020;146(5):1153–67.
Chung J, Karkhanis V, Baiocchi RA, Sif S. Protein arginine methyltransferase 5 (PRMT5) promotes survival of lymphoma cells via activation of WNT/β-catenin and AKT/GSK3β proliferative signaling. J Biol Chem. 2019;294(19):7692–710.
Zhang Y, Wang X. Targeting the Wnt/β-catenin signaling pathway in cancer. J Hematol Oncol. 2020;13(1):165.
Chen JF, Luo X, Xiang LS, Li HT, Zha L, Li N, He JM, Xie GF, Xie X, Liang HJ. EZH2 promotes colorectal cancer stem-like cell expansion by activating p21cip1-Wnt/β-catenin signaling. Oncotarget. 2016;7(27):41540–58.
Yang L, Shi P, Zhao G, Xu J, Peng W, Zhang J, Zhang G, Wang X, Dong Z, Chen F, et al. Targeting cancer stem cell pathways for cancer therapy. Signal Transduct Target Ther. 2020;5(1):8.
Andersson ER, Lendahl U. Therapeutic modulation of Notch signalling–are we there yet? Nat Rev Drug Discov. 2014;13(5):357–78.
Muñoz P, Iliou MS, Esteller M. Epigenetic alterations involved in cancer stem cell reprogramming. Mol Oncol. 2012;6(6):620–36.
Bhagat TD, Zou Y, Huang S, Park J, Palmer MB, Hu C, Li W, Shenoy N, Giricz O, Choudhary G, et al. Notch Pathway Is Activated via Genetic and Epigenetic Alterations and Is a Therapeutic Target in Clear Cell Renal Cancer. J Biol Chem. 2017;292(3):837–46.
Lu B, He Y, He J, Wang L, Liu Z, Yang J, Gao Z, Lu G, Zou C, Zhao W. Epigenetic Profiling Identifies LIF as a Super-enhancer-Controlled Regulator of Stem Cell-like Properties in Osteosarcoma. Mol Cancer Res. 2020;18(1):57–67.
Kohnken R, Wen J, Mundy-Bosse B, McConnell K, Keiter A, Grinshpun L, Hartlage A, Yano M, McNeil B, Chakravarti N, et al. Diminished microRNA-29b level is associated with BRD4-mediated activation of oncogenes in cutaneous T-cell lymphoma. Blood. 2018;131(7):771–81.
Lima-Fernandes E, Murison A, da Silva MT, Wang Y, Ma A, Leung C, Luciani GM, Haynes J, Pollett A, Zeller C, et al. Targeting bivalency de-represses Indian Hedgehog and inhibits self-renewal of colorectal cancer-initiating cells. Nat Commun. 2019;10(1):1436.
Samadani AA, Nikbakhsh N, Taheri H, Shafaee S, Fattahi S, Pilehchian Langroudi M, Hajian K, Akhavan-Niaki H. CDX1/2 and KLF5 Expression and Epigenetic Modulation of Sonic Hedgehog Signaling in Gastric Adenocarcinoma. Pathol Oncol Res. 2019;25(3):1215–22.
Pak E, Segal RA. Hedgehog Signal Transduction: Key Players, Oncogenic Drivers, and Cancer Therapy. Dev Cell. 2016;38(4):333–44.
Kim S, Kim Y, Kong J, Kim E, Choi JH, Yuk HD, Lee H, Kim HR, Lee KH, Kang M, et al. Epigenetic regulation of mammalian Hedgehog signaling to the stroma determines the molecular subtype of bladder cancer. Elife. 2019;30(8):e43024.
Lamar JM, Xiao Y, Norton E, Jiang ZG, Gerhard GM, Kooner S, Warren JSA, Hynes RO. SRC tyrosine kinase activates the YAP/TAZ axis and thereby drives tumor growth and metastasis. J Biol Chem. 2019;294(7):2302–17.
Chang C, Goel HL, Gao H, Pursell B, Shultz LD, Greiner DL, Ingerpuu S, Patarroyo M, Cao S, Lim E, et al. A laminin 511 matrix is regulated by TAZ and functions as the ligand for the α6Bβ1 integrin to sustain breast cancer stem cells. Genes Dev. 2015;29(1):1–6.
Sougawa N, Miyagawa S, Fukushima S, Yokoyama J, Kitahara M, Harada A, Mochizuki-Oda N, Sato-Nishiuchi R, Sekiguchi K, Sawa Y. Laminin-511 Supplementation Enhances Stem Cell Localization With Suppression in the Decline of Cardiac Function in Acute Infarct Rats. Transplantation. 2019;103(5):e119–27.
Goel HL, Gritsko T, Pursell B, Chang C, Shultz LD, Greiner DL, Norum JH, Toftgard R, Shaw LM, Mercurio AM. Regulated splicing of the α6 integrin cytoplasmic domain determines the fate of breast cancer stem cells. Cell Rep. 2014;7(3):747–61.
Varzavand A, Hacker W, Ma D, Gibson-Corley K, Hawayek M, Tayh OJ, Brown JA, Henry MD, Stipp CS. α3β1 Integrin Suppresses Prostate Cancer Metastasis via Regulation of the Hippo Pathway. Cancer Res. 2016;76(22):6577–87.
Nakada M, Nambu E, Furuyama N, Yoshida Y, Takino T, Hayashi Y, Sato H, Sai Y, Tsuji T, Miyamoto KI, et al. Integrin α3 is overexpressed in glioma stem-like cells and promotes invasion. Br J Cancer. 2013;108(12):2516–24.
Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015;25(4):234–40.
Cooper J, Giancotti FG. Integrin Signaling in Cancer: Mechanotransduction, Stemness, Epithelial Plasticity, and Therapeutic Resistance. Cancer Cell. 2019;35(3):347–67.
Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10(1):9–22.
Xiao W, Ma W, Wei S, Li Q, Liu R, Carney RP, Yang K, Lee J, Nyugen A, Yoneda KY, et al. High-affinity peptide ligand LXY30 for targeting α3β1 integrin in non-small cell lung cancer. J Hematol Oncol. 2019;12(1):56.
Wang Y, Xiao W, Zhang Y, Meza L, Tseng H, Takada Y, Ames JB, Lam KS. Optimization of RGD-Containing Cyclic Peptides against αvβ3 Integrin. Mol Cancer Ther. 2016;15(2):232–40.
Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H, et al. An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat Cell Biol. 2014;16(5):457–68.
Lathia JD, Gallagher J, Heddleston JM, Wang J, Eyler CE, Macswords J, Wu Q, Vasanji A, McLendon RE, Hjelmeland AB, et al. Integrin alpha 6 regulates glioblastoma stem cells. Cell Stem Cell. 2010;6(5):421–32.
Krebsbach PH, Villa-Diaz LG. The Role of Integrin α6 (CD49f) in Stem Cells: More than a Conserved Biomarker. Stem Cells Dev. 2017;26(15):1090–9.
Yu KR, Yang SR, Jung JW, Kim H, Ko K, Han DW, Park SB, Choi SW, Kang SK, Schöler H, et al. CD49f enhances multipotency and maintains stemness through the direct regulation of OCT4 and SOX2. Stem Cells. 2012;30(5):876–87.
Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012;21(3):283–96.
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45.
Korkaya H, Kim GI, Davis A, Malik F, Henry NL, Ithimakin S, Quraishi AA, Tawakkol N, D’Angelo R, Paulson AK, et al. Activation of an IL6 inflammatory loop mediates trastuzumab resistance in HER2+ breast cancer by expanding the cancer stem cell population. Mol Cell. 2012;47(4):570–84.
Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468–76.
Hoey T, Yen WC, Axelrod F, Basi J, Donigian L, Dylla S, Fitch-Bruhns M, Lazetic S, Park IK, Sato A, et al. DLL4 blockade inhibits tumor growth and reduces tumor-initiating cell frequency. Cell Stem Cell. 2009;5(2):168–77.
Fessler E, Borovski T, Medema JP. Endothelial cells induce cancer stem cell features in differentiated glioblastoma cells via bFGF. Mol Cancer. 2015;19(14):157.
Borovski T, Verhoeff JJ, ten Cate R, Cameron K, de Vries NA, van Tellingen O, Richel DJ, van Furth WR, Medema JP, Sprick MR. Tumor microvasculature supports proliferation and expansion of glioma-propagating cells. Int J Cancer. 2009;125(5):1222–30.
Wang R, Chadalavada K, Wilshire J, Kowalik U, Hovinga KE, Geber A, Fligelman B, Leversha M, Brennan C, Tabar V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature. 2010;468(7325):829–33.
Ricci-Vitiani L, Pallini R, Biffoni M, Todaro M, Invernici G, Cenci T, Maira G, Parati EA, Stassi G, Larocca LM, et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature. 2010;468(7325):824–8.
Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A, Sotiropoulou PA, Loges S, Lapouge G, Candi A, et al. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature. 2011;478(7369):399–403.
Müller L, Tunger A, Plesca I, Wehner R, Temme A, Westphal D, Meier F, Bachmann M, Schmitz M. Bidirectional Crosstalk Between Cancer Stem Cells and Immune Cell Subsets. Front Immunol. 2020;5(11):140.
Clara JA, Monge C, Yang Y, Takebe N. Targeting signalling pathways and the immune microenvironment of cancer stem cells - a clinical update. Nat Rev Clin Oncol. 2020;17(4):204–32.
Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, et al. Periostin secreted by glioblastoma stem cells recruits M2 tumour-associated macrophages and promotes malignant growth. Nat Cell Biol. 2015;17(2):170–82.
Jinushi M. Role of cancer stem cell-associated inflammation in creating pro-inflammatory tumorigenic microenvironments. Oncoimmunology. 2014;15(3):e28862.
Zhang Q, Cai DJ, Li B. Ovarian cancer stem-like cells elicit the polarization of M2 macrophages. Mol Med Rep. 2015;11(6):4685–93.
Liu L, Zhang L, Yang L, Li H, Li R, Yu J, Yang L, Wei F, Yan C, Sun Q, et al. Anti-CD47 Antibody As a Targeted Therapeutic Agent for Human Lung Cancer and Cancer Stem Cells. Front Immunol. 2017;21(8):404.
Theocharides AP, Jin L, Cheng PY, Prasolava TK, Malko AV, Ho JM, Poeppl AG, van Rooijen N, Minden MD, Danska JS, et al. Disruption of SIRPα signaling in macrophages eliminates human acute myeloid leukemia stem cells in xenografts. J Exp Med. 2012;209(10):1883–99.
Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Priebe W, et al. Glioblastoma cancer-initiating cells inhibit T-cell proliferation and effector responses by the signal transducers and activators of transcription 3 pathway. Mol Cancer Ther. 2010;9(1):67–78.
Schatton T, Schütte U, Frank NY, Zhan Q, Hoerning A, Robles SC, Zhou J, Hodi FS, Spagnoli GC, Murphy GF, et al. Modulation of T-cell activation by malignant melanoma initiating cells. Cancer Res. 2010;70(2):697–708.
Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. 2015;16(3):225–38.
Sistigu A, Musella M, Galassi C, Vitale I, De Maria R. Tuning Cancer Fate: Tumor Microenvironment’s Role in Cancer Stem Cell Quiescence and Reawakening. Front Immunol. 2020;21(11):2166.
Hinshaw DC, Shevde LA. The Tumor Microenvironment Innately Modulates Cancer Progression. Cancer Res. 2019;79(18):4557–66.
Zhong M, Zhong C, Cui W, Wang G, Zheng G, Li L, Zhang J, Ren R, Gao H, Wang T, et al. Induction of tolerogenic dendritic cells by activated TGF-β/Akt/Smad2 signaling in RIG-I-deficient stemness-high human liver cancer cells. BMC Cancer. 2019;19(1):439.
Lu JM, Jiang XL, Liu JL, Wang HF, Li XL, Song XJ. Murine corneal stroma cells suppress bone marrow-derived dendritic cells maturation in vitro. Chin Med J (Engl). 2012;125(11):2041–7.
Spranger S, Dai D, Horton B, Gajewski TF. Tumor-Residing Batf3 Dendritic Cells Are Required for Effector T Cell Trafficking and Adoptive T Cell Therapy. Cancer Cell. 2017;31(5):711-723.e4.
Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, Goode EL, Kalli KR, Knutson KL. Tumor-infiltrating programmed death receptor-1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol. 2011;186(12):6905–13.
Grange C, Tapparo M, Tritta S, Deregibus MC, Battaglia A, Gontero P, Frea B, Camussi G. Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC Cancer. 2015;24(15):1009.
Liang S, Ristich V, Arase H, Dausset J, Carosella ED, Horuzsko A. Modulation of dendritic cell differentiation by HLA-G and ILT4 requires the IL-6–STAT3 signaling pathway. Proc Natl Acad Sci U S A. 2008;105(24):8357–62.
Hira VV, Ploegmakers KJ, Grevers F, Verbovšek U, Silvestre-Roig C, Aronica E, Tigchelaar W, Turnšek TL, Molenaar RJ, Van Noorden CJ. CD133+ and Nestin+ Glioma Stem-Like Cells Reside Around CD31+ Arterioles in Niches that Express SDF-1α, CXCR4, Osteopontin and Cathepsin K. J Histochem Cytochem. 2015;63(7):481–93.
Hsu YL, Chen YJ, Chang WA, Jian SF, Fan HL, Wang JY, Kuo PL. Interaction between Tumor-Associated Dendritic Cells and Colon Cancer Cells Contributes to Tumor Progression via CXCL1. Int J Mol Sci. 2018;19(8):2427.
Chakrabarti R, Celià-Terrassa T, Kumar S, Hang X, Wei Y, Choudhury A, Hwang J, Peng J, Nixon B, Grady JJ, et al. Notch ligand Dll1 mediates cross-talk between mammary stem cells and the macrophageal niche. Science. 2018;360(6396):eaan4153.
Jinushi M, Chiba S, Yoshiyama H, Masutomi K, Kinoshita I, Dosaka-Akita H, Yagita H, Takaoka A, Tahara H. Tumor-associated macrophages regulate tumorigenicity and anticancer drug responses of cancer stem/initiating cells. Proc Natl Acad Sci U S A. 2011;108(30):12425–30.
Fan QM, Jing YY, Yu GF, Kou XR, Ye F, Gao L, Li R, Zhao QD, Yang Y, Lu ZH, et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352(2):160–8.
Cioffi M, Trabulo S, Hidalgo M, Costello E, Greenhalf W, Erkan M, Kleeff J, Sainz B Jr, Heeschen C. Inhibition of CD47 Effectively Targets Pancreatic Cancer Stem Cells via Dual Mechanisms. Clin Cancer Res. 2015;21(10):2325–37.
Lee TK, Cheung VC, Lu P, Lau EY, Ma S, Tang KH, Tong M, Lo J, Ng IO. Blockade of CD47-mediated cathepsin S/protease-activated receptor 2 signaling provides a therapeutic target for hepatocellular carcinoma. Hepatology. 2014;60(1):179–91.
Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD Jr, van Rooijen N, Weissman IL. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–99.
Lei MML, Lee TKW. Cancer Stem Cells: Emerging Key Players in Immune Evasion of Cancers. Front Cell Dev Biol. 2021;21(9):692940.
Welte T, Kim IS, Tian L, Gao X, Wang H, Li J, Holdman XB, Herschkowitz JI, Pond A, Xie G, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18(6):632–44.
Shidal C, Singh NP, Nagarkatti P, Nagarkatti M. MicroRNA-92 Expression in CD133+ Melanoma Stem Cells Regulates Immunosuppression in the Tumor Microenvironment via Integrin-Dependent Activation of TGFβ. Cancer Res. 2019;79(14):3622–35.
Gao L, Yu S, Zhang X. Hypothesis: Tim-3/galectin-9, a new pathway for leukemia stem cells survival by promoting expansion of myeloid-derived suppressor cells and differentiating into tumor-associated macrophages. Cell Biochem Biophys. 2014;70(1):273–7.
Ai L, Mu S, Sun C, Fan F, Yan H, Qin Y, Cui G, Wang Y, Guo T, Mei H, et al. Myeloid-derived suppressor cells endow stem-like qualities to multiple myeloma cells by inducing piRNA-823 expression and DNMT3B activation. Mol Cancer. 2019;18(1):88.
Wang Y, Yin K, Tian J, Xia X, Ma J, Tang X, Xu H, Wang S. Granulocytic Myeloid-Derived Suppressor Cells Promote the Stemness of Colorectal Cancer Cells through Exosomal S100A9. Adv Sci (Weinh). 2019;6(18):1901278.
Kuroda H, Mabuchi S, Yokoi E, Komura N, Kozasa K, Matsumoto Y, Kawano M, Takahashi R, Sasano T, Shimura K, et al. Prostaglandin E2 produced by myeloid-derived suppressive cells induces cancer stem cells in uterine cervical cancer. Oncotarget. 2018;9(91):36317–30.
Wei J, Barr J, Kong LY, Wang Y, Wu A, Sharma AK, Gumin J, Henry V, Colman H, Sawaya R, et al. Glioma-associated cancer-initiating cells induce immunosuppression. Clin Cancer Res. 2010;16(2):461–73.
Xu Y, Dong X, Qi P, Ye Y, Shen W, Leng L, Wang L, Li X, Luo X, Chen Y, et al. Sox2 Communicates with Tregs Through CCL1 to Promote the Stemness Property of Breast Cancer Cells. Stem Cells. 2017;35(12):2351–65.
Liu S, Zhang C, Wang B, Zhang H, Qin G, Li C, Cao L, Gao Q, Ping Y, Zhang K, et al. Regulatory T cells promote glioma cell stemness through TGF-β-NF-κB-IL6-STAT3 signaling. Cancer Immunol Immunother. 2021;70(9):2601–16.
Oh E, Hong J, Yun CO. Regulatory T Cells Induce Metastasis by Increasing Tgf-β and Enhancing the Epithelial-Mesenchymal Transition. Cells. 2019;8(11):1387.
Rezalotfi A, Ahmadian E, Aazami H, Solgi G, Ebrahimi M. Gastric Cancer Stem Cells Effect on Th17/Treg Balance; A Bench to Beside Perspective. Front Oncol. 2019;5(9):226.
Mahic M, Yaqub S, Johansson CC, Taskén K, Aandahl EM. FOXP3+CD4+CD25+ adaptive regulatory T cells express cyclooxygenase-2 and suppress effector T cells by a prostaglandin E2-dependent mechanism. J Immunol. 2006;177(1):246–54.
Lee Y, Shin JH, Longmire M, Wang H, Kohrt HE, Chang HY, Sunwoo JB. CD44+ Cells in Head and Neck Squamous Cell Carcinoma Suppress T-Cell-Mediated Immunity by Selective Constitutive and Inducible Expression of PD-L1. Clin Cancer Res. 2016;22(14):3571–81.
Reim F, Dombrowski Y, Ritter C, Buttmann M, Häusler S, Ossadnik M, Krockenberger M, Beier D, Beier CP, Dietl J, et al. Immunoselection of breast and ovarian cancer cells with trastuzumab and natural killer cells: selective escape of CD44high/CD24low/HER2low breast cancer stem cells. Cancer Res. 2009;69(20):8058–66.
Golan H, Shukrun R, Caspi R, Vax E, Pode-Shakked N, Goldberg S, Pleniceanu O, Bar-Lev DD, Mark-Danieli M, Pri-Chen S, et al. In Vivo Expansion of Cancer Stemness Affords Novel Cancer Stem Cell Targets: Malignant Rhabdoid Tumor as an Example. Stem Cell Reports. 2018;11(3):795–810.
Bruttel VS, Wischhusen J. Cancer stem cell immunology: key to understanding tumorigenesis and tumor immune escape? Front Immunol. 2014;29(5):360.
Mennonna D, Maccalli C, Romano MC, Garavaglia C, Capocefalo F, Bordoni R, Severgnini M, De Bellis G, Sidney J, Sette A, et al. T cell neoepitope discovery in colorectal cancer by high throughput profiling of somatic mutations in expressed genes. Gut. 2017;66(3):454–63.
Hsu JM, Xia W, Hsu YH, Chan LC, Yu WH, Cha JH, Chen CT, Liao HW, Kuo CW, Khoo KH, et al. STT3-dependent PD-L1 accumulation on cancer stem cells promotes immune evasion. Nat Commun. 2018;9(1):1908.
Meder L, Schuldt P, Thelen M, Schmitt A, Dietlein F, Klein S, Borchmann S, Wennhold K, Vlasic I, Oberbeck S, et al. Combined VEGF and PD-L1 Blockade Displays Synergistic Treatment Effects in an Autochthonous Mouse Model of Small Cell Lung Cancer. Cancer Res. 2018;78(15):4270–81.
Wang C, Li Y, Jia L, Kim JK, Li J, Deng P, Zhang W, Krebsbach PH, Wang CY. CD276 expression enables squamous cell carcinoma stem cells to evade immune surveillance. Cell Stem Cell. 2021;28(9):1597–613.
Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, Singh B, Rosenblum MD, Fuchs E. Adaptive Immune Resistance Emerges from Tumor-Initiating Stem Cells. Cell. 2019;177(5):1172–86.
Caputo S, Grioni M, Brambillasca CS, Monno A, Brevi A, Freschi M, Piras IS, Elia AR, Pieri V, Baccega T, et al. Galectin-3 in Prostate Cancer Stem-Like Cells Is Immunosuppressive and Drives Early Metastasis. Front Immunol. 2020;10(11):1820.
Agudo J, Park ES, Rose SA, Alibo E, Sweeney R, Dhainaut M, Kobayashi KS, Sachidanandam R, Baccarini A, Merad M, et al. Quiescent Tissue Stem Cells Evade Immune Surveillance. Immunity. 2018;48(2):271–85.
Castriconi R, Daga A, Dondero A, Zona G, Poliani PL, Melotti A, Griffero F, Marubbi D, Spaziante R, Bellora F, et al. NK cells recognize and kill human glioblastoma cells with stem cell-like properties. J Immunol. 2009;182(6):3530–9.
Tallerico R, Todaro M, Di Franco S, Maccalli C, Garofalo C, Sottile R, Palmieri C, Tirinato L, Pangigadde PN, La Rocca R, et al. Human NK cells selective targeting of colon cancer-initiating cells: a role for natural cytotoxicity receptors and MHC class I molecules. J Immunol. 2013;190(5):2381–90.
Jewett A, Tseng HC, Arasteh A, Saadat S, Christensen RE, Cacalano NA. Natural killer cells preferentially target cancer stem cells; role of monocytes in protection against NK cell mediated lysis of cancer stem cells. Curr Drug Deliv. 2012;9(1):5–16.
Akhter MZ, Sharawat SK, Kumar V, Kochat V, Equbal Z, Ramakrishnan M, Kumar U, Mathur S, Kumar L, Mukhopadhyay A. Aggressive serous epithelial ovarian cancer is potentially propagated by EpCAM+CD45+ phenotype. Oncogene. 2018;37(16):2089–103.
Zhong Y, Guan K, Guo S, Zhou C, Wang D, Ma W, Zhang Y, Li C, Zhang S. Spheres derived from the human SK-RC-42 renal cell carcinoma cell line are enriched in cancer stem cells. Cancer Lett. 2010;299(2):150–60.
Özgül Özdemir RB, Özdemir AT, Oltulu F, Kurt K, Yiğittürk G, Kırmaz C. A comparison of cancer stem cell markers and nonclassical major histocompatibility complex antigens in colorectal tumor and noncancerous tissues. Ann Diagn Pathol. 2016;25:60–3.
Malladi S, Macalinao DG, Jin X, He L, Basnet H, Zou Y, de Stanchina E, Massagué J. Metastatic Latency and Immune Evasion through Autocrine Inhibition of WNT. Cell. 2016;165(1):45–60.
Laughney AM, Hu J, Campbell NR, Bakhoum SF, Setty M, Lavallée VP, Xie Y, Masilionis I, Carr AJ, Kottapalli S, et al. Regenerative lineages and immune-mediated pruning in lung cancer metastasis. Nat Med. 2020;26(2):259–69.
Chen P, Hsu WH, Han J, Xia Y, DePinho RA. Cancer Stemness Meets Immunity: From Mechanism to Therapy. Cell Rep. 2021;34(1):108597.
Gomez KE, Wu F, Keysar SB, Morton JJ, Miller B, Chimed TS, Le PN, Nieto C, Chowdhury FN, Tyagi A, et al. Cancer Cell CD44 Mediates Macrophage/Monocyte-Driven Regulation of Head and Neck Cancer Stem Cells. Cancer Res. 2020;80(19):4185–98.
Peng D, Tanikawa T, Li W, Zhao L, Vatan L, Szeliga W, Wan S, Wei S, Wang Y, Liu Y, et al. Myeloid-Derived Suppressor Cells Endow Stem-like Qualities to Breast Cancer Cells through IL6/STAT3 and NO/NOTCH Cross-talk Signaling. Cancer Res. 2016;76(11):3156–65.
Komura N, Mabuchi S, Shimura K, Yokoi E, Kozasa K, Kuroda H, Takahashi R, Sasano T, Kawano M, Matsumoto Y, et al. The role of myeloid-derived suppressor cells in increasing cancer stem-like cells and promoting PD-L1 expression in epithelial ovarian cancer. Cancer Immunol Immunother. 2020;69(12):2477–99.
Li X, Wang J, Wu W, Gao H, Liu N, Zhan G, Li L, Han L, Guo X. Myeloid-derived suppressor cells promote epithelial ovarian cancer cell stemness by inducing the CSF2/p-STAT3 signalling pathway. FEBS J. 2020;287(23):5218–35.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348(6230):62–8.
Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365(8):725–33.
Badieyan ZS, Hoseini SS. Adverse Effects Associated with Clinical Applications of CAR Engineered T Cells. Arch Immunol Ther Exp (Warsz). 2018;66(4):283–8.
Wang Y, Qi K, Cheng H, Cao J, Shi M, Qiao J, Yan Z, Jing G, Pan B, Sang W, et al. Coagulation Disorders after Chimeric Antigen Receptor T Cell Therapy: Analysis of 100 Patients with Relapsed and Refractory Hematologic Malignancies. Biol Blood Marrow Transplant. 2020;26(5):865–75.
Srivastava S, Riddell SR. Engineering CAR-T cells: Design concepts. Trends Immunol. 2015;36(8):494–502.
Gzil A, Zarębska I, Bursiewicz W, Antosik P, Grzanka D, Szylberg Ł. Markers of pancreatic cancer stem cells and their clinical and therapeutic implications. Mol Biol Rep. 2019;46(6):6629–45.
Yang D, Sun B, Dai H, Li W, Shi L, Zhang P, Li S, Zhao X. T cells expressing NKG2D chimeric antigen receptors efficiently eliminate glioblastoma and cancer stem cells. J Immunother Cancer. 2019;7(1):171.
Ahmed N, Salsman VS, Kew Y, Shaffer D, Powell S, Zhang YJ, Grossman RG, Heslop HE, Gottschalk S. HER2-specific T cells target primary glioblastoma stem cells and induce regression of autologous experimental tumors. Clin Cancer Res. 2010;16(2):474–85.
Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan CT, Song H, et al. Recognition of glioma stem cells by genetically modified T cells targeting EGFRvIII and development of adoptive cell therapy for glioma. Hum Gene Ther. 2012;23(10):1043–53.
Badrinath N, Yoo SY. Recent Advances in Cancer Stem Cell-Targeted Immunotherapy. Cancers (Basel). 2019;11(3):310.
Hu W, Zi Z, Jin Y, Li G, Shao K, Cai Q, Ma X, Wei F. CRISPR/Cas9-mediated PD-1 disruption enhances human mesothelin-targeted CAR T cell effector functions. Cancer Immunol Immunother. 2019;68(3):365–77.
Ai L, Chen J, Yan H, He Q, Luo P, Xu Z, Yang X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des Devel Ther. 2020;8(14):3625–49.
Zheng F, Dang J, Zhang H, Xu F, Ba D, Zhang B, Cheng F, Chang AE, Wicha MS, Li Q. Cancer Stem Cell Vaccination With PD-L1 and CTLA-4 Blockades Enhances the Eradication of Melanoma Stem Cells in a Mouse Tumor Model. J Immunother. 2018;41(8):361–8.
Ames E, Canter RJ, Grossenbacher SK, Mac S, Chen M, Smith RC, Hagino T, Perez-Cunningham J, Sckisel GD, Urayama S, et al. NK Cells Preferentially Target Tumor Cells with a Cancer Stem Cell Phenotype. J Immunol. 2015;195(8):4010–9.
Wang B, Wang Q, Wang Z, Jiang J, Yu SC, Ping YF, Yang J, Xu SL, Ye XZ, Xu C, et al. Metastatic consequences of immune escape from NK cell cytotoxicity by human breast cancer stem cells. Cancer Res. 2014;74(20):5746–57.
Paczulla AM, Rothfelder K, Raffel S, Konantz M, Steinbacher J, Wang H, Tandler C, Mbarga M, Schaefer T, Falcone M, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572(7768):254–9.
Cristiani CM, Turdo A, Ventura V, Apuzzo T, Capone M, Madonna G, Mallardo D, Garofalo C, Giovannone ED, Grimaldi AM, et al. Accumulation of Circulating CCR7+ Natural Killer Cells Marks Melanoma Evolution and Reveals a CCL19-Dependent Metastatic Pathway. Cancer Immunol Res. 2019;7(5):841–52.
Avril T, Vauleon E, Hamlat A, Saikali S, Etcheverry A, Delmas C, Diabira S, Mosser J, Quillien V. Human glioblastoma stem-like cells are more sensitive to allogeneic NK and T cell-mediated killing compared with serum-cultured glioblastoma cells. Brain Pathol. 2012;22(2):159–74.
Wei F, Rong XX, Xie RY, Jia LT, Wang HY, Qin YJ, Chen L, Shen HF, Lin XL, Yang J, et al. Cytokine-induced killer cells efficiently kill stem-like cancer cells of nasopharyngeal carcinoma via the NKG2D-ligands recognition. Oncotarget. 2015;6(33):35023–39.
Leuci V, Casucci GM, Grignani G, Rotolo R, Rossotti U, Vigna E, Gammaitoni L, Mesiano G, Fiorino E, Donini C, et al. CD44v6 as innovative sarcoma target for CAR-redirected CIK cells. Oncoimmunology. 2018;7(5):e1423167.
Guo X, Zheng H, Luo W, Zhang Q, Liu J, Yao K. 5T4-specific chimeric antigen receptor modification promotes the immune efficacy of cytokine-induced killer cells against nasopharyngeal carcinoma stem cell-like cells. Sci Rep. 2017;7(1):4859.
Chen X, Han J, Chu J, Zhang L, Zhang J, Chen C, Chen L, Wang Y, Wang H, Yi L, et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget. 2016;7(19):27764–77.
Sahm C, Schönfeld K, Wels WS. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol Immunother. 2012;61(9):1451–61.
Hu Y, Lu L, Xia Y, Chen X, Chang AE, Hollingsworth RE, Hurt E, Owen J, Moyer JS, Prince ME, et al. Therapeutic Efficacy of Cancer Stem Cell Vaccines in the Adjuvant Setting. Cancer Res. 2016;76(16):4661–72.
Guo M, Luo B, Pan M, Li M, Zhao F, Dou J. MUC1 plays an essential role in tumor immunity of colorectal cancer stem cell vaccine. Int Immunopharmacol. 2020;85:106631.
El-Ashmawy NE, Salem ML, Khedr EG, El-Zamarany EA, Ibrahim AO. Dual-targeted therapeutic strategy combining CSC-DC-based vaccine and cisplatin overcomes chemo-resistance in experimental mice model. Clin Transl Oncol. 2020;22(7):1155–65.
Cheema TA, Wakimoto H, Fecci PE, Ning J, Kuroda T, Jeyaretna DS, Martuza RL, Rabkin SD. Multifaceted oncolytic virus therapy for glioblastoma in an immunocompetent cancer stem cell model. Proc Natl Acad Sci U S A. 2013;110(29):12006–11.
Liu TC, Galanis E, Kirn D. Clinical trial results with oncolytic virotherapy: a century of promise, a decade of progress. Nat Clin Pract Oncol. 2007;4(2):101–17.
Friedman GK, Langford CP, Coleman JM, Cassady KA, Parker JN, Markert JM, Yancey GG. Engineered herpes simplex viruses efficiently infect and kill CD133+ human glioma xenograft cells that express CD111. J Neurooncol. 2009;95(2):199–209.
Zhang W, Fulci G, Wakimoto H, Cheema TA, Buhrman JS, Jeyaretna DS, Stemmer Rachamimov AO, Rabkin SD, Martuza RL. Combination of oncolytic herpes simplex viruses armed with angiostatin and IL-12 enhances antitumor efficacy in human glioblastoma models. Neoplasia. 2013;15(6):591–9.
Alemany R, Balagué C, Curiel DT. Replicative adenoviruses for cancer therapy. Nat Biotechnol. 2000;18(7):723–7.
Eriksson M, Guse K, Bauerschmitz G, Virkkunen P, Tarkkanen M, Tanner M, Hakkarainen T, Kanerva A, Desmond RA, Pesonen S, et al. Oncolytic adenoviruses kill breast cancer initiating CD44+CD24-/low cells. Mol Ther. 2007;15(12):2088–93.
Bach P, Abel T, Hoffmann C, Gal Z, Braun G, Voelker I, Ball CR, Johnston IC, Lauer UM, Herold-Mende C, et al. Specific elimination of CD133+ tumor cells with targeted oncolytic measles virus. Cancer Res. 2013;73(2):865–74.
Devaux P, Hodge G, McChesney MB, Cattaneo R. Attenuation of V- or C-defective measles viruses: infection control by the inflammatory and interferon responses of rhesus monkeys. J Virol. 2008;82(11):5359–67.
Marcato P, Dean CA, Giacomantonio CA, Lee PW. Oncolytic reovirus effectively targets breast cancer stem cells. Mol Ther. 2009;17(6):972–9.
Wang H, Chen NG, Minev BR, Szalay AA. Oncolytic vaccinia virus GLV-1h68 strain shows enhanced replication in human breast cancer stem-like cells in comparison to breast cancer cells. J Transl Med. 2012;17(10):167.
Guse K, Cerullo V, Hemminki A. Oncolytic vaccinia virus for the treatment of cancer. Expert Opin Biol Ther. 2011;11(5):595–608.
Liu X, Liu L, Ren Z, Yang K, Xu H, Luan Y, Fu K, Guo J, Peng H, Zhu M, et al. Dual Targeting of Innate and Adaptive Checkpoints on Tumor Cells Limits Immune Evasion. Cell Rep. 2018;24(8):2101–11.
van den Berg TK, van der Schoot CE. Innate immune “self” recognition: a role for CD47-SIRPalpha interactions in hematopoietic stem cell transplantation. Trends Immunol. 2008;29(5):203–6.
Yang L, Zhang Y. Tumor-associated macrophages: from basic research to clinical application. J Hematol Oncol. 2017;10(1):58.
Lindberg FP, Gresham HD, Schwarz E, Brown EJ. Molecular cloning of integrin-associated protein: an immunoglobulin family member with multiple membrane-spanning domains implicated in alpha v beta 3-dependent ligand binding. J Cell Biol. 1993;123(2):485–96.
Campbell IG, Freemont PS, Foulkes W, Trowsdale J. An ovarian tumor marker with homology to vaccinia virus contains an IgV-like region and multiple transmembrane domains. Cancer Res. 1992;52(19):5416–20.
Naujokat C. Monoclonal antibodies against human cancer stem cells. Immunotherapy. 2014;6(3):290–308.
Chao MP, Weissman IL, Majeti R. The CD47-SIRPα pathway in cancer immune evasion and potential therapeutic implications. Curr Opin Immunol. 2012;24(2):225–32.
van Beek EM, Cochrane F, Barclay AN, van den Berg TK. Signal regulatory proteins in the immune system. J Immunol. 2005;175(12):7781–7.
Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50.
van den Berg TK, Yoder JA, Litman GW. On the origins of adaptive immunity: innate immune receptors join the tale. Trends Immunol. 2004;25(1):11–6.
Matlung HL, Szilagyi K, Barclay NA, van den Berg TK. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol Rev. 2017;276(1):145–64.
Soltanian S, Matin MM. Cancer stem cells and cancer therapy. Tumour Biol. 2011;32(3):425–40.
Ribas A. Adaptive Immune Resistance: How Cancer Protects from Immune Attack. Cancer Discov. 2015;5(9):915–9.
Dong Y, Sun Q, Zhang X. PD-1 and its ligands are important immune checkpoints in cancer. Oncotarget. 2017;8(2):2171–86.
Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol. 2015;33(17):1974–82.
Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11(11):3887–95.
Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25(21):9543–53.
Alsaab HO, Sau S, Alzhrani R, Tatiparti K, Bhise K, Kashaw SK, Iyer AK. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front Pharmacol. 2017;23(8):561.
Sznol M, Chen L. Antagonist antibodies to PD-1 and B7–H1 (PD-L1) in the treatment of advanced human cancer. Clin Cancer Res. 2013;19(5):1021–34.
Turley SJ, Cremasco V, Astarita JL. Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol. 2015;15(11):669–82.
Ohaegbulam KC, Assal A, Lazar-Molnar E, Yao Y, Zang X. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol Med. 2015;21(1):24–33.
Li C, Li W, Xiao J, Jiao S, Teng F, Xue S, Zhang C, Sheng C, Leng Q, Rudd CE, et al. ADAP and SKAP55 deficiency suppresses PD-1 expression in CD8+ cytotoxic T lymphocytes for enhanced anti-tumor immunotherapy. EMBO Mol Med. 2015;7(6):754–69.
Lu P, Youngblood BA, Austin JW, Mohammed AU, Butler R, Ahmed R, Boss JM. Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J Exp Med. 2014;211(3):515–27.
Massari F, Santoni M, Ciccarese C, Santini D, Alfieri S, Martignoni G, Brunelli M, Piva F, Berardi R, Montironi R, et al. PD-1 blockade therapy in renal cell carcinoma: current studies and future promises. Cancer Treat Rev. 2015;41(2):114–21.
Almozyan S, Colak D, Mansour F, Alaiya A, Al-Harazi O, Qattan A, Al-Mohanna F, Al-Alwan M, Ghebeh H. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Int J Cancer. 2017;141(7):1402–12.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93.
Darvin P, Sasidharan Nair V, Elkord E. PD-L1 Expression in Human Breast Cancer Stem Cells Is Epigenetically Regulated through Posttranslational Histone Modifications. J Oncol. 2019;21(2019):3958908.
Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, Honjo T. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8(5):765–72.
Scalapino KJ, Daikh DI. CTLA-4: a key regulatory point in the control of autoimmune disease. Immunol Rev. 2008;223:143–55.
Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG, Golstein P. A new member of the immunoglobulin superfamily–CTLA-4. Nature. 1987;328(6127):267–70.
Jago CB, Yates J, Câmara NO, Lechler RI, Lombardi G. Differential expression of CTLA-4 among T cell subsets. Clin Exp Immunol. 2004;136(3):463–71.
Jain N, Nguyen H, Chambers C, Kang J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci U S A. 2010;107(4):1524–8.
Grosso JF, Jure-Kunkel MN. CTLA-4 blockade in tumor models: an overview of preclinical and translational research. Cancer Immun. 2013;13:5.
Codony-Servat J, Verlicchi A, Rosell R. Cancer stem cells in small cell lung cancer. Transl Lung Cancer Res. 2016;5(1):16–25.
Freeman GJ, Casasnovas JM, Umetsu DT, DeKruyff RH. TIM genes: a family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol Rev. 2010;235(1):172–89.
Chae SC, Song JH, Pounsambath P, Yuan HY, Lee JH, Kim JJ, Lee YC, Chung HT. Molecular variations in Th1-specific cell surface gene Tim-3. Exp Mol Med. 2004;36(3):274–8.
Gorman JV, Colgan JD. Regulation of T cell responses by the receptor molecule Tim-3. Immunol Res. 2014;59(1–3):56–65.
Anderson AC. Tim-3: an emerging target in the cancer immunotherapy landscape. Cancer Immunol Res. 2014;2(5):393–8.
Das M, Zhu C, Kuchroo VK. Tim-3 and its role in regulating anti-tumor immunity. Immunol Rev. 2017;276(1):97–111.
Du W, Yang M, Turner A, Xu C, Ferris RL, Huang J, Kane LP, Lu B. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int J Mol Sci. 2017;18(3):645.
Zhu C, Anderson AC, Schubart A, Xiong H, Imitola J, Khoury SJ, Zheng XX, Strom TB, Kuchroo VK. The Tim-3 ligand galectin-9 negatively regulates T helper type 1 immunity. Nat Immunol. 2005;6(12):1245–452.
Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka-Akita H, Fujioka Y, Ohba Y, Gorman JV, Colgan JD, et al. Tumor-infiltrating DCs suppress nucleic acid-mediated innate immune responses through interactions between the receptor TIM-3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832–42.
Huang YH, Zhu C, Kondo Y, Anderson AC, Gandhi A, Russell A, Dougan SK, Petersen BS, Melum E, Pertel T, et al. CEACAM1 regulates TIM-3-mediated tolerance and exhaustion. Nature. 2015;517(7534):386–90.
Jan M, Chao MP, Cha AC, Alizadeh AA, Gentles AJ, Weissman IL, Majeti R. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci U S A. 2011;108(12):5009–14.
Kikushige Y, Shima T, Takayanagi S, Urata S, Miyamoto T, Iwasaki H, Takenaka K, Teshima T, Tanaka T, et al. TIM-3 is a promising target to selectively kill acute myeloid leukemia stem cells. Cell Stem Cell. 2010;7(6):708–17.
Anderson AC. Tim-3, a negative regulator of anti-tumor immunity. Curr Opin Immunol. 2012;24(2):213–6.
Kikushige Y, Miyamoto T. TIM-3 as a novel therapeutic target for eradicating acute myelogenous leukemia stem cells. Int J Hematol. 2013;98(6):627–33.
Wang JM, Ma CJ, Li GY, Wu XY, Thayer P, Greer P, Smith AM, High KP, Moorman JP, Yao ZQ. Tim-3 alters the balance of IL-12/IL-23 and drives TH17 cells: role in hepatitis B vaccine failure during hepatitis C infection. Vaccine. 2013;31(18):2238–45.
Ma CJ, Li GY, Cheng YQ, Wang JM, Ying RS, Shi L, Wu XY, Niki T, Hirashima M, Li CF, Moorman JP, Yao ZQ. Cis association of galectin-9 with Tim-3 differentially regulates IL-12/IL-23 expressions in monocytes via TLR signaling. PLoS ONE. 2013;8(8):e72488.
Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, Hsu CY, Huang CT, Su WT, Chu YY, et al. Apoptosis of tumor infiltrating effector TIM-3+CD8+ T cells in colon cancer. Sci Rep. 2015;23(5):15659.
Kikushige Y, Miyamoto T, Yuda J, Jabbarzadeh-Tabrizi S, Shima T, Takayanagi S, Niiro H, Yurino A, Miyawaki K, Takenaka K, et al. A TIM-3/Gal-9 Autocrine Stimulatory Loop Drives Self-Renewal of Human Myeloid Leukemia Stem Cells and Leukemic Progression. Cell Stem Cell. 2015;17(3):341–52.
Gupta S, Thornley TB, Gao W, Larocca R, Turka LA, Kuchroo VK, Strom TB. Allograft rejection is restrained by short-lived TIM-3+PD-1+Foxp3+ Tregs. J Clin Invest. 2012;122(7):2395–404.
Gautron AS, Dominguez-Villar M, de Marcken M, Hafler DA. Enhanced suppressor function of TIM-3+ FoxP3+ regulatory T cells. Eur J Immunol. 2014;44(9):2703–11.
Khademi M, Illés Z, Gielen AW, Marta M, Takazawa N, Baecher-Allan C, Brundin L, Hannerz J, Martin C, Harris RA, et al. T Cell Ig- and mucin-domain-containing molecule-3 (TIM-3) and TIM-1 molecules are differentially expressed on human Th1 and Th2 cells and in cerebrospinal fluid-derived mononuclear cells in multiple sclerosis. J Immunol. 2004;172(11):7169–76.
Ndhlovu LC, Lopez-Vergès S, Barbour JD, Jones RB, Jha AR, Long BR, Schoeffler EC, Fujita T, Nixon DF, Lanier LL. Tim-3 marks human natural killer cell maturation and suppresses cell-mediated cytotoxicity. Blood. 2012;119(16):3734–43.
Xu L, Huang Y, Tan L, Yu W, Chen D, Lu C, He J, Wu G, Liu X, Zhang Y. Increased Tim-3 expression in peripheral NK cells predicts a poorer prognosis and Tim-3 blockade improves NK cell-mediated cytotoxicity in human lung adenocarcinoma. Int Immunopharmacol. 2015;29(2):635–41.
Dardalhon V, Anderson AC, Karman J, Apetoh L, Chandwaskar R, Lee DH, Cornejo M, Nishi N, Yamauchi A, Quintana FJ, et al. Tim-3/galectin-9 pathway: regulation of Th1 immunity through promotion of CD11b+Ly-6G+ myeloid cells. J Immunol. 2010;185(3):1383–92.
Triebel F, Jitsukawa S, Baixeras E, Roman-Roman S, Genevee C, Viegas-Pequignot E, Hercend T. LAG-3, a novel lymphocyte activation gene closely related to CD4. J Exp Med. 1990;171(5):1393–405.
Andrews LP, Marciscano AE, Drake CG, Vignali DA. LAG3 (CD223) as a cancer immunotherapy target. Immunol Rev. 2017;276(1):80–96.
He Y, Rivard CJ, Rozeboom L, Yu H, Ellison K, Kowalewski A, Zhou C, Hirsch FR. Lymphocyte-activation gene-3, an important immune checkpoint in cancer. Cancer Sci. 2016;107(9):1193–7.
Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44(5):989–1004.
Hemon P, Jean-Louis F, Ramgolam K, Brignone C, Viguier M, Bachelez H, Triebel F, Charron D, Aoudjit F, Al-Daccak R, et al. MHC class II engagement by its ligand LAG-3 (CD223) contributes to melanoma resistance to apoptosis. J Immunol. 2011;186(9):5173–83.
Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, Jaffee E. Galectin-3 Shapes Antitumor Immune Responses by Suppressing CD8+ T Cells via LAG-3 and Inhibiting Expansion of Plasmacytoid Dendritic Cells. Cancer Immunol Res. 2015;3(4):412–23.
Maccalli C, De Maria R. Cancer stem cells: perspectives for therapeutic targeting. Cancer Immunol Immunother. 2015;64(1):91–7.
Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, Betts MR, Freeman GJ, Vignali DA, Wherry EJ. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29–37.
Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, Guo B, Herbert DR, Bulfone A, Trentini F, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. 2013;19(6):739–46.
Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A, Corbett M, Rankin AL, Dibella R, Campbell L, et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature. 2019;574(7779):565–70.
Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;17(7):10501.
McLaughlin J, Han G, Schalper KA, Carvajal-Hausdorf D, Pelekanou V, Rehman J, Velcheti V, Herbst R, LoRusso P, Rimm DL. Quantitative Assessment of the Heterogeneity of PD-L1 Expression in Non-Small-Cell Lung Cancer. JAMA Oncol. 2016;2(1):46–54.
Brady L, Kriner M, Coleman I, Morrissey C, Roudier M, True LD, Gulati R, Plymate SR, Zhou Z, Birditt B, et al. Inter- and intra-tumor heterogeneity of metastatic prostate cancer determined by digital spatial gene expression profiling. Nat Commun. 2021;12(1):1426.
Yonesaka K, Haratani K, Takamura S, Sakai H, Kato R, Takegawa N, Takahama T, Tanaka K, Hayashi H, Takeda M, et al. B7–H3 Negatively Modulates CTL-Mediated Cancer Immunity. Clin Cancer Res. 2018;24(11):2653–64.
Su W, Han HH, Wang Y, Zhang B, Zhou B, Cheng Y, Rumandla A, Gurrapu S, Chakraborty G, Su J, et al. The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression. Cancer Cell. 2019;36(2):139–55.
Jia L, Zhang W, Wang CY. BMI1 Inhibition Eliminates Residual Cancer Stem Cells after PD1 Blockade and Activates Antitumor Immunity to Prevent Metastasis and Relapse. Cell Stem Cell. 2020;27(2):238–53.
Mortezaee K, Shabeeb D, Musa AE, Najafi M, Farhood B. Metformin as a Radiation Modifier; Implications to Normal Tissue Protection and Tumor Sensitization. Curr Clin Pharmacol. 2019;14(1):41–53.
Scharping NE, Menk AV, Whetstone RD, Zeng X, Delgoffe GM. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol Res. 2017;5(1):9–16.
Mortezaee K, Majidpoor J. (Im)maturity in Tumor Ecosystem. Front Oncol. 2022;25(11):813897.
Blum Y, Meiller C, Quetel L, Elarouci N, Ayadi M, Tashtanbaeva D, Armenoult L, Montagne F, Tranchant R, Renier A, et al. Dissecting heterogeneity in malignant pleural mesothelioma through histo-molecular gradients for clinical applications. Nat Commun. 2019;10(1):1333.
Tang M, Zhao Y, Zhao J, Wei S, Liu M, Zheng N, Geng D, Han S, Zhang Y, Zhong G, et al. Liver cancer heterogeneity modeled by in situ genome editing of hepatocytes. Sci Adv. 2022;8(25):eabn5683.
Cheng W, Li HL, Xi SY, Zhang XF, Zhu Y, Le X, Mo YX, Li MM, Kong FE, Zhu WJ, et al. Growth differentiation factor 1-induced tumour plasticity provides a therapeutic window for immunotherapy in hepatocellular carcinoma. Nat Commun. 2021;12(1):7142.
Molina-Sánchez P, Ruiz de Galarreta M, Yao MA, Lindblad KE, Bresnahan E, Bitterman E, Martin TC, Rubenstein T, Nie K, Golas J, et al. Cooperation Between Distinct Cancer Driver Genes Underlies Intertumor Heterogeneity in Hepatocellular Carcinoma. Gastroenterology. 2020;159(6):2203-2220.e14.
Wang X, Li M. Correlate tumor mutation burden with immune signatures in human cancers. BMC Immunol. 2019;20(1):4.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348(6230):124–8.
Liu S, Cong Y, Wang D, Sun Y, Deng L, Liu Y, Martin-Trevino R, Shang L, McDermott SP, Landis MD, et al. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Reports. 2013;2(1):78–91.
Mortezaee K, Majidpoor J. Key promoters of tumor hallmarks. Int J Clin Oncol. 2022;27(1):45–58.
Lou Y, Diao L, Cuentas ER, Denning WL, Chen L, Fan YH, Byers LA, Wang J, Papadimitrakopoulou VA, Behrens C, et al. Epithelial-Mesenchymal Transition Is Associated with a Distinct Tumor Microenvironment Including Elevation of Inflammatory Signals and Multiple Immune Checkpoints in Lung Adenocarcinoma. Clin Cancer Res. 2016;22(14):3630–42.
Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–95.
Chen L, Gibbons DL, Goswami S, Cortez MA, Ahn YH, Byers LA, Zhang X, Yi X, Dwyer D, Lin W, et al. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat Commun. 2014;28(5):5241.
Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, Pracht M, Yokosuka O, Rosmorduc O, Breder V, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;389(10064):56–66.
Abou-Alfa GK, Meyer T, Cheng AL, El-Khoueiry AB, Rimassa L, Ryoo BY, Cicin I, Merle P, Chen Y, Park JW, et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N Engl J Med. 2018;379(1):54–63.
Zhu AX, Park JO, Ryoo BY, Yen CJ, Poon R, Pastorelli D, Blanc JF, Chung HC, Baron AD, Pfiffer TE, et al. REACH Trial Investigators. Ramucirumab versus placebo as second-line treatment in patients with advanced hepatocellular carcinoma following first-line therapy with sorafenib (REACH): a randomised, double-blind, multicentre, phase 3 trial. Lancet Oncol. 2015;16(7):859–70.
Zhu AX, Kang YK, Yen CJ, Finn RS, Galle PR, Llovet JM, Assenat E, Brandi G, Pracht M, Lim HY, et al. REACH-2 study investigators. Ramucirumab after sorafenib in patients with advanced hepatocellular carcinoma and increased α-fetoprotein concentrations (REACH-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2019;20(2):282–96.
Bhoori S, Mazzaferro V. Combined immunotherapy and VEGF-antagonist in hepatocellular carcinoma: a step forward. Lancet Oncol. 2020;21(6):740–1.
Finn RS, Qin S, Ikeda M, Galle PR, Ducreux M, Kim TY, Kudo M, Breder V, Merle P, Kaseb AO, et al. IMbrave150 Investigators. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. N Engl J Med. 2020;382(20):1894–905.
Fukuoka S, Hara H, Takahashi N, Kojima T, Kawazoe A, Asayama M, Yoshii T, Kotani D, Tamura H, Mikamoto Y, et al. Regorafenib Plus Nivolumab in Patients With Advanced Gastric or Colorectal Cancer: An Open-Label, Dose-Escalation, and Dose-Expansion Phase Ib Trial (REGONIVO, EPOC1603). J Clin Oncol. 2020;38(18):2053–61.
Hirata E, Sahai E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb Perspect Med. 2017;7(7):a026781.
D’Arcangelo E, Wu NC, Cadavid JL, McGuigan AP. The life cycle of cancer-associated fibroblasts within the tumour stroma and its importance in disease outcome. Br J Cancer. 2020;122(7):931–42.
Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP, Cox TR, Timpson P. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer. 2019;5(11):724–41.
Oshimori N, Guo Y, Taniguchi S. An emerging role for cellular crosstalk in the cancer stem cell niche. J Pathol. 2021;254(4):384–94.
Zhang J, Song Q, Wu M, Zheng W. The Emerging Roles of Exosomes in the Chemoresistance of Hepatocellular Carcinoma. Curr Med Chem. 2021;28(1):93–109.
Raudenska M, Balvan J, Masarik M. Crosstalk between autophagy inhibitors and endosome-related secretory pathways: a challenge for autophagy-based treatment of solid cancers. Mol Cancer. 2021;20(1):140.
Kim YH, Kwak MS, Lee B, Shin JM, Aum S, Park IH, Lee MG, Shin JS. Secretory autophagy machinery and vesicular trafficking are involved in HMGB1 secretion. Autophagy. 2021;17(9):2345–62.
Brun S, Pascussi JM, Gifu EP, Bestion E, Macek-Jilkova Z, Wang G, Bassissi F, Mezouar S, Courcambeck J, Merle P, et al. GNS561, a New Autophagy Inhibitor Active against Cancer Stem Cells in Hepatocellular Carcinoma and Hepatic Metastasis from Colorectal Cancer. J Cancer. 2021;12(18):5432–8.
Pan T, Xu J, Zhu Y. Self-renewal molecular mechanisms of colorectal cancer stem cells. Int J Mol Med. 2017;39(1):9–20.
López de Andrés J, Griñán-Lisón C, Jiménez G, Marchal JA. Cancer stem cell secretome in the tumor microenvironment: a key point for an effective personalized cancer treatment. J Hematol Oncol. 2020;13(1):136.
Bebelman MP, Janssen E, Pegtel DM, Crudden C. The forces driving cancer extracellular vesicle secretion. Neoplasia. 2021;23(1):149–57.
Su C, Zhang J, Yarden Y, Fu L. The key roles of cancer stem cell-derived extracellular vesicles. Signal Transduct Target Ther. 2021;6(1):109.
Maruyama M, Nakano Y, Nishimura T, Iwata R, Matsuda S, Hayashi M, Nakai Y, Nonaka M, Sugimoto T. PC3-Secreted Microprotein Is Expressed in Glioblastoma Stem-Like Cells and Human Glioma Tissues. Biol Pharm Bull. 2021;44(7):910–9.
Wang MJ, Chen JJ, Song SH, Su J, Zhao LH, Liu QG, Yang T, Chen Z, Liu C, Fu ZR, et al. Inhibition of SIRT1 Limits Self-Renewal and Oncogenesis by Inducing Senescence of Liver Cancer Stem Cells. J Hepatocell Carcinoma. 2021;29(8):685–99.
Phiboonchaiyanan PP, Puthongking P, Chawjarean V, Harikarnpakdee S, Sukprasansap M, Chanvorachote P, Priprem A, Govitrapong P. Melatonin and its derivative disrupt cancer stem-like phenotypes of lung cancer cells via AKT downregulation. Clin Exp Pharmacol Physiol. 2021;48(12):1712–23.
Tang D, Xu X, Ying J, Xie T, Cao G. Transfer of metastatic traits via miR-200c in extracellular vesicles derived from colorectal cancer stem cells is inhibited by atractylenolide I. Clin Transl Med. 2020;10(4):e139.
Brossa A, Fonsato V, Grange C, Tritta S, Tapparo M, Calvetti R, Cedrino M, Fallo S, Gontero P, Camussi G, et al. Extracellular vesicles from human liver stem cells inhibit renal cancer stem cell-derived tumor growth in vitro and in vivo. Int J Cancer. 2020;147(6):1694–706.
Zhang K, Dong C, Chen M, Yang T, Wang X, Gao Y, Wang L, Wen Y, Chen G, Wang X, et al. Extracellular vesicle-mediated delivery of miR-101 inhibits lung metastasis in osteosarcoma. Theranostics. 2020;10(1):411–25.
Waclaw B, Bozic I, Pittman ME, Hruban RH, Vogelstein B, Nowak MA. A spatial model predicts that dispersal and cell turnover limit intratumour heterogeneity. Nature. 2015;525(7568):261–4.
Hwang WL, Lan HY, Cheng WC, Huang SC, Yang MH. Tumor stem-like cell-derived exosomal RNAs prime neutrophils for facilitating tumorigenesis of colon cancer. J Hematol Oncol. 2019;12(1):10.
Mirzaei R, Sarkar S, Dzikowski L, Rawji KS, Khan L, Faissner A, Bose P, Yong VW. Brain tumor-initiating cells export tenascin-C associated with exosomes to suppress T cell activity. Oncoimmunology. 2018;7(10):e1478647.
Su CY, Li JQ, Zhang LL, Wang H, Wang FH, Tao YW, Wang YQ, Guo QR, Li JJ, Liu Y, et al. The Biological Functions and Clinical Applications of Integrins in Cancers. Front Pharmacol. 2020;11(11):579068.
Domenis R, Cesselli D, Toffoletto B, Bourkoula E, Caponnetto F, Manini I, Beltrami AP, Ius T, Skrap M, Di Loreto C, et al. Systemic T Cells Immunosuppression of Glioma Stem Cell-Derived Exosomes Is Mediated by Monocytic Myeloid-Derived Suppressor Cells. PLoS ONE. 2017;12(1):e0169932.
Gabrusiewicz K, Li X, Wei J, Hashimoto Y, Marisetty AL, Ott M, Wang F, Hawke D, Yu J, Healy LM, et al. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology. 2018;7(4):e1412909.
Yang J, Sun G, Hu Y, Yang J, Shi Y, Liu H, Li C, Wang Y, Lv Z, Niu J, et al. Extracellular Vesicle lncRNA Metastasis-Associated Lung Adenocarcinoma Transcript 1 Released From Glioma Stem Cells Modulates the Inflammatory Response of Microglia After Lipopolysaccharide Stimulation Through Regulating miR-129-5p/High Mobility Group Box-1 Protein Axis. Front Immunol. 2020;7(10):3161.
Cheng WC, Liao TT, Lin CC, Yuan LE, Lan HY, Lin HH, Teng HW, Chang HC, Lin CH, Yang CY, et al. RAB27B-activated secretion of stem-like tumor exosomes delivers the biomarker microRNA-146a-5p, which promotes tumorigenesis and associates with an immunosuppressive tumor microenvironment in colorectal cancer. Int J Cancer. 2019;145(8):2209–24.
He X, Smith SE, Chen S, Li H, Wu D, Meneses-Giles PI, Wang Y, Hembree M, Yi K, Zhao X, et al. Tumor-initiating stem cell shapes its microenvironment into an immunosuppressive barrier and pro-tumorigenic niche. Cell Rep. 2021;36(10):109674.
Arabi L, Badiee A, Mosaffa F, Jaafari MR. Targeting CD44 expressing cancer cells with anti-CD44 monoclonal antibody improves cellular uptake and antitumor efficacy of liposomal doxorubicin. J Control Release. 2015;220(Pt A):275–86.
Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P, Chu PW, Lam CT, Poon RT, Fan ST. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13(2):153–66.
Annett S, Robson T. Targeting cancer stem cells in the clinic: Current status and perspectives. Pharmacol Ther. 2018;187:13–30.
Lytle NK, Barber AG, Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat Rev Cancer. 2018;18(11):669–80.
Wang J, Zheng Y, Zhao M. Exosome-Based Cancer Therapy: Implication for Targeting Cancer Stem Cells. Front Pharmacol. 2017;12(7):533.
Ha D, Yang N, Nadithe V. Exosomes as therapeutic drug carriers and delivery vehicles across biological membranes: current perspectives and future challenges. Acta Pharm Sin B. 2016;6(4):287–96.
Kibria G, Ramos EK, Wan Y, Gius DR, Liu H. Exosomes as a Drug Delivery System in Cancer Therapy: Potential and Challenges. Mol Pharm. 2018;15(9):3625–33.
Steinbichler TB, Dudás J, Skvortsov S, Ganswindt U, Riechelmann H, Skvortsova II. Therapy resistance mediated by exosomes. Mol Cancer. 2019;18(1):58.
Yong T, Zhang X, Bie N, Zhang H, Zhang X, Li F, Hakeem A, Hu J, Gan L, Santos HA, et al. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat Commun. 2019;10(1):3838.
Zhao L, Gu C, Gan Y, Shao L, Chen H, Zhu H. Exosome-mediated siRNA delivery to suppress postoperative breast cancer metastasis. J Control Release. 2020;318:1–15.
Tian Y, Li S, Song J, Ji T, Zhu M, Anderson GJ, Wei J, Nie G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials. 2014;35(7):2383–90.
Allahverdiyev AM, Parlar E, Dinparvar S, Bagirova M, Abamor EŞ. Current aspects in treatment of breast cancer based of nanodrug delivery systems and future prospects. Artif Cells Nanomed Biotechnol. 2018;46(sup3):S755–62.
Acknowledgements
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China (No.82073286,No.81970466), China Postal Science Foundation (No.2018M641743,No.2019M661168), Natural Science Foundation of Liaoning Province(N0.2019-ZD-0784) and Liaoning Baiqianwan Talents Program(No.XLYC1905002).
Author information
Authors and Affiliations
Contributions
BW and HX-L offered direction and guidance of the manuscript. BW, XS and MX-J drafted the initial manuscript. HX-L illustrated the figures for the manuscript. All authors approved the final manuscript.
Corresponding author
Ethics declarations
Consent for publication
All authors agree to submit the article for publication.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated in a credit line to the data.
About this article

Cite this article
Wu, B., Shi, X., Jiang, M. et al. Cross-talk between cancer stem cells and immune cells: potential therapeutic targets in the tumor immune microenvironment. Mol Cancer 22, 38 (2023). https://doi.org/10.1186/s12943-023-01748-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12943-023-01748-4